
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ SAXS investigation of vinyl acetate polymerization-induced self-assembly†
Fabrice
Brunel
 *,
Paul
Galanopoulo
,
Edgar
Espinosa Rodriguez
,
Muriel
Lansalot
and
Franck
D'Agosto
*,
Paul
Galanopoulo
,
Edgar
Espinosa Rodriguez
,
Muriel
Lansalot
and
Franck
D'Agosto
Univ Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, CP2M (UMR 5128), 43 Bvd. du 11 Novembre 1918, F-69616 Villeurbanne, France. E-mail: fabrice.brunel@univ-lyon1.fr
First published on 7th February 2024
Abstract
In situ small angle X-ray scattering (SAXS) was used to investigate the formation of higher-order morphologies in vinyl acetate (VAc) polymerization-induced self-assembly (PISA). Poly(ethylene glycol) (PEG) bearing a xanthate chain end was used as a hydrophilic macromolecular chain transfer agent in the macromolecular design via interchange of xanthates (MADIX)-mediated aqueous emulsion polymerization of VAc (see P. Galanopoulo, et al., Poly. Chem., 2020, 11, 3922–3930). In situ SAXS monitoring confirmed that the PISA process occurs in three steps: the nucleation of PEG45-b-PVAc200 block copolymer primary particles, a rapid morphological transition, and a final step in which the nano-objects grow slowly to their final dimensions. The transition step appears to be critical for the formation of higher-order morphologies through the aggregation of primary particles into larger clusters and their subsequent reorganization. Our results show that the transition step is extremely fast, making it highly sensitive to small perturbations. This explains why higher-order morphologies are usually obtained within a narrow window of conditions and often as a mixture. Furthermore, the morphological evolution observed in parallel static experiments appears to be more gradual than in in situ SAXS experiments, suggesting that in our system the sampled latexes were not thermodynamically stable and continued to evolve after sampling. Therefore, the sampling method may not provide a realistic view of the morphological transition during the reaction process in this system.
Introduction
Polymerization-induced self-assembly (PISA) has attracted considerable attention over the past two decades for its ability to yield ordered structures in a wide range of morphologies.1 Briefly, a solvophilic living polymer precursor prepared by a controlled polymerization such as reversible deactivation radical polymerization is extended with a solvophobic second block, resulting in amphiphilic block copolymers that self-assemble in situ into self-stabilized nano-objects with different morphologies.2–4 PISA is typically implemented using reversible addition–fragmentation chain transfer (RAFT) polymerization as an emulsion or dispersion polymerisation.1,5,6 Polymeric nano-objects with higher-order morphology (e.g. worms, vesicles) are particularly interesting from a fundamental point of view and because they can extend the range of more morphology dependent applications.7–9 The change in morphology can be rationalized by the concept of the packing parameter, P, which is given by equation:where ν0 and lc are the volume and length of the solvophobic block, respectively, and a is the effective interfacial area of the block junction. This concept, originally introduced by Israelachvili and co-workers10 to explain the self-assembly of surfactants, was later extended to the self-assembly of diblock copolymers by Antonietti and Förster.11 It is generally accepted that spherical micelles are favored when P ≤ 1/3, cylindrical micelles are produced when 1/3 < P ≤ 1/2, and vesicles are formed when 1/2 < P ≤ 1. Therefore, the particle morphology in PISA, as in the self-assembly of preformed block copolymers, is influenced by the various parameters that affect the packing parameter, such as the molar mass,3,4,12–14 the chemical nature15–21 and the architecture of the block copolymer,22–24 the solvent quality25–29 and, in aqueous media, the pH14,30 and/or ionic strength.31–33 However, the packing parameter only applies to morphologies in thermodynamic equilibrium, but higher-order morphologies are not necessarily in equilibrium and kinetic aspects must also be considered. Temperature,9,17 initiating system, solid content, monomer concentration and conversion30,34,35 and stirring32 also play a role by modifying the polymerization kinetics or the shear flow. In addition, higher-order morphologies, especially worms, are generally obtained in a narrow experimental window by PISA. This often results in the formation of particles with multiple morphologies.1 Morphology prediction in PISA remains thus difficult and the detailed mechanism of the higher-order morphology formation still deserves further investigation.
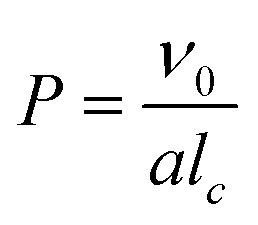
It is generally accepted that PISA starts with the nucleation of primary particles (i.e., spherical micelles), followed by their aggregation and their fusion into higher-order morphology. Recent investigations of PISA in aqueous emulsions were performed in our group using molecular dynamics (MD) simulations.36 Using an umbrella sampling algorithm, the interaction free energies between different self-assembled copolymer micelles were calculated. Depending on the position of the poly(ethylene glycol) (PEG) branches carried by the hydrophilic block under consideration, the attractive interaction between the copolymer micelles is in some cases followed by a repulsive barrier that controls the aggregation kinetics: a strong repulsive barrier leads to a lower aggregation rate and thus to larger and denser clusters (i.e. reaction limited cluster aggregation). These clusters then coalesce into large vesicles due to the presence of interstitial water molecules in the cluster. Conversely, a weak repulsive barrier causes a rapid aggregation resulting in loose and ramified clusters (i.e. diffusion limited cluster aggregation), which coalesce after swelling with hydrophobic monomer, resulting in worms and small vesicles. This new mechanistic approach can explain the formation of higher-order morphology depending on the polymer architecture by revealing the crucial role of the aggregation state of the primary particles. This mechanism also demonstrates the importance of a slight increase in hydrophobicity within the hydrophilic block (i.e. a combination of an attractive well and a repulsive barrier) for the formation of higher-order morphologies in emulsion PISA, which is often provided by the hydrophobic character of PEG upon temperature increase.2,14,30–32,37–40 Recently, our group used a linear PEG bearing a xanthate chain end as a hydrophilic macromolecular chain transfer agent (macro-CTA) in the macromolecular design via interchange of xanthates (MADIX)-mediated aqueous emulsion polymerization of vinyl acetate (VAc).41 MADIX is indeed a polymerization based on a RAFT process. Stable dispersions of self-assembled amphiphilic diblock copolymers of PEG45-b-PVAc were obtained, and the molar mass of the PVAc block was varied to induce the formation of different morphologies. Due to the relatively low repulsion between the PEG corona and to the high water solubility of VAc (0.565 M at 65 °C),42 this system yields a wide variety of morphologies, including spheres, vesicles and large compound vesicles. Although the solubility of VAc is relatively high, we still consider it to be in the case of an emulsion polymerization since the since two insoluble phases (namely, VAc and water) were present in the starting medium.
The electron density contrast between the water and the PEG is very small.43–45 This allows for relevant SAXS analyses since we can then follow the organization of only the lipophilic phase (i.e. VAc and PVAc). Therefore, this system is perfectly suited for in situ synchrotron-SAXS studies to investigate the formation of higher-order morphologies. In situ SAXS can provide insights into the formation mechanism of the different morphologies. Brotherton et al. performed a in situ SAXS analysis of the RAFT aqueous emulsion polymerization of 2-methoxyethyl methacrylate using a water-soluble poly(glycerol monomethacrylate) macro-molecular chain transfer agent and observed a morphological evolution from spheres to worms to vesicles.46 They observed a nucleation step (from 0 to 25 min) during which the primary particles are formed, followed by a transition step between 25 and 30 min where the morphology rapidly changes into worms, and finally after 70 min, the formation of vesicles. However, they only provide rather rudimentary data analyses (based solely on the slope of the scattering intensities between 0.005 and 0.015 Å−1), which do not allow conclusion to be drawn about the precise mechanism at work during the transition step. Liao et al. used this technique to monitor the polymerization kinetic during the synthesis of spherical nanoparticles,47 while Czajka et al. were able to monitor the evolution of copolymer particle morphology, in both cases during PISA in dispersion.48 In the latter study, the authors performed the RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate using a poly(glycerol monomethacrylate) steric stabilizer and recorded a three-step process: (1) the nucleation of spherical micelles (from 0 to 30 min), followed by (2) a transition step (from 30 to 50 min) during which the growth of worms by 1D fusion of multiple spheres are observed (between 40–44 min), and (3) a final step, during which the vesicles gradually grow into their final size through an “inward growth mechanism”. In the present study, in situ synchrotron-SAXS has been used to monitor the morphological transitions that occur during the emulsion polymerization of VAc using a PEG macro-CTA. A form and a structure factors were implemented at each stage of the PISA process, facilitating precise tracking of the morphological transition through the incorporation of the structure factor. Thanks to the new high brilliance source of the ESRF, we were able to monitor the self-assembly of copolymer micelles and their transition into higher-order morphologies through aggregation and coalescence, confirming the morphological transition mechanism previously highlighted by MD simulations.49
Materials and methods
Materials
Vinyl acetate (VAc, 99%, Aldrich), 2,2′-azobis(2-amidinopropane) dihydrochloride (AIBA, 97%, Aldrich), poly(ethylene glycol) methyl ether (PEG-OH, Mn = 2000 g mol−1, Aldrich), O-ethyl xanthic acid potassium salt (98%, Alfa Aesar), triethylamine (99%, Aldrich), 2-bromopropionyl bromide (97%, Aldrich), dichloromethane (ACS reagent, Aldrich), ammonium chloride (99.5%, ACS reagent, Aldrich), sodium hydrogen carbonate (ACS reagent, Aldrich), sodium chloride (99%, ACS reagent, Aldrich), and anhydrous magnesium sulfate (99.5%, Alfa Aesar) were used as received. Water was deionized before use (PURELAB Classic UV, Elga LabWater).Vinyl acetate (VAc) PISA in emulsion
PEG45-b-PVAc200 amphiphilic diblock copolymers were synthesized in water through MADIX polymerization. A PEG with a dithiocarbonate (xanthate) end-group (PEG45-X) was prepared according to a previously reported method,41 and then chain extended in water using VAc to produce PEG45-b-PVAc200 amphiphilic diblock copolymers (Scheme 1). Briefly, VAc, PEG-X, AIBA and deionized water were introduced in the reactor, either a 50 mL flask or a 200 mL flask connected to a flow-through cell for static or in situ synchrotron-SAXS experiments, respectively. After degassing of the medium for 15 min under argon, the flask was placed in an oil bath previously heated at 70 °C. | ||
| Scheme 1 Reaction scheme of the vinyl acetate PISA in emulsion using PEG45-X as macro-CTA. | ||
Size-exclusion chromatography (SEC)
SEC analyses were performed using a Viscotek system (Malvern Instruments) including a four-capillary differential viscometer, a differential refractive index (RI) detector and a UV detector. THF was used as the mobile phase at a flow rate of 1 mL min−1 at 35 °C. All samples were injected at a concentration of 3 to 5 mg mL−1 after filtration through a 0.45 μm PTFE membrane. The separation was carried out on three Polymer Standard Service columns (SDVB, 5 μm, 300 × 7.5 mm) and a guard column. The average molar masses (number-average molar mass, Mn, and weight-average molar mass, Mw) and the dispersity, Đ = Mw/Mn, were calculated from the RI signal with a calibration curve based on polystyrene standards (Polymer Standards Service).Cryogenic-transmission electron microscopy (cryo-TEM)
The diluted latex samples were dropped onto 300 mesh holey carbon films (Quantifoil R2/1) and immediately quench-frozen in liquid ethane. The grid was then mounted on a precooled Gatan 626 specimen holder and transferred to a Philips CM120 microscope operating at an accelerating voltage of 120 kV (Centre Technologique des Microstructures (CTμ), platform of the Université Claude Bernard Lyon 1, Villeurbanne, France).Small-angle X-ray scattering (SAXS)
SAXS technique consist of measuring the scattered intensity of an X-ray beam as a function of the scattering vector q = (4π/λ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ), where λ is the wavelength of the radiation and θ is one half of the scattering angle. SAXS measurements were carried out at the BM02 beamline of the European Synchrotron Radiation Facilities (ESRF, Grenoble, France). Experiments were carried out at an energy of 17.5 keV (λ = 0.07 nm) with a sample to detector distance of 175 cm, using a fast photon counting pixel detectors (ImXpad D5) allowing q-range between 0.005 and 0.5 Å−1. The recorded images were first corrected from dark current and flat field and normalized with respect to the intensity of the incident beam. The radial integration of the corrected images gave the scattering curves. Finally, the obtained scattering curves were corrected for sample transmission and background subtraction and calibrated using silver behenate (q-axis) and glassy carbon (absolute intensity). To study morphological changes during the polymerization, the reaction medium was pumped (2 mL min−1) in a closed loop through a flow cell equipped with a glass capillary (50 mm long and 3 mm in internal diameter) placed in front of the beam. SAXS data were analyzed using SASfit 0.94.11 software. The form and structure-factor equations used to fit the curves are given below for the different situations.
sinθ), where λ is the wavelength of the radiation and θ is one half of the scattering angle. SAXS measurements were carried out at the BM02 beamline of the European Synchrotron Radiation Facilities (ESRF, Grenoble, France). Experiments were carried out at an energy of 17.5 keV (λ = 0.07 nm) with a sample to detector distance of 175 cm, using a fast photon counting pixel detectors (ImXpad D5) allowing q-range between 0.005 and 0.5 Å−1. The recorded images were first corrected from dark current and flat field and normalized with respect to the intensity of the incident beam. The radial integration of the corrected images gave the scattering curves. Finally, the obtained scattering curves were corrected for sample transmission and background subtraction and calibrated using silver behenate (q-axis) and glassy carbon (absolute intensity). To study morphological changes during the polymerization, the reaction medium was pumped (2 mL min−1) in a closed loop through a flow cell equipped with a glass capillary (50 mm long and 3 mm in internal diameter) placed in front of the beam. SAXS data were analyzed using SASfit 0.94.11 software. The form and structure-factor equations used to fit the curves are given below for the different situations.
At large q values a generalized-Porod's law predicts the scattering intensity:
| I(q) = Cq−α | (1) |
Quantitative analysis of particle size and shape starts with the Guinier approximation. For three-dimensional objects the Guinier approximation is given by:
 | (2) |
During the morphological transition some spectra are characterized by a broad peak even though they are from amorphous soft materials. The following simple functional form reproduces the broad peak feature:
 | (3) |
During the transition step, a hard sphere structure factor was included using a monodisperse approach. This approach simply multiplies the size averaged form factor with the structure factor. Here, it is assumed that the interaction potential between particles is spherical symmetric and independent of the particle size. Structure factor (SHS(q)) for a hard sphere having a radius RHS and a volume fraction fp is given below:
 | (4.1) |
 | (4.2) |
 | (4.3) |
The structure factor of the fractal aggregate was also used during the transition step and was calculated from:
 | (5) |
with r0 the characteristic dimension of individual scattering objects, ξ the cut-off length for the fractal correlations (i.e. the size of an aggregate) and D the fractal dimension.
Finally, during the final step, the form factor of a vesicle was used, according to the cryo-TEM observation of the final morphology.
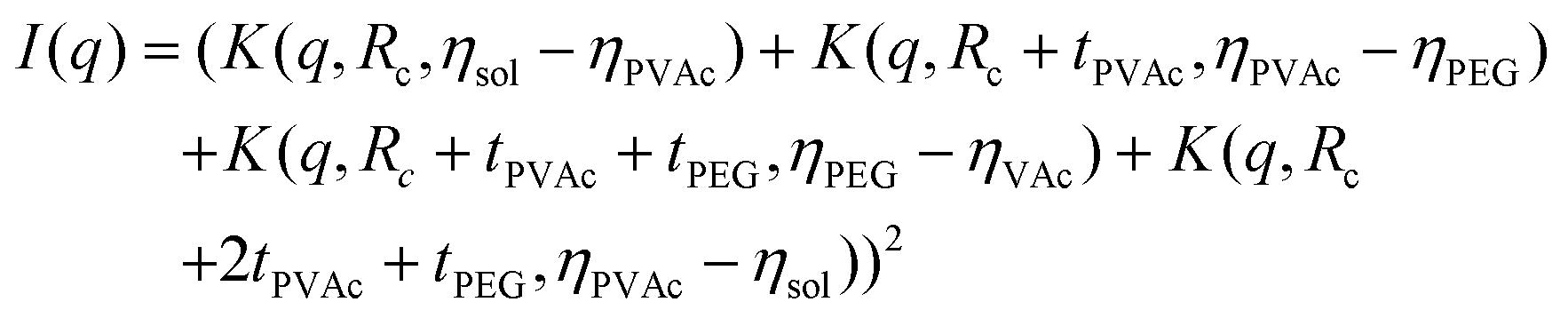 | (6.1) |
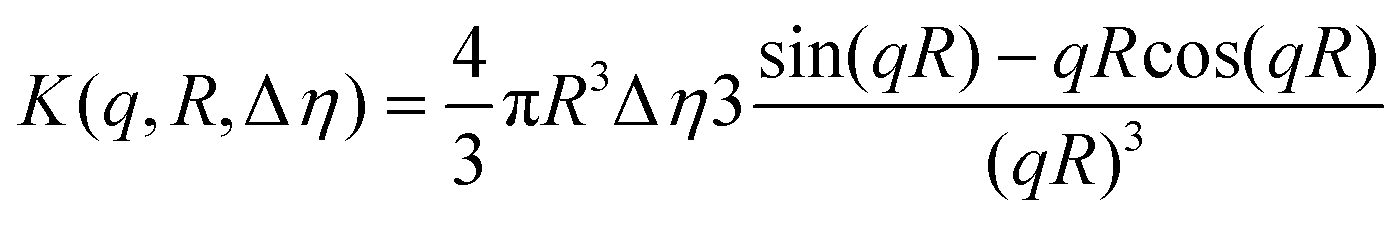 | (6.2) |
| Rg = l × DPν | (7) |
Results and discussion
The aqueous MADIX-mediated emulsion polymerization of vinyl acetate using PEG45-X as a hydrophilic macro-CTA led to the formation of stable spherical particles for a degree of polymerization (DP) for the PVAc block lower than 50, to a mixture of spheres and vesicles for DP comprised between 50 and 100, and eventually to higher-order morphologies, notably large compound vesicles for a DP ≥ 100.41 The formation of higher-order morphologies was interpreted as the result of (1) the high water solubility of VAc compared to other hydrophobic monomers classically employed in emulsion polymerization, (2) the low glass transition temperature of the hydrophobic block (Tg ≈ 30 °C) compared to the polymerization temperature that increases mobility of the polymer chains, thus facilitating rearrangements, and (3) the relatively low repulsion between the PEG corona compared to other hydrophilic macro-CTAs, that enhances the effective collision of the assemblies and thus promotes their rearrangement.41 Thus, although the mechanism associated with the formation of higher-order morphologies is still not fully addressed,53 all the hypotheses formulated above suggest that the aggregation and rearrangement of primary particles play a crucial roleas shown theoretically by our group using MD simulations.49 Therefore, in situ synchrotron-SAXS measurements were performed to monitor the nucleation and the morphological transitions of PEG-b-PVAc block copolymer nanoparticles obtained via RAFT/MADIX, targeting a polymerization degree close to 200 for the PVAc block.Comparison between static and in situ SAXS experiments
First, cryo-TEM images of the final particle morphology obtained using either in situ (see ESI† for the details of this experiment) or static41 reactor step-up were compared (Fig. S2 and S3†). The cryo-TEM images showed mostly small vesicles and large compound vesicles for both reaction processes. For the in situ experiment, small fiber-like objects are also visible although it does not seem to be the main morphology. Indeed, the presence of a significant number of elongated objects should result in an increase of the viscosity which was not observed. Furthermore, such elongated objects are not observed on SAXS, probably because their concentration is too low. Therefore, the vesicles appear to be the most abundant morphology. However, the in situ experiment yields smaller multi-compartment vesicles and more densely packed compared with the static experiment. Scattering intensities of the final morphologies obtained with in situ and static experiments also confirmed this observation (Fig. S4†). The form factor of polydisperse vesicles with the hard sphere structure factor plus an additional Porod slope was used to fit either in situ or static experiments. Smaller characteristic dimensions (see Table S2†) are observed for the in situ compared with the static experiment, in good agreement with cryo-TEM images.Brotherton et al. also obtained smaller vesicles during the in situ SAXS experiments compared to those produced in the laboratory-based synthesis. They attributed this difference in morphology to a difference in mechanical agitation or perhaps to a polymerization rate enhancement induced by X-ray irradiation which could produce radicals. However, an order of magnitude calculation proves that this phenomenon is probably negligible. According to ESRF specifications, the beamline used here has a photon flux of about 1011 ph s−1.54 Thus, considering a 10% absorption coefficient (i.e., the average absorption coefficient for soft matter according to the NIST calculator)55 and a photochemical yield of 100%, a rate of X-rays produced radicals of 1.66 × 10−14 mol s−1 would be calculated. On the other hand, 0.103 g of AIBA initiator (4.79 × 10−5 mol, Table S1†), with a decomposition rate of 1.2 × 10−4 s−1 at 70 °C56 gives a rate of chemically produced radicals of 9.1 × 10−8 mol s−1, which if 5000000 times higher than for X-ray photoinitiation. The difference of morphology between the static and in situ experiments is thus more likely due to the reactor hydrodynamics rather than to a side initiation from the X-ray beam. The formation of large compound vesicles is likely hindered by the high shear caused by the pump in the flow-through cell setup. The formation of vesicles is often reported through a shear-induced processes,57,58 which is suspected to happen here. This is also supported by Boissé et al. who clearly demonstrated the impact of stirring on morphology in a PISA emulsion polymerization system.32 Thereby, the main limitation of our SAXS setup concerns the formation of larger compound vesicles, which are most likely impacted by the change of shear flow. Moreover, the formation of large objects cannot be observed in our working q-range. Therefore, the mechanism of formation of large compound vesicles will not be discussed and this article will focus on the formation of small vesicle assemblies. Finally, it is interesting to compare the morphological evolution observed between the samples collected during a static experiment and those measured using in situ SAXS for the same reaction time (see Fig. S5†). During the in situ experiment the morphological transition appears between 20 and 23 min (Fig. 1). On the contrary, the morphological evolution appears more gradually in the case of the static experiment (Fig. S5†). This can be explained by the fact that the sampled latexes were cooled in the static experiment, while in situ measurements were performed at constant reaction temperature (70 °C). This difference also suggests that the sampled latexes are not thermodynamically stable and continue to evolve after sampling (i.e. the cooling did not completely freeze the morphology formed in the reactor). Only after 40 min and 80% conversion both in situ and static experiments show similar scattering profile, indicating that above this point kinetically frozen morphologies are obtained. Anyhow, the sampling method might not provide a realistic view of the morphological transition during the reaction process.
 | ||
| Fig. 1 The three-step evolution of the VAc conversion during the PISA process regarding morphological transition of the formed nano-objects. The data were obtained from an experiment carried out in the laboratory under identical conditions to the in situ SAXS experiment (VAc conversion was determined by gravimetric analysis). | ||
In situ SAXS experiments
Over the course of the reaction 377 SAXS spectra were collected. They were converted into a video file, available online59 that allows to rapidly visualize the evolution of the scattering intensity during the PISA process. The morphological evolution observed can be divided into three steps (see Fig. 1): (1) the nucleation (first 20 min), (2) the transition step (between 20 and 23 min) where the critical morphological transition occurs, and (3) the final step (from 23 min to the end), that could be called the curing or ripening step, where the nano-objects slowly grow to reach their final dimension. | ||
| Fig. 2 (a) SAXS-scattering intensities from t = 10 to 18.3 min. Black dots and red curves are experimental points and fitting curves, respectively. (b) and (c) are the evolution of the fitting parameters corresponding to the lower q-range Porod's law and the higher q-range Guinier model, respectively. | ||
The size of the primary particles (corresponding to the Guinier plateau) decreases at the very beginning (see Fig. 2c), which could be an artifact due to the very low scattering intensity. Hence, this early trend might not be significant. The q−4 slope increases in intensity during the first few minutes (see Fig. 2b), which probably corresponds to a decrease of the droplet size making them more visible in the observed q-range (i.e. shifted to lower q). The scattering intensity in the Porod region reaches a maximum for around 14 min, then it decreases. At the same time, the intensity of the Guinier plateau increases corresponding to an increased number of primary particles while the size of those remains constant. These observations are consistent with a nucleation step; the diffusion of the monomer from larger droplets through the water, where it can polymerize. This forms amphiphilic copolymers which then self-assemble into small primary particles (i.e. nucleation). The swelling of the primary particles by the monomer is visible between 14 and 18 min in Fig. 2c (their radius increases from 4 to 5.5 nm). This swelling is rather limited which could be due to the solubility of vinyl acetate in water which prevents its sequestration into the particle hydrophobic core.
At the end of this first step (Fig. 3), the Guinier plateau, observed at a high q-range and corresponding to the primary particles (i.e. the copolymer micelles), maintains a consistent radius of approximately 5 nm, while their count marginally increases. At the same time, the Porod's slope, observed at low q-range, is gradually transformed into a Guinier plateau. This evolution of the scattering intensity reflects the decrease in size of large objects from the micrometer range to the nanometer range, specifically depleted monomer droplets. By the end of the nucleation phase, the number of monomer droplets decreases along with their size, from 25 to 10 nm (see fitting parameter in the table in Fig. 3). From the value of both the forward scattering (I0) and the radius of gyration (Rg) we can estimate the number of particles (I0 ∝ N × V ∝ N × Rg3) with N and V the number and volume of the particles, respectively. According to these calculations the number of primary particles from self-assembly nucleation would be ten-fold the number of monomer nano-droplets. Therefore, prior to the morphological transition, the system consists mainly of primary particles from self-assembly nucleation and a small population of residual monomer nano-droplets. This confirms that primary particle nucleation and simultaneous monomer droplet depletion are the main mechanisms driving the early stages of the PISA process.60
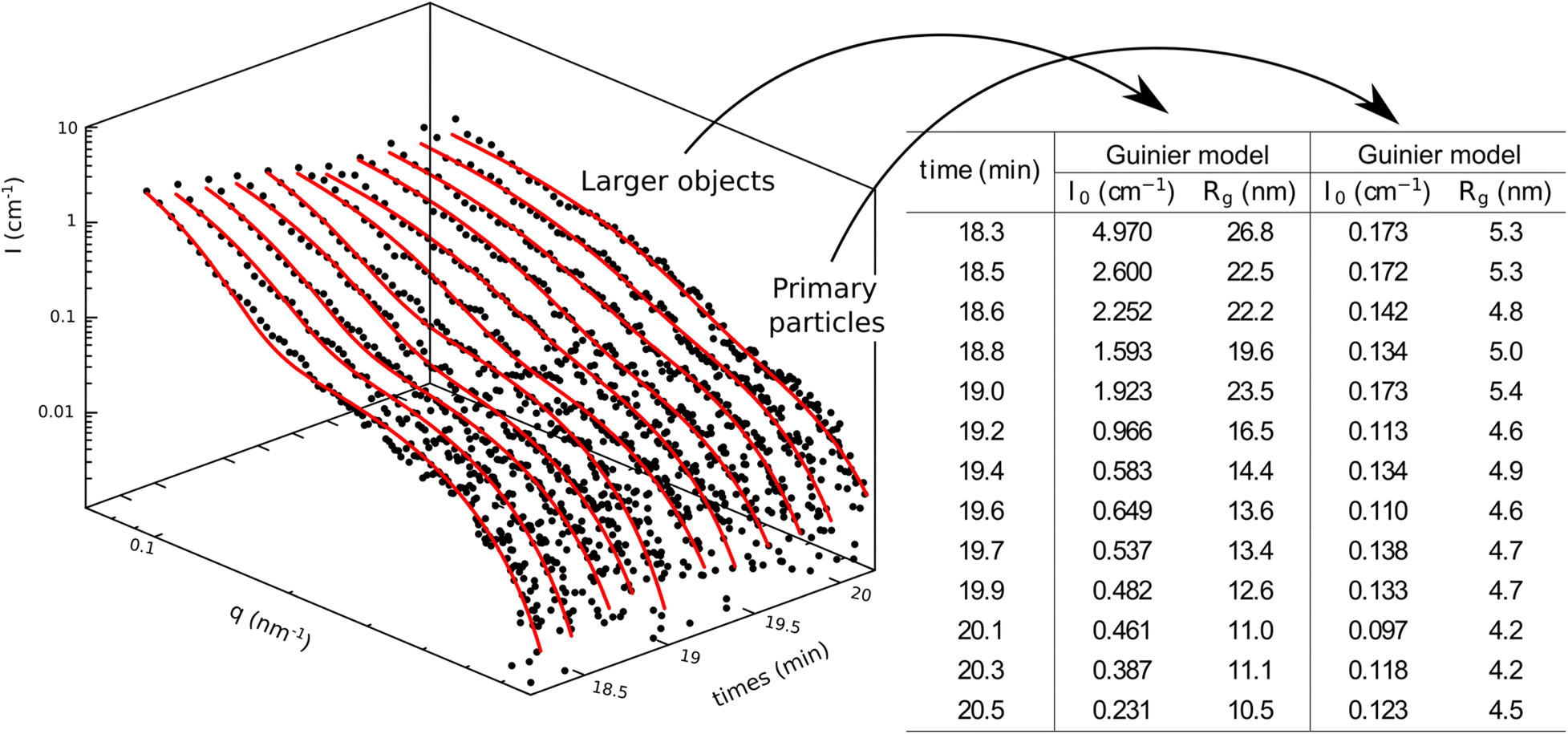 | ||
| Fig. 3 SAXS-scattering intensities from t = 18.3 to 20.2 min. Black dots and red curves are experimental points and fitting curves, respectively. The corresponding fitting parameters (Guinier model) are given in the table alongside the graph. | ||
 | ||
| Fig. 4 SAXS-scattering intensities: (a) from t = 20.2 to 21.8 min and (b) from t = 21.9 to 23.2 min. Black dots and red curves are experimental points and fitting curves, respectively. The corresponding fitting parameters are given in the tables (c) and (d) below the graph. | ||
In the high q-range, the transition from Guinier to a broad peak reveals the high concentration of particles. As particles get closer due to a lack of space a broad peak at around 0.5 nm−1 arises in SAXS curves (Fig. 4a). A hard sphere structure factor was added to the Guinier form factor to fit this broad peak. Here, the hard sphere radius is equal to the Guinier radius of gyration and the volume fraction (fp) was determined at around 0.3, which corresponds to a high concentration of small particles.
In the low q-range, the increased intensity can be attributed to the particle aggregation. A mass fractal structure factor was added to the previous Guinier model to fit the increase of the scattering intensity at the lowest q-range (cf. Table (c) in Fig. 4). At around 22 min, a Porod regime (q−4 slope) is observed through the merger of the spherical form factor and the structure factor suggesting that the aggregated particles are coalescing into a larger structure (see Fig. 4b). Between 22 and 23 min, the scattering intensities are rapidly evolving (see Fig. 4b). The corresponding fitting parameters are summarized in Table (d) in Fig. 4. The Porod slope at low q-range increases in intensity, while the Guinier form factor corresponding to primary particle disappears. This evolution is characteristic of the disappearance of the small particles because of their coagulation. Simultaneously, a broad peak appears around 0.2–0.3 nm−1 that probably corresponds to structural reorganization within the cluster of aggregated particles.
During this step, the aggregation of the copolymer particles formed during the previous nucleation step was observed. Indeed, the two Guinier plateaus (corresponding to small and large particles) disappear with a simultaneous increase of the intensities at lower q-range, which is a typical feature of the aggregation process. Furthermore, at the end of this step, the final morphology is formed. Indeed, at 23 min, the scattering intensities can be fitted with the form factor of polydisperse vesicles with an additional structure factor (hard sphere) plus a Porod slope at very low q-range. This fitting model, used in the next step and until the end, agrees well with the vesicular assembly observed in cryo-TEM (see Fig. S2†). This step can therefore be seen as a rapid morphological transition driven by the aggregation of the primary particles. Such mechanism has already been discussed using MD simulations.37 The coalescence of the copolymer particles within clusters of aggregated particles can lead to the formation of vesicles because of the interstitial water trapped within these clusters, which will reorganize to form the core of the vesicles (see Fig. 6).
 | ||
| Fig. 5 (a) SAXS-scattering intensities from t = 23 to 60 min. Black dots and red curves are experimental points and fitting curves, respectively. The evolutions of the fitting parameters are plotted below: (b) the polydisperse vesicle form factor and (c) the hard sphere structure factor. | ||
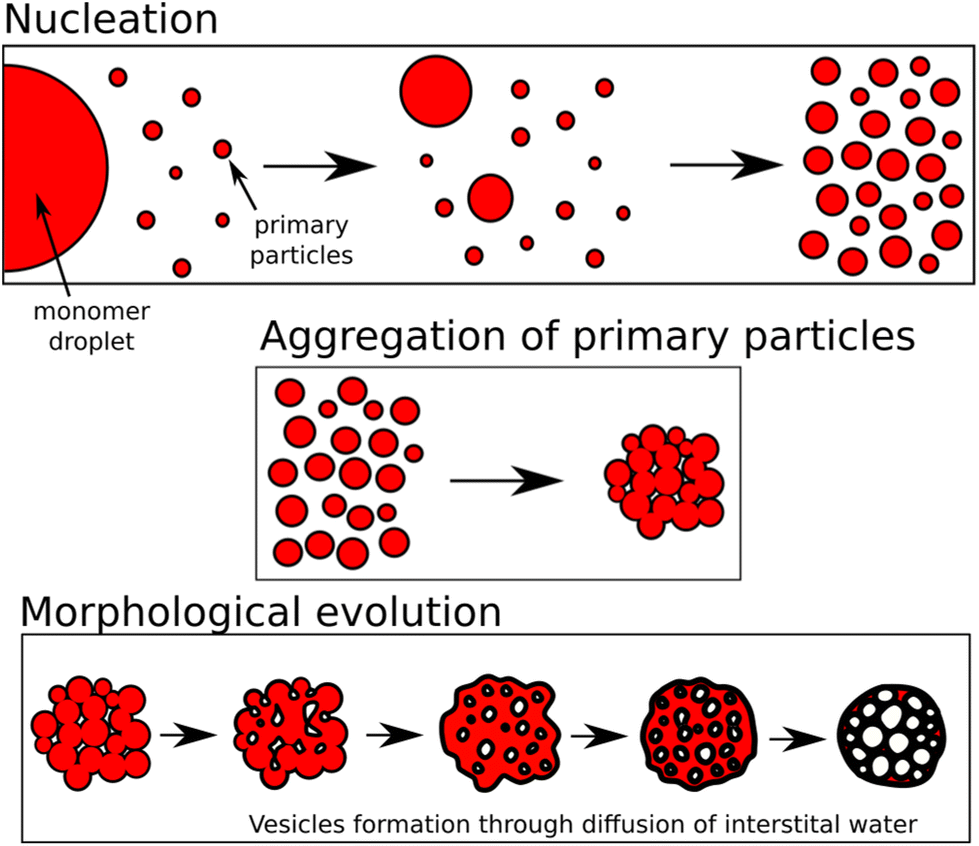 | ||
| Fig. 6 Illustration of the morphological transition. From top to bottom: the nucleation, the aggregation, and the morphological reorganization. | ||
Discussion
In a previous paper,37 we discussed the crucial role of the aggregation kinetics of the primary particles on the final morphology. We showed that the presence of an attracto-repulsive interaction between primary particles can influence their aggregation rate leading to larger clusters that can later reorganize into more complex architectures depending on their packing density. Here, we present a direct observation of this mechanism. Indeed, the aggregation of the primary particles is the first step that triggers the morphological transition through the structural reorganization within these clusters. Moreover, this mechanism could explain why higher-order morphologies are obtained in a very narrow window of conditions. According to this mechanism, obtaining a higher-order morphology requires an intermediate degree of colloidal stability (i.e. a combination of an attractive well and a repulsive barrier), a criterion that is met in this PEG-stabilized system. This causes a rapid aggregation but without complete destabilization (i.e. limited coalescence) and thus the formation of large and poorly organized clusters, which then evolve into a higher-order morphology. This rapid aggregation occurring during the transition step presents a chaotic aspect; a small perturbation during this step may lead to significantly different future behavior, and thus to the formation of a mixture of morphologies. For all these reasons and as already suspected, the sole concept of packing parameter (which only considers geometric/steric constraint) may not be sufficient to understand the formation of higher-order morphologies by PISA. The physicochemical interactions between the copolymer primary particles and their aggregation kinetics should also be considered when trying to predict the PISA morphology.Conclusion
Using time-resolved synchrotron SAXS, morphological transitions occuring during VAc emulsion PISA mediated by a xanthate functionalized PEG were studied. We observed that the PISA process occurs in three distinct steps: (i) the nucleation of copolymer primary particles, (ii) a rapid morphological transition in which the primary particles aggregate into clusters that rearrange themselves into higher-order morphologies, and (iii) a final curing step in which the nano-objects grow slowly to reach their final dimensions. Previous studies of PISA using in situ SAXS (Brotherton et al. and Czajka et al.)46,48 also observed three distinct steps: (i) the nucleation of copolymer primary particles, followed by (ii) an intermediate step where worm-like morphologies are observed, which then (iii) evolve into vesicles.However, in our study, the duration of the transition stage appears to be much shorter than previously reported and worm-like morphologies are not observed. These differences may be due to the use of a different PISA formulation as well as to our fitting strategy. Previous studies interpret the SAXS data in terms of pure morphology, namely spheres, worms, or vesicles, while we use a combination of a form and a structure factors, which leads to a slightly different description of the morphological transition. In other word, a loosely packed and ramified aggregate of particles may resemble worm-like morphologies. In our system, this transition step appears to be critical for the formation of higher-order morphologies through the aggregation of the primary particles created during the nucleation step. This observation agrees well with the mechanism predicted by MD simulations, highlighting the importance of the aggregation kinetics in obtaining higher-order morphologies by PISA.36
Synchrotron SAXS is a powerful method to study the mechanism of PISA. However, the final morphologies obtained are slightly different from the classical (i.e. static) synthesis probably due to the shear caused by the pumping of the liquid into the glass capillary placed in front of the X-ray beam. This shear could be detrimental to the formation of large vesicles, favoring the formation of smaller and more compact vesicular assemblies. Indeed, vesicles are often formed from preexisting planar bilayers that breakup under shear before reaching sufficient size.61 The theoretical understanding of the formation of large vesicles during the PISA processes will be the subject of further research. Finally, the morphological evolution appears much more gradual in the case of the static experiment. This difference suggests that the sampled latexes were not thermodynamically stable and continued to evolve. Therefore, it seems that the sampling method may not provide a realistic view of the morphological transition during the reaction process.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Agence Nationale de la Recherche (ANR-18-CE06–0014 CKAPART) is acknowledged for financial support. The authors thank Isabelle Morfin for her help on the ESRF: BM02 – D2AM French CRG Beamline. The assistance of Pierre-Yves Dugas and the members of “Centre Technologique des Microstructures, Université Lyon 1” (CTμ) for the generation of TEM images is also gratefully acknowledge.References
- F. D'Agosto, J. Rieger and M. Lansalot, RAFT-Mediated Polymerization-Induced Self-Assembly, Angew. Chem., Int. Ed., 2020, 59(22), 8368–8392 CrossRef PubMed.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, One-pot synthesis of poly(methacrylic acid-co-poly(ethylene oxide) methyl ether methacrylate)-b-polystyrene amphiphilic block copolymers and their self-assemblies in water via RAFT-mediated radical emulsion polymerization. A kinetic study, Macromolecules, 2011, 44(19), 7584–7593 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Polymerization-induced self-assembly: From soluble macromolecules to block copolymer nano-objects in one step, Macromolecules, 2012, 45(17), 6753–6765 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Predictive phase diagrams for RAFT aqueous dispersion polymerization: effect of block copolymer composition, molecular weight, and copolymer concentration, Macromolecules, 2012, 45(12), 5099–5107 CrossRef CAS.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polymerization techniques in polymerization-induced self-assembly (PISA), Polym. Chem., 2020, 11(22), 3673–3689 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, Emerging trends in polymerization-induced self-assembly, ACS Macro Lett., 2019, 8(8), 1029–1054 CrossRef CAS PubMed.
- X. Zhu, C. Vo, M. Taylor and B. R. Smith, Non-spherical micro-and nanoparticles in nanomedicine, Mater. Horiz., 2019, 6(6), 1094–1121 RSC.
- N. Daum, C. Tscheka, A. Neumeyer and M. Schneider, Novel approaches for drug delivery systems in nanomedicine: effects of particle design and shape, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4(1), 52–65 CAS.
- R. A. Meyer and J. J. Green, Shaping the future of nanomedicine: anisotropy in polymeric nanoparticle design, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8(2), 191–207 CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- S. Förster, M. Zisenis, E. Wenz and M. Antonietti, Micellization of strongly segregated block copolymers, J. Chem. Phys., 1996, 104(24), 9956–9970 CrossRef.
- J. Rieger, Guidelines for the synthesis of block copolymer particles of various morphologies by RAFT dispersion polymerization, Macromol. Rapid Commun., 2015, 36(16), 1458–1471 CrossRef CAS PubMed.
- M. Lansalot, J. Rieger and F. D'Agosto, Polymerization-Induced Self-Assembly: The Contribution of Controlled Radical Polymerization to The Formation of Self-Stabilized Polymer Particles of Various Morphologies. Macromolecular Self–assembly, John Wiley & Sons, Ltd, 2016, pp. 33–82. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/9781118887813.ch2 Search PubMed.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Toward a better understanding of the parameters that lead to the formation of nonspherical polystyrene particles via RAFT-mediated one-pot aqueous emulsion polymerization, Macromolecules, 2012, 45(10), 4075–4084 CrossRef CAS.
- X. Wang, S. Man, J. Zheng and Z. An, Alkyl α-Hydroxymethyl Acrylate Monomers for Aqueous Dispersion Polymerization-Induced Self-Assembly, ACS Macro Lett., 2018, 7(12), 1461–1467 CrossRef CAS PubMed.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh and R. T. Mathers, et al., Predicting Monomers for Use in Polymerization-Induced Self-Assembly, Angew. Chem., 2018, 130(48), 15959–15963 CrossRef.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams and M. W. Murray, et al., Effect of Monomer Solubility on the Evolution of Copolymer Morphology during Polymerization-Induced Self-Assembly in Aqueous Solution, Macromolecules, 2017, 50(3), 796–802 CrossRef CAS PubMed.
- P. Shi, H. Zhou, C. Gao, S. Wang, P. Sun and W. Zhang, Macro-RAFT agent mediated dispersion copolymerization: A small amount of solvophilic co-monomer leads to a great change, Polym. Chem., 2015, 6(27), 4911–4920 RSC.
- J. Zhou, W. Zhang, C. Hong and C. Pan, Promotion of morphology transition of di-block copolymer nano-objects via RAFT dispersion copolymerization, Polym. Chem., 2016, 7(19), 3259–3267 RSC.
- C. A. Figg, R. N. Carmean, K. C. Bentz, S. Mukherjee, D. A. Savin and B. S. Sumerlin, Tuning hydrophobicity to program block copolymer assemblies from the inside out, Macromolecules, 2017, 50(3), 935–943 CrossRef CAS.
- W. Zhang, F. D. Agosto, P. Y. Dugas, J. Rieger, B. Charleux and F. D'Agosto, et al., RAFT-mediated one-pot aqueous emulsion polymerization of methyl methacrylate in presence of poly(methacrylic acid-co-poly(ethylene oxide) methacrylate) trithiocarbonate macromolecular chain transfer agent, Polymer, 2013, 54(8), 2011–2019, DOI:10.1016/j.polymer.2012.12.028 . Available from: DOI.
- J. de la Haye, X. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, et al., The Effect of Hydrophile Topology in RAFT-Mediated Polymerization-Induced Self-Assembly, Angew. Chem., Int. Ed., 2016, 55(11), 3739–3743, DOI:10.1002/anie.201511159 . Available from: DOI.
- M. Huo, Z. Xu, M. Zeng, P. Chen, L. Liu and L.-T. Yan, et al., Controlling vesicular size via topological engineering of amphiphilic polymer in polymerization-induced self-assembly, Macromolecules, 2017, 50(24), 9750–9759 CrossRef CAS.
- M. Huo, M. Zeng, D. Wu, Y. Wei and J. Yuan, Topological engineering of amphiphilic copolymers via RAFT dispersion copolymerization of benzyl methacrylate and 2-(perfluorooctyl) ethyl methacrylate for polymeric assemblies with tunable nanostructures, Polym. Chem., 2018, 9(7), 912–919 RSC.
- X. Zhang, J. Rieger and B. Charleux, Effect of the solvent composition on the morphology of nano-objects synthesized via RAFT polymerization of benzyl methacrylate in dispersed systems, Polym. Chem., 2012, 3(6), 1502–1509 RSC.
- Y. Zhang, G. Han, M. Cao, T. Guo and W. Zhang, Influence of solvophilic homopolymers on RAFT polymerization-induced self-assembly, Macromolecules, 2018, 51(11), 4397–4406 CrossRef CAS.
- Y. Wu, M. Tan, Z. Su, Y. Shi, Z. Fu and W. Yang, In situ synthesis of PAA-b-PSt nano-assemblies via dispersion RAFT polymerization: effects of PEG in the medium, New J. Chem., 2018, 42(24), 19353–19356 RSC.
- X. Chen, L. Liu, M. Huo, M. Zeng, L. Peng and A. Feng, et al., Direct Synthesis of Polymer Nanotubes by Aqueous Dispersion Polymerization of a Cyclodextrin/Styrene Complex, Angew. Chem., Int. Ed., 2017, 56(52), 16541–16545 CrossRef CAS PubMed.
- P. Yang, Y. Ning, T. J. Neal, E. R. Jones, B. R. Parker and S. P. Armes, Block copolymer microparticles comprising inverse bicontinuous phases prepared via polymerization-induced self-assembly, Chem. Sci., 2019, 10(15), 4200–4208 RSC.
- S. Y. Khor, N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polymerization-Induced Self-Assembly: The Effect of End Group and Initiator Concentration on Morphology of Nanoparticles Prepared via RAFT Aqueous Emulsion Polymerization, ACS Macro Lett., 2017, 6(9), 1013–1019 CrossRef CAS PubMed.
- S. Boissé, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier and M. H. Li, et al., Amphiphilic block copolymer nano-fibers via RAFT-mediated polymerization in aqueous dispersed system, Chem. Commun., 2010, 46(11), 1950–1952 RSC.
- S. Boissé, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, Influence of the stirring speed and CaCl2 concentration on the nano-object morphologies obtained via RAFT-mediated aqueous emulsion polymerization in the presence of a water-soluble macroRAFT agent, J. Polym. Sci., Part A: Polym. Chem., 2011, 49(15), 3346–3354 CrossRef.
- D. Zhou, S. Dong, R. P. Kuchel, S. Perrier and P. B. Zetterlund, Polymerization induced self-assembly: tuning of morphology using ionic strength and pH, Polym. Chem., 2017, 8(20), 3082–3089 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Comparison of photo-and thermally initiated polymerization-induced self-assembly: a lack of end group fidelity drives the formation of higher order morphologies, Polym. Chem., 2017, 8(18), 2860–2871 RSC.
- W.-M. Wan and C.-Y. Pan, Formation of polymeric yolk/shell nanomaterial by polymerization-induced self-assembly and reorganization, Macromolecules, 2010, 43(6), 2672–2675 CrossRef CAS.
- F. Brunel, J. Lesage De La Haye, M. Lansalot and F. D'Agosto, New Insight into Cluster Aggregation Mechanism during Polymerization-Induced Self-Assembly by Molecular Dynamics Simulation, J. Phys. Chem. B, 2019, 123(30), 6609–6617 CrossRef CAS PubMed.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto and J. Rieger, et al., Well-defined amphiphilic block copolymers and nano-objects formed in situ via RAFT-mediated aqueous emulsion polymerization, Macromolecules, 2011, 44(11), 4149–4158 CrossRef CAS.
- X. G. Qiao, M. Lansalot, E. Bourgeat-Lami and B. Charleux, Nitroxide-mediated polymerization-induced self-assembly of poly(poly(ethylene oxide) methyl ether methacrylate-co-styrene)-b-poly(n-butyl methacrylate-co-styrene) amphiphilic block copolymers, Macromolecules, 2013, 46(11), 4285–4295 CrossRef CAS.
- N. P. Truong, J. F. Quinn, A. Anastasaki, M. Rolland, M. N. Vu and D. M. Haddleton, et al., Surfactant-free RAFT emulsion polymerization using a novel biocompatible thermoresponsive polymer, Polym. Chem., 2017, 8(8), 1353–1363 RSC.
- N. P. Truong, C. Zhang, T. A. H. Nguyen, A. Anastasaki, M. W. Schulze and J. F. Quinn, et al., Overcoming Surfactant-Induced Morphology Instability of Noncrosslinked Diblock Copolymer Nano-Objects Obtained by RAFT Emulsion Polymerization, ACS Macro Lett., 2018, 7(2), 159–165 CrossRef CAS PubMed.
- P. Galanopoulo, P.-Y. Dugas, M. Lansalot and F. D'Agosto, Poly (ethylene glycol)-b-poly (vinyl acetate) block copolymer particles with various morphologies via RAFT/MADIX aqueous emulsion PISA, Polym. Chem., 2020, 11(23), 3922–3930 RSC.
- X.-S. Chai, F. J. Schork, A. DeCinque and K. Wilson, Measurement of the solubilities of vinylic monomers in water, Ind. Eng. Chem. Res., 2005, 44(14), 5256–5258 CrossRef CAS.
- Z. Varga, A. Wacha, U. Vainio, J. Gummel and A. Bóta, Characterization of the PEG layer of sterically stabilized liposomes: a SAXS study, Chem. Phys. Lipids, 2012, 165(4), 387–392 CrossRef CAS PubMed.
- D. Kilburn, J. H. Roh, L. Guo, R. M. Briber and S. A. Woodson, Molecular crowding stabilizes folded RNA structure by the excluded volume effect, J. Am. Chem. Soc., 2010, 132(25), 8690–8696 CrossRef CAS PubMed.
- E. Kostyurina, J. U. De Mel, A. Vasilyeva, M. Kruteva, H. Frielinghaus and M. Dulle, et al., Controlled LCST Behavior and Structure Formation of Alternating Amphiphilic Copolymers in Water, Macromolecules, 2022, 55(5), 1552–1565 CrossRef CAS.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka and E. J. Cornel, et al., In situ small-angle X-ray scattering studies during reversible addition–fragmentation chain transfer aqueous emulsion polymerization, J. Am. Chem. Soc., 2019, 141(34), 13664–13675 CrossRef CAS PubMed.
- A. G. Liao, M. J. Derry, A. J. Smith and P. Steven, Determination of Reaction Kinetics by Time-Resolved Small-angle X-ray Scattering during Polymerization-Induced Self-Assembly: Direct Evidence for Monomer-Swollen Nanoparticles, Angew. Chem., Int. Ed., 2023, 63(2), e202312119 CrossRef PubMed.
- A. Czajka and S. P. Armes, In situ SAXS studies of a prototypical RAFT aqueous dispersion polymerization formulation: monitoring the evolution in copolymer morphology during polymerization-induced self-assembly, Chem. Sci., 2020, 11(42), 11443–11454 RSC.
- F. Brunel, J. Lesage De La Haye, M. Lansalot and F. D’Agosto, New Insight into Cluster Aggregation Mechanism during Polymerization-Induced Self-Assembly by Molecular Dynamics Simulation, J. Phys. Chem. B, 2019, 123(30), 6609–6617 CrossRef CAS PubMed.
- G. B. McKenna, Looking at the glass transition: Challenges of extreme time scales and other interesting problems, Rubber Chem. Technol., 2020, 93(1), 79–120 CrossRef CAS.
- C. Cruje and D.B. Chithrani, Polyethylene Glycol Density and Length Affects Nanoparticle Uptake by Cancer Cells, J. Nanomed. Res., 2014, 1(1), 8 Search PubMed.
- F. Piguet, H. Ouldali, F. Discala, M.-F. Breton, J. C. Behrends and J. Pelta, et al., High temperature extends the range of size discrimination of nonionic polymers by a biological nanopore, Sci. Rep., 2016, 6(1), 38675 CrossRef CAS PubMed.
- C. Debrie, N. Coudert, J. M. Guigner, T. Nicolai, F. Stoffelbach and O. Colombani, et al., Unimer Exchange Is not Necessary for Morphological Transitions in Polymerization-Induced Self-Assembly, Angew. Chem., Int. Ed., 2023, 62(8), e202215134 CrossRef CAS PubMed.
- ESRF. D2AM – BM02 overview. Available from: https://f-crg.fr/page-d-exemple/d2am-bm02/.
- J. H. Hubbell and S. M. Seltzer, X-Ray mass attenuation coefficients: NIST standard reference database 126, National Institute of Standards and Technology, Gaithersburg, Maryland, USA, 2004 Search PubMed.
- FUJIFILM Wako Chemicals. Azo Polymerization Initiators Comprehensive Catalog. Available from: https://specchem-wako.fujifilm.com/europe/en/water-soluble-azo-initiators/V-50.htm.
- F. Nettesheim, J. Zipfel, U. Olsson, F. Renth, P. Lindner and W. Richtering, Pathway of the shear-induced transition between planar lamellae and multilamellar vesicles as studied by time-resolved scattering techniques, Langmuir, 2003, 19(9), 3603–3618 CrossRef CAS.
- B. Demé, M. Dubois, T. Gulik-Krzywicki and T. Zemb, Giant collective fluctuations of charged membranes at the lamellar-to-vesicle unbinding transition. 1. Characterization of a new lipid morphology by SANS, SAXS, and electron microscopy, Langmuir, 2002, 18(4), 997–1004 CrossRef.
- https://myvideo.univ-lyon1.fr/videos/scattering-intensity-during-the-pisa-process/ .
- H. J. Kim, F. Ishizuka, S. Chatani, H. Niino and P. B. Zetterlund, Aqueous RAFT Polymerization-Induced Self-Assembly (PISA): Amphiphilic MacroRAFT Self-Assembly vs Monomer Droplet Nucleation (Miniemulsion Polymerization), Polym. Chem., 2023, 14(6), 687–696 RSC.
- D. D. Lasic, The mechanism of vesicle formation, Biochem. J., 1988, 256(1), 1–11 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental conditions for in situ SAXS monitoring of PEG45-xanthate mediated vinyl acetate emulsion polymerization. SEC traces of PEG45-b-PVAc200. Cryo-TEM images of the obtained particles. SAXS spectra and fitting parameter (in situ vs. static experiments). Calculation of the photo and chemical initiation rates. See DOI: https://doi.org/10.1039/d3py01381b |
| This journal is © The Royal Society of Chemistry 2024 |