
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiTEMPS methacrylate dynamic covalent cross-linker providing rapid reprocessability and extrudability of covalent adaptable networks: high-yield synthesis with strong selectivity for disulfide linkages†
Tapas
Debsharma‡
 a,
Nathan S.
Purwanto‡
b,
Logan M.
Fenimore
a,
Sarah
Mitchell
c,
Jayme
Kennedy
c and
John M.
Torkelson
*ab
a,
Nathan S.
Purwanto‡
b,
Logan M.
Fenimore
a,
Sarah
Mitchell
c,
Jayme
Kennedy
c and
John M.
Torkelson
*ab
aDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: j-torkelson@northwestern.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
cBraskem America, 550 Technology Drive, Pittsburgh, Pennsylvania 15219, USA
First published on 3rd May 2024
Abstract
The dialkylamino disulfide-based dynamic cross-linker bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide, also known as BiTEMPS methacrylate (BTMA), has been of recent interest in the preparation of various reprocessable cross-linked polymers, otherwise known as covalent adaptable networks (CANs), by free-radical polymerization or free-radical reactive processing. Here, we revised the synthesis of BTMA to produce “BTMA-S2”, i.e., BTMA with a significantly higher yield, higher purity, and less color, with ≥95% disulfide linkages compared to “BTMA-Sn”, i.e., a previous version of BTMA that was synthesized with a relatively low yield, lower purity, and more color with a mixture of oligosulfide linkages. We used a low level (5 mol%) of this BTMA-S2 to synthesize CANs with n-hexyl methacrylate (BTMA-S2-HMA CANs). The BTMA-S2-HMA CANs recover their original cross-link densities after reprocessing. Additionally, compared to the BTMA-Sn-HMA CANs, the BTMA-S2-HMA CANs exhibit much faster stress relaxation at elevated temperatures, which manifests in rapid reprocessability. Specifically, BTMA-S2-HMA CANs made with 5 mol% BTMA-S2 can be reprocessed by compression molding at 130 °C for 5 min with full recovery of cross-link density. This is a factor of six faster reprocessing than analogous BTMA-Sn-CANs made with BTMA-Sn. Additionally, we demonstrate facile melt extrusion at 180 °C of the BTMA-S2-HMA CAN material with full recovery of cross-link density after extrusion.
Introduction
Relative to thermoplastics, traditional thermoset polymers with three-dimensional cross-linked networks exhibit superior dimensional stability at the expense of malleability.1 The presence of percolated permanent covalent cross-links makes conventional thermosets non-reshapeable, non-recyclable, and non-repairable.2–4 Due to this inherent unrecyclability of thermosets, they typically meet their end-of-life fate in the form of landfilling, incineration, or as devalued filler materials.5,6 To tackle this issue of unrecyclability, a new class of polymer materials called covalent adaptable networks (CANs)7–12 was introduced, providing both cross-linked structures at use conditions and malleability owing to their external stimuli-based reversible covalent bonds.13,14 A range of functional groups, such as esters,15 silyl ethers,16,17 siloxanes,18,19 amides,20 β-amino esters,21 and others,22–28 have been used as reversible or dynamic covalent bonds in CANs.Dialkylamino disulfide compounds have a dynamic covalent bond that has been of recent scientific interest because of the catalyst-free activation of the disulfide dynamic chemistry, commercially available building blocks, and amenability for free-radical processing of existing commodity polymers.29–38 Upon thermal triggering, a dialkylamino disulfide compound dissociates into two sulfur radical moieties, thereby initiating the dynamic nature. This chemistry was first reported by Otsuka and coworkers in the context of CAN materials.29 Over the last several years, this chemistry has been utilized for the preparation of CANs based on polyurethanes30,31 and polymethacrylates32–35 as well as traditional commodity polymers such as low-density and high-density polyethylene36 and ethylene/1-octene copolymers.37,38 Recently, Torkelson and coworkers have shown that this chemistry can be used to produce melt-extrudable CANs made from ethylene/1-octene copolymers cross-linked with bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide, also known as BiTEMPS methacrylate (BTMA), which makes it a potentially attractive choice for industrial applications.37 However, the current methods to synthesize dialkylamino-disulfide compounds present significant drawbacks. For instance, dialkylamino-disulfide compounds have been synthesized either by a three-step protocol via protection–deprotection group chemistry in the presence of a strong base, followed by laborious chromatographic purification techniques, as reported by Otsuka and coworkers,29 or by a simpler one-step approach reported by Torkelson and coworkers33–39 that is, however, lower yielding and impure. The CANs previously synthesized or prepared with BTMA also typically exhibited a yellow tint, possibly due to the presence of some polysulfide impurities.40 Therefore, there is a need for a simple, high purity, and high-yielding approach for the synthesis of BTMA.
Otsuka and coworkers have shown that the synthesis of dialkylamino disulfides can lead not only to disulfide but also trisulfide and tetrasulfide linkages as well.41 They showed that dialkylamino compounds with trisulfide linkages disproportionate into disulfide and tetrasulfide compounds,41,42 with all compounds having dynamic covalent character. They also showed that CANs made with trisulfide BiTEMPS displayed different relaxation timescales than those of disulfide BiTEMPS.40 Through optimization experiments to produce BTMA, we undertook studies to determine whether a variety of sulfide linkages were present in our previous method. Potential sources of the trisulfide and tetrasulfide linkages are impurities in the commercial sulfur monochloride (S2Cl2) and the reaction of S2Cl2 with moisture during handling. The latter is of concern because sulfur monochloride can produce trisulfide and tetrasulfide linkages in the presence of moisture, necessitating air-/moisture-free reaction conditions.40 Further, we sought to elucidate the differences in dynamic characteristics of materials containing predominantly dialkylamino-disulfide-based dynamic covalent cross-linker and those made with a mixture of oligosulfide-based dynamic cross-linkers.
Here, we show an in-depth analysis of BTMA synthesized using our previously reported method33–39 and identify the presence of oligosulfide linkages (BTMA-Sn). Through purification and optimization, we describe an alternative single-step protocol for the synthesis of BTMA. This optimized procedure consistently produces BTMA at very high isolated yields that comprises ≥95% exclusively disulfide linkages (BTMA-S2) (Fig. 1). The structure of the BTMA-S2 is confirmed through single-crystal X-ray crystallography, and the purity of the compound is analyzed using liquid chromatography-tandem mass spectrometry (LC-MS). To further explore BTMA-S2, we prepare CANs made via free-radical polymerization of n-hexyl methacrylate (HMA) and characterize a variety of properties: glass-transition temperature (Tg), tensile storage modulus, elevated-temperature stress relaxation (including average stress relaxation times and apparent activation energy for stress relaxation), thermal stability, and UV-Vis absorbance characteristics. Importantly, HMA-based CANs made from BTMA-S2 exhibit much shorter stress relaxation times than CANs made from BTMA-Sn, which contains a mix of disulfides, trisulfides, and tetrasulfides. The faster elevated-temperature stress relaxation correlates with faster processing times. The BTMA-S2-HMA CANs made with 5 mol% BTMA-S2 may be reprocessed with full recovery of cross-link density by compression molding for 5 min at 130 °C, a short (re)processing time scale for compression molding rarely associated with CANs. In contrast, the analogous CANs made with BTMA-Sn containing a mix of disulfides, trisulfides, and tetrasulfides require the use of compression molding for 30 min at 130 °C to achieve a similar recovery of cross-link density after reprocessing. Such improvements in melt-processing speed are reflected in the extrudability of the BTMA-S2-HMA CAN at 180 °C, generating a robust, transparent, and defect-free network with full recovery of cross-link density after extrusion.
 | ||
| Fig. 1 (A) Syntheses of BTMA-Sn (BTMA-Sn) and BTMA-S2 from 2,2,6,6-tetramethyl-4-piperidyl methacrylate (TMPM) and S2Cl2. (B) Optimization of BTMA-S2 synthesis by varying solvent and temperature. (C) BTMA-HMA CAN synthesis by free radical polymerization using 5 mol% BTMA. | ||
Experimental
Materials
All chemicals were commercially available and used as received unless otherwise noted. 2,2,6,6-Tetramethyl-4-piperidyl methacrylate (TMPM) was from TCI America. Anhydrous dichloromethane (DCM), anhydrous methyl tetrahydrofuran (MeTHF), anhydrous petroleum ether, and methanol (99.9%) were from Fisher Scientific. Sulfur monochloride (98%), n-hexyl methacrylate (HMA, 98%), N,N-dimethylacetamide (DMAc, anhydrous, 99.8%), toluene (99.9%) and chloroform-d (99.8 atom% D) were from Sigma-Aldrich. HMA monomer was de-inhibited using inhibitor remover (Sigma Aldrich, 311340) in the presence of calcium hydride (Sigma Aldrich, 90%).Synthesis – previous procedure for bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide
Bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide was prepared using the previously reported procedure.33,34 2,2,6,6-tetramethyl-4-piperidyl methacrylate (8.81 g, 39.09 mmol) was dissolved in a pre-dried petroleum ether (∼90 mL). The solution was then cooled to −70 °C using a dry ice/acetone bath, and then sulfur monochloride (1.31 g, 9.72 mmol) dissolved in pre-dried petroleum ether (1.30 mL) was added dropwise to the solution with continuous stirring. The solution was stirred at −70 °C for 15 min and then at room temperature for an additional 30 min. The reaction mixture was then poured into distilled water and stirred at room temperature overnight. The formed precipitates were filtered off, washed with distilled water, and dried in a vacuum oven at 40 °C for 24 h to give BiTEMPS methacrylate (2.35 g, 48% yield). Anal. Calcd for C26H44N2O4S2: N, 5.46; S, 12.51. Found: N, 5.40; S, 16.35.Synthesis – new procedure for bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide
The new procedure for bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide is a modification of the procedure previously reported in ref. 33 and 34. Sulfur monochloride was distilled under vacuum at room temperature using a kügelrohr distillation apparatus and stored in a septum-sealed vial under a nitrogen environment. 2,2,6,6-Tetramethyl-4-piperidyl methacrylate (TMPM) (7.50 g, 16.66 mmol) was taken in a dry and septum-sealed round-bottom flask fitted with a nitrogen balloon. A dry solvent of choice (∼50 mL) was added to dissolve the TMPM. The solution was then cooled to a desired temperature if needed, and then sulfur monochloride (1.00 g, 3.70 mmol) was dissolved in the same reaction solvent (∼10 mL) and added dropwise to the solution with continuous stirring (see Fig. 1 for solvent and reaction conditions). The solution was stirred at the previously determined temperature for 3 h and then at room temperature for an additional 30 min. The reaction mixture was then poured into distilled water and stirred at room temperature overnight. In the case of the use of MeTHF as a solvent, the reaction mixture was evaporated to dryness in a rotary evaporator, and DCM was added to it. The crude reaction mixture was then poured into distilled water (∼250 mL) and stirred vigorously overnight. The formed precipitates were filtered off, washed with distilled water two times, and dried in a vacuum oven at 50 °C overnight to give BTMA. Anal. Calcd for C26H44N2O4S2: N, 5.46; S, 12.51. Found: N, 5.42; S, 12.58.Synthesis – BTMA-containing networks
The BTMA-HMA networks with 5 mol% BTMA were prepared using the method described in a previous report with AIBN as radical initiator and performing free-radical polymerization at 70 °C overnight to achieve full conversion.39Molding and reprocessing of networks
Networks were cut into millimeter-sized pieces and processed using a PHI press (Model 0230C-X1). To mold the samples used in dynamic mechanical analysis and stress relaxation experiments, network pieces were hot pressed into ∼1-mm-thick films at 130 °C with a 10-ton ram force for 60 min. After molding, samples were cooled to room temperature in a cold compression mold with a 4-ton ram force for 5 min. Such films are considered the 1st-molded sample. 2nd-molded samples were prepared in a similar way where a 1st-molded sample was cut into small pieces and pressed again to obtain a 2nd-molded sample. In cases where remolding speed was being assessed, the reprocessing continued to be done at 130 °C with a 10-ton ram force but for shorter time scales to assess the shortest reprocessing time at which cross-link density could be fully recovered.Dynamic mechanical analysis (DMA)
A TA Instruments RSA-G2 Solids Analyzer was used to measure the tensile storage modulus (E′), tensile loss modulus (E′′), and the damping ratio (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ = E′′/E′) of the samples as functions of temperature upon heating rectangular specimens (8 mm × 3 mm × 1 mm) from −30 °C to 150 °C with a heating rate of 3 °C min−1 under nitrogen atmosphere. The instrument was operated in tension mode at a frequency of 1.00 Hz and 0.03% oscillatory strain. Reported values are the averages of three measurements.
δ = E′′/E′) of the samples as functions of temperature upon heating rectangular specimens (8 mm × 3 mm × 1 mm) from −30 °C to 150 °C with a heating rate of 3 °C min−1 under nitrogen atmosphere. The instrument was operated in tension mode at a frequency of 1.00 Hz and 0.03% oscillatory strain. Reported values are the averages of three measurements.
NMR spectroscopy
1H NMR spectroscopy was performed at room temperature using a Bruker Avance III 500 MHz NMR spectrometer. Deuterated chloroform (CDCl3) was used as a solvent, and the spectrum was reported relative to tetramethylsilane.Electrospray ionization-mass spectrometry (ESI-MS)
ESI-MS data were collected using a Bruker AmaZon-SL configured with an Agilent 1100 Series HPLC module, an ESI source, and a 3D ion trap mass analyzer. A 20% methanol/80% DCM mobile phase composition was utilized to collect positive ion spectra of samples.Liquid chromatography-mass spectrometry (LC-MS)
Diluted samples were injected on a 1290 Infinity II UHPLC System (Agilent Technologies Inc., Santa Clara, California, USA) onto an Acquity Premier C18 column (1.9 μm, 150 × 2.1 mm) (Waters Corporation, Milford, Massachusetts, USA) for reversed-phase chromatography which was maintained at 50 °C with a constant flow rate at 0.200 mL min−1, using a gradient of mobile phase A (9:1 water/methanol, 10 mM ammonium acetate, 0.2 mM ammonium fluoride) and mobile phase B (2:3:5 acetonitrile/methanol/isopropanol, 10 mM ammonium acetate, 0.2 mM ammonium fluoride). The gradient was programmed as follows: 0–1 min, 95%B; 1–3 min, 95–96%B; 3–6 min, 96–100%B; 6–10 min, hold 100%B; 10–10.10 min, 100–95%B; 10.10–13 min, hold 95%B. After C18 chromatography, the eluents passed through a 1260 Infinity II multi-wavelength detector (Agilent Technologies Inc., Santa Clara, California, USA) prior to MS detection. The absorbance wavelength was set at 214 nm with a 10-nm bandwidth. “MS-Only”, positive ion mode acquisition was conducted on the samples on an Agilent 6545 quadrupole time-of-flight mass spectrometer (Q-TOF LC-MS) equipped with a JetStream ionization source. The source conditions were as follows: gas temperature, 200 °C; drying gas flow, 12 L min−1; nebulizer, 50 psi; sheath gas temperature, 300 °C; sheath gas flow, 12 L min−1; VCap, 2500 V; fragmentor, 120 V; skimmer, 65 V; and oct 1 RF, 750 V. The acquisition rate in MS-Only mode was 5 spectra per sec between 40–1700 m/z range. The reference ion mass solution was introduced in the ion source using a separate quaternary HPLC pump containing m/z 121.050873 and m/z 922.009798 as reference masses in positive ion mode. Agilent (.d) files were imported to MassHunter Qualitative Analysis software (v. 10) where peak areas and mass spectra were extracted for each dataset.
Differential scanning calorimetry (DSC)
A Mettler Toledo DSC822e was used to characterize the glass transition temperatures. Samples were first heated to 150 °C at a rate of 20 °C min−1 and maintained for 5 min, followed by cooling to −60 °C at a rate of −20 °C min−1. The Tg values were obtained from a second heating ramp from −60 °C to 150 °C at a 10 °C min−1 rate using the one-half ΔCp method.Thermogravimetric analysis (TGA)
TGA was performed using a Mettler Toledo TGA/DSC3+. Samples were heated under an airflow of 50 min mL−1 from 25 °C to 800 °C at a rate of 20 °C min−1. The weight change was recorded as a function of temperature.Stress relaxation
Tensile stress relaxation was characterized using a TA Instruments RSA-G2 Solids Analyzer. Rectangular specimens were mounted on the fixture and allowed to equilibrate at the test temperature for 10 min before starting the test. Once thermal equilibrium was reached, samples were subjected to an instantaneous 5% strain, which was maintained throughout the test. The stress relaxation modulus was recorded until it had relaxed to 20% of its initial value.X-ray crystallography
Single crystals from BTMA samples were produced by recrystallization from methanol. For each structure, a suitable crystal was selected, and the crystal was mounted on a MITIGEN holder in paratone oil on a XtaLAB Synergy R, DW system, HyPix diffractometer. Each crystal was maintained at ∼100 K during data collection. Using Olex2,43 the structure was solved with the SHELXT44 structure solution program using Intrinsic Phasing and refined with the XL45 refinement package using least squares minimization.Crystal structure determination of unreacted TMPM in BTMA-Sn
Crystal Data for C13H23NO2 (M = 225.32): triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 6.28690(10) Å, b = 9.3906(2) Å, c = 11.5393(2) Å, α = 89.579(2)°, β = 78.811(2)°, γ = 86.145(2)°, V = 666.78(2) Å3, Z = 2, T = 100.1(3) K, μ (Cu Kα) = 0.589 mm−1, Dcalc = 1.122 g mm−3, 50438 reflections measured (7.81 ≤ 2Θ ≤ 157.446), 2769 unique (Rint = 0.0555, Rsigma = 0.0171) which were used in all calculations. The final R1 was 0.0413 (I > 2σ(I)) and wR2 was 0.1084 (all data).
(no. 2), a = 6.28690(10) Å, b = 9.3906(2) Å, c = 11.5393(2) Å, α = 89.579(2)°, β = 78.811(2)°, γ = 86.145(2)°, V = 666.78(2) Å3, Z = 2, T = 100.1(3) K, μ (Cu Kα) = 0.589 mm−1, Dcalc = 1.122 g mm−3, 50438 reflections measured (7.81 ≤ 2Θ ≤ 157.446), 2769 unique (Rint = 0.0555, Rsigma = 0.0171) which were used in all calculations. The final R1 was 0.0413 (I > 2σ(I)) and wR2 was 0.1084 (all data).
Crystal structure determination of BTMA-S2
Crystal Data for C26H44N2O4S2 (M = 512.75): monoclinic, space group P21/c (no. 14), a = 12.9420(2) Å, b = 31.1363(6) Å, c = 14.2239(2) Å, β = 90.9430(10)°, V = 5730.97(16) Å3, Z = 8, T = 100.00(11) K, μ (Cu Kα) = 1.936 mm−1, Dcalc = 1.189 g mm−3, 82193 reflections measured (5.676 ≤ 2Θ ≤ 152.092), 11460 unique (Rint = 0.0802, Rsigma = 0.0373) which were used in all calculations. The final R1 was 0.0681 (I > 2σ(I)) and wR2 was 0.1599 (all data).
Crystal structure determination of BTMA-S3
Crystal Data for C26H44N2O4S3 (M = 544.84): monoclinic, space group P21/n (no. 14), a = 12.49888(12) Å, b = 12.16712(12) Å, c = 19.36765(17) Å, β = 91.5744(8)°, V = 2944.23(5) Å3, Z = 4, T = 99.99(13) K, μ (Cu Kα) = 2.559 mm−1, Dcalc = 1.229 g mm−3, 58106 reflections measured (8.316 ≤ 2Θ ≤ 156.69), 6160 unique (Rint = 0.0354, Rsigma = 0.0167) which were used in all calculations. The final R1 was 0.0497 (I > 2σ(I)) and wR2 was 0.0818 (all data).
Crystal structure determination of BTMA-S4
Crystal Data for C26H44N2O4S4 (M = 576.87): monoclinic, space group P21/c (no. 14), a = 13.9546(4) Å, b = 20.1055(6) Å, c = 11.0995(3) Å, β = 101.725(3)°, V = 3049.14(15) Å3, Z = 4, T = 99.99(13) K, μ (Cu Kα) = 3.123 mm−1, Dcalc = 1.257 g mm−3, 58595 reflections measured (6.468 ≤ 2Θ ≤ 153.326), 6266 unique (Rint = 0.0701, Rsigma = 0.0319) which were used in all calculations. The final R1 was 0.0497 (I > 2σ(I)) and wR2 was 0.1382 (all data).
UV-vis diffuse reflectance spectroscopy
The UV-vis-NIR spectrum in diffuse reflectance mode was collected on lightly grounded powder using a Cary 5000 UV-vis-NIR double-beam spectrophotometer with a monochromator. BaSO4 powder was used for the baseline collection, and a mixture of sample powder with BaSO4 was used for the data collection at room temperature. Absorbance data were converted from reflectance data in software using the Kubelka–Munk equation, α/S = (1 − R)2/(2R), where α and S are the absorption and scattering coefficients, respectively, and R is the reflectance.Melt extrusion
A Thermo Scientific Haake MiniLab 3 twin-screw extruder was used to perform melt extrusion of the BTMA-S2-HMA CAN samples that had been cut to small pieces. Approximately 3 g of the pieces were fed into the hopper, circulated through the screws at 180 °C and 7 rpm, flushed through a 1-mm-thick die, and subsequently air-cooled.Results and discussion
We begin by reconsidering the composition of the previously reported BTMA cross-linker,33,34,39 which we call BTMA-Sn here, with elemental analysis reported in ref. 34 of 5.36% N and 12.64% S (theoretical values of C26H44N2O4S2 — 5.46% N and 12.51% S). It is important to appreciate that elementary analysis is not a confirmatory test, with any experimentally determined composition being achievable from different compositions of an array of components. However, in ref. 34, the absence of a TMPM peak by FTIR spectroscopy after the synthesis of BTMA-Sn (see Fig. 1 for the synthesis from TMPM and S2Cl2) was taken to indicate that TMPM had been fully converted within experimental uncertainty to BTMA. That interpretation along with the elemental analysis led in ref. 34 to the judgment that the BTMA contained mostly disulfides. Since the publication of ref. 34 and prior to the current focused study, ESI-MS results (Fig. S1†) indicated that BTMA-Sn contains protonated TMPM (TMPH-H+). Unlike TMPM, the presence of protonated TMPM in the sample would not be apparent from FTIR spectroscopy, in agreement with the “after-reaction” FTIR spectrum reported in ref. 34. However, the previously unacknowledged presence of protonated TMPM in the sample brought into question the judgment from ref. 34 that the BTMA was mostly disulfide. The ESI-MS results (Fig. S1†) also showed the presence of BTMA-S3 in both samples tested, with BTMA-S4 also clearly present in one sample and possibly present in the second sample. Finally, recent X-ray crystallography results on two single crystals from a sample of BTMA-Sn were consistent with the presence of TMPM and trisulfide compounds (see Fig. S2A and B†). Taken together, these results indicated that it was important to undertake a study aimed at more fully characterizing BTMA-Sn and leading to a synthesis route to achieve pure or nearly pure BTMA-S2.For the current, focused study, we resynthesized BTMA-Sn and characterized it by LC-MS, which displayed three peaks denoting nearly equal contents of BTMA-S2, BTMA-S3, and BTMA-S4 molecules which correspond to disulfide-, trisulfide-, and tetrasulfide-containing versions of BTMA, respectively (see Fig. 2). Additionally, LC analysis also showed considerable levels of protonated TMPM (TMPM-H+) impurity, and elemental analysis also showed higher percentages of sulfur content compared to theoretical predictions, indicating the presence of significant levels of higher-order sulfur connectivities. (Anal. calcd for C26H44N2O4S2: N, 5.46; S, 12.51. Found: N, 5.40; S, 16.35.) It is notable that the 16.35% S content in this BTMA-Sn sample is much higher than the 12.64% S content for the BTMA-Sn sample reported in ref. 34. However, these differences are easily understood to arise from somewhat different levels of the four components in the BTMA-Sn. A sample containing 31% BTMA-S2, 31% BTMA-S3, 31% BTMA-S4, and 7% TMPM(HCl) would have a 16.2% S content, close to that obtained for the BTMA-Sn sample synthesized in this study. In contrast, a sample containing 24% BTMA-S2, 24% BTMA-S3, 24% BTMA-S4, and 28% TMPM(HCl) would have a 12.6% S content, close to that reported for the sample in ref. 34.
 | ||
| Fig. 2 (A) Liquid chromatography (LC) chromatogram of a blank (methanol) sample. LC chromatogram and mass spectrometry (MS) spectra for (B) BTMA-S2, and (C) BTMA-Sn, showing individual fractions of TMPM-H+ and BTMA species with different sulfide linkages. | ||
The BTMA-Sn obtained in the current study was crystallized from methanol, and single-crystal X-ray crystallography of a random crystal confirmed the presence of BTMA-S4 molecules (see Fig. S2C†). We hypothesize that the source of trisulfide and tetrasulfide linkages is due to impure commercial S2Cl2, as indicated by a reddish color. As described in literature, S2Cl2 is a bright golden color; however, through reaction with moisture, impurities such as SCl2, S3Cl2, and S4Cl2 can be formed which are darker in color.40 To ensure disulfide-only linkages, S2Cl2 was purified by vacuum distillation under a high vacuum at room temperature in a kügelrohr distillation apparatus. This distillation produced a bright golden colored compound matching the color of S2Cl2 described in the literature.40 Distilled S2Cl2 was subsequently stored in a septum-sealed vial under an inert atmosphere and was used for the synthesis of the optimized BTMA-S2.
For the optimized synthesis of BTMA, TMPM was dissolved in various dry solvents, including petroleum ether, MeTHF, and DCM, in a septum-sealed, dry round-bottom flask fitted with a nitrogen balloon to avoid atmospheric moisture. We note that even purified S2Cl2 can transform into a mixture of S3Cl2 and S4Cl2 through the presence of moisture during reaction.40 Then, purified S2Cl2, dissolved in a chosen reaction solvent, was added to a solution of TMPM at various temperatures (see results in Fig. 1). As seen in Scheme S1,† 1 mol of S2Cl2 reacts with 2 moles of TMPM to produce 1 mol of BTMA as well as 2 moles of HCl. The HCl further reacts with TMPM to produce 2 moles of [TMPM-H+][Cl−] species, hence, the need to maintain 4 equiv. of TMPM during the reaction. When petroleum ether and MeTHF were used as solvents, TMPM and TMPM-H+ precipitated from the solution during cooling. Such precipitation leads to a stoichiometric imbalance with S2Cl2, causing unreacted S2Cl2 to react with water during subsequent work-up steps, allowing the generation of oligosulfide side products. Meanwhile, DCM was found to dissolve TMPM and TMPM-H+ even at temperatures as low as −70 °C. Hence, the use of DCM resulted in the highest isolated yield of BTMA (up to 97%) compared to other solvents used in this study.
The dissolved mixtures were then poured into distilled water and stirred vigorously overnight to dissolve the protonated TMPM formed during the reaction (see Scheme S1†) and the remaining excess TMPM. Crude BTMA floated on top of the water phase, which was then vacuum-filtered and washed two times with distilled water. The collected compound was dried in a vacuum oven at 50 °C overnight. The 1H NMR spectrum (Fig. S3†) for this BTMA-S2 was similar to that of the BTMA-Sn, which we attribute to the symmetry of the molecule. To gain further insight, this BTMA-S2 compound was then subjected to LC-MS46 which primarily showed a single peak that corresponds to BTMA-S2 in the mass spectrum (Fig. 2). Moreover, elemental analysis of BTMA-S2 also indicated sulfur content that is in very good agreement with theoretical predictions (Anal. calcd for C26H44N2O4S2: N, 5.46; S, 12.51. Found: N, 5.42; S, 12.58). Interestingly, this BTMA-S2 is more thermally stable than BTMA-Sn (Fig. S4†), with a 1 wt% decomposition temperature (Td1%) of 208 °C for BTMA-S2 and 188 °C for BTMA-Sn. BTMA-S2 was then crystallized in methanol and subjected to single-crystal X-ray crystallography which confirms the BTMA-S2 structure (Fig. 3).
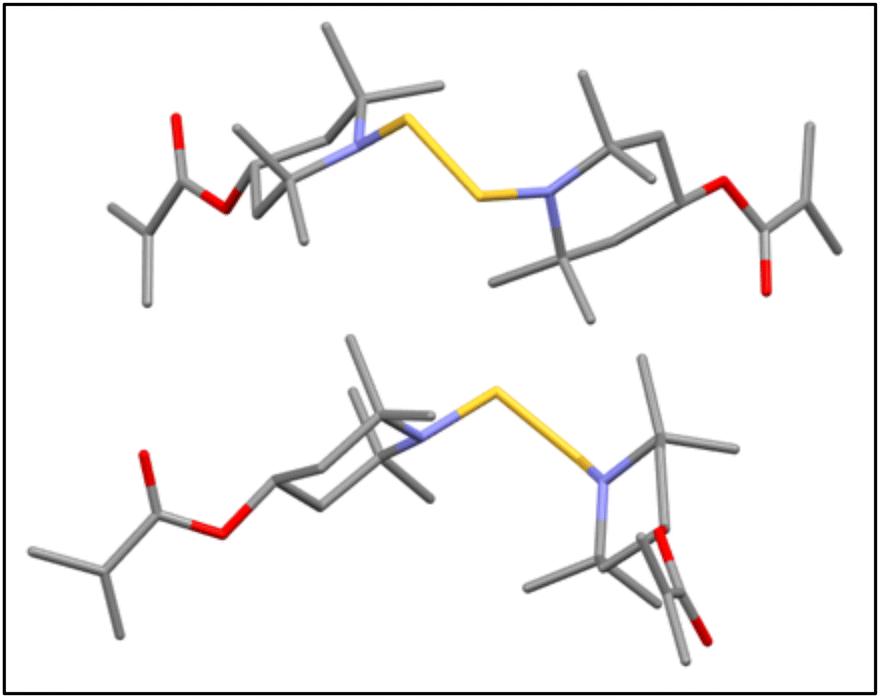 | ||
| Fig. 3 A unit cell of a random crystal from BTMA-S2 measured in single-crystal X-ray crystallography showing two BTMA molecules with exclusively disulfide linkages. Grey, red, purple, and yellow represent carbon, oxygen, nitrogen, and sulfur atoms, respectively. Hydrogen atoms are omitted for clarity. | ||
With the structure and composition of the BTMA-S2 having been verified, we sought to compare the dynamic characteristics of BTMA-HMA CANs made with 5 mol% of BTMA-S2 as compared with 5 mol% of BTMA-Sn. BTMA-Sn has been the subject of extensive studies by Torkelson and co-workers,33–39 showing robust dynamic covalent characteristics for reprocessing, stress relaxation, and elevated-temperature creep resistance of CANs containing BTMA. To study the effects of pure disulfide BTMA and oligosulfide-containing BTMA, HMA was cross-linked with either BTMA-Sn or BTMA-S2 by free-radical polymerization using AIBN at 70 °C to produce BTMA-Sn-HMA CANs and BTMA-S2-HMA CANs (see Fig. 1). Polymerizations were initiated at 70 °C, and gelation was observed within 5 h for both samples. The reaction was continued overnight to ensure complete monomer conversion within experimental uncertainty. After drying in a vacuum oven at 40 °C for 3 days, the materials were reprocessed at 130 °C under 10-ton ram force for 1 h, yielding the 1st mold sample.
It is important to note that samples of the BTMA-Sn-HMA CANs had a more discernible yellow tint compared to samples of the BTMA-S2-HMA CANs (see Fig. S5†). We spectroscopically assessed such observation by UV-vis diffuse reflectance spectroscopy, which showed that BTMA-Sn-HMA CAN absorb at higher wavelengths (onset wavelength: 427 nm) than BTMA-S2-HMA CAN (onset wavelength: 390 nm) (see Fig. S6†). We also note that both BTMA-Sn-HMA and BTMA-S2-HMA CANs absorb at higher wavelengths than linear poly(n-hexyl methacrylate), which absorbs at much lower wavelength (∼320 nm). According to the Kubelka–Munk theory,47 materials with a darker or deeper color absorb at longer wavelengths, consistent with the higher coloration in the BTMA-Sn-HMA CAN due to the impurities.
DSC analysis showed that BTMA-S2-HMA CAN materials have a Tg value of ∼17 °C (see Fig. S7†), in agreement (within experimental error) with the Tg of BTMA-Sn-HMA CAN as reported in the literature.33,34,39 This similarity in Tg suggests that there is no significant difference in network structure in both BTMA-S2-HMA CAN and BTMA-Sn-HMA CAN.
DMA analysis of the compression-molded BTMA-S2-HMA CAN showed excellent overlap of the quasi-rubbery plateau values (at temperatures exceeding ∼70 °C in Fig. 4) of tensile storage modulus for 1st and 2nd molds. According to Flory's theory of ideal rubber elasticity,47 this indicates complete recovery of cross-link densities after reprocessing of both BTMA-Sn-HMA CANs and BTMA-S2-HMA CANs. Notably, temperature-sweep DMA analysis indicated that, at a given temperature in the quasi-rubbery plateau region, the tensile storage modulus for the BTMA-S2-HMA CAN is ∼80% as large as that for the BTMA-Sn-HMA CAN (see Fig. 4 and Table S1†). Interestingly, as seen in Fig. 4A, the quasi-rubbery plateau tensile storage modulus of the BTMA-Sn-HMA CAN decreases somewhat more sharply with increasing temperature than that of BTMA-S2-HMA CAN. This difference may be attributed to differences in the temperature sensitivity of the extent of dissociation between BTMA-S2 and BTMA-Sn containing oligosulfides. Nevertheless, in both the old and new CANs, the tensile storage modulus values of the 2nd mold materials are, within error, equal to those of the 1st mold materials, indicating full recovery of cross-link density after reprocessing of both CANs.
 | ||
| Fig. 4 Storage modulus as a function of temperature for (A) 1st-molded samples produced by compression molding of both BTMA-S2-HMA CAN and BTMA-Sn-HMA CAN materials, (B) 1st-molded and 2nd-molded (reprocessed) samples produced by compression molding of BTMA-Sn-HMA CAN and (C) 1st-molded and 2nd-molded (reprocessed) samples produced by compression molding of BTMA-S2-HMA CAN. Based on Flory's theory of ideal rubbery elasticity,48 the excellent reproduction of the storage modulus in the quasi-rubbery plateau regions (as well as at all studied temperatures) in (B) and (C) indicates the full recovery of cross-link density after reprocessing. | ||
To compare the dynamic characteristics of the BTMAs, both BTMA-Sn-HMA and BTMA-S2-HMA CANs were subjected to stress relaxation experiments (Fig. 5). These curves were fitted with the Kohlrausch–Williams–Watts (KWW) stretched exponential decay function which generally has been found to provide a much better fit for CANs than the single-exponential-decay Maxwell model.49,50 The KWW decay function can be written as follows:
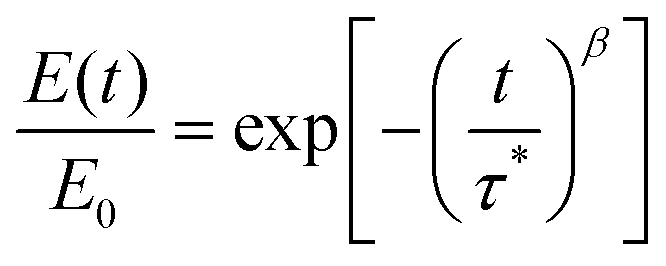 | (1) |
 | (2) |
 | ||
| Fig. 5 (A) Normalized stress-relaxation data of BTMA-Sn-HMA CAN at various temperatures. (B) Normalized stress-relaxation data of BTMA-S2-PHMA at various temperatures. (C) Arrhenius-type plots relating the natural logarithm of average relaxation time to inverse temperature and yielding apparent stress-relaxation activation energies of BTMA-Sn-HMA and BTMA-S2-HMA CANs. | ||
With CANs, shorter stress relaxation times indicate faster dynamic chemistry. Other things being equal, that should correlate with faster processability. Here, we observed that compression molding at 130 °C with 10-ton ram force of the BTMA-S2-HMA CANs required only 5 min of compression molding time to achieve final rubbery plateau modulus (equal to the same values at 30–60 min remolding times) and, thus, cross-link density. In contrast, using the same compression molding temperature and 10-ton ram force, the BTMA-Sn-HMA CANs required 30 min of compression molding time to reach the final rubbery plateau modulus (see Fig. 6 and Fig. S5, and S8†). Additionally, we see excellent reproduction of the rubbery plateau modulus (and, thus, cross-link density) for BTMA-S2-HMA CAN samples after 1st and 2nd moldings (see Fig. S9†) at 130 °C and 5 min. This makes BTMA-S2-HMA CAN among the fastest CAN materials ever to be reprocessed by compression molding.37,51–57
 | ||
| Fig. 6 Storage modulus as a function of temperature for samples that were compression molded for different times at 130 °C and 10-ton ram force: (A) BTMA-Sn-HMA CAN; (B) BTMA-S2-HMA CAN. | ||
Such fast processability of BTMA-S2 CAN materials provide opportunities for realistic processing and reprocessing by melt extrusion, an important polymer processing method for commercial applications. A previous extrusion of a CAN made from an ethylene-based copolymer and BTMA-Sn was performed at 200 °C.37 Here, we extruded BTMA-S2-HMA CAN at 180 °C and obtained a robust, transparent, and defect-free extrudate, as shown in Fig. 7. DMA of the extrudate revealed that the temperature-dependent storage modulus values of the extrudate fully recover those of the 1st-molded BTMA-S2-HMA CAN obtained via compression molding (Fig. 7c). This includes the rubbery plateau region, indicating that the extruded sample fully recovers the cross-link density of the compression-molded sample. This demonstration of extrudability with complete cross-link density recovery further accentuates the potential for dialkylamino disulfide dynamic chemistry to be implemented in developing melt-processable and recyclable polymer networks for commercial application.
 | ||
| Fig. 7 Demonstration of extrudability of BTMA-S2-HMA CAN. (A) Image of BTMA-S2-HMA CAN being extruded out of the die. (B) A section of BTMA-S2-HMA CAN extrudate after cooling. (C) Comparison of the storage moduli of BTMA-S2-HMA CAN (1st mold and extrudate) as a function of temperature, with the rubbery plateau modulus (and, hence, cross-link density) of the extrudate showing full recovery in comparison with the molded sample. | ||
Conclusions
We developed a new synthesis to produce a BTMA dynamic cross-linker with a high isolated yield of up to 97% BTMA-S2 and ≥95% disulfide linkages. LC-MS characterization showed that the old method of synthesizing BTMA-Sn produces substantial levels of disulfide, trisulfide, and tetrasulfide linkages in addition to some other traces of unidentified impurities. In contrast, LC-MS and single-crystal X-ray crystallography showed that BTMA-S2 (synthesized with the new method) was highly pure and consisted of nearly exclusively disulfide linkages. Both BTMA-S2 and BTMA-Sn produced robust CANs when reacted with n-hexyl methacrylate by free-radical polymerization, and both types of CANs could be reprocessed with full recovery of cross-link density. The CANs made from 5 mol% BTMA-Sn and 5 mol% BTMA-S2 possessed similar properties in the cross-linked state but dramatic differences in their dynamic character. Although the materials have similar activation energies for elevated-temperature stress relaxation, at 130 °C, the BTMA-S2-HMA CANs exhibited a factor of 3.5 shorter average stress relaxation time, i.e., much faster stress relaxation, than the BTMA-Sn-HMA CANs. We attribute this difference to nearly exclusive disulfide linkages in the BTMA-S2-HMA CANs. Such fast dynamic chemistry translated into complete cross-link density recovery after only 5 min of reprocessing by compression molding at 130 °C and 10-ton ram force. Moreover, melt extrusion of BTMA-S2-HMA CANs at 180 °C produced robust, transparent, and defect-free networks with full recovery of cross-link density. These results suggest that CANs made with BTMA-S2 have promising potential for melt processing and recycling at large scales.Conflicts of interest
One U. S. patent application has been filed related to the research described in this manuscript.Acknowledgements
This project was funded by Braskem. We further acknowledge the support of Northwestern University via discretionary funds associated with a Walter P. Murphy Professorship (J. M. T.) and from an NSF Graduate Research Fellowship (L. M. F.). We also thank Dr Fernando “Ralph” Tobias, Dr Indrajit Paul, Dr Chris D. Malliakas, and Charlotte Stern for their contributions and Subeen Kim and Molly Sun for productive discussions and training on the twin-screw extruder. This work made use of the MatCI Facility at Northwestern University, which receives support from the IMRSEC Program (NSF DMR 1720139) of the Materials Research Center at Northwestern University. This work also made use of the IMSERC Mass Spectrometry Facility, Crystallography Facility, and NMR Facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSFECCS 2025633), the State of Illinois, and the International Institute for Nanotechnology (IIN).References
- W. Post, A. Susa, R. Blaauw, K. Molenveld and R. J. I. Knoop, Polym. Rev., 2020, 60, 359–388 CrossRef CAS.
- E. Chabert, J. Vial, J.-P. Cauchois, M. Mihaluta and F. Tournilhac, Soft Matter, 2016, 12, 4838–4845 RSC.
- T. Debsharma, V. Amfilochiou, A. A. Wróblewska, I. De Baere, W. Van Paepegem and F. E. Du Prez, J. Am. Chem. Soc., 2022, 144, 12280–12289 CrossRef CAS PubMed.
- T. Debsharma, S. Engelen, I. De Baere, W. Van Paepegem and F. Du Prez, Macromol. Rapid Commun., 2023, 44, 2300020 CrossRef CAS PubMed.
- E. Morici and N. T. Dintcheva, Polymer, 2022, 14, 4153 CAS.
- D. S. Cousins, Y. Suzuki, R. E. Murray, J. R. Samaniuk and A. P. Stebner, J. Cleaner Prod., 2019, 209, 1252–1263 CrossRef CAS.
- A. E. Crolais, N. D. Dolinski, N. R. Boynton, J. M. Radhakrishnan, S. A. Snyder and S. J. Rowan, J. Am. Chem. Soc., 2023, 145, 14427–14434 CrossRef CAS PubMed.
- J. Liu and K. V. Bernaerts, Chem. Eng. J., 2023, 477, 147299 CrossRef CAS.
- A. L. Santefort, P. A. Yuya and D. A. Shipp, Polymer, 2023, 281, 126120 CrossRef CAS.
- T. Yan, A. H. Balzer, K. M. Herbert, T. H. Epps and L. T. J. Korley, Chem. Sci., 2023, 14, 5243–5265 RSC.
- S. Wang and M. W. Urban, Chem, 2023, 9, 1362–1377 CAS.
- C. W. H. Rajawasam, O. J. Dodo, M. A. S. N. Weerasinghe, I. O. Raji, S. V. Wanasinghe, D. Konkolewicz and N. De Alwis Watuthanthrige, Polym. Chem., 2024, 15, 219–247 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- C. A. Tretbar, J. A. Neal and Z. Guan, J. Am. Chem. Soc., 2019, 141, 16595–16599 CrossRef CAS PubMed.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- C. Tretbar, J. Castro, K. Yokoyama and Z. Guan, Adv. Mater., 2023, 35, 2303280 CrossRef CAS PubMed.
- X. Wu, X. Yang, R. Yu, X.-J. Zhao, Y. Zhang and W. Huang, J. Mater. Chem. A, 2018, 6, 10184–10188 RSC.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS PubMed.
- C. Bowman, F. Du Prez and J. Kalow, Polym. Chem., 2020, 11, 5295–5296 RSC.
- M. Podgórski, N. Spurgin, S. Mavila and C. N. Bowman, Polym. Chem., 2020, 11, 5365–5376 RSC.
- Y. Chen, B. Chen and J. M. Torkelson, Macromolecules, 2023, 56, 3687–3702 CrossRef CAS.
- N. S. Purwanto, Y. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2023, 5, 6651–6661 CrossRef CAS.
- A. Takahashi, R. Goseki and H. Otsuka, Angew. Chem., Int. Ed., 2017, 56, 2016–2021 CrossRef CAS PubMed.
- A. Tsuruoka, A. Takahashi, D. Aoki and H. Otsuka, Angew. Chem., Int. Ed., 2020, 59, 4294–4298 CrossRef CAS PubMed.
- A. Takahashi, R. Goseki, K. Ito and H. Otsuka, ACS Macro Lett., 2017, 6, 1280–1284 CrossRef CAS PubMed.
- S. Kataoka, A. Tsuruoka, D. Aoki and H. Otsuka, ACS Appl. Polym. Mater., 2021, 3, 888–895 CrossRef CAS.
- M. A. Bin Rusayyis and J. M. Torkelson, Polym. Chem., 2021, 12, 2760–2771 RSC.
- M. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2020, 53, 8367–8373 CrossRef CAS.
- L. M. Fenimore, M. J. Suazo and J. M. Torkelson, Macromolecules, 2024, 57, 2756–2772 CrossRef CAS.
- L. M. Fenimore, B. Chen and J. M. Torkelson, J. Mater. Chem. A, 2022, 10, 24726–24745 RSC.
- B. Chen, L. M. Fenimore, Y. Chen, S. M. Barbon, H. A. Brown, E. Auyeung, C. L. P. Shan and J. M. Torkelson, Polym. Chem., 2023, 14, 3621–3637 RSC.
- L. M. Fenimore, B. Chen, Y. Chen, S. M. Barbon, H. A. Brown, E. Auyeung, C. L. P. Shan and J. M. Torkelson, Eur. Polym. J., 2024, 202, 112661 CrossRef CAS.
- M. A. Bin Rusayyis, L. M. Fenimore, N. S. Purwanto and J. M. Torkelson, Polym. Chem., 2023, 14, 3519–3534 RSC.
- G. Brauer, Handbook of Preparative Inorganic Chemistry, Elseier, 2012 Search PubMed.
- M. Aiba, T.-A. Koizumi, M. Futamura, K. Okamoto, M. Yamanaka, Y. Ishigaki, M. Oda, C. Ooka, A. Tsuruoka, A. Takahashi and H. Otsuka, ACS Appl. Polym. Mater., 2020, 2, 4054–4061 CrossRef CAS.
- T. L. Pickering, K. J. Saunders and A. V. Tobolsky, J. Am. Chem. Soc., 1967, 89, 2364–2367 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Kruve, R. Rebane, K. Kipper, M.-L. Oldekop, H. Evard, K. Herodes, P. Ravio and I. Leito, Anal. Chim. Acta, 2015, 870, 29–44 CrossRef CAS PubMed.
- L. Yang and B. Kruse, J. Opt. Soc. Am. A, 2004, 21, 1933–1941 CrossRef PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press Ithaca, N.Y., Ithaca, N.Y., 1953 Search PubMed.
- X. Chen, L. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- Q. Li, S. Ma, P. Li, B. Wang, Z. Yu, H. Feng, Y. Liu and J. Zhu, Macromolecules, 2021, 54, 8423–8434 CrossRef CAS.
- X. Xu, S. Ma, S. Wang, B. Wang, H. Feng, P. Li, Y. Liu, Z. Yu and J. Zhu, Macromol. Rapid Commun., 2022, 43, 2100777 CrossRef CAS PubMed.
- X. Xu, S. Ma, H. Feng, J. Qiu, S. Wang, Z. Yu and J. Zhu, Polym. Chem., 2021, 12, 5217–5228 RSC.
- G. Li, J. Huang, H. S. Soo, Y. Zhao, T. Li, Y. Wang, S. Wang and W. Dong, Eur. Polym. J., 2023, 194, 112165 CrossRef CAS.
- G. Lee, H. Y. Song, S. Choi, C. B. Kim, K. Hyun and S.-K. Ahn, Macromolecules, 2022, 55, 10366–10376 CrossRef CAS.
- S. Engelen, A. A. Wróblewska, K. De Bruckyer, R. Aksakal, V. Ladmiral, S. Caillol and F. E. Du Prez, Polym. Chem., 2021, 13, 2665–2673 RSC.
- K. Hu, B. Wang, X. Xu, Y. Su, W. Zhang, S. Zhou, C. Zhang, J. Zhu and S. Ma, Macromol. Rapid Commun., 2022, 44, 2200726 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Balanced equation for synthesis of BTMA-S2; ESI-MS spectra of BTMA-Sn; X-ray crystallography results of various crystals from BTMA-Sn samples; 1H NMR spectrum of BTMA-S2; TGA curves of BTMA-S2 and BTMA-Sn; images of BTMA-HMA CANs molded at 130 °C for 3 min and 5 min; UV-vis spectra of BTMA-HMA CANs; DSC thermograms of reprocessed BTMA-S2-HMA CANs; DMA results for BTMA-HMA CANs as functions of compression molding times; storage modulus values for BTMA-HMA CANs as a function of temperature; image of twin-screw extruder; KWW fitting parameters for BTMA-HMA CANs. CCDC 2332023, 2332024, 2346357 and 2346358. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4py00160e |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |