
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInverse vulcanization employing epoxy compounds as crosslinking agents for elemental sulfur in the preparation of sulfur-rich epoxy resins†
Yue-Sheng
Lai
and
Ying-Ling
Liu
 *
*
Department of Chemical Engineering, National Tsing Hua University, Hsinchu 300044, Taiwan. E-mail: liuyl@mx.nthu.edu.tw; Fax: +886-3-5715408; Tel: +886-3-5711450
First published on 26th March 2024
Abstract
Inverse vulcanization is an effective approach for the utilization of waste sulfur as a feedstock in the preparation of sulfur-rich polymers. The organic compounds employed in inverse vulcanization play a key role in the properties of the sulfur-rich polymers. In this work, epoxy compounds are demonstrated to be effective reagents for inverse vulcanization and synthesis of sulfur-rich epoxy resins. The epoxy-sulfur inverse vulcanization is performed through the formation of thiol-containing polysulfanes via hydrogen abstraction by sulfur radicals and thiol-epoxide addition reactions. Hence, the highest sulfur contents of the resulting sulfur-epoxy resins are majorly dependent on epoxy values (epoxy equivalent weights) of reagents and minorly affected by the miscibility between molten sulfur and epoxy reagents. Aliphatic glycidyl ethers exhibit relatively higher reaction rates than aromatic analogues. The wide scope of epoxy compounds contributes to the high flexibility of the molecular designs of sulfur-rich epoxy polymers. Consequently, the properties of sulfur-epoxy polymers could be tailored conveniently and feasibly. Within 4,4′-methylene bis(N,N-diglycidylaniline) (MDGA) as the reagent, the obtained sulfur-rich epoxy resin (containing 55 wt% sulfur) demonstrates a storage modulus of about 2000 MPa at 50 °C and a glass transition temperature (Tg) above 197 °C, a record-high Tg for sulfur polymers from inverse vulcanization. This work brings about significant progress in the chemistry of inverse vulcanization and preparation of sulfur-rich polymers.
Introduction
Inverse vulcanization involves a reaction between elemental sulfur and organic compounds (crosslinking agents) to provide an innovative pathway for preparing sulfur-rich polymers.1 The utilization of waste sulfur as the feedstock in inverse vulcanization addresses “green” issues for chemistry and materials. The obtained sulfur-rich polymers have demonstrated wide applications in many fields and targets.2 The first crosslinking agent used in inverse vulcanization was 1,3-diisopropenylbenzne (DIB).1 Later studies demonstrated other suitable crosslinking agents, including styrenic compounds,3–8 cycloalkenes,9–13 biomass-based vegetable oils,14–18 and various unsaturated molecules.5,19–27 Based on the proposed mechanism, crosslinking reactions in inverse vulcanization systems are mainly performed between unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and elemental sulfur. All the above-mentioned crosslinking agents possess unsaturated CC bonds to be involved in inverse vulcanization reactions. Moreover, other functional groups, like benzoxazine, have also been employed as crosslinking agents in inverse vulcanization, to integrate inverse vulcanization and conventional crosslinking reactions in one system.25,26 Different crosslinking agents might alter the properties of the corresponding sulfur-rich polymers, making them suitable for different fields.10,14,27–33
C bonds and elemental sulfur. All the above-mentioned crosslinking agents possess unsaturated CC bonds to be involved in inverse vulcanization reactions. Moreover, other functional groups, like benzoxazine, have also been employed as crosslinking agents in inverse vulcanization, to integrate inverse vulcanization and conventional crosslinking reactions in one system.25,26 Different crosslinking agents might alter the properties of the corresponding sulfur-rich polymers, making them suitable for different fields.10,14,27–33
The initially proposed reaction mechanism of inverse vulcanization consisted of the formation of sulfur radicals through a ring-opening reaction of S8, addition of the sulfur radicals to the CC groups of the crosslinking agents, and a coupling reaction between carbon and the sulfur radicals.1 The reactions resulted in bis-sulfurated units of each CC group.1 Nevertheless, the formation of hydrogen sulfide (H2S) in inverse vulcanization processes was observed in some cases.2,26,34–36 The H2S byproduct was formed from the sequential hydrogen-abstraction reactions of the sulfur radicals and thiol groups. Based on these results, studies by Pyun et al. reported that linear bis-thiocumyl units were the major microstructures of the product of sulfur-DIB inverse vulcanization, and the hydrogen-abstraction reaction played a crucial role in inverse vulcanization reaction pathways (Fig. 1).37 In addition to the addition reaction between the sulfur radicals and CC bonds, the hydrogen-abstraction reaction of the sulfur radicals converted the sulfur radicals to thiol-containing polysulfanes. Subsequent reactions of thiol-containing polysulfanes resulted in bis-thiocumyl structures. The hydrogen-abstraction reactions in the inverse vulcanization process suggested the feasibility of employing crosslinking agents without CC groups in inverse vulcanization processes. Our previous work employed benzoxazine groups as crosslinking sites to react with elemental sulfur in inverse vulcanization.38 Hydrogen abstraction by the sulfur radicals performed at the –CH2– groups of benzoxazine rings resulted in carbon radicals and –SH groups. With 1,3,5-triisopropylbenzene (TIPB) as an example,39 our recent studies further extended the crosslinking agents for inverse vulcanization to saturated compounds. In the reaction between TIPB and elemental sulfur, hydrogen abstraction took place between the sulfur radicals and isopropyl groups. The reaction is similar to the radical transfer reaction from the sulfur radicals to isopropyl groups with the formation of the carbon radicals. Subsequent reactions included radical coupling reactions and an H2S formation reaction between –SH and isopropyl groups.39
 | ||
| Fig. 1 Chemical reactions and structures in inverse vulcanization proposed in 20131 and amended in 2023.37 | ||
The wide scope of crosslinking agents for inverse vulcanization is highly attractive for preparing and tailoring the properties of the corresponding sulfur polymers. The integration of conventional crosslinked polymers and the sulfur polymers from inverse vulcanization is attractive, since conventional thermosetting polymers usually have good thermal and mechanical properties. One example was the utilization of benzoxazine-containing allyl compounds as crosslinking agents for inverse vulcanization.25,26 Considering that epoxy resins have wide applications, combining epoxy resins and sulfur-polymers in one system is worthy of study. A pioneering example was reported40 before the concept of inverse vulcanization was revealed. An allyl-containing epoxy compound was reacted with sulfur, where sulfur was utilized as a crosslinking agent for epoxy (in a relatively small fraction).40 Hydrogen abstraction between the sulfur radicals and allyl groups generated the –SH groups at polysulfide ends. The following addition reaction between –SH and epoxy groups contributed to the crosslinking of epoxy compounds. Further studies on sulfur-epoxy reactions were conducted with trinary systems composed of elemental sulfur, an allyl compound, and a bifunctional epoxy compound.41,42 Later studies on trinary systems involved 2-step reactions. In the first step elemental sulfur was reacted with an allyl compound to result in a sulfur-rich polymer. The second step employed the thiol-containing sulfur-rich polymer as a curing agent for epoxy compounds through an –SH/epoxy addition reaction.43–45 Epoxy resins with high sulfur content were prepared in the process. Moreover, Yan et al.46 utilized an –OH-containing vinyl compound as the crosslinking agent in an inverse vulcanization system. With the addition of an epoxy compound, sulfur-vinyl and hydroxyl-epoxy reactions were performed independently in the crosslinking reactions. Jin and coworkers47 reported a similar study employing carboxylic acid as a crosslinking group for epoxy compounds. Conventional crosslinking reactions and inverse vulcanization chemistry were integrated in one system.
Although epoxy groups were involved in inverse vulcanization reactions in previous studies,40–47 a reaction between elemental sulfur and epoxy groups was not observed or discussed. Lian et al.42 reported that mixtures of elemental sulfur and epoxy compound showed an exothermic peak at temperatures above 230 °C in differential scanning calorimetry (DSC) measurements. This exothermic behavior was correlated with the ring-opening anionic polymerization of epoxy groups initiated by sulfur anions from heterolytic S8 rings at a high temperature. Hence, with the continuing interest in studying new crosslinking agents for elemental sulfur in inverse vulcanization,38,39 this work reports the first example in the literature of the direct utilization of epoxy compounds as crosslinking agents for elemental sulfur. The scientific basis concerns the hydrogen-abstraction reaction of sulfur radicals in inverse vulcanization generating a carbon radical at the crosslinking agent. Hence, a compound possessing chemical structures tending to form stable carbon radicals could be reactive toward sulfur in inverse vulcanization. This issue has been demonstrated with previously reported crosslinking agents, which have a stabilization effect on the carbon radicals with π-bond resonance (agents with CC bonds) and neighboring electron-denoting atoms (benzoxazine group38).
Accordingly, epoxy compounds possessing glycidyl ether units were examined in this work to serve as crosslinking agents in inverse vulcanization. The α-methylene (–CH2–) units of ether linkages are reactive sites for hydrogen abstraction by the sulfur radicals, as the corresponding carbon radicals are stabilized by the neighboring electro-donating O atom. In inverse vulcanization systems employing epoxy compounds as crosslinking agents, the –SH groups formed with the hydrogen-abstraction reaction would react with epoxy groups in the following crosslinking reaction. Moreover, glycidyl amine compounds (amine-based epoxy) are more effective crosslinking agents for inverse vulcanization as amine groups serve as activators for the sulfur ring-opening reaction. This provides an efficient approach for the preparation of epoxy resins with high sulfur content. The prepared sulfur-epoxy resins showed a wide range of thermal and mechanical properties based on the large family of epoxy compounds.
Results and discussion
To illustrate the feasibility of employing epoxy compounds as crosslinking agents for inverse vulcanization, diglycidyl ether of bisphenol A (DGEBA) was first utilized as the reactant (Fig. 2). DGEBA and sulfur in equal weight fractions (w/w: 1/1) were reacted at 180 °C in a molten manner without solvents or catalysts. DGEBA gradually dissolved in the molten sulfur, similar to the reaction between TIPB and sulfur.39 The reaction system became vitrified after 12 h of reaction. At this stage, the vitrified sample still possessed some unreacted epoxide groups and crystalline sulfur. A post-reaction process was conducted at 140 °C to enable the high conversion of the reaction. Moreover, the reaction system might exhibit an abrupt expansion while being post-reacted at 180 °C due to an auto-acceleration effect. The obtained polymer product is coded as poly(S-DGEBA-50), where the number 50 denotes the weight fraction of sulfur in the feed composition (Fig. S1†). For a reaction system with 10 wt% sulfur, the reaction mixture became homogenous soon after the reaction was performed, due to the molten sulfur being dissolved in liquid DGEBA. Vitrification was not observed with this reaction system even after 24 h of reaction at 180 °C. Poly(S-DGEBA-10) is fully soluble in tetrahydrofuran (THF), acetone, and chloroform. Nevertheless, poly(S-DGEBA-50) has gel fractions of 48 wt% in THF, 68 wt% in acetone, and 51 wt% in chloroform. These results indicated the high extent of the reaction between DGEBA and sulfur (Fig. S1†). The number-averaged molecular weight and polydispersity index of the soluble fraction of poly(S-DGEBA-50) in THF, being measured by gel permeation chromatography (GPC), are 2230 Da and 1.28, respectively. Since DGEBA did not show any features of reaction under these reaction conditions (180 °C, 24 h),42 the obtained product is formed from the reaction between DGEBA and elemental sulfur. Moreover, the mixture of DGEBA and S8 heated at 150 °C (below the temperature of 159 °C for the S8 ring-opening reaction) did not demonstrate any reaction or changes in appearance (Fig. S1†). The results indicate that the ring-opening reaction of S8 and generation of the sulfur radicals are critical steps for the inverse vulcanization of DGEBA and elemental sulfur. | ||
| Fig. 2 Epoxy compounds used as crosslinking agents in inverse vulcanization and basic data for the corresponding sulfur polymers. EEW: epoxy equivalent weight. | ||
The mixture of elemental sulfur and DGEBA (w/w 1/1) was reacted at 180 °C. Samples taken at reaction times of 0.5, 1, and 2 h underwent proton nuclear magnetic resonance (1H NMR) instrumental measurement using deuterated chloroform (CDCl3) as a solvent (Fig. S2†). The sample taken at 0.5 h of reaction exhibited an 1H NMR spectrum similar to that recorded for pure DGEBA. After 2 h of reaction, the resonance peaks of epoxide groups disappeared, indicating that the epoxide groups were involved in the reactions between S8 and DGEBA. The changes in the chemical structure of DGEBA after the reactions were also observed with the appearance of some additional resonance peaks. The relatively low intensity of the resonance peaks associated with reaction products was attributed to the poor solubility of the reaction products in CDCl3. Hence, phenyl glycidyl ether (PGE) was utilized as a model compound to react with elemental sulfur, as their reaction product was fully soluble in CDCl3. The model reaction was conducted with a mixture of S8/PGE (w/w 1/1) at 180 °C for 24 h. The collected product (S-PGE-50) was dissolved in CDCl3 for NMR measurement (Fig. 3). The resonance peaks of the cyclic oxirane group (δ = 2.7 ppm, δ = 2.9 ppm, and δ = 3.3 ppm) appeared in the 1H NMR spectrum of PGE and were not observed in the spectrum of S-PGE-50. The results indicated that the epoxide groups were involved in the reactions between S8 and PGE with high conversion. Moreover, S-PGE-50 exhibited some new peaks associated with sulfur-linked and ring-opened epoxide structures at δ = 3.1–3.3 ppm (–S–C![[H with combining low line]](https://www.rsc.org/images/entities/b_i_char_0048_0332.gif) 2–) and δ = 3.4–4.5 ppm (–O–C2–C(OH)–).48–52 The addition of sulfur segments to the PGE structure also resulted in shifts of the peaks of aromatic protons. More information from 13C NMR measurements provided supporting results, in which the peak at δ = 43 ppm was assigned to the secondary carbon of –CH2–S groups formed in the reaction between polysulfanes and epoxy groups. The –CH2–S groups still demonstrated a secondary carbon signal in the 13C-DEPT (distortionless enhancement by polarization transfer) spectrum (Fig. S3†) and Ha′–CA′ coupling in the HSQC (heteronuclear single quantum coherence) spectrum (Fig. S4†). The oxygen-linked secondary (–O–CH2–) and tertiary (–CH(OH)–) carbons, formed in the thiol-addition/ring-opening reaction of oxirane groups, contributed to signals at δ = 60–80 ppm. As the hydrogen-abstraction reaction of sulfur radicals with glycidyl ether groups (formation of thiol groups) could be the first step for S8/PGE reactions, a radical coupling reaction between the carbon and sulfur radicals might still occur in the reactions. Although all the C–H bonds of the glycidyl groups of PGE could be reactive sites for the hydrogen-abstraction reaction, the methylene units, –O–
2–) and δ = 3.4–4.5 ppm (–O–C2–C(OH)–).48–52 The addition of sulfur segments to the PGE structure also resulted in shifts of the peaks of aromatic protons. More information from 13C NMR measurements provided supporting results, in which the peak at δ = 43 ppm was assigned to the secondary carbon of –CH2–S groups formed in the reaction between polysulfanes and epoxy groups. The –CH2–S groups still demonstrated a secondary carbon signal in the 13C-DEPT (distortionless enhancement by polarization transfer) spectrum (Fig. S3†) and Ha′–CA′ coupling in the HSQC (heteronuclear single quantum coherence) spectrum (Fig. S4†). The oxygen-linked secondary (–O–CH2–) and tertiary (–CH(OH)–) carbons, formed in the thiol-addition/ring-opening reaction of oxirane groups, contributed to signals at δ = 60–80 ppm. As the hydrogen-abstraction reaction of sulfur radicals with glycidyl ether groups (formation of thiol groups) could be the first step for S8/PGE reactions, a radical coupling reaction between the carbon and sulfur radicals might still occur in the reactions. Although all the C–H bonds of the glycidyl groups of PGE could be reactive sites for the hydrogen-abstraction reaction, the methylene units, –O–![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/b_char_0032_0332.gif) —oxirane, could be possible sites for the hydrogen-abstraction reaction due to the formed carbon radicals being stabilized by the neighboring phenyl–oxygen group. Moreover, although radical-induced epoxide ring-opening polymerization could be chemically feasible, it could be excluded from the discussion about inverse vulcanization systems because of the presence of a sulfur-rich composition for radical capture and transfer.53,54
—oxirane, could be possible sites for the hydrogen-abstraction reaction due to the formed carbon radicals being stabilized by the neighboring phenyl–oxygen group. Moreover, although radical-induced epoxide ring-opening polymerization could be chemically feasible, it could be excluded from the discussion about inverse vulcanization systems because of the presence of a sulfur-rich composition for radical capture and transfer.53,54
 | ||
| Fig. 3 1H NMR (a and b) and 13C NMR (c) spectra recorded on tracing the model reaction of phenyl glycidyl ether (PGE) and elemental sulfur. | ||
The formation of the –SH groups from hydrogen abstraction by the sulfur radicals was further examined with the model reaction between S8 and butyl phenyl ether (BPE, without an epoxide group) in a weight ratio of 9/1. The reaction product (S-BPE) was characterized with 1H NMR (Fig. S5†). In addition to the resonance peaks of BPE, the additional resonance peak at δ = 1.6 ppm is noteworthy. This peak is assigned to the thiol groups of polysulfanes,34 supporting the formation of the –SH groups from the hydrogen-abstraction reaction. Hydrogen abstraction taking place at α-methylene of butyl groups was supported by the decreased intensity of the α-methylene signal (δ = 3.9 ppm) and new resonance peak of –CH–Sx– at δ = 4.7 ppm. The addition of a sulfur segment to BPE also resulted in new signals at about δ = 6.8 ppm associated with the aromatic protons affected by the sulfur segments. Moreover, water in the reaction system might react with elemental sulfur to form –SH groups55 contributing to the following thiol-epoxide addition reaction. To study the effect of water in this work, the reaction between S8 and 4,4′-methylene bis(N,N-diglycidylaniline) (MDGA, w/w 1/1) under common and dried (pretreated at 100 °C under vacuum for 2 h) conditions were conducted. The common and dried reaction systems gave vitrification times (the time at which the reaction system lost fluidity) of 9.4 min and 8.6 min, respectively. Because the pretreatment for drying the reactants (removal of water) did not alter the reaction rate, the presence of a trace amount of water in the reaction system did not significantly affect the inverse vulcanization between elemental sulfur and epoxide compounds. As the –SH/epoxy addition is a stepwise reaction, the small amount of the –SH groups possibly formed with trace water did not play a significant role in the reaction system.
Based on previous studies and spectral characterization of model reactions in this work, the reactions between elemental sulfur and epoxy compounds are proposed to be (Fig. 4):
(a) Sulfur radicals generated from the ring-opening reaction of elemental sulfur above the floor temperature of S8,
(b) Hydrogen-abstraction reaction of the sulfur radicals with epoxy molecules to form thiol-terminated polysulfanes and the carbon radicals (radical transfer),
(c) Thiol-epoxide addition reactions between the epoxide groups and terminal thiol groups of polysulfanes,
(d) Radical coupling reactions between the carbon and sulfur radicals.
 | ||
| Fig. 4 Reaction scheme between epoxy compounds and elemental sulfur. | ||
Although the proposed mechanism is similar to the reactions reported for sulfur/allyl-epoxy systems,24,40 the critical issue newly reported in this work concerns the lack of CC groups, which are essential units for conventional inverse vulcanization systems. Epoxy compounds themselves could act as effective crosslinking agents for elemental sulfur in inverse vulcanization systems, providing an effective approach for the preparation of sulfur-rich epoxy materials.
Fig. 5 collects some instrumental characterization results for poly(S-DGEBA-50). In the electron paramagnetic resonance (EPR) spectrum, the signal at g = 2.0054 (ΔBpp = 8.1 Gauss) was assigned to carbon-centered radicals generated by the hydrogen-abstraction reaction by the sulfur radicals on the epoxy compounds. The sulfur-centered radicals were not observed in measurements, similar to results reported for conventional inverse vulcanization.34,39,56 In the Fourier transform infrared spectroscopic (FTIR) analysis, the reactions between S8 and DGEBA resulted in the disappearance of the asymmetric C–O–C stretching bands of epoxy groups at 915 cm−1 and appearance of broad O–H stretching bands at 3430 cm−1, indicating the occurrence of the ring-opening reaction of the epoxy groups. Poly(S-DGEBA-50) exhibited obvious S signals in the wide-scan X-ray photoelectron spectrum (XPS). The formation of C–S linkages was supported by signals at 285.3 eV and 165.2 eV in C1s and S2p core-level spectra, respectively. XPS characterization supports sulfur being involved in the ring-opening reaction of the epoxide groups with the formation of C–S linkages through the proposed mechanism discussed above. In X-ray diffraction (XRD) spectra, elemental sulfur exhibited a diffraction pattern with many sharp peaks associated with its crystalline structure. Only a broad diffraction peak was observed with poly(S-DGEBA-50) associated with its amorphous structure. The lack of crystalline diffraction peaks observed for poly(S-DGEBA-50) suggested that there was no unreacted or crystalline sulfur remaining in the sample (Fig. 6). The prepared poly(S-DGEBA-50) material is a sulfur-epoxy polymer rather than a physical mixture of the 2 reaction components. The sulfur content of poly(S-DGEBA-50) determined by an elemental analysis method is about 44.5 wt% (Table S1†). The S weight fraction of poly(S-DGEBA-50) is somewhat lower than the expected value from the sulfur (50 wt%) loaded into the reaction system. The result could be attributed to the evolved H2S byproduct with the hydrogen abstraction of thiol groups.35,57 The evolved H2S byproduct still brought out some hydrogen atoms from the reaction system, resulting in a decreased H/C atomic ratio (1.09 found with poly(S-DGEBA-50) compared to the theoretical value of 1.14 for DGEBA). Catalytic inverse vulcanization with lower reaction temperature was reported to reduce the formation of H2S byproduct,57 which could be interesting for further studies.
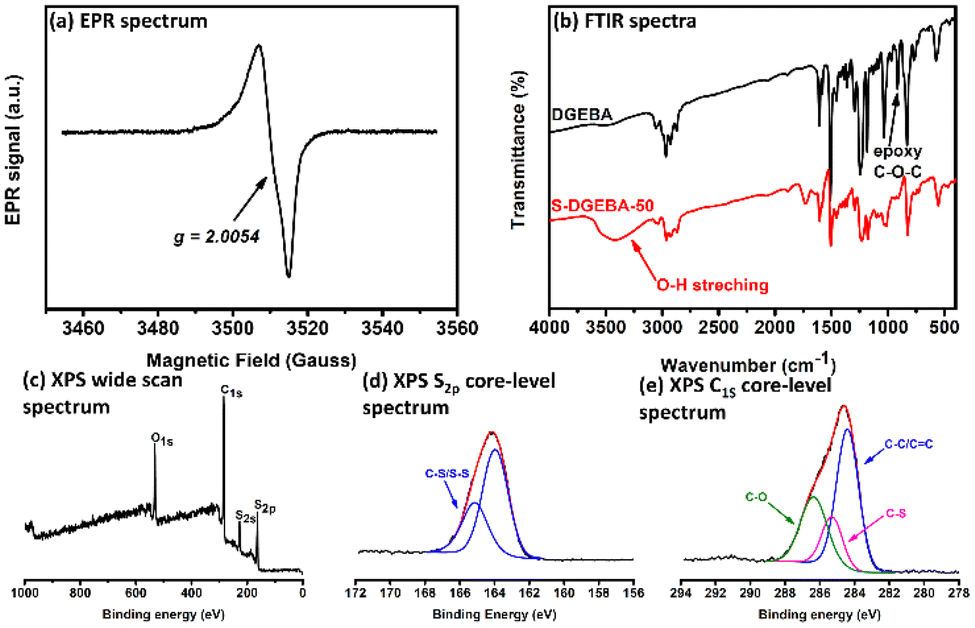 | ||
| Fig. 5 Characterization of poly(S-DGEBA-50): (a) EPR spectrum, (b) FTIR spectra, (c) XPS wide-scan spectrum, (d) XPS S2p core-level spectrum, and (e) XPS C1s core-level spectrum. | ||
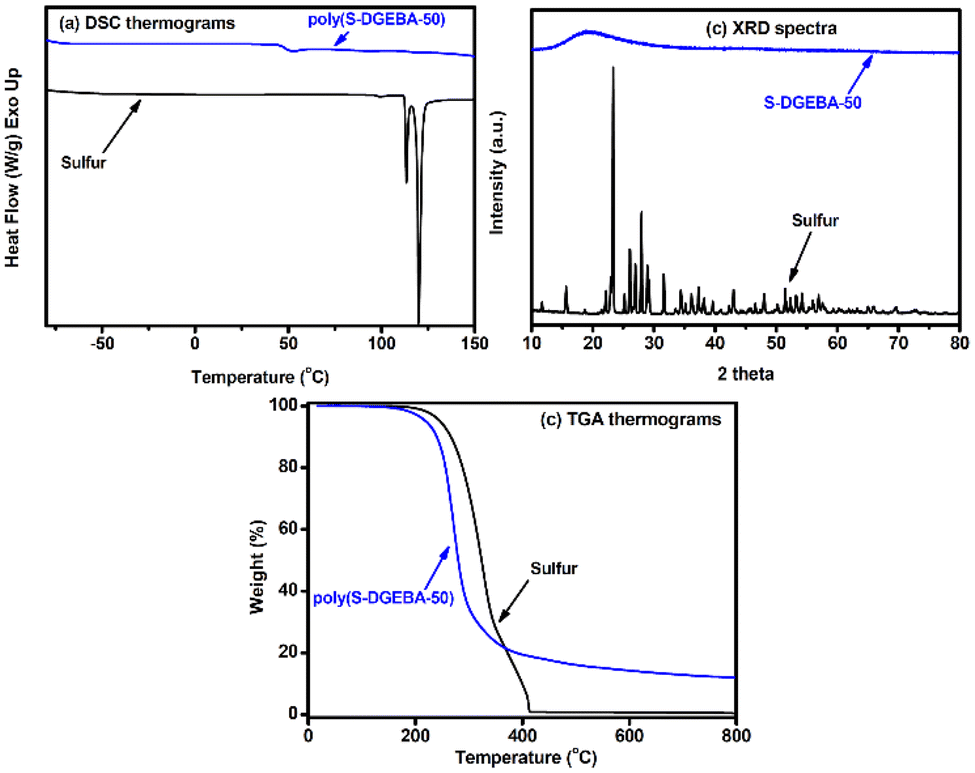 | ||
| Fig. 6 (a) DSC, (b) XRD, and (c) TGA measurements on poly(S-DGEBA-50). | ||
Compared to the melting behavior of crystalline sulfur in the heating scan of differential scanning calorimetric (DSC) measurement, poly(S-DGEBA-50) did not show similar endothermic behavior in its DSC thermogram to support it being free of crystalline sulfur (Fig. 6). The temperature (about 47 °C) of the baseline shift in the thermogram is assigned as the glass transition temperature (Tg) of poly(S-DGEBA-50), which is higher than the Tg (28 °C) reported for poly(S-DIB-50)1 (the sulfur polymer from inverse vulcanization with 50 wt% DIB as a reagent). The relatively high Tg of poly(S-DGEBA-50) could be attributed to the rigid structure of bisphenol A units and hydrogen bonding between –OH groups. The crystalline sulfur-free feature of poly(S-DGEBA-50) was further verified with the disappearance of the crystalline diffraction peaks of poly(S-DGEBA-50) in the XRD measurement. In thermogravimetric analysis (TGA), elemental sulfur and DGEBA exhibited rapid weight losses at about 300 °C and did not show obvious residuals at temperatures above 500 °C in an N2 atmosphere. Poly(S-DGEBA-50) exhibited the onset of degradation and rapid weight loss at about 245 °C and 275 °C, respectively. It is noteworthy that a residual of about 13 wt% was observed with poly(S-DGEBA-50) at 800 °C, which suggests that sulfur might be involved in the degradation reactions of poly(S-DGEBA-50) to form some thermally stable sulfur-containing carbon char. This fact is similar to what has been reported for sulfur-containing flame retardants for polymers.58 Moreover, time-resolved rheological analysis on poly(S-DGEBA-50) was conducted.8,39 The storage modulus of poly(S-DGEBA-50) was first determined at 130 °C by the application of a low-strain amplitude (1%, frequency: 100 rad s−1). Mechanical damage was applied to the sample with it enduring a high strain amplitude (100%) for 300 s through inducing S–S bond scission. The storage modulus of the sample decreased accordingly. After being annealed at 100 °C for 300 s (without oscillation), the recovered storage modulus was recorded and attributed to dynamic S–S debonding and reformation. Poly(S-DGEBA-50) showed a significant self-healing feature in tests for 3 cycles. The dynamic characteristics of S–S bonds provide recycling feasibility to poly(S-DGEBA-50). Hence, poly(S-DGEBA-50) was ground into powder and further thermally pressed at 180 °C and 10 MPa for 1 h. The powder was recycled and reprocessed into a dense bulk sample, demonstrating the recyclability and reprocessing feature of poly(S-DGEBA-50) (Fig. S6†).
The inverse vulcanization of S8/DGEBA with high sulfur content was also conducted. Nevertheless, all the obtained samples (poly(S-DGEBA-X), X > 60) showed obvious endothermic peaks at 116 °C in DSC measurements (Fig. S7†), indicating that the samples possessed unreacted crystalline sulfur. The upper limit of sulfur content (Sul) of poly(S-DGEBA) is about 50–60 wt%, which is much smaller than the sulfur content of 90 wt% reported for poly(S-DIB)1 and other sulfur polymers from inverse vulcanization.3 The limited S content might be correlated with the concentration of the epoxide groups of the crosslinking agents, as the epoxide groups are involved in the reactions through the addition reaction toward the –SH groups. Hence, epoxy agents with relatively small EEW values (data shown in Fig. 2) might provide more epoxide groups in inverse vulcanization to result in sulfur polymers with high Sul values. Conversely, molten sulfur and DGEBA are initially immiscible with each other. The reaction product became soluble in DGEBA. This fact limits the reaction rate between sulfur and DGEBA and Sul values for sulfur/DGEBA reaction systems.
Sulfur/epoxy inverse vulcanization has been investigated by employing various epoxy compounds as crosslinking agents (Fig. 2). The crystalline sulfur-free products containing the highest sulfur contents for each epoxy reagent were subjected to thermal and mechanical tests. For the 3 aromatic epoxy compounds (DGEBA, N,N-diglycidyl-4-glycidyloxyaniline (DGGA), and MDGA), the Sul values were reasonably increased with decreases in the EEW values of the epoxy reagents. DGGA has the lowest EEW value of 92 g mol−1 epoxide among the 3 reagents; consequently, the poly(S-DGGA-60) polymer has the highest sulfur content (60 wt%) among the 3 corresponding sulfur polymers. The relationship between the EEW values of the epoxy reagents and sulfur contents of the corresponding sulfur polymers is still valid for the 4 aliphatic epoxy compounds. Moreover, the aromatic epoxy compounds show relatively high reaction conversion in inverse vulcanization, which might be attributed to the resonance-stabilization effect on carbon radicals. Moreover, DGGA and MDGA have relatively high reactivity to sulfur, needing a relatively short reaction time to reach a monophasic mixture and vitrification. The tertiary amine could be considered as an activator to bring about a catalytic effect on inverse vulcanization.22 More investigation could be of interest in future studies. All the sulfur polymers have similar temperatures at 5% weight loss (Td5) in TGA measurements (Fig. S8†). Td5 values might be somewhat dependent on aromatic/aliphatic structures and the sulfur content. Similar to poly(S-DGEBA-50), the other 2 aromatic epoxy-based sulfur polymers (poly(S-DGGA-60) and poly(S-MDGA-55)) showed a certain amount of residual char at 800 °C in the TGA measurements. A high sulfur content results in a lower amount of residual char. In contrast, all 4 sulfur polymers from aliphatic epoxy compounds had residual char of about 20 wt% at 400 °C. Nevertheless, thermal degradation of the residual char took place at higher temperatures for poly(S-TGETMP-50) and poly(S-PGEDGE-35). The second stage of thermal degradation is attributed to the oxidation of the residual char, which is widely observed for polymer degradation in an air environment. As the TGA measurements were performed in a nitrogen atmosphere, the oxidation degradation could be due to the high oxygen content of the 2 reagents (TGETMP and PEGDGE). It is noteworthy that although DGEG also has a relatively high oxygen content, the corresponding poly(S-DGEG-50) sample did not exhibit weight loss from oxidation degradation. The reason for this is the dehydration reaction of the –OH groups of DGEG to remove oxygen in a relatively low-temperature region. The mechanical properties of the sulfur polymers from tensile tests are also included in Fig. 2 and Fig. S9.† The sulfur polymers from aromatic epoxy agents are brittle within very small strain at break. Poly(S-MDGA-55) showed a tensile strength and elongation at break of 4.95 MPa and 2.2%, respectively. On the contrary, the sulfur polymers from aliphatic epoxy reagents showed high elongation at break and low tensile strength (data shown in Fig. 2). For example, poly(S-DGENG-40) showed a tensile strength and elongation at break of 0.11 MPa and 36.6%, respectively. The highest strain of 59% was found with poly(S-PGEDGE-35), which exhibited a low tensile strength of 0.01 MPa. The wide scope of epoxy compounds as crosslinking agents for inverse vulcanization provides convenience and flexibility for adjusting the properties of the resulting sulfur polymers.
Poly(S-MDGA-55), which has the highest mechanical property among the prepared samples, is suitable for dynamic mechanical analysis (DMA). As shown in Fig. 7, poly(S-MDGA-55) had a storage modulus of about 2000 MPa at 50 °C, which is comparable to the values reported for typical crosslinked epoxy resins.59 The Tg of poly(S-MDGA-55) read from tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ could be above 197 °C. The measurement was interrupted at about 200 °C due to approaching the thermal degradation temperature of the sample. It is noteworthy that the Tg of poly(S-MDGA-55) is higher than the values of Tg for DGEBA-based epoxy resins59 and other sulfur polymers from inverse vulcanization.1,2,23,38,39,60 The thermal and mechanical properties of aromatic epoxy-based sulfur polymers from inverse vulcanization are attractive for extending their scope of application.
δ could be above 197 °C. The measurement was interrupted at about 200 °C due to approaching the thermal degradation temperature of the sample. It is noteworthy that the Tg of poly(S-MDGA-55) is higher than the values of Tg for DGEBA-based epoxy resins59 and other sulfur polymers from inverse vulcanization.1,2,23,38,39,60 The thermal and mechanical properties of aromatic epoxy-based sulfur polymers from inverse vulcanization are attractive for extending their scope of application.
 | ||
| Fig. 7 DMA thermograms recorded on poly(–MDGA-55). | ||
Experimental
Materials
Sulfur (S8, sublimed powder, 99.5%, Alfa Aesar), bisphenol A diglycidyl ether (DGEBA, EEW: 172–176 g mol−1, Sigma-Aldrich), N,N-diglycidyl-4-glycidyloxyaniline (DGGA, EEW: 92 g mol−1, Sigma-Aldrich), 4,4′-methylenebis(N,N-diglycidylaniline) (MDGA, EEW: 106 g mol−1, Sigma-Aldrich), diglycidyl ether of glycerol (DGEG, EEW: 106 g mol−1, Sigma-Aldrich), diglycidyl ether of neopentyl glycol (DGENG, EEW 135–165 g mol−1, Sigma-Aldrich), triglycidyl ether of trimethylolpropane (TGETMP, EEW 138–154 g mol−1, Sigma-Aldrich), and poly(ethylene glycol) diglycidyl ether (PEGDGE, EEW about 250 g mol−1, Sigma-Aldrich) were used as received for the preparation of sulfur-rich epoxy resins. Phenyl glycidyl ether (PGE, 99%, Sigma-Aldrich) and butyl phenyl ether (BPE, 99%, Sigma-Aldrich) were used as received for model reactions. Chloroform (CHCl3, 99.8%, Macron Fine Chemicals), tetrahydrofuran (THF, HPLC grade, TEDIA), and acetone (99.5%, Macron Fine Chemicals) were used as solvents in the experimental work. Deuterated chloroform (CDCl3, 0.05% v/v tetramethylsilane, atom D %: 99.8%, Sigma-Aldrich) was employed as a solvent for nuclear magnetic resonance spectroscopic measurements.Instrumental methods
A Fourier transform infrared (FTIR) spectrometer equipped with an attenuated total reflectance (ATR) accessory from PerkinElmer Instrument Co. (FTIR Spectrum Two) was applied for FTIR measurement. Nuclear magnetic resonance (NMR) measurements, including 1H NMR spectra, 13C NMR spectra, heteronuclear single quantum coherence (HSQC) spectra and distortionless enhancement by polarization transfer (DEPT) spectra, were conducted with a Brüker Avance 500 MHz NMR spectrometer. Molecular weights of sulfur polymers were measured with a gel permeation chromatography (GPC) system equipped with a Waters 1515 isocratic pump, Waters Styragel HR4 series columns, and a Waters 2414 differential refractometer. The elution was performed with THF as a mobile phase at a flow rate of 1 mL min−1. The molar weights of the measured samples were calculated using Waters Breeze 2 software and calibrated against low polydispersity linear polystyrene standards. Differential scanning calorimetry (DSC) thermograms were recorded with a TA-Q20 instrument (Thermal Analysis Co.) in a temperature range from −90 to 200 °C and at a temperature ramp rate of 10 °C min−1. Thermogravimetric analysis (TGA) was performed using a TA-Q50 instrument under a nitrogen or air atmosphere from 50 to 800 °C at a ramp rate of 10 °C min−1. Dynamic mechanical analysis (DMA) was carried out on a TA-Q800 DMA instrument within a three-point bending mode. The measurements were conducted at a heating rate of 3 °C min−1 with an applied force of 0.1 N, an amplitude of 5 μm, and a frequency of 1 Hz. X-ray diffraction (XRD) spectra were collected using a Brüker D8 ADVANCE (Cu Kα radiation of 1.5418 Å, 40 kV, 25 mA) in the range of 10 to 80° (2θ) scale. X-ray photoelectron spectroscopy (XPS) spectra were recorded on an ULVAC-PHI PHI 5000 Versaprobe III (monochromatized Al Kα radiation), calibrated on the S2p3/2 peak at a binding energy of 164.0 eV. Elemental analysis was performed using an Elementar UNICUBE. Rheological characterizations were collected using an Anton Paar MCR 302 instrument. The mechanical property was measured using an Instron 3343 universal testing system with a 10 mm min−1 crosshead speed and a 50 N load cell. Electron paramagnetic resonance (EPR) experiments were conducted on a continuous-wave X-band EPR spectrometer (Brüker Elexsys E580) equipped with a variable temperature controller. The EPR experiments were performed at 298 K with a microwave frequency of 9.85 GHz, microwave power of 299.3 μW, a modulation amplitude of 0.16 mT, and a modulation frequency of 100 kHz.Preparation of sulfur-rich epoxy resins
The method for the preparation of poly(S-DGEBA-50) is illustrated as an example. Sulfur (S8) and DGEBA (w/w 1/1) were charged into a sample vial with a magnetic stir bar. The mixture was heated to 180 °C. Phase separation occurred between molten polymeric sulfur and DGEBA. After about 12 h of reaction, the reaction mixture turned from phase separation to a homogeneous phase. Then, vitrification behavior was observed. The reaction system was cooled to 140 °C and later held isothermally for 12 h. The product (poly(S-DGEBA-50)) was obtained. Sulfur polymers of S8 and DGEBA containing different sulfur contents were prepared in the same manner.All other sulfur-rich epoxy resins were prepared by similar methods, except S8/DGGA and S8/MDGA reactions were carried out at 170 °C.
Samples for mechanical tests were obtained by a different route. Sulfur and epoxy compound were added into a sample vial and reacted at 180 °C (or 170 °C) for 45 min to afford a black viscous liquid. The liquid was poured into a silicone mold, thermally treated at 180 °C (or 170 °C) for 30 min and later at 140 °C for 23 h. The obtained specimen was applied for mechanical measurements.
Conclusions
Taking advantage of inverse vulcanization, which utilizes waste sulfur as a feedstock for the synthesis of sulfur polymers, this work extends the chemistry of inverse vulcanization using epoxy compounds as effective reagents. The reactions between elemental sulfur and epoxy compounds start with hydrogen abstraction by sulfur radicals on methylene groups to result in carbon radicals at the epoxy compounds and polysulfanes with thiol groups. A thiol-epoxide addition reaction builds up chemical linkages between sulfur segments and epoxy compounds to result in corresponding crosslinked structures. The big family of epoxy compounds warrants the high flexibility of molecular designs and tailoring of the properties of the sulfur-epoxy resins from the reported approach. An attractive reaction route for inverse vulcanization and preparation of sulfur-rich epoxy resins has been demonstrated.Author contributions
Yue-Sheng Lai. Data curation, formal analysis, investigation, methodology, visualization, writing – original draft. Ying-Ling Liu. Conceptualization, funding acquisition, methodology, resources, supervision, validation, writing – review & editing.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
The authors thank the National Science and Technology Council (NSTC) of Taiwan for the financial supports on this work (NSTC 111-2221-E-007-012-MY3).References
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524, DOI:10.1038/nchem.1624.
- J. M. Chalker, M. J. H. Worthington, N. A. Lundquist and L. J. Esdaile, Synthesis and Applications of Polymers Made by Inverse Vulcanization, in Sulfur Chemistry. Topics in Current Chemistry Collections, ed. X. Jiang, Springer, Cham. 2019, DOI:10.1007/978-3-030-25598-5_4.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116, DOI:10.1002/pola.28266.
- S. Diez, A. Hoefling, P. Theato and W. Pauer, Polymers, 2017, 9, 59, DOI:10.3390/polym9020059.
- M. Arslan, B. Kiskan, E. C. Cengiz, R. Demir-Cakan and Y. Yagci, Eur. Polym. J., 2016, 80, 70–77, DOI:10.1016/j.eurpolymj.2016.05.007.
- Y. Zhang, K. M. Konopka, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2017, 8, 5167–5173, 10.1039/c7py00587c.
- Y. Zhang, T. S. Kleine, K. J. Carothers, D. D. Phan, R. S. Glass, M. E. Mackey, K. Char and J. Pyun, Polym. Chem., 2018, 9, 2290–2294, 10.1039/c8py00270c.
- T. S. Kleine, N. A. Nguyen, L. E. Anderson, S. Namnabat, E. A. Lavilla, S. A. Showghi, P. T. Dirlam, C. B. Arrington, M. S. Manchester, J. Schwiegerling, R. S. Glass, K. Char, R. A. Norwood, M. E. Mackay and J. Pyun, ACS Macro Lett., 2016, 5, 1152–1156, DOI:10.1021/acsmacrolett.6b00602.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtar and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692, 10.1039/c6ta09862b.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718, DOI:10.1002/anie.201508708.
- M. Y. Omeir, V. S. Wadi and S. M. Alhassan, Mater. Lett., 2020, 259, 126887, DOI:10.1016/j.matlet.2019.126887.
- I. Gomez, O. Loenet, J. A. Blazquez and D. Mecerreyes, ChemSusChem, 2016, 9, 3419–3425, DOI:10.1002/cssc.201601474.
- D. J. Parker, S. T. Chong and T. Hasell, RSC Adv., 2018, 8, 27892–27899, 10.1039/c8ra04446e.
- M. J. H. Worthington, R. L. Kucera, I. S. Albuquerque, C. T. Gibson, A. Sibley, A. D. Slattery, J. A. Campbell, S. F. K. Alboaiji, K. A. Muller, J. Young, N. Adamson, J. R. Gascooke, D. Jampaiah, Y. M. Sabri, S. K. Bhargava, S. J. Ippolito, D. A. Lewis, J. S. Quinton, A. V. Ellis, A. Johs, G. J. L. Bernardes and J. M. Chalker, Chem. – Eur. J., 2017, 23, 16219–16230, DOI:10.1002/chem.201702871.
- J. A. Smith, S. J. Green, S. Petcher, D. J. Parker, B. Zhang, M. J. H. Worthington, X. Wu, C. A. Kelly, T. Baker, C. T. Gibson, J. A. Campbell, D. A. Lewis, M. J. Jenkins, H. Willock, J. M. Chalker and T. Hasell, Chem. – Eur. J., 2019, 25, 10433–10440, DOI:10.1002/chem.201901619.
- A. Hoefling, Y. J. Lee and P. Theato, Macromol. Chem. Phys., 2017, 218, 1600303, DOI:10.1002/macp.201600303.
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Polym. Sci., 2020, 58, 2259–2266, DOI:10.1002/pol.20200292.
- C. Herrera, K. J. Ysinja and C. L. Jenkins, ACS Appl. Mater. Interfaces, 2019, 11, 35312–35318, DOI:10.1021/acsami.9b11204.
- P. T. Dirlam, A. G. Simmonds, T. S. Kleine, N. A. Nguyen, L. E. Anderson, A. O. Klever, A. Florian, P. J. Costanzo, P. Theato, M. E. Mackey, R. S. Glass, K. Char and J. Pyun, RSC Adv., 2015, 5, 24718–24722, 10.1039/c5ra01188d.
- A. Hoefling, D. T. Nguyen, Y. J. Lee, S. W. Song and P. Theato, Mater. Chem. Front., 2017, 1, 1818–1822, 10.1039/c7qm00083a.
- H. Kang, H. Kim and M. J. Park, Adv. Energy Mater., 2018, 8, 1802423, DOI:10.1002/aenm.201802423.
- Y. Zhang, N. G. Pavlopoulos, T. S. Kleine, M. Karayilan, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 7–12, DOI:10.1002/pola.29266.
- H. K. Lin, Y. S. Lai and Y. L. Liu, ACS Sustainable Chem. Eng., 2019, 7, 4515–4522, DOI:10.1021/acssuschemeng.8b06815.
- S. Park, M. Chung, A. Lamprou, K. Seidel, S. Song, C. Schade, J. Lim and K. Char, Chem. Sci., 2022, 13, 566–572, 10.1039/d1sc05896g.
- S. Shukla, A. Ghosh, U. K. Sen, P. K. Roy, S. Mitra and B. Lochab, ChemistrySelect, 2016, 1, 594–600, DOI:10.1002/slct.201600050.
- M. Arslan, B. Kiskan and Y. Yagci, Macromolecules, 2016, 49, 767–773, DOI:10.1021/acs.macromol.5b02791.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647, DOI:10.1038/s41467-019-08430-8.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232, DOI:10.1021/mz400649w.
- P. T. Dirlam, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1635–1668, DOI:10.1002/pola.28551.
- J. J. Griebel, N. A. Nguyen, A. V. Astashkin, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 1258–1261, DOI:10.1021/mz500678m.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K.-J. Kim, J. Van Der Laan, N. A. Nguyen, E. L. Dereniak, M. E. Mackay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018, DOI:10.1002/adma.201305607.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866, DOI:10.1021/acsmacrolett.5b00502.
- J. A. Smith, R. Mulhall, S. Goodman, G. Fleming, H. Allison, R. Raval and T. Hasell, ACS Omega, 2020, 5, 5229–5234, DOI:10.1021/acsomega.9b04267.
- V. K. Shankarayya Wadi, K. K. Jena, S. Z. Khawaja, K. Yannakopoulou, M. Fardis, G. Mitrikas, M. Karagianni, G. Papavassiliou and S. M. Alhassan, ACS Omega, 2018, 3, 3330–3339, DOI:10.1021/acsomega.8b00031.
- L. J. Dodd, Ö. Omar, X. Wu and T. Hasell, ACS Catal., 2021, 11, 4441–4455, DOI:10.1021/acscatal.0c05010.
- K. Orme, A. H. Fistrovich and C. L. Jenkins, Macromolecules, 2020, 53, 9353–9361, DOI:10.1021/acs.macromol.0c01932.
- J. Bao, K. P. Martin, E. Cho, K.-S. Kang, R. S. Glass, V. Coropceanu, J.-L. Bredas, W. O. Parker, J. T. Njardarson and J. Pyun, J. Am. Chem. Soc., 2023, 145, 12386–12397, DOI:10.1021/jacs.3c03604.
- H. K. Lin and Y. L. Liu, Macromol. Rapid Commun., 2018, 39, 1700832, DOI:10.1002/marc.201700832.
- Y. S. Lai and Y. L. Liu, Macromol. Rapid Commun., 2023, 44, 2300014, DOI:10.1002/marc.202300014.
- G. Zhang, J. Cheng, L. Shi, X. Lin and J. Zhang, Thermochim. Acta, 2012, 538, 36–42, DOI:10.1016/j.tca.2012.03.012.
- Q. Lian, Y. Li, T. Yang, K. Li, Y. Xu, L. Liu, J. Zhao, J. Zhang and J. Cheng, J. Mater. Sci., 2016, 51, 7887–7898, DOI:10.1007/s10853-016-0044-z.
- Q. Lian, Y. Li, K. Li, J. Cheng and J. Zhang, Macromolecules, 2017, 50, 803–810, DOI:10.1021/acs.macromol.6b02335.
- L. Sun, S. Gao, X. Gui, L. Liu, K. Xu and H. Liu, Eur. Polym. J., 2020, 123, 109440, DOI:10.1016/j.eurpolymj.2019.109440.
- T. Zhang, F. Hu, W. Shao, S. Liu, H. Peng, Z. Song, C. Song, N. Li and X. Jian, ACS Nano, 2021, 15, 15027–15038, DOI:10.1021/acsnano.1c05330.
- Y. Jin, C. Hu, Z. Wang, Z. Xia, R. Li, S. Shi, S. Xu and L. Yuan, ACS Sustainable Chem. Eng., 2023, 11, 11259–11268, DOI:10.1021/acssuschemeng.3c02478.
- P. Yan, W. Zhao, S. J. Tonkin, J. M. Chalker, T. L. Schiller and T. Hasell, Chem. Mater., 2022, 34, 1167–1178, DOI:10.1021/acs.chemmater.1c03662.
- Y. Jin, Z. Wang, C. Hu, J. Wang, K. Yan, J. He, Z. Wang, Z. Wang and L. Yuan, Green Chem., 2023, 25, 1157–1168, 10.1039/D2GC04744F.
- M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070, DOI:10.1002/pola.28195.
- Q. Zhang, A. Anastasaki, G.-Z. Li, A. J. Haddleton, P. Wilson and D. M. Haddleton, Polym. Chem., 2014, 5, 3876–3883, 10.1039/c4py00320a.
- S. De and A. Khan, Chem. Commun., 2012, 48, 3130–3132, 10.1039/c2cc30434a.
- C. T. Huynh, F. Liu, Y. Cheng, K. A. Coughlin and E. Alsberg, ACS Appl. Mater. Interfaces, 2018, 10, 25936–25942, DOI:10.1021/acsami.8b07167.
- T. J. Wallace and R. J. Gritter, Tetrahedron, 1963, 19, 657–665, DOI:10.1016/S0040-4020(01)98551-5.
- T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997, DOI:10.1021/ja00082a021.
- D. Wang, Z. Tang, Y. Liu and B. Guo, Green Chem., 2020, 22, 7337–7342, 10.1039/d0gc02660c.
- J. J. Dale, J. Stanley, R. A. Dop, G. Chronowska-Bojczuk, A. J. Fielding, D. R. Neill and T. Hasell, Eur. Polym. J., 2023, 195(17), 112198, DOI:10.1016/j.eurpolymj.2023.112198.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 1–9, DOI:10.1038/s41467-019-08430-8.
- J. H. Hwang, J. M. Lee, J. H. Seo, G. Y. Noh, W. Byun, S. Kim, W. Lee, S. Park, D. G. Kim and Y. S. Kim, Green Chem., 2023, 25, 4641–4646, 10.1039/D3GC01102J.
- A. Battig, J. C. Markwart, F. R. Wurm and B. Schartel, Eur. Polym. J., 2020, 122, 109390, DOI:10.1016/j.eurpolymj.2019.109390.
- D. Y. Hung, J. J. Lee and Y. L. Liu, Polym. Chem., 2023, 14, 5004–5013, 10.1039/d3py00957b.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105, 10.1039/C9PY00636B.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00074a |
| This journal is © The Royal Society of Chemistry 2024 |