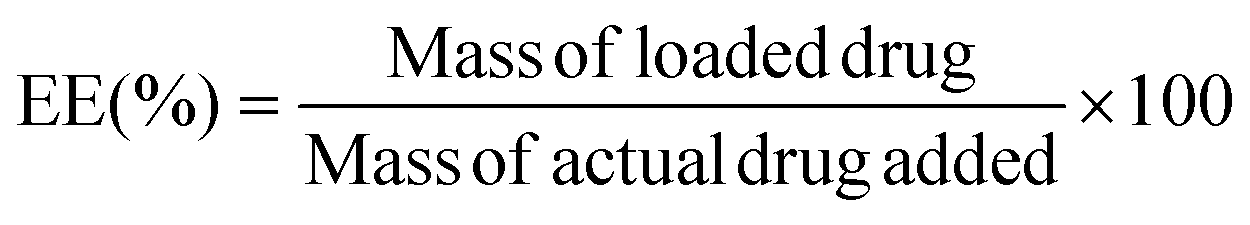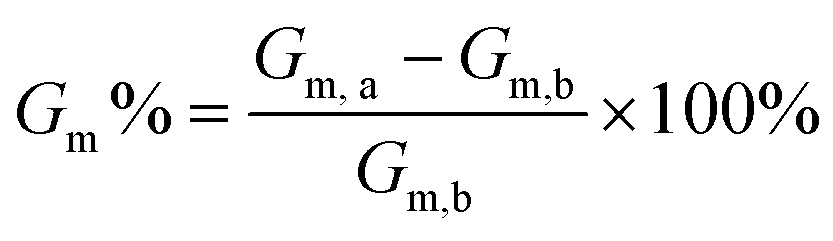DOI:
10.1039/D4PY00155A
(Paper)
Polym. Chem., 2024,
15, 1648-1659
Loop to linear: exploring the impact of corona topology on the properties of self-assembled polymer nanoparticles†
Received
8th February 2024
, Accepted 24th March 2024
First published on 26th March 2024
Abstract
Macromolecular architecture plays a pivotal role in endowing distinct properties to polymer nanomaterials. We introduce a synthesis approach to produce cyclic polystyrene-b-poly(acrylic acid) block copolymers featuring UV-cleavable motifs by combining atom transfer radical polymerisation and copper-catalyzed azide–alkyne cycloaddition. The resulting cyclic copolymers could self-assemble into discrete nanoparticles. Their coronal topology could be altered from looped to linear poly(acrylic acid) chains upon UV irradiation while maintaining the original nanoparticle morphology, therefore achieving the post-assembly modification of polymer nanoparticles. Small molecule release profiles were markedly different for self-assemblies with looped or linear corona, as was their interaction with model cell membranes in electrochemical impedance spectroscopy assays. Compared to their linear counterparts, cyclic copolymer assemblies exhibited slower release and weakened membrane interactions.
Introduction
Self-assembled nanomaterials with the capability of modulating their properties by responding to specific stimuli constitute a rapidly growing area of polymer research.1–5 Recent advancements in stimuli-responsive polymers, with dynamic-covalent (e.g., Diels–Alder and aliphatic azo)6 and supramolecular (e.g., rotaxane molecular switch)7 bonds introduced new opportunities to alter polymer architecture and topology post assembly, that is after the polymers had formed a self-assembled superstructure. Post-assembly modification (PAM) has seen significant growth in recent years, enabling it to become a versatile tool for introducing new features to a complex structure after it has been formed and, more importantly, preserving its self-assembled superstructure. To achieve PAM, polymers must carry reactive sites that can be selectively addressed, either by covalent bond-forming or bond-breaking reactions. Among many stimuli-responsive systems, light has been rediscovered as a favourable stimulus due to its convenience and controllability.2,8–11 Unlike other stimuli, light offers the advantage of being readily accessible and can be employed without additional reagents and under mild reaction conditions. Various light-responsive systems, such as anthracene dimerisation,12,13 azobenzene,11,14 and o-nitrobenzyl (o-NB),15–18 have been developed, enabling precise control over molecules by adjusting parameters such as light intensity, wavelength, and irradiation time. Such photo-responsive reactions are highly selective and preserve interactions that grant the integrity of self-assembled nanomaterials.
Macromolecular architecture plays a fundamental role in determining polymer properties and those of their self-assembled superstructures. Topology transformations in polymer building blocks, such as brush-to-linear transitions, typically also change the polymer chemistry and composition, which often results in concurrent morphological transformations of assemblies.14,19,20 The impact of distinct in situ architectural variations within polymer assemblies on their properties remains relatively underexplored. This is connected to an inherent characteristic that polymer topology is typically static and fixed within a macromolecule. While it is possible to control how polymers are made up chemically, their covalent nature makes it rather challenging to selectively break bonds to switch between polymer topologies. Cyclic polymers, characterised by their unique looped topology, can be synthesised via various methods.21–26 Compared to their linear analogues, cyclic polymers possess the same chemical composition and molecular weight yet exhibit distinctively different physical properties.27–29 One of the most significant differences lies in their hydrodynamic behaviours, which indicate how macromolecules interact with and move through the solvent system. This property is directly influenced by the shape, size, and extent of interactions with the solvent of macromolecules. Due to their absence of end groups and looped structure, cyclic polymers have a more compact topology, leading to the reduced resistance against their motions, moving better through the solvent, and exhibiting smaller hydrodynamic volumes than extended linear polymers.30–32 These unique characteristics pave the way for investigating how topology alterations may affect polymer behaviours, especially when induced in situ and within self-assembled nanomaterials.
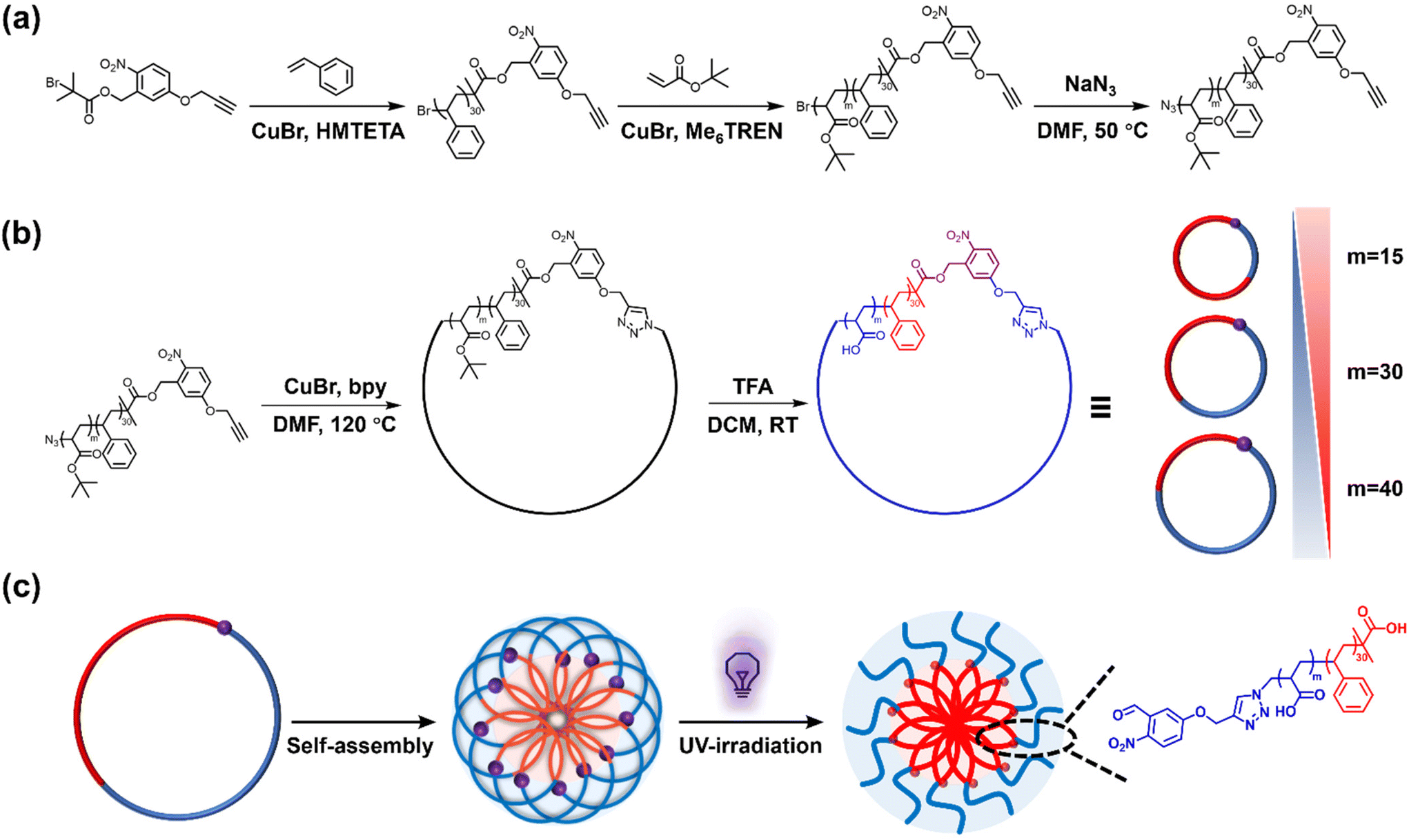
In this work, we present a synthesis approach for UV-cleavable cyclic polystyrene-block-poly(acrylic acid) (C-NB-PS-b-PAA) block copolymers from an o-nitrobenzyl ester containing ATRP initiator. The UV-cleavable moiety in the cyclic polymer can open the macrocycle to switch the polymer topology to its linear analogue (L(UV)-PS-b-PAA). Self-assembled flower micelles from C-NB-PS-b-PAA could be equally altered via UV irradiation, switching their looped corona to linear chains. At the same time, the high glass transition temperature of PS segments ensured the structural integrity of PS-b-PAA assemblies, preventing morphology transitions during PAM (Scheme 1). The near-identical self-assembled nanomaterials with distinctly different coronal topologies were further compared for their drug encapsulation efficiencies and drug release profiles, displaying noticeably altered properties. In addition, the two systems exhibited different interactions with cell membrane mimics in an electrochemical impedance spectroscopy (EIS) assay revealing topology-dependent nanoparticle–membrane interactions.
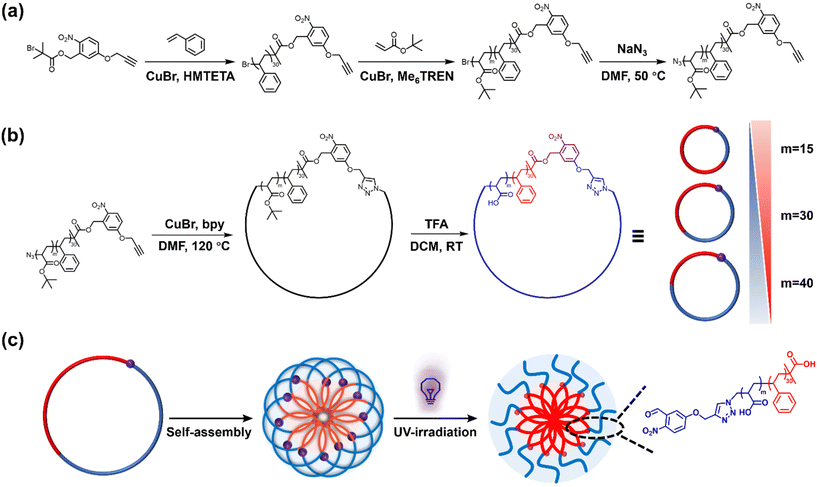 |
| | Scheme 1 (a) Synthesis of NB-PS30-b-PtBAm (m = 15, 30, and 40) linear block copolymers; (b) synthesis of C-NB-PS30-b-PtBAm and C-NB-PS30-b-PAAm cyclic block copolymers; (c) schematic representation of transforming polymer assemblies with looped corona to linear corona through UV-irradiation. | |
Experimental section
Materials
5-Hydroxy-2-nitrobenzyl alcohol (98%), potassium carbonate (K2CO3, ≥99%), propargyl bromide (80 wt% in toluene), α-bromoisobutyryl bromide (98%), triethylamine (TEA, ≥99%), styrene (≥99%), copper(I) bromide (CuBr, 98%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%), tert-butyl acrylate (tBA, 98%), 2,2′-bipyridine (bpy, ≥99%), methyl ethyl ketone (MEK, ≥99%), Dulbecco's phosphate buffered saline (DPBS, no calcium or magnesium) were sourced from Sigma-Aldrich. Sodium azide (NaN3, pure solid), trifluoroacetic acid (TFA), and methanol (MeOH, 98%) were sourced from Ajax. Ethyl acetate (≥99%), tetrahydrofuran (THF, ≥99.9%), and dichloromethane (DCM, ≥99%) were sourced from Merck. Anhydrous N,N-dimethyl formamide (DMF, 99.8% by gas chromatography (GC)) was sourced from RCI LabScan. 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) was sourced from SDX Tethered Membranes. All chemicals were used as received. Monomers were passed through basic alumina before use. Tris[2-(dimethylamino)ethyl]amine (Me6TREN) was synthesised according to literature.33
Characterisation
Nuclear magnetic resonance (NMR).
NMR spectra were recorded at the University of Sydney using Bruker AVIII 400 MHz (DOSY) and Bruker NEO 300 MHz NMR spectrometers. 1H NMR experiments were carried out using a zg pulse program (90° pulse) with a recycle delay (D1) of 2–5 s. 1H NMR spectra are referenced to the residual solvent peak for CDCl3 (7.26 ppm), DMSO-d6 (2.50 ppm), or acetone-d6 (2.05 ppm) as appropriate. Deuterated solvents were obtained from Sigma Aldrich and Cambridge Isotope Laboratories (via Novachem Australia) and used without further purification.
Size exclusion chromatography (SEC).
SEC was performed using a Shimadzu Prominence UFLC (ultra-fast liquid chromatography) system fitted with a Shim-pack GPC-800DP guard column followed by two in-series Phenogel columns (5 μm, 104 Å and 105 Å). The system eluent was HPLC grade tetrahydrofuran, eluting at a flow rate of 1 mL min−1. The column assembly was incubated at 40 °C, and retention times were calibrated using PS narrow standards from PSS.
Fourier-transform infrared spectroscopy (FTIR).
FTIR spectra were recorded on a Bruker ALPHA II Platinum ATR spectrometer fitted with a monolithic diamond crystal. Spectra were recorded with 16 scans at 4 cm−1 resolution and with atmospheric background subtraction.
Dynamic light scattering (DLS).
DLS measurements were performed on a Malvern Zetasizer Ultra equipped with a He–Ne (633 nm) laser. The hydrodynamic diameters of self-assembled polymer samples were directly measured on DLS with concentrations of 0.2 mg mL−1 without any further treatment.
Transmission electron microscopy (TEM).
TEM was performed on a JEM-2100CR instrument equipped with a 5k × 4k CMOS camera (EMSIS). Images were collected in bright-field mode with a spot size of 3 with diffraction contrast enhanced by using an objective lens with an aperture size of 20 μm, at an accelerating voltage of 200 kV. TEM samples were prepared by adding 4 μL of the polymer self-assembly solution onto a carbon-coated copper grid. After drying in air for 10 min, the remained solution was removed by touching the edge of the grid with a filter paper. The samples were then stained using 10 μL of 2% uranyl acetate (UA) solution. First, UA solution was dropped on a parafilm to form a droplet. The grid was then stained for 30 seconds by inverting the grids on the droplet. After removing the UA solution with a filter paper, the grid was left to dry in the air.
UV-Vis absorption.
UV-vis absorption spectra were recorded using a Cary 60 UV-Vis spectrophotometer (Agilent Technologies). The data was recorded with a data interval of 1.0 nm and scan speed at 60 nm min−1.
Fluorescence spectroscopy.
Fluorescence spectroscopy was recorded on a RF-6000 Fluorescence Spectrophotometer (Shimadzu). The data was recorded with a data interval of 2.0 nm and scan speed at 200 nm min−1. An excitation and emission slit size of 5 nm was used.
Electrical impedance spectroscopy (EIS) measurements.
EIS measurements were performed using the Tethapod system (SDX Tethered Membranes, Australia). The instrument is used for EIS to measure the impedance properties of lipid bilayers. We used the T10 electrode chips that are supplied for use in the Tethapod. The T10 electrode chips had a pattern of adhesive laminate and methoxy PEG-coated gold counter electrodes that were supplied with a pre-coating of a stable monolayer that comprised a mixture of ester-free DLP and BnSS TEG molecules in the molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 provided by SDX Tethered Membranes.
:90 provided by SDX Tethered Membranes.
Synthetic procedure
Synthesis of NB-PS30-Br macroinitiator.
In a typical procedure, 5-propargylether-2-nitrobenzyl bromoisobutyrate (100 mg, 0.28 mmol, 1 eq.), HMTETA (152 μL 0.56 mmol, 2 eq.), styrene (6.43 mL, 56.15 mmol, 200 eq.) were added into a Schlenk flask. The mixture was degassed through three freeze–pump–thaw cycles and CuBr (40 mg, 0.28 mmol, 1 eq.) was added to start the polymerisation. After stirring at 100 °C for 190 min, the polymerisation was quenched by putting the flask into liquid nitrogen, followed by exposing to air, diluting with THF, and passing through a neutral Al2O3 column to remove the copper. The obtained solution was concentrated and precipitated into cold methanol/water (7/3 v/v) mixture. The procedure of dissolution with THF and precipitation into methanol/water was repeated twice more. The dried product was obtained as a white solid.
Synthesis of NB-PS30-b-PtBAm-Br.
In a typical process for synthesis NB-PS30-b-PtBA15-Br, NB-PS30-Br (100 mg, 0.028 mmol, 1 eq.), Me6TREN (30 μL, 0.11 mmol, 4 eq.), tert-butyl acrylate (1.67 mL, 11.51 mmol, 400 eq.), and 3.34 mL methyl ethyl ketone (MEK) were added into a Schlenk flask. The mixture was degassed through three freeze–pump–thaw cycles, and CuBr (8 mg, 0.057 mmol, 2 eq.) was then added to start the polymerisation. After stirring at 70 °C for 60 min, the polymerisation was quenched by putting the flask into liquid nitrogen, followed by exposing to air, diluting with THF, and passing through a neutral Al2O3 column to remove the copper. The obtained solution was concentrated and precipitated into cold methanol/water (7/3 v/v) mixture. The procedure of dissolution with THF and precipitation into methanol/water was repeated twice more. The dried product was obtained as a white solid. For NB-PS30-b-PtBA30-Br and NB-PS30-b-PtBA40-Br, adjust the reaction time to 90 min and 120 min, respectively.
General procedure for azidation
The azidation of bromine end groups on NB-PS30-b-PtBAm-Br was carried out in DMF with sodium azide. In a typical azidation process, NB-PS30-b-PtBA15-Br (100 mg, 0.020 mmol, 1 eq.) was dissolved in 2 mL DMF and stirred at 50 °C. Sodium azide (7 mg, 0.12 mmol, 6 eq.) was added to the polymer solution to start the azidation. After running for 24 hours, the reaction was stopped and the DMF was removed under reduced pressure. The solid was dissolved into ethyl acetate and washed with deionised water for three times to remove the unreacted sodium azide. The products from the organic phases were collected and dried as NB-PS30-b-PtBA15-N3. The same reactant equivalent and procedure were adopted to prepare NB-PS30-b-PtBA30-N3 and NB-PS30-b-PtBA40-N3.
General procedure for cyclisation
The cyclic polymers were synthesised using CuAAC click reaction under high dilution conditions. In a typical process to prepare cyclic polymer C-NB-PS30-b-PtBA15, 2,2′-bipyridyl (93 mg, 0.59 mmol, 100 eq.) and CuBr (43 mg, 0.30 mmol, 50 eq.) were added to degassed DMF (80 mL) in a Schlenk flask stirred at 120 °C. NB-PS30-b-PtBA15-N3 (30 mg, 0.006 mmol, 1 eq.) dissolved in 8 mL of DMF was degassed through three freeze–pump–thaw cycles and subsequently slowly added into the catalyst solution using a syringe pump at the speed of 0.2 mL h−1. After finishing the addition of polymer solutions in the syringe, the reaction continued for another 24 hours. The reaction was stopped by exposing to air and cooling down to room temperature. The solution was concentrated under reduced pressure and redissolved into THF, followed by passing through a neutral Al2O3 column to remove the copper. The collected solution was concentrated and precipitated into cold methanol/water = 7/3 to yield off-white solids. The same reactant equivalent and procedure were adopted to prepare C-NB-PS30-b-PtBA30 and C-NB-PS30-b-PtBA40.
General procedure for amphiphilic cyclic polymers
The amphiphilic cyclic C-NB-PS30-b-PAAm were prepared from cyclic C-NB-PS30-b-PtBAm by deprotecting the tert-butyl groups. Typically, C-NB-PS30-b-PAA15 (30 mg, 0.006 mmol, 1 eq.) was dissolved in DCM (2 mL), followed by the addition of trifluoroacetic acid (TFA) (100 μL, 1.31 mmol). After stirring at room temperature overnight, polymers were observed to precipitate out from the solution. The DCM and TFA were removed under gentle nitrogen flow. To remove the remaining TFA, the polymer was dissolved in DMF and transferred to a dialysis bag (MWCO = 1000 Da). The dialysis was conducted against Milli-Q water for 10 hours by replacing the dialysis solvent every 3 hours. The polymer solution was freeze-dried and collected as an off-white sticky solid.
Preparation of polymer assemblies with looped and linear corona
To prepare homogeneous self-assembled nanoparticles (NPs), first, 5 mg C-NB-PS30-b-PAAm were dissolved in 100 μL of DMF and stirred vigorously for 24 hours. 4.9 mL of PBS buffer was subsequently added to the polymer solution through a syringe pump at 1 mL h−1 to prepare polymer assemblies. After stirring for 24 hours to stabilise, the solution was transferred to a dialysis bag (MWCO = 1000 Da). The dialysis was conducted against PBS buffer for 10 hours by replacing the dialysis solvent every 3 hours. The solution was finally transferred to a 21 mL vial and diluted to the desired concentration using PBS buffer. Such cyclic assemblies with looped PAA corona were then irradiated with UV light (365 nm, 12 W) for 5 min to open the ring structure and form assemblies with linear corona topology.
Preparation of Nile red (NR) loaded assemblies
In a typical process, 20 μL of 0.1 mg mL−1 NR solution in DCM was added to a 2 mL vial. The solvent was stirred and evaporated to form a thin film of NR. 1 mL of above prepared assembly solution (0.2 mg mL−1) was then added to the vial with NR. The solution was then stirred for 24 hours to incorporate NR molecules into the assemblies. To investigate and compare the encapsulation efficiency (EE) of polymer NPs with looped and linear corona, the NR loaded solutions were passed through a 0.45 μm syringe filter, freeze-dried, and redissolved in DMF. The amount of NR molecules encapsulated within polymer NPs was quantified using fluorometer as follows:
The following parameters were used: excitation wavelength = 540 nm, emission wavelength range = 560–750 nm, and data interval = 2 nm.
Interactions of polymer assemblies with model cell membrane
In this study, we chose DOPC prepared as supported lipid bilayers to serve as a model membrane system. To start, 8 μL of a 3 mM DOPC solution was added to each track via the circular sample addition port. After 2 minutes, the tracks were flushed with 100 μL of PBS. This flushing step was repeated twice more, with PBS buffer being added through the sample addition port and removed from the oval waste reservoir. Finally, the cartridge was inserted into the reader for electrochemical impedance spectroscopy (EIS) measurements. Upon stabilisation of their spectroscopy readings, 100 μL of a 0.2 mg mL−1 polymer NP solution in PBS buffer was then introduced into each track. Triplicate membrane conductance (Gm) profiles were obtained for all six polymer assembly solutions. By comparing the membrane conductance data before (Gm,b) and after (Gm,a) the addition of polymer assembly solutions, we could quantitively evaluate the interactions between the assemblies and membranes, as given by the equation:
Results and discussion
Polymer synthesis
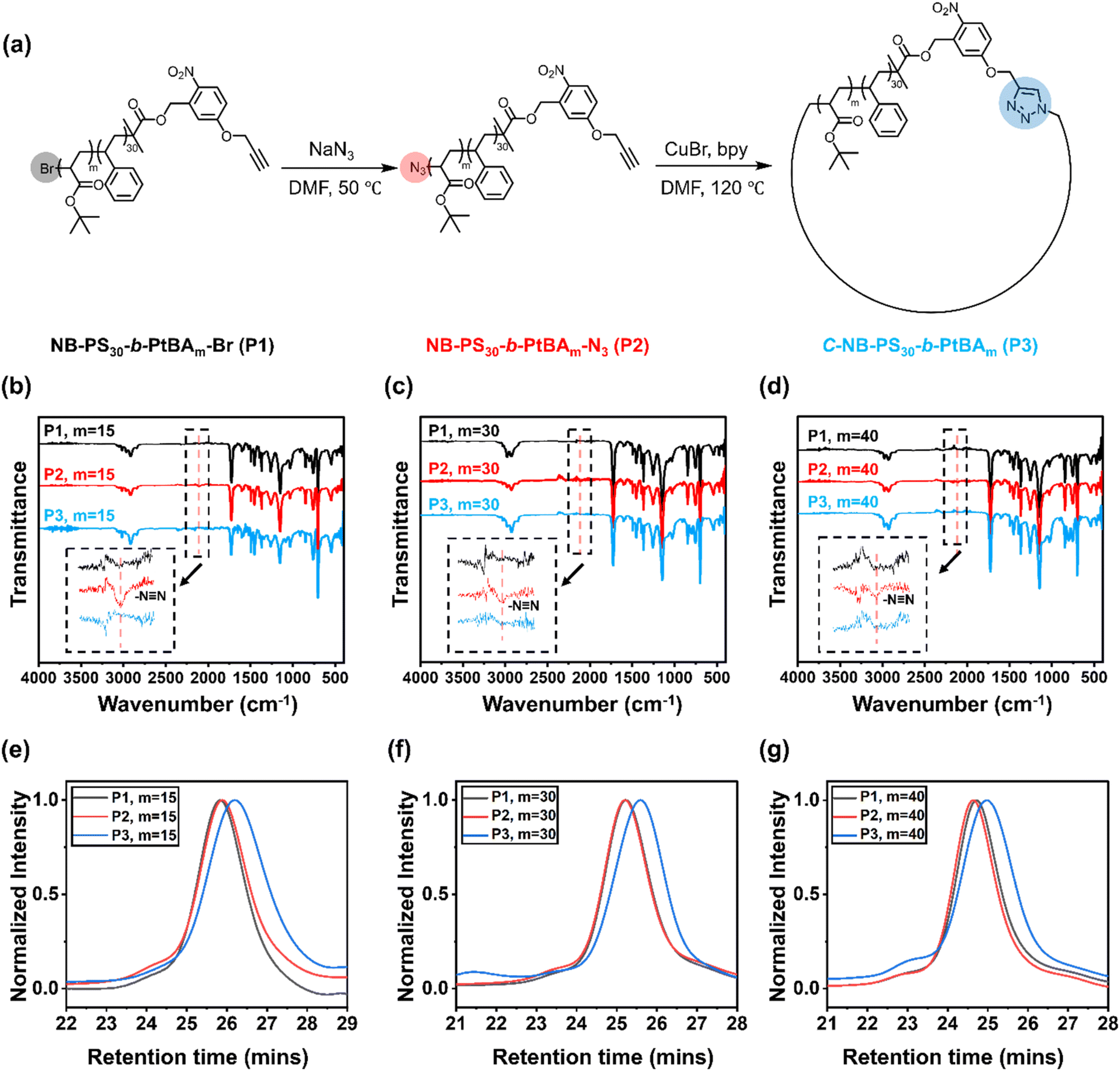
The UV-responsive cyclic polymers were synthesised in a facile procedure (Fig. 1a). First, o-NB containing ATRP initiator was prepared according to a previously reported method (Fig. S1 and S2†).16 The UV-responsive initiator was used to prepare a PS macroinitiator (Fig. S3†), NB-PS30-Br, which was subsequently chain-extended with tert-butyl acrylate. NB-PS30-b-PtBAm-Br with different block lengths (m = 15, 30, 40) were prepared (Fig. S4–S6†), as summarised in Table 1. The polymers were then reacted with sodium azide to convert the bromine end groups to azide groups. Successful azidation was verified by Fourier-transform infrared (FTIR) spectroscopy, where the appearance of a new signal at 2109 cm−1 corresponded to the azide functional groups (Fig. 1b–d).
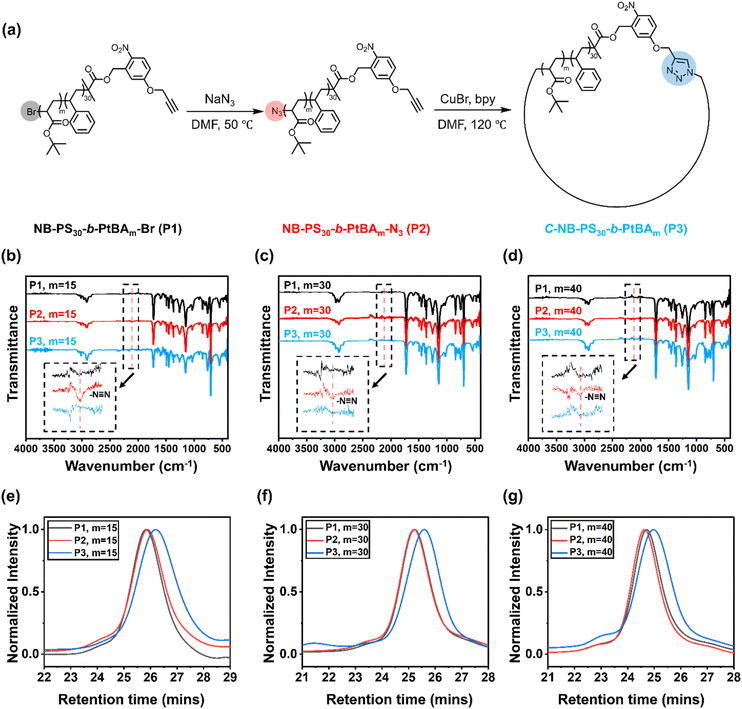 |
| | Fig. 1 (a) Schematic representation for the synthesis of C-NB-PS30-b-PtBAm cyclic polymers from NB-PS30-b-PtBAm-Br (m = 15, 30, and 40); (b)–(d) FTIR spectroscopy of NB-PS30-b-PtBAm-Br (P1), NB-PS30-b-PtBAm-N3 (P2, azidation products), C-NB-PS30-b-PtBAm (P3, cyclisation products) (m = 15, 30, and 40); (e)–(g) SEC elugrams obtained in THF (40 °C and 1 mL min−1) of NB-PS30-b-PtBAm-Br, NB-PS30-b-PtBAm-N3, C-NB-PS30-b-PtBAm (m = 15, 30, and 40). | |
Table 1 Reaction conditions, molecular characteristics, and degree of polymerisation of linear block copolymers synthesised using ATRP
| Samples |
[M]/[Initiator]/[CuBr]/[Ligand] |
Solvent |
v[M]/v[Solvent] |
Reaction time |
M
n,NMRa (g mol−1) |
M
n,SECb (g mol−1) |
Đ
|
|
Number-averaged molecular weight of polymers determined by 1H NMR spectroscopy using end group analysis.
Number-averaged molecular weight and dispersity of polymers determined by THF-SEC using PS standards.
|
| NB-PS30-Br |
200/1/1/2 |
Bulk |
— |
190 min |
3500 |
3100 |
1.07 |
| NB-PS30-b-PtBA15-Br |
400/1/2/4 |
MEK |
1/2 |
60 min |
5400 |
5600 |
1.14 |
| NB-PS30-b-PtBA30-Br |
400/1/2/4 |
MEK |
1/2 |
90 min |
7300 |
7200 |
1.13 |
| NB-PS30-b-PtBA40-Br |
400/1/2/4 |
MEK |
1/2 |
120 min |
8600 |
8900 |
1.14 |
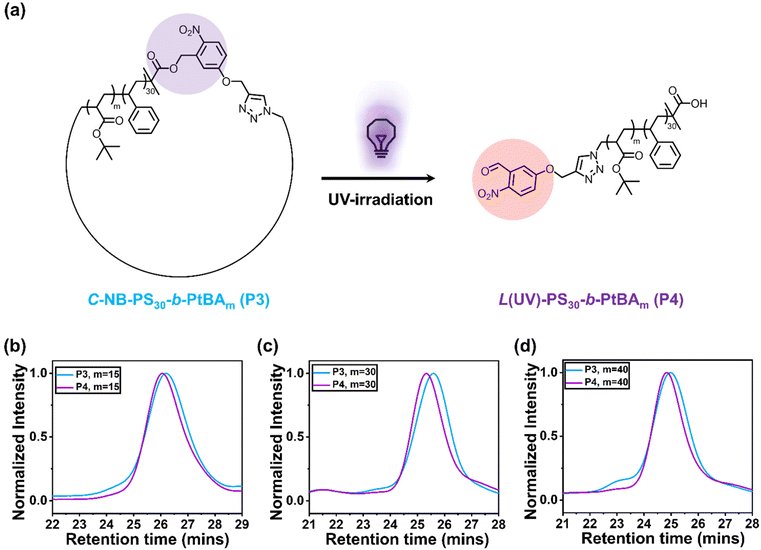
After preparing the α,ω-heterotelechelic block copolymers with alkyne and azide end groups, we used CuAAC click reactions under high dilution conditions to favour intramolecular ring-closure over step-growth to prepare cyclic block copolymers.34,35 No obvious degradation and unwanted ring-opening were observed during the synthesis (Fig. S7†). Successful cyclisation was inferred from FTIR (Fig. 1b–d) and SEC (Fig. 1e–g), whereby FTIR spectroscopy indicated the disappearance of the azide signal and SEC demonstrated an expected decrease in apparent molecular weight. Finally, we used trifluoroacetic acid (TFA) to deprotect the tert-butyl groups on the cyclic C-NB-PS-b-PtBA block copolymers to yield amphiphilic, cyclic C-NB-PS-b-PAA copolymers. Deprotection of PtBA was confirmed via1H NMR, which marked a clear disappearance of the proton environment around 1.43 ppm (Fig. S8–S10†). Noteworthy, when the polymer chain length reaches a certain threshold, a minor proportion of polymers engaged in intermolecular step-growth instead of cyclisation (m = 40). This reaction between terminal azide and alkyne functional groups led to the formation of dimers, contributing to a higher molecular weight shoulder (5% in integration ratio) in the SEC elugrams (Fig. 2d, P3). Upon subsequent UV-irradiation, such dimers could be effectively reverted to single polymer constituents (Fig. S11d†).
 |
| | Fig. 2 (a) Schematic representation of the photo-cleavage of the C-NB-PS30-b-PtBAm (m = 15, 30, and 40) cyclic polymers; SEC elugrams obtained from THF (40 °C and 1 mL min−1) of (b) C-NB-PS30-b-PtBA15, (c) C-NB-PS30-b-PtBA30, and (d) C-NB-PS30-b-PtBA40 before and after treated with UV light. | |
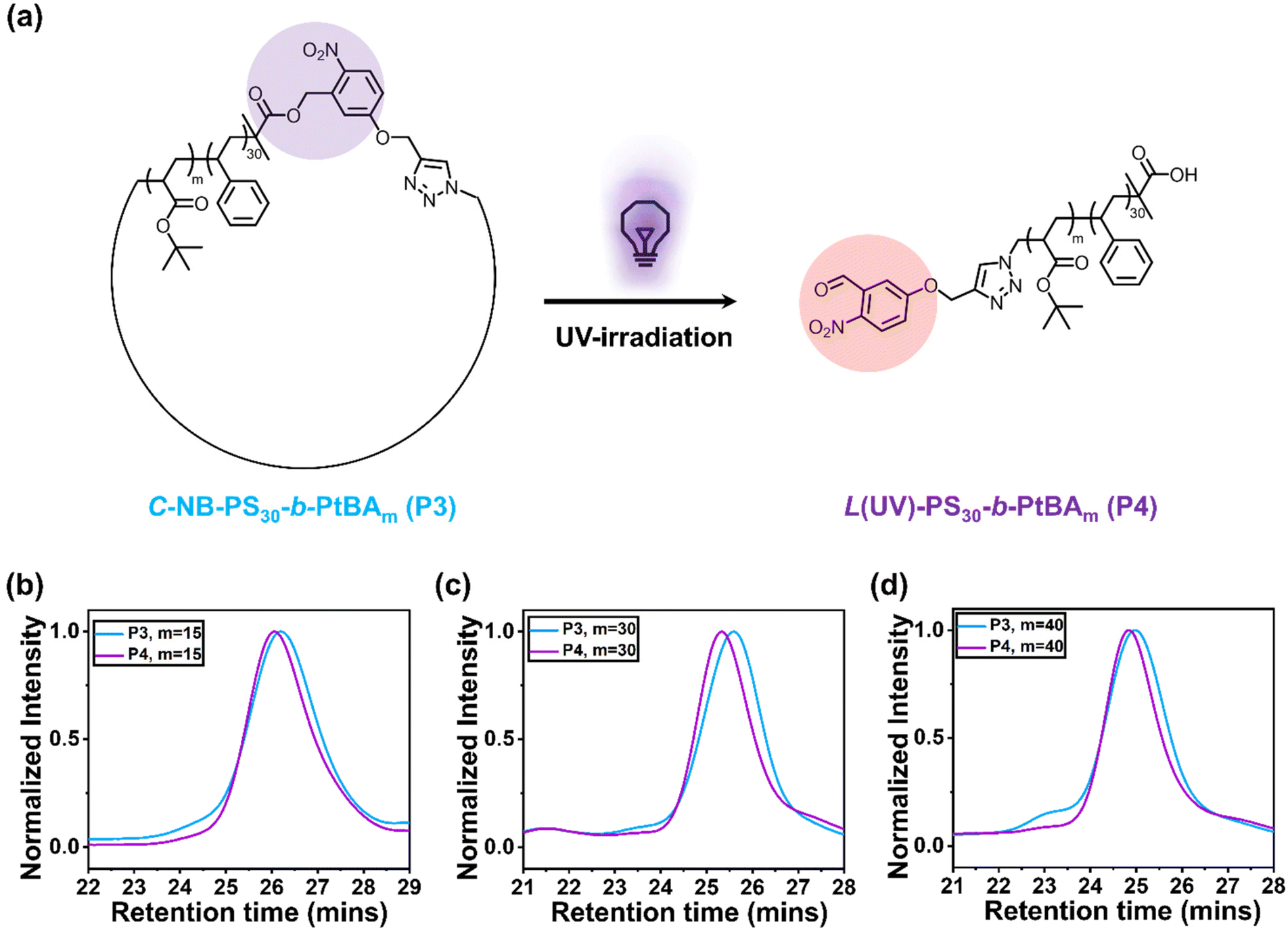
Photo-cleavage of cyclic polymers
To explore the photocleavage of cyclic polymers, we first dissolved C-NB-PS30-b-PtBAm in DCM. The polymer solution was subsequently irradiated with UV light for 5 min. After removing DCM under nitrogen flow, the UV-treated polymers were characterised using SEC (Fig. 2b–d). The apparent molecular weight of cyclic polymers increased after treated with UV light, indicating the successful ring-opening process. Interestingly, when compared to NB-PS30-b-PtBAm-N3, L(UV)-PS30-b-PtBAm exhibited a slightly smaller hydrodynamic volume (Fig. S11†). This phenomenon is likely attributed to the different end groups between P2 and P4, which caused different polymer-solvent interactions and hydrodynamic volume.28
Polymer assemblies with looped and linear corona
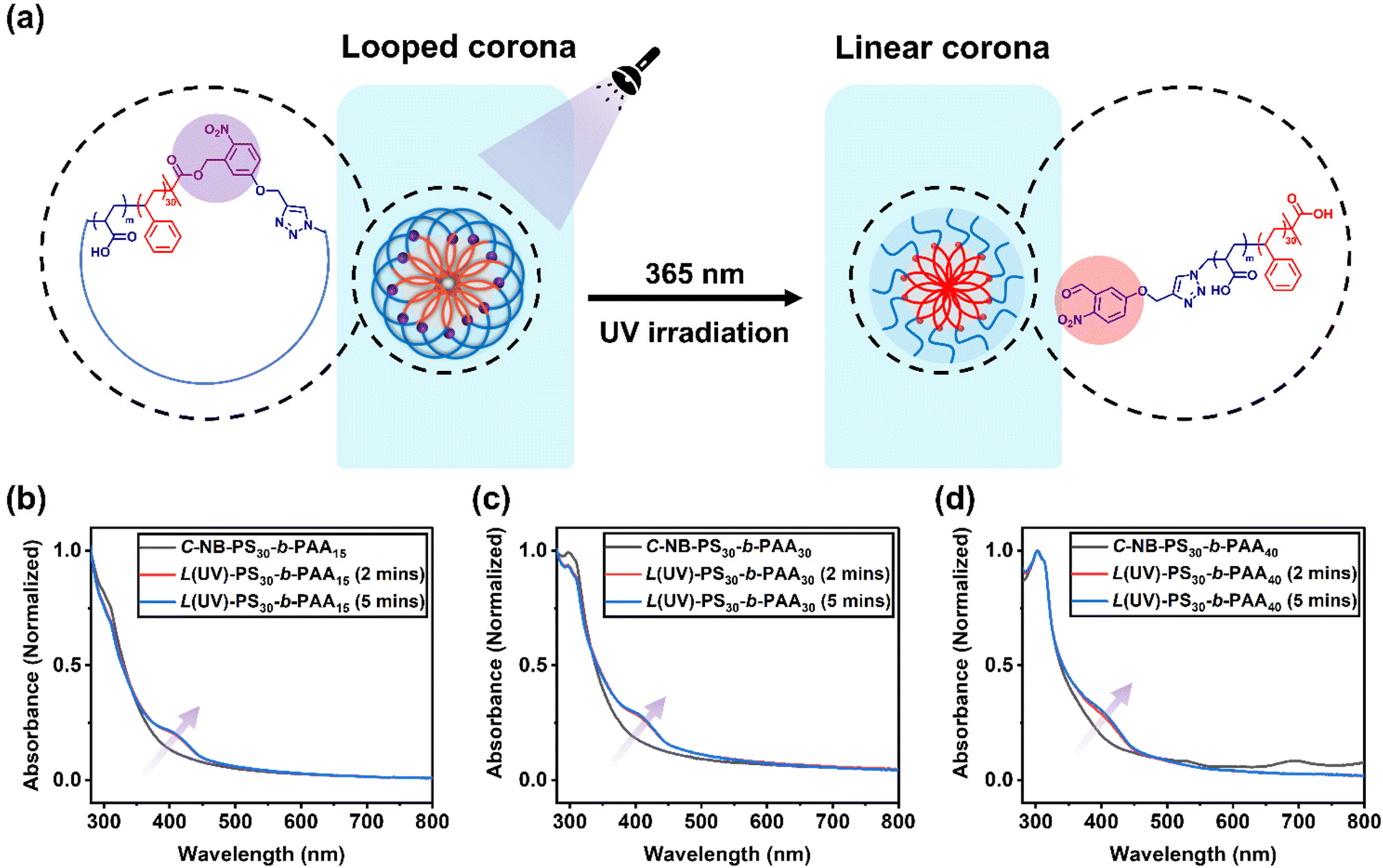
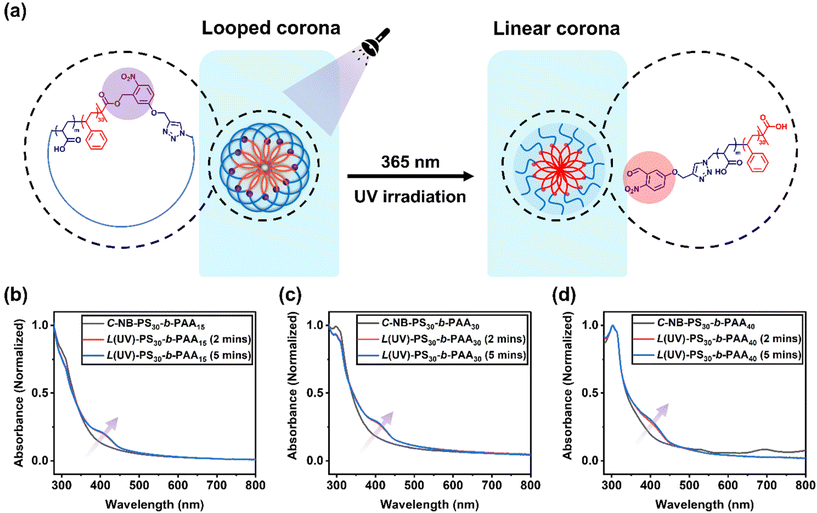
To assess the penetration of UV light through the aqueous environment and its effect on the architecture within C-NB-PS30-b-PAAm assemblies, UV-vis absorption spectroscopy of the irradiated polymer self-assembly solutions was measured at specific time points (Fig. 3). Notably, a new peak at 400 nm, corresponding to o-nitrosobenzaldehyde, emerged after 2 minutes of UV exposure, indicating the breakage of cyclic structure and successful conversion to linear architecture. However, extending the irradiation time revealed that the UV-vis absorption profile of the solution remained almost unchanged from 2 to 5 minutes, implying a rapid cyclic-to-linear transition within the initial 2 minutes.
 |
| | Fig. 3 (a) Schematic representation for the preparation of linear assemblies from cyclic assemblies through UV irradiation; UV-vis absorption spectroscopy of (b) C-NB-PS30-b-PAA15 assemblies, (c) C-NB-PS30-b-PAA30 assemblies, and (d) C-NB-PS30-b-PAA40 assemblies before and after treated with UV light (2 min and 5 min). | |
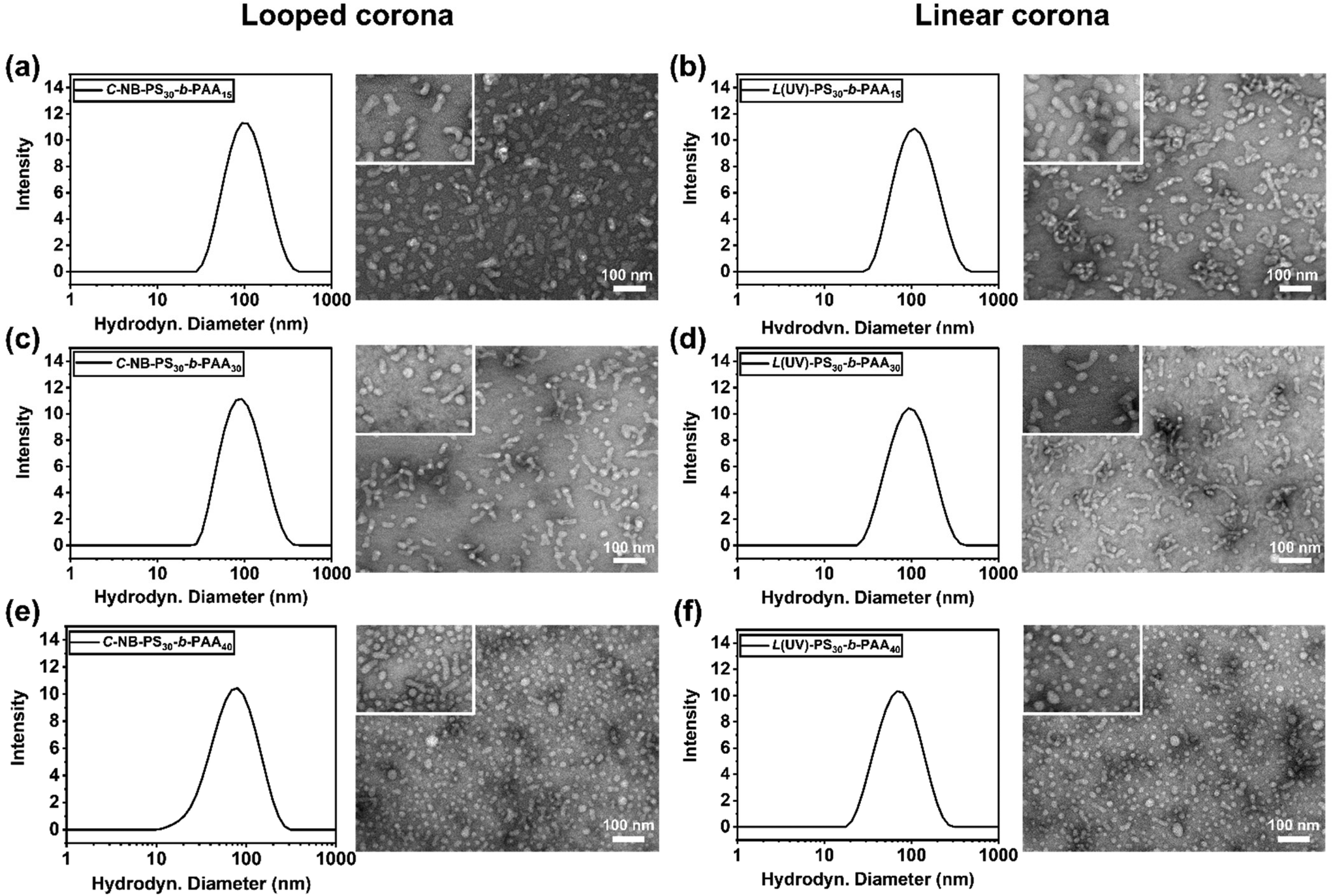
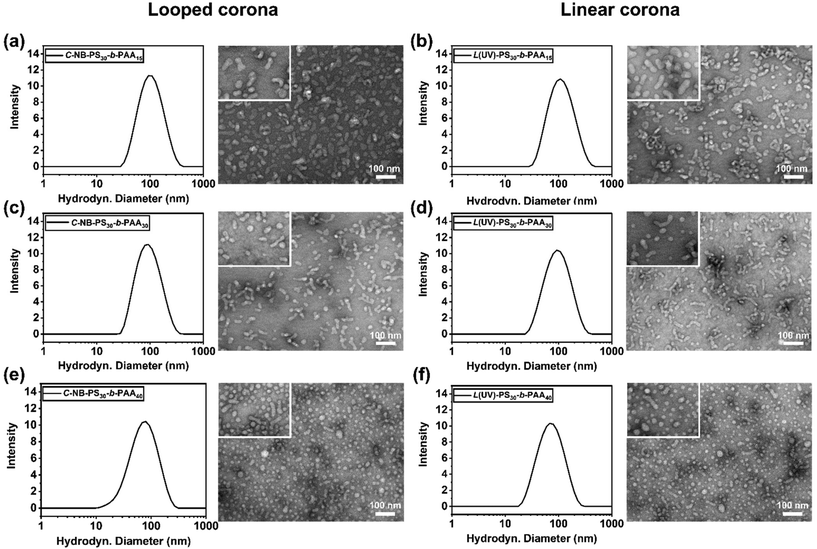
Subsequently, the impact of ring-opening the cyclic polymer on the morphology of polymer assemblies was investigated. The self-assembly behaviours of cyclic C-NB-PS30-b-PAAm in PBS buffer were analysed before and after UV irradiation using DLS and TEM. The sizes of the cyclic and UV-induced linear assemblies were measured on DLS to determine an average diameter (Fig. S12†), while TEM was used to observe the overall morphology of the assemblies (Table 2). By varying the length of PAA segments, polymer assemblies with different sizes and morphologies were prepared (Fig. 4). With increasing the chain length of PAA segments, we have observed the NPs evolve from rod-like micelles to spherical micelles. In all cases, DLS data affirmed that assemblies that had undergone UV-induced ring-opening had a slightly larger diameter than the cyclic assemblies. We attribute this primarily to the extension of PAA corona segments after the UV-cleavage process. Yet, no morphological change was observed after UV irradiation of the cyclic assembly sample from TEM. The results underscore that the UV-induced cyclic-to-linear topology transformation must have occurred within the polymer assemblies, without leading to morphological changes.
 |
| | Fig. 4 DLS measurements of the size distribution and overall morphologies of (a) C-NB-PS30-b-PAA15, (b) L(UV)-PS30-b-PAA15, (c) C-NB-PS30-b-PAA30, (d) L(UV)-PS30-b-PAA30, (e) C-NB-PS30-b-PAA40, (f) L(UV)-PS30-b-PAA40 assemblies in PBS with concentration at 0.2 mg mL−1. | |
Table 2 Characteristics of polymer assemblies with looped and linear coronas
| Samples |
Corona topology |
f
PAA (wt%) |
Diametera (nm) |
PDIa |
Shapeb |
|
Intensity weighted hydrodynamic diameter as determined by DLS. The results were expressed as means ± SD, n = 3.
Morphologies of self-assembled polymer nanoparticles as observed by TEM.
|
|
C-NB-PS30-b-PAA15 |
Loop |
25.7 |
92.3 ± 0.6 |
0.1878 ± 0.0092 |
Rod-like micelles |
|
L(UV)-PS30-b-PAA15 |
Linear |
25.7 |
99.4 ± 0.6 |
0.1949 ± 0.0087 |
Rod-like micelles |
|
C-NB-PS30-b-PAA30 |
Loop |
40.9 |
85.5 ± 0.9 |
0.2051 ± 0.0006 |
Rod-like micelles |
|
L(UV)-PS30-b-PAA30 |
Linear |
40.9 |
98.9 ± 0.2 |
0.2390 ± 0.0101 |
Rod-like micelles |
|
C-NB-PS30-b-PAA40 |
Loop |
48.0 |
64.6 ± 0.3 |
0.2516 ± 0.0032 |
Spherical micelles |
|
L(UV)-PS30-b-PAA40 |
Linear |
48.0 |
68.1 ± 0.6 |
0.2585 ± 0.0076 |
Spherical micelles |
Encapsulation efficiency of polymer assemblies with looped and linear corona
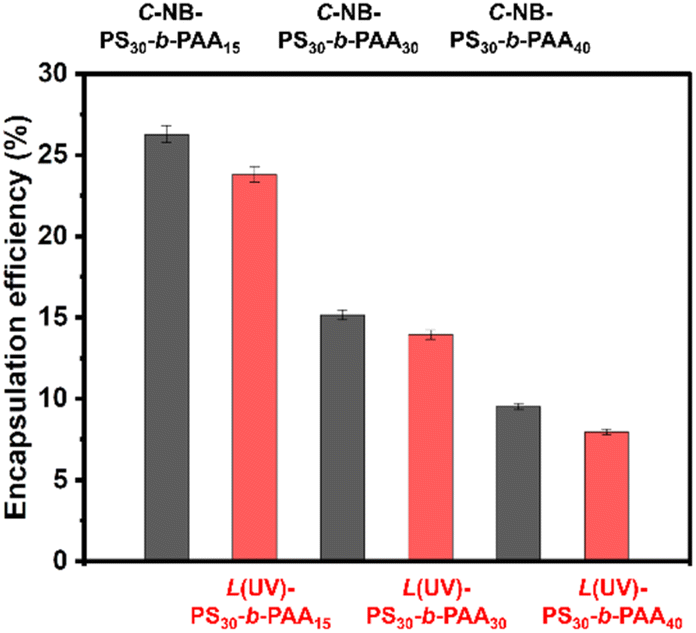
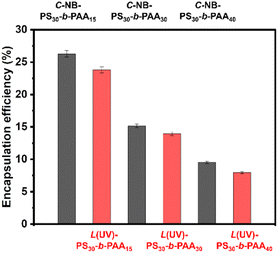
Polymer assemblies have gained substantial attention as drug carriers due to their capability to encapsulate hydrophobic drugs, enhancing their solubility and stability.36,37 Furthermore, they can also improve drug bioavailability and offer controlled or targeted release,38,39 which is indispensable for mitigating side effects and amplifying therapeutic efficacy. Therefore, it is important to study their drug encapsulation efficiency. In this study, we used Nile red (NR) as the model hydrophobic drug molecule. As shown in Fig. 5, the encapsulation efficiencies of NR in prepared polymer nanoparticles were ∼26.3% (C-NB-PS30-b-PAA15), ∼23.8% (L(UV)-PS30-b-PAA15), ∼15.1% (C-NB-PS30-b-PAA30), ∼13.9% (L(UV)-PS30-b-PAA30), ∼9.5% (C-NB-PS30-b-PAA40), and 7.9% (L(UV)-PS30-b-PAA40) respectively (Fig. 5 and Fig. S13†). The increasing ratio of PAA segments has enhanced the hydrophilicity of polymer NPs and, therefore, led to a decrease in their encapsulation efficiencies. Furthermore, NPs with looped corona have all shown slightly higher encapsulation efficiencies than their linear counterparts. We conclude the phenomenon is primarily due to the dense core structures such cyclic polymer NPs have, which could provide a more effective encapsulation environment for hydrophobic drug molecules, trapping them more efficiently within the polymer matrix.
 |
| | Fig. 5 Encapsulation efficiency of Nile red in 0.2 mg mL−1C-NB-PS30-b-PAA15, L(UV)-PS30-b-PAA15, C-NB-PS30-b-PAA30, L(UV)-PS30-b-PAA30, C-NB-PS30-b-PAA40, and L(UV)-PS30-b-PAA40 polymer NPs in PBS. The results were expressed as means ± SD, n = 3. | |
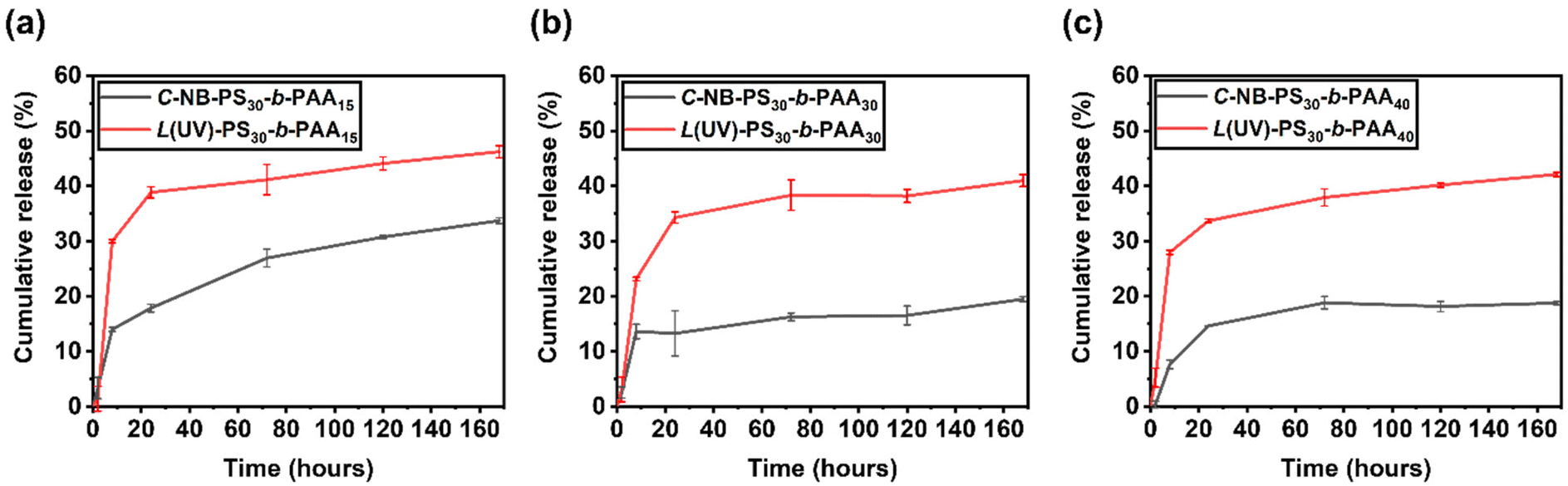
Drug release profile of polymer assemblies with looped and linear corona
To further study the drug release properties of polymer assemblies and assess their potentials as drug delivery systems, we recorded the release profile of encapsulated NR molecules by measuring the fluorescence of polymer assembly solutions at selected time points (Fig. S14–S16†), primarily focusing on assessing the total release of the encapsulated NR molecules. A stark contrast was observed between the release profiles of assemblies with linear and looped coronas (Fig. 6). Specifically, the linear polymer assemblies, L(UV)-PS30-b-PAAm, facilitated the release of approximately 40% of the encapsulated NR over the period of 7 days, whereas the cyclic assemblies, C-NB-PS30-b-PAAm, demonstrated a more conservative release, with about 20% of NR released in the same timeframe. This comparison underscores the significant impact of polymer topology on the drug release dynamics of self-assembled NPs. While end groups may also influence these dynamics, their role was not the primary focus of this study and requires further investigation in the future. The results predominantly suggest that the architecture of cyclic polymers contributes to a more regulated release pattern, indicating the importance of polymer structure in the development of effective drug delivery systems.
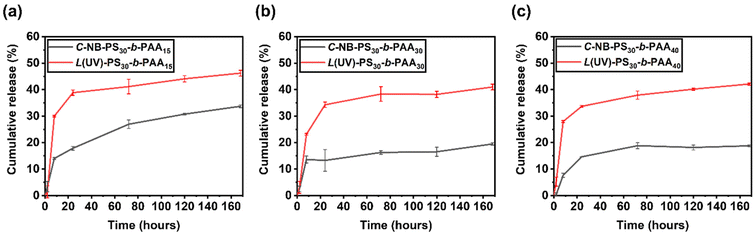 |
| | Fig. 6 Release profile of Nile red loaded (a) C-NB-PS30-b-PAA15 and L(UV)-PS30-b-PAA15; (b) C-NB-PS30-b-PAA30 and L(UV)-PS30-b-PAA30; (c) C-NB-PS30-b-PAA40 and L(UV)-PS30-b-PAA40 polymer nanoparticles in PBS buffer obtained from fluorescence spectroscopy. The results were expressed as means ± SD, n = 3. | |
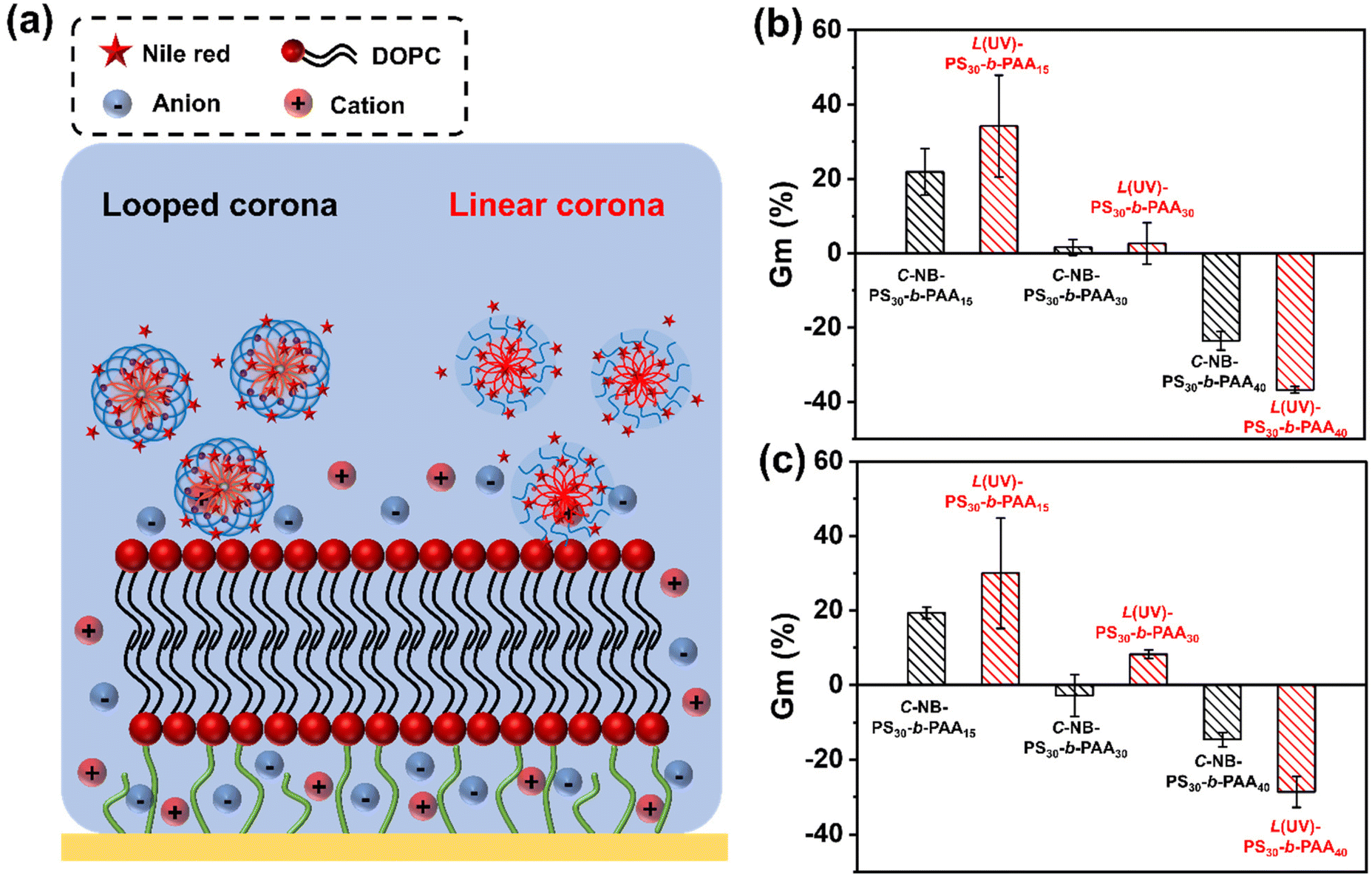
Interactions of polymer assemblies with model cell membrane
When using polymer assemblies as drug carriers, it is crucial to understand the interactions between these nanoparticles and phospholipid membranes, as such interactions are central to targeted delivery and nanotoxicology considerations.40–42 Despite their importance, the molecular interactions between nanoparticles and biomembranes are not well understood. Hence, investigating polymer assemblies with biomimetic membranes is not only of fundamental importance but also holds practical value for their downstream applications in drug delivery systems. Tethered bilayer lipid membrane (tBLM) represents a biomembrane mimetic system that closely mimics the natural biological membranes. These synthetic membranes consist of two lipid layers, where the inner layer is chemically anchored to a solid substrate, and the outer layer is free to interact with external entities. This configuration provides a stable and representative model of cell membranes, making tBLMs useful for studying the interactions between nanoparticles, including polymer assemblies, and lipid bilayers.43,44 Electrochemical impedance spectroscopy (EIS) can be used to measure changes in the electrical properties of a tBLM (i.e. resistance and conductance), which can serve as a insightful platform to determine the dynamics of interactions in the interfacial regions of materials and membranes. Therefore, we chose DOPC prepared in the form of supported lipid bilayers as a model membrane. By introducing our polymer assemblies to the system, the interactions between nanoparticles and membranes could be examined by measuring the impedance change using the Tethapod system from SDX Tethered Membranes, Australia (Fig. S17†).
As shown in Fig. 7, rod-like micelles, particularly PS30-b-PAA15 assemblies, have led to a noticeable increase in Gm value (Fig. S18†). This contrasted with particles with more spherical morphologies, such as PS30-b-PAA40 assemblies, which caused a notable decline in Gm value. The difference in these interactions can be largely attributed to the varied sizes and morphologies of the assemblies. For instance, the rod-like micelles have the capacity to infiltrate the bilayer membrane and induce defect formation, which consequently increases the conductance of the membrane. Similar attributes have been seen for other 1D polymers.45,46 Conversely, for more globular structures, the spherical hydrophobic cores of the nanoparticles acted as barriers on the membrane surface, obstructing ion flow and thereby reducing the conductance value.
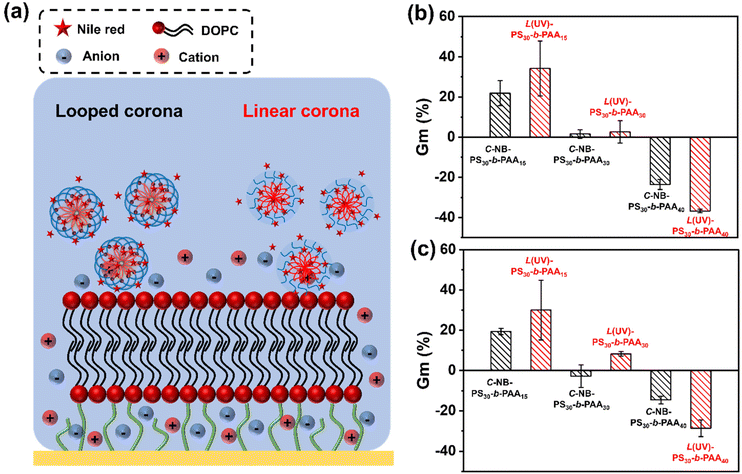 |
| | Fig. 7 (a) Schematic representation of measuring the interactions of cyclic and linear polymer assemblies with the DOPC membrane; (b) Gm% calculated from the Gm profiles of cyclic and linear polymer assemblies with DOPC membrane in PBS buffer; (c) Gm% calculated from the Gm profiles of Nile red loaded cyclic and linear polymer assemblies with DOPC membrane in PBS buffer. The results were expressed as means ± SD, n = 3. | |
A comparative analysis between looped and linear corona assemblies was subsequently conducted to disclose their respective interactions with the membrane (Fig. 7b). Intriguingly, the looped species have exhibited reduced interactions with the model membrane. Noteworthy, such interactions could be switched on on-demand through UV irradiation. Specifically, in the case of C-NB-PS30-b-PAA15, their linear counterparts displayed a 13% higher increase in Gm%, whereas, for C-NB-PS30-b-PAA30 and C-NB-PS30-b-PAA40, their corresponded linear assemblies showed a 1% higher increase and a 13% higher decrease in Gm% respectively. A similar trend was observed with NR-loaded assembly samples (Fig. 7c and Fig. S19†), where all linear corona samples displayed a significant increase in their Gm% values compared to the looped corona samples. Such results suggest that linear assemblies engage in stronger interactions with the membrane, which can be attributed to their corona topologies and the presence of end groups.47,48 An on-demand presence of end groups could allow nanoparticles to have more dynamic interactions with membrane lipids. Furthermore, the inherent flexibility of linear polymer chains offers greater conformational adaptability, enabling nanoparticles to conform more closely to the membrane surface, allowing larger contact areas and stronger interactions. This observation underscores the significant role of the corona topology of nanoparticles in modulating membrane interactions, which is pivotal for tailoring polymer assemblies for future drug delivery applications.
Conclusions
Our study has highlighted the impact of corona topology on the properties of self-assembled polymer nanoparticles. The synthesis of photo-cleavable cyclic block copolymers provided a robust post-assembly modification method to rapidly alter the surface properties of polymer assemblies via switching the corona conformation from looped to linear chains. Using NR as the cargo molecule, the cyclic assemblies (looped corona) showed higher encapsulation efficiencies but less cumulative release than UV-induced linear assemblies (linear corona), indicating their potential as sustained release drug carriers. Furthermore, the weaker interactions of cyclic assemblies with a DOPC model membrane may indicate a lower potential for adverse effects or toxicity when interacting with biological systems, making cyclic assemblies a potentially safer alternative for prolonged drug delivery applications. On the other hand, the study shed light on the distinctive behaviour of UV-induced linear assemblies. Specifically, such linear assemblies have demonstrated facilitated drug release profiles and more pronounced interactions with the DOPC membrane than their cyclic precursors. Overall, the discussed properties underscore the divergent behaviours induced by different macromolecular architectures and may better inform tye development of controlled drug release strategies.
Author contributions
H. Z. and M. M. conceived the idea, M. M. supervised the research, H. Z. and M. M. planned and developed the experimental setup. H. Z. performed all the experiments and analysed the data. H. Z. and M. M. discussed the results. H. Z. and M. M. co-wrote the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research was facilitated by access to Sydney Analytical, a core research facility at the University of Sydney. The authors acknowledge the technical and scientific assistance from Sydney Microscopy & Microanalysis, The University of Sydney node of Microscopy Australia. The authors thank Associate Professor Ronald Clarke for providing access to the Tethapod system from SDX Tethered Membranes and the Key Centre for Polymers and Colloids (KCPC) for access to equipment. H. Z. is a grateful recipient of Postgraduate Research Scholarship from the University of Sydney. M. M. acknowledges the Australian Research Council for a Future Fellowship (FT200100185) and Discovery Project (DP220100452), respectively. M. M. is a grateful recipient of a University of Sydney Research Accelerator (SOAR) Prize.
References
- A. Kumar, A. Srivastava, I. Y. Galaev and B. Mattiasson, Prog. Polym. Sci., 2007, 32, 1205–1237 CrossRef CAS.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- P. Theato, B. S. Sumerlin, R. K. O′reilly and T. H. Epp, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- X. Zhang, L. Chen, K. H. Lim, S. Gonuguntla, K. W. Lim, D. Pranantyo, W. P. Yong, W. J. T. Yam, Z. Low, W. J. Teo, H. P. Nien, Q. W. Loh and S. Soh, Adv. Mater., 2019, 31, 1804540 CrossRef PubMed.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61–75 CrossRef CAS.
- T. Takata, ACS Cent. Sci., 2020, 6, 129–143 CrossRef CAS PubMed.
- H. Liu, Z. Guo, W. Ma, S. Li, D. Wang, Z. Zheng, Y. Liu, C.-Y. Yu and H. Wei, ACS Macro Lett., 2023, 12, 1025–1030 CrossRef CAS PubMed.
- O. Bertrand and J.-F. Gohy, Polym. Chem., 2017, 8, 52–73 RSC.
- X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1322–1349 CrossRef CAS.
- P. G. Maschmeyer, X. Liang, A. Hung, O. Ahmadzai, A. L. Kenny, Y. C. Luong, T. N. Forder, H. Zeng, E. R. Gillies and D. A. Roberts, Polym. Chem., 2021, 12, 6824–6831 RSC.
- J. Van Damme and F. Du Prez, Prog. Polym. Sci., 2018, 82, 92–119 CrossRef CAS.
- Z. Hou, W. M. Nau and R. Hoogenboom, Polym. Chem., 2021, 12, 307–315 RSC.
- Q. Ye, M. Huo, M. Zeng, L. Liu, L. Peng, X. Wang and J. Yuan, Macromolecules, 2018, 51, 3308–3314 CrossRef CAS.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723–1736 CrossRef CAS.
- J.-M. Schumers, J.-F. Gohy and C.-A. Fustin, Polym. Chem., 2010, 1, 161–163 RSC.
- Y. Zhang, C. Y. Ang, M. Li, S. Y. Tan, Q. Qu, Z. Luo and Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 18179–18187 CrossRef CAS PubMed.
- H. Zeng, X. Liang, D. A. Roberts, E. R. Gillies and M. Müllner, Angew. Chem., Int. Ed., 2024, 63, e202318881 CrossRef CAS PubMed.
- J. Kim, K. Baek, D. Shetty, N. Selvapalam, G. Yun, N. Hoon Kim, Y. Ho Ko, K. Min Park, I. Hwang and K. Kim, Angew. Chem., Int. Ed., 2015, 54, 2693–2697 CrossRef CAS PubMed.
- B. Mo, H. Liu, X. Zhou and Y. Zhao, Polym. Chem., 2015, 6, 3489–3501 RSC.
- R. J. Williams, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2015, 6, 2998–3008 RSC.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
- R. Liénard, J. De Winter and O. Coulembier, J. Polym. Sci., 2020, 58, 1481–1502 CrossRef.
- C. Chen and T. Weil, Nanoscale Horiz., 2022, 7, 1121–1135 RSC.
- K. Hakobyan, C. S. P. McErlean and M. Müllner, Macromolecules, 2021, 54, 7732–7742 CrossRef CAS.
- R.-T. Gao, L. Xu, S.-Y. Li, N. Liu, Z. Chen and Z.-Q. Wu, Chem. – Eur. J., 2023, 29, e202300916 CrossRef CAS PubMed.
- T. Yamamoto and Y. Tezuka, Soft Matter, 2015, 11, 7458–7468 RSC.
- J. Kim, H. Y. Jung and M. J. Park, Macromolecules, 2020, 53, 746–763 CrossRef CAS.
- C. Chen, M. K. Singh, K. Wunderlich, S. Harvey, C. J. Whitfield, Z. Zhou, M. Wagner, K. Landfester, I. Lieberwirth, G. Fytas, K. Kremer, D. Mukherji, D. Y. W. Ng and T. Weil, Nat. Commun., 2021, 12, 3959 CrossRef CAS PubMed.
- G. Hadziioannou, P. M. Cotts, G. Ten Brinke, C. C. Han, P. Lutz, C. Strazielle, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 493–497 CrossRef CAS.
- T. E. Hogen-Esch, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2139–2155 CrossRef CAS.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed.
- D. M. Eugene and S. M. Grayson, Macromolecules, 2008, 41, 5082–5084 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. del P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- W. Xu, P. Ling and T. Zhang, J. Drug Delivery, 2013, 2013, 340315 Search PubMed.
- P. Abasian, S. Ghanavati, S. Rahebi, S. Nouri Khorasani and S. Khalili, Polym. Adv. Technol., 2020, 31, 2939–2954 CrossRef CAS.
- P. Abasian, S. Shakibi, M. S. Maniati, S. Nouri Khorasani and S. Khalili, Polym. Adv. Technol., 2021, 32, 931–944 CrossRef CAS.
- C. Contini, M. Schneemilch, S. Gaisford and N. Quirke, J. Exp. Nanosci., 2018, 13, 62–81 CrossRef CAS.
- E. Rascol, J. M. Devoisselle and J. Chopineau, Nanoscale, 2016, 8, 4780–4798 RSC.
- A. G. Rodríguez-Hernández, R. Vazquez-Duhalt and A. Huerta-Saquero, Curr. Med. Chem., 2018, 27, 3330–3345 CrossRef PubMed.
- S. Rebaud, O. Maniti and A. P. Girard-Egrot, Biochimie, 2014, 107, 135–142 CrossRef CAS PubMed.
- M. Gavutis, E. Schulze-Niemand, H. H. Lee, B. Liedberg, M. Stein and R. Valiokas, Nanoscale, 2023, 15, 9759–9774 RSC.
- M. Müllner, Chem. Commun., 2022, 58, 5683–5716 RSC.
- M. Müllner, K. Yang, A. Kaur and E. J. New, Polym. Chem., 2018, 9, 3461–3465 RSC.
- M. Schulz, A. Olubummo and W. H. Binder, Soft Matter, 2012, 8, 4849–4864 RSC.
- B. Golba, E. M. Benetti and B. G. De Geest, Biomaterials, 2021, 267, 120468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ATRP initiator synthesis and supporting results, including polymer characterisation (NMR and SEC), drug release studies (fluorescence spectroscopy), and nanoparticle-membrane studies (experiment setup and EIS). The authors have cited additional references within the supporting information. See DOI: https://doi.org/10.1039/d4py00155a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Markus
Müllner
a and
Markus
Müllner