
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transformation of poly(L-lactide) crystals composed of linear chains into crystals composed of cycles†
Steffen M.
Weidner
 a,
Andreas
Meyer
b and
Hans R.
Kricheldorf
*c
a,
Andreas
Meyer
b and
Hans R.
Kricheldorf
*c
aBundesanstalt für Materialforschung und -prüfung (BAM), Richard-Willstätter-Strasse 11, D-12489 Berlin, Germany
bUniversität Hamburg, Institut für Physikalische Chemie, Grindelallee 117, D-20146 Hamburg, Germany
cUniversität Hamburg, Institut für Technische and Makromolekulare Chemie der Universität, Bundesstr. 45, D-20146 Hamburg, Germany. E-mail: hrkricheldorf@aol.de
First published on 15th February 2024
Abstract
A poly(L-lactide) with a trifluoro ethyl ester end group and an average degree of polymerization (DP) of 50 was synthesized by ROP of L-lactide initiated with trifluoroethanol. Small-angle X-ray scattering (SAXS) in combination with differential scanning calorimetry (DSC) measurements revealed an average crystal thickness of 13 nm, corresponding to 45 repeat units. This suggests that most crystallites were formed by extended PLA chains, and both flat surfaces were covered by CF3 groups. The crystalline PLAs were annealed at 140 or 160 °C in the presence of two catalysts: tin(II) 2-ethylhexanoate, (SnOct2) or dibutyltin bis(pentafluorophenoxide) (BuSnPhF). The chemical reactions, such as polycondensation and cyclization, proceeded in the solid state and were monitored by matrix-assisted laser desorption/ionization time-of-flight (MALDI TOF) mass spectrometry and gel permeation chromatography (GPC) measurements. Under optimal conditions a large fraction of linear chains was transformed into crystallites composed of extended cycles. Additionally, MALDI TOF MS analysis of GPC fractions from samples annealed for 28 or 42 days detected chain elongation of the linear species up to a factor of 20.
Introduction
Over the past fifty years, poly(L-lactide), PLA, has been widely used as a biodegradable and resorbable material and has found numerous applications.1–5 Starting from a few tons per year, the technical production of PLA has now reached the level of 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year. This growth has led to extensive research in synthetic methods, characterization, and potential applications. The technical production is based on an alcohol-initiated ring-opening polymerization (ROP) catalyzed by tin(II) 2-ethylhexanoate (SnOct2). Alcohol-initiated ROPs have attracted much interest and have been studied by numerous research groups. These studies have explored variations in the structure of monofunctional alcohols, as well as the use of di- and multifunctional alcohols as initiators. Additionally, the influence of catalysts, including their concentration, time, and temperature, has been examined.6,7
000 tons per year. This growth has led to extensive research in synthetic methods, characterization, and potential applications. The technical production is based on an alcohol-initiated ring-opening polymerization (ROP) catalyzed by tin(II) 2-ethylhexanoate (SnOct2). Alcohol-initiated ROPs have attracted much interest and have been studied by numerous research groups. These studies have explored variations in the structure of monofunctional alcohols, as well as the use of di- and multifunctional alcohols as initiators. Additionally, the influence of catalysts, including their concentration, time, and temperature, has been examined.6,7
The addition of alcohols as initiators has resulted in four potential advantages or disadvantages, depending on the intended application of the resulting PLA. First, the alcohol accelerates the polymerization process, at least at temperatures below 120 °C.6 Second, the molecular mass can be controlled by the lactide/alcohol (LA/In) ratio, at least for LA/In ratios < 200/1.8–10 Third, low dispersities (D < 1.3) can be achieved at polymerization temperatures around or below 120 °C, because in the case of primary alcohols the initiation step is faster than the propagation steps for steric and electronic reasons (Scheme 1). Fourth, the reactivity of the end group resulting from the incorporation of the initiator can be varied by the structure of the alcohol.11 These last three points provide the background for the present study.
 | ||
| Scheme 1 Alcohol-initiated ROP of lactide catalyzed by SnOct2. | ||
In a recent publication, the authors demonstrated that the OH and ethyl ester end groups of an ethyl lactate-initiated PLA allow for numerous solid-state transesterification reactions in the presence of reactive polymerization (transesterification) catalysts.12 These reactions occur on the surface of the crystallites and can lead to chain growth reactions, cyclization and modification of end groups. These results open a new field of work, because crystallites with well-defined surfaces can be prepared when the length of the PLA chains does not significantly exceed the thickness of the crystallites. In this case, the crystallites are almost exclusively formed by extended chains, and only a few long chains will fold upon crystallization. The two flat surfaces of the lamellar crystallites are then covered by an alternating arrangement of the two different end groups, as illustrated in Scheme 2 for a trifluoroethanol-initiated PLA. This scheme is, of course, a simplification, as not all chain ends sticking out from the surface will have identical chain lengths. In this context, a review dealing with end-to-end cyclization of polymers including tin-catalyzed polymerizations of lactide should be mentioned.13
 | ||
| Scheme 2 Schemes of crystallites based on PLAs initiated with trifluoroethanol, and chain growth via formation of loops by condensation of neighboring end groups (alcoholytic transesterification). | ||
It was known from ethyl lactate-initiated ROPs that the crystallite thickness is in the range of 12.5–13.5 nm. It was also known from the X-ray studies of Wasanasuk and Tashiro that ten lactyl units forming a 103 helix in the α modification have a length of 2.9 nm.14 Therefore, PLAs with a polymerization degree of approximately 40–45 lactyl units can fit into a crystal thickness of 12.5–13.5 nm. In the present work, PLAs with an average DP of 50 were prepared using an LA/initiator ratio of 25/1, taking into account that some lactyl units will protrude from the surface of the crystallites. In this context, the present work served three purposes. First, to determine whether trifluoro-ethanol and hexafluoro-isopropanol could be used as initiators to achieve quantitative incorporation of these alcohols and low dispersities of the resulting PLAs. The success of these experiments was not predictable, as it was found that neither quantitative incorporation nor low dispersities were achieved when acidic phenols were used as initiators. Second, the sensitivity of the fluorinated surfaces to transesterification reactions should be elucidated. One objective of this study was to determine whether the higher electrophilicity of the fluorinated ester end groups allows for the formation of extended ring crystallites by efficiently forming loops over both surfaces (see Scheme 3). First results in this direction, albeit with low efficiency, were obtained by annealing PLA ethyl esters in the presence of different tin catalysts. Third, two different catalysts were compared, namely tin(II) 2-ethylhexanoate (SnOct2) and dibutyltin bis(pentafluoro-phenoxide) (BuSnPhF). In recent experiments on transesterification with poly(L-lactide) ethyl esters, BuSnPhF was found to be the superior catalyst for alcoholytic transesterification (condensation, Scheme S1†), while SnOct2 was the better catalyst for ester–ester interchange reactions (Scheme S2†).
 | ||
| Scheme 3 Formation of crystallites composed of extended cycles via formation of loops on both sides of a crystallite and via the wandering of loops across the surface. | ||
Finally, it should be mentioned that the acid-catalyzed polycondensation and solid-state polycondensation of free lactic acid has been studied by several research groups.15–18 However, none of these groups have formulated or discussed individual reaction steps, nor have they used MALDI-TOF mass spectrometry to gain insight into the chain growth mechanisms.
Experimental
Materials
L-Lactide was supplied by Thyssen-Uhde SE (Berlin, Germany). It was recrystallized from toluene (99.89% extra dry, Thermo-Scientific, Germany). Anhydrous pyridine and anhydrous dichloromethane were also purchased from Thermo-Scientific Fisher (Schwerte, Germany). Trifluoroethanol, and tin(II) 2-ethylhexanoate (SnOct2) were purchased from Alfa Aesar (Kandel, Germany) and used as received. Dibutyltin-bis(pentafluorophenoxide), BuSnPhF, was prepared as described previously.19ROP of LA with 2,2,2-trifluoroethanol as initiator (LA/In = 25/1; LA/Cat = 200/1)
(A) TFE (4 mmol) and lactide (100 mmol) were weighed into a flame-dried 100 mL Erlenmeyer flask under a blanket of argon. SnOct2 (0.5 mmol) was injected in the form of a 1 M solution in toluene and a magnetic bar was added and toluene (90 mL, roughly corresponding to a 1 M solution). The reaction vessel was immersed into an oil bath thermostated at 80 °C and after 22 h the reaction mixture was precipitated into ligroin (1 L). The precipitated PLA was isolated by filtration and dried at 50 °C in vacuo. Yield: 95%. Mn = 7800, Mw = 9500 (Đ = 1.2) The 1H NMR spectrum (Fig. S1†) displays the CH–OH quadruplet at 4.35 ppm as usual and two OCHCF3 multiplet signals at 4.45 and 4.6 ppm (along with traces of unreacted LA at 5.05 ppm).Measurements
The MALDI TOF mass spectra were measured with an Autoflex Max mass spectrometer (Bruker Daltonik, Bremen, Germany). All spectra were recorded in the positive ion linear mode. The MALDI stainless steel targets were prepared from chloroform (HPLC grade, Carl Roth, Germany) solutions of poly(L-lactide) (3–5 mg mL−1) doped with potassium trifluoroacetate (Sigma-Aldrich, 2 mg mL−1 in THF). Typically, 20 μL of the sample solution, 2 μL of the potassium salt solution and 50 μL of the matrix solution (DCTB – trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile, Sigma-Aldrich, 20 mg mL−1 in CHCl3) were pre-mixed in an Eppendorf vial. A droplet (1 μL) of this solution was deposited on the MALDI target and, after evaporation of the solvent, inserted in the mass spectrometer. 8000 single spectra were recorded and accumulated from 4 different places of each spot.The GPC measurements were performed in chloroform (HPLC grade, Carl Roth, Germany) in a LC 1200 (Agilent, USA) instrument kept at 40 °C. Three Phenogel (Phenomenex, Aschaffenburg, Germany) columns (1 × 105 Å; 1 × 103 Å; 1 × 100 Å, 5 μm pore size, 7.8 × 300 mm) were used for separation along with a SecurityGuard cartride (5 μm, 4 × 3 mm). The flow rate was 1 mL min−1. A refractive index detector was used for detection. Samples were automatically injected (100 μL, 2–4 mg mL−1 in chloroform). For instrument control and data calculation Win GPC software (Polymer Standards Service – PSS, Mainz, Germany) was applied. The calibration was performed using polystyrene standard sets (Polymer Standards Service – PSS, Mainz). The number average (Mn) and weight average (Mw) masses listed in tables are uncorrected. The GPC elution curves of all samples listed in Table 1 are displayed in Fig. S2–S4 (ESI part†). Fractionation experiments were done manually by collecting the eluents at the end of the capillary using glass vials. 20 μL of these solutions were taken without further concentration and premixed with 20 μL of the matrix/salt solution before spotting onto the target plate.
| Exp. no. | LA/Cat. | Temp. (°C) | Time (d) | M n | M w | T m (°C) | ΔHm (J g−1) | Cryst.b (%) |
|---|---|---|---|---|---|---|---|---|
a Synthesis and annealing of the starting material.
b Calculated from ΔHm with  . .
|
||||||||
| 0a | 200/1 | 80/120 | 1/1 | 7800 | 9500 | — | — | — |
| 1A | 1000/1 | 140 | 14 | 16300 |
29300 |
177.1 | 86.7 | 74 |
| 1B | 1000/1 | 140 | 28 | 17600 |
32500 |
177.3 | 97.5 | 85 |
| 2A | 1000/1 | 160 | 7 | 19000 |
37500 |
181.3 | 92.5 | 80 |
| 3A | 500/1 | 160 | 7 | 21500 |
40000 |
169.5 | 71,6 | 62 |
| 3 | 500/1 | 160 | 14 | 23000 |
43500 |
180.3 | 86.4 | 84 |
| 3C | 500/1 | 160 | 28 | 28500 |
53000 |
186.3 | 92.6 | 80 |
| 4A | 250/1 | 160 | 7 | 22500 |
42000 |
179.1 | 93.4 | 81 |
| 4B | 250/1 | 160 | 14 | 33500 |
64000 |
183.8 | 97.5 | 84 |
| 4C | 250/1 | 160 | 28 | 35500 |
67000 |
185.2 | 92.3 | 80 |
| 4D | 250/1 | 160 | 42 | 31000 |
66500 |
185.5 | 93.7 | 82 |
The SAXS measurements were performed using our in-house SAXS/WAXS apparatus equipped with an Incoatec™ X ray source IμS and Quazar Montel optics. The wavelength of the X-ray beam was 0.154 nm and the focal spot size at the sample position was 0.6 mm2. The samples were measured in transmission geometry and were recorded with a Rayonix™ SX165 CCD-Detector. The SAXS measurements were performed at sample-detector distance of 1.6 m and the accumulation was 20 minutes. DPDAK, a customizable software for reduction and analysis of X-ray scattering data sets was used for gathering 1D scattering curves.20 The SAXS curves were converted into Kratky plots. The long periods of the lamellar domains were determined by the q values of the reflection maxima.
Results and discussion
Synthesis of PLAs having trifluoro-ethyl ester end groups
A first attempt to prepare a PLA with trifluoro-ethyl ester end groups (PLA-TFE) by means of trifluoro-ethanol and SnOct2 in bulk at 120 °C failed. The isolated PLA was bare of any trifluoro-ethyl end groups. Apparently, the evaporation of the low boiling trifluoroethanol (b.p. 75 °C) was much faster than its consumption by the initiation reaction. Therefore, a second attempt was performed in toluene at 80 °C, which was successfully used by Jalabert et al. for the ROP of LA initiated with alkanols.9 This experiment yielded the desired linear PLA-TFE with low dispersity (Đ = 1.2) as demonstrated by the MALDI-TOF mass spectrum in Fig. 1A. Repetition of this experiment proved its reproducibility, but an analogous experiment with hexafluoro-isopropanol failed, because no polymerization occurred. The SEC measurements of the PLA-TEF in chloroform resulted in a number average molecular weight (Mn) of 7500 which was higher than the intended value of 3700 (corresponding to a DP of 50), even when it is taken into account that polystyrene calibrated SEC measurements in chloroform overestimate the real Mn's of PLA by 50–55%. Obviously, the ROP in toluene was accompanied by several polycondensation steps. The small fraction of longer chains did not appear in the MALDI TOF mass spectrum, which showed a maximum at the expected Mn around m/z 3700 (Fig. 1A). This assumption is confirmed by the quantification of the 1H NMR end group signals (Fig. S1†) which indicates a degree of polymerization around 33–35, rather than 25. The GPC elution curve exhibited a weak shoulder at higher molecular masses (Fig. 1B) which could be attributed to polycondensation reactions. This shoulder is responsible for the relatively high molecular masses measured by GPC. | ||
| Fig. 1 PLA initiated with trifluoroethanol: LA/In = 25/1 (A), MALDI TOF mass spectrum (B) GPC elution curve. | ||
Annealing with addition of SnOct2
Considering the recent experiments with PLA ethyl ester, the first two annealing experiments were performed with a LA/Cat ratio of 1000/1 at 140 °C. PLA samples were isolated and characterized after 14 and 28 h. A first interesting result was a significant increase of the molecular masses accompanied by a broadening of the molecular mass distribution (1A and B, Table 1). The GPC curves (Fig. S2, ESI part†) demonstrate a broadening, and a tendency towards a multimodal character resulting from a coupling of chains having the length of the starting material (see discussion of fractionation experiments below). The 1H NMR spectra confirmed the consumption of end groups, showing a loss of end groups by a factor of 4 after annealing at 140 °C for 14 days and a loss by a factor of 6 after 28 days. The apparent DPs based on the end group signals do not reflect the average chain lengths due to the inability of 1H NMR spectra to distinguish between cycles and linear chains. As a result, the apparent DPs are higher than the Mn values determined by GPC measurements.The MALDI TOF mass spectra showed the formation of a weak new maximum around m/z 7000, which doubled the molecular mass of the starting material (Fig. 2A). However, the peaks of this new maximum originate from cycles and display a “saw-tooth pattern”. When, as described recently,21 a PLA ethyl ester (prepared with an LA/In ratio of 30/1) is annealed with SnOct2 under the same conditions, again a new weak maximum is formed around m/z 8000, as shown in Fig. 2B.
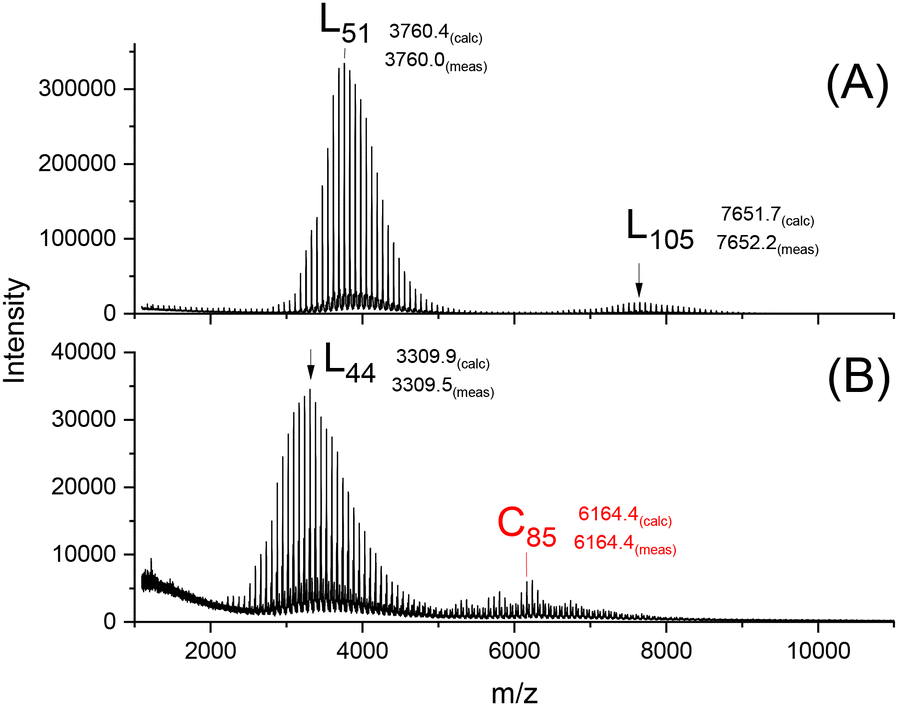 | ||
| Fig. 2 MALDI-TOF mass spectra of PLA-esters annealed with SnOct2 (LA/Cat = 1000/1) at 140 °C for 28 d: (A) ethyl ester end group (from ref. 12), (B) trifluoro ethyl ester end group (1B, Table 1). | ||
Yet, this maximum exclusively consists of linear chains. Therefore, these experiments demonstrate, that the formation of loops through condensation reactions, as outlined in Scheme 3, is much more efficient, when trifluoro-ethyl ester end groups are involved. Based on this result, additional annealing experiments were performed at 160 °C with variations in time and catalyst concentration. The maximum temperature that can be used for a long-term annealing of PLA is 160 °C, to avoid side reactions, particularly racemization, which can affect the results. At first, the LA/Cat ratio of 1000/1 was maintained, and the temperature was varied from 7 to 14, and finally 28 days (2A–C, Table 1). As expected, the fraction of cyclic PLAs with masses around m/z 7000 significantly increased. In seven more experiments the concentration of SnOct2 was enhanced by a factor of 2 (LA/Cat = 500/1, 3A–C, Table 1), and finally by a factor of 4 (LA/Cat = 250/1, 4A–C, Table 1). The experiments with an LSA/Cat ratio of 500/1 revealed higher fractions of the cyclic species compared to those conducted with LA/Cat = 1000/1. Further progress was achieved with a LA/Cat ratio of 250/1 (Fig. 3 and 4).
 | ||
| Fig. 3 MALDI TOF mass spectra of PLA-TFE annealed with SnOct2 (LA/Cat = 250/1) at 160 °C: (A) after 7 d (4A, Table 1), (B) after 14 d (4B, Table 1). | ||
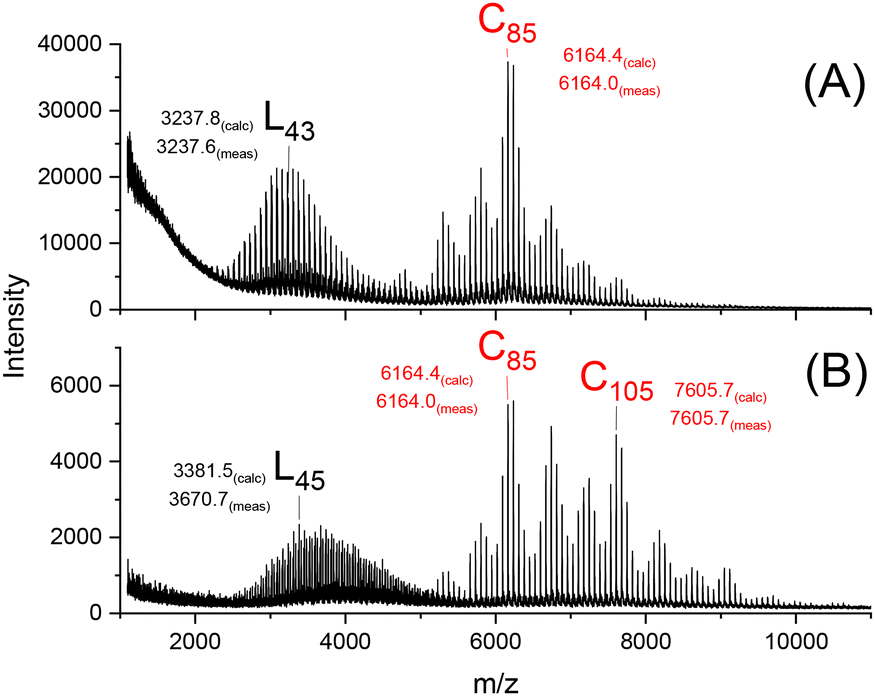 | ||
| Fig. 4 MALDI TOF mass spectra of PLAs annealed with SnOct2 (LA/Cat = 250/1) at 160 °C: (A) after 28 d (4C, Table 1), (B) after 42 d (4D, Table 1). | ||
A comparison of Fig. 4A with Fig. 2A demonstrates the enormous progress in the transformation of “linear crystals” into “cyclic crystals” by the combination of higher temperature and higher catalyst concentration. The spectra in Fig. 3 and 4 also illustrate the strong influence of the reaction time. The transformation of the “linear crystallites” into the crystallites composed of extended cycles proceeds even after 28 d, but the limited thermal stability of PLA hindered a complete transformation at reaction times above 42 d.
To better understand whether and to what extent polycondensation reactions occur at 160 °C and above m/z 10000, the sample with the highest catalyst concentration and the longest reaction time (4D, Table 1) was subjected to fractionation by GPC, followed by characterization of the individual fractions by mass spectrometry. The elution curve with the fractionation scheme (12 fractions) is shown in Fig. S5 (ESI†). As shown in Fig. 5 and 6, this fractionation revealed the existence of several maxima in the molecular mass distribution, and even a flat maximum was detected around m/z 63000 (fraction 3). Up to fraction 9, the maxima clearly represent a doubling or tripling of the original Mn of the starting material.
 | ||
| Fig. 5 MALDI TOF mass spectra of GPC fractions of PLA-TFE annealed with SnOct2 at 160 °C for 28 d (LA/Cat = 250/1): fractions 12–9 showing peak resolution. | ||
 | ||
| Fig. 6 MALDI TOF mass spectra of GPC fractions of PLA-TFE annealed with SnOct2 at 160 °C for 28 d (LA/Cat = 250/1): fractions 7–3 with lower mass resolution and fitted Mn values. | ||
Such a trend was also found in the case of PLA ethyl ester in the mass range below m/z 20000. This finding can be explained by chain growth via formation of loops over the surface of the crystallites. The maximum at m/z 63000 indicates that the Mn of the starting material (3700 Da) has been multiplied by a factor of 17, demonstrating that the polycondensation process over the surface of the crystallites is highly efficient when the trifluoro ethyl ester group forms the chain end. When the 42-day annealed PLA became available, it was also fractionated, into 20 fractions and characterized by mass spectrometry, as documented in Fig. S5–S7.† This fractionation confirmed the previous results and revealed a flat maximum around m/z 80000–82000, indicating a 22(±1) increase in the polycondensation of the starting material. The usefulness of this fractionation for constructing a calibration curve is discussed below.
The fractionations revealed some more interesting results about the nature and origin of the cyclic PLAs. Firstly, the STP is only detectable in the mass range between m/z 5000 and 10000. Secondly, linear and cyclic PLAs crystallized separately, as evidenced by the different shapes of their distribution curves. Separate crystallization of cyclic and linear PLAs has already been demonstrated in recent publications for PLAs prepared under quite different reaction conditions. The origin and significance of the STP will be discussed in more detail below.
Annealing with addition of BuSnPhF
A second set of annealing experiments was performed with BuSnPhF (Table 2). Again, two experiments were conducted at 140 °C with a LA/Cat ratio of 1000/1 for comparison with the SnOct2 experiments and previous studies of PLA ethyl ester. The mass spectra of the resulting PLAs were nearly identical to those obtained in the SnOct2 catalyzed experiments, as shown by the comparison of Fig. 7A with the mass spectrum of Fig. 2B. Therefore, the results obtained at 140 °C do not require further discussion. The experiments carried out at 160 °C with an LA/Cat ratio of 1000/1 (2, Table 2) or with a ratio of 250/1 (3, Table 2) yielded results that differed significantly from those obtained for SnOct2-catalyzed annealing under identical conditions. A comparison of Fig. 7B with Fig. 4A illustrates the difference between BuSnPhF and SnOct2. | ||
| Fig. 7 MALDI TOF mass spectra of PLAs annealed with BuSnPhF (LA/Cat = 1000/1): (A) 140 °C per 28 d (1B, Table 2), (B) 160 °C per 7 d (2, Table 2). | ||
| Exp no. | La/Cat. | Temp. (°C) | Time (d) | M n | M w | T m (°C) | ΔHm (J g−1) | Cryst.a (%) |
|---|---|---|---|---|---|---|---|---|
a Calculated from ΔHm with.  . .
|
||||||||
| 0 | 200/1 | 80/120 | 1/1 | 7800 | 9500 | — | — | — |
| 1A | 1000/1 | 140 | 14 | 15600 |
26800 |
177.2 | 74.0 | 66 |
| 1B | 1000/1 | 140 | 28 | 17000 |
30500 |
177.0 | 93.7 | 82 |
| 2 | 1000/1 | 160 | 7 | 24300 |
50000 |
184.3 | 66.6 | 58 |
| 3 | 250/1 | 160 | 7 | 22200 |
47000 |
183,6 | 62.6 | 54 |
When BusnPhF was added as a catalyst at 160 °C, decomposition of the PLA-TFE occurred, as indicated by a brownish discoloration after 7 days and by the formation of a brownish syrup when the experiment was extended to 14 days. According to the mass spectrum (see mass region below m/z 4000 in Fig. 7B), this brown syrup consisted mainly of low molar mass degradation products whose end groups could not be identified (see also mass region below m/z 4000 in Fig. 7B). The early loss of crystallinity is most likely caused by partial racemization. According to the work of Bigg et al., 7% of D-units in a poly(L-lactide) chain can lower the Tm below 160 °C, and at a content of 20% the crystallinity disappears entirely.22 A more comprehensive study of degradation reactions was not within the scope of this work. These results demonstrated that SnOct2 was the superior catalyst in this study, contrary to previous investigations of PLA ethyl esters.
Origin and meaning of the “saw tooth pattern”
Recently, numerous PLAs have been synthesized, which exhibit STP in MALDI-TOF mass spectra. The origin and significance of this unusual mass distribution have been intensively discussed.23–27 To ensure a plausible and consistent interpretation of the present study, a brief summary of these previous results is necessary. It should be noted that STPs are unique to cyclic PLAs and have never been observed in linear PLAs. STPs were not observed when cyclic PLAs were precipitated from solution or quenched from the melt. The formation of STPs requires annealing of crystallized cyclic PLAs for one or more days. STPs are limited to masses below m/z 20000 and usually cover the mass range of m/z 4000–15000. STPs result from thermodynamically controlled surface modification of extended-ring crystals. To fully understand the results presented in this work, it is crucial to note that extended-ring crystallites are the thermodynamically optimal form of crystalline PLA for masses below m/z 20000. This conclusion is supported by both experimental findings and theoretical arguments. Firstly, the ring size and length of the extended rings align with the thickness of the crystallites, as determined by SAXS measurements. Secondly, STPs are always found together with a new maximum in the mass distribution of cyclic PLAs. This indicates a thermodynamically controlled rearrangement of the mass distribution in favor of cyclic PLAs with masses ranging from m/z 4000 to 15000. The three theoretical arguments are as follows: first, the linear segments of the cycles necessarily adopt the thermodynamically most stable antiparallel orientation. Second, there are no defects inside the crystallites due to buried end groups. Third, a surface consisting of loops of nearly equal size is the smoothest and thermodynamically most favorable surface. Scheme 4 presents a simplified comparison between extended-ring crystallites (A) and crystallites that are formed by linear chains (B).
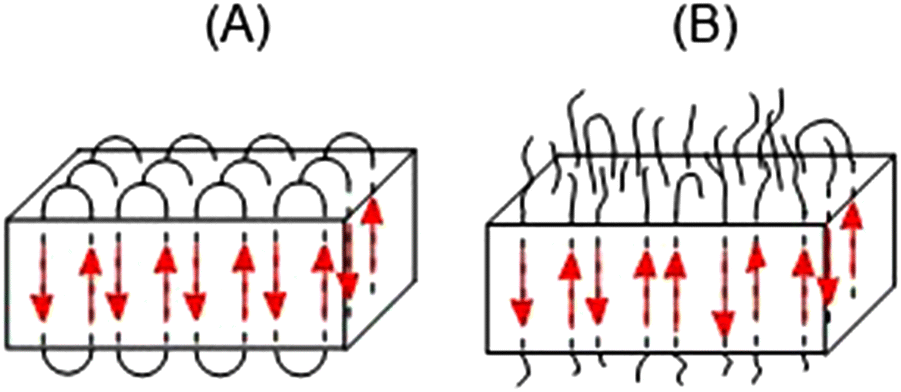 | ||
| Scheme 4 Supposed structure of extended ring crystal (A) and crystals composed of linear chains (B). | ||
Each tooth of an STP represents a population of crystallites defined by the same ring size and ring size distribution. The most prominent mass peak indicates the most abundant species in this population. It is accompanied by rings having one or two lactyl units more or less in the same cycle. Therefore, all loops have nearly the same size, resulting in a smooth surface. The formation of a STP is a secondary step that follows the formation of extended-ring crystals. It has the unique analytical advantage of allowing for the unambiguous identification of extended-ring crystals. Since all transesterification reactions are reversible in the presence of an active catalyst, the thermodynamic properties of the system determine the overriding trend, which is the formation of extended-ring crystals with a smooth surface.
As schematically illustrated in Scheme 3 the reversibility of transesterification reactions enables a wandering of loops across the surface of the crystallites, so that cycles can accumulate in a corner of the crystallite. This nucleus grows with time until the entire crystallite consists of extended cycles. One may hypothesize that the amorphous contributes to the growth of extended-ring crystals. Linear chains can dissociate laterally from the crystallites and cyclize in the amorphous phase. The cycles can then join the small surfaces and continue the lateral growth of the extended-ring crystallites. This sequence of physical and chemical steps is a complementary route to the formation of extended-ring crystals at the expense of the thermodynamically less stable “linear crystals” (Scheme 4B).
Finally, it should be noted that several research groups have reported that low molar cyclic poly(ethylene oxide)s, cyclic poly(ε-caprolactone), and poly(L-lactide) nucleate and crystallize faster than their linear counterparts with similar molecular masses. Therefore, the formation of extended-ring crystallites is not only thermodynamically favored but also kinetically favored. One consequence of this scenario is that cyclic and linear PLAs crystallize separately from the same reaction mixture, regardless of whether the polymerization proceeds in solution or in the melt. These findings support the conclusion that it is possible to transform crystallites consisting of linear PLAs into crystallites composed of extended cycles.
GPC measurements
All the GPC elution curves recorded from the annealed PLA samples show significant chain growth of the starting material, consistent with the fractionation message. Furthermore, the elution curves of all annealed PLA samples display a bimodal structure, as shown in Fig. S5 and S8,† along with the fractionation scheme based on the Mp (molecular mass at the maximum of the distribution) values from the mass spectra depicted in Fig. 5, 6 and S6, S7.† The multimodal character observed in the recently reported annealed PLA ethyl ester was never found in this work. As shown in Fig. 5 and 6, the fractionation allowed the identification of individual maxima in the MWD and the determination of their exact masses. This information was then used to construct a calibration curve based on GPC. This calibration curve is illustrated in Fig. S9(B)† together with the calibration curve based on commercial polystyrene standards. The two curves differ because polystyrene calibration overestimates the molecular masses of aliphatic polyesters. This is due to the lower hydrodynamic volume of the more compact coils of polystyrene chains. In the case of linear PLAs, Kowalski et al. reported a correction factor of 0.68 for calculating the true molecular masses from polystyrene-calibrated Mn and Mw data in dichloromethane.28 Comparison of the calibration curves shown in Fig. S9† gives a correction factor of 0.67 for measurements in chloroform, which is consistent with the literature value.SAXS measurements
SAXS measurements were performed on PLA samples 4A–D (Table 1) prepared with SnOct2 due to the continuous and significant change in topology and molecular mass distribution of PLA observed in their mass spectra. Surprisingly, only diffuse scattering was observed, in contrast to the sharp reflections seen in the SAXS curves of annealed PLA ethyl esters, which include a second and third order reflection. These higher order reflections indicate a high degree of 3D order within the spherulites. Clear reflections including a second order reflection were also observed for annealed cyclic PLAs. In contrast to these published PLAs, the reaction products obtained in this work contain three different types of crystallites: crystallites composed of extended linear chains with Mn around 3700 Da, crystallites containing log-folded linear chains, and crystallites composed of extended cycles. Apparently, this mixture of different crystallites cannot form an ordered layered structure with the amorphous phase inside the spherulites.In summary, the results obtained by annealing PLA trifluoro ethyl ester with BuSnPhF at 160 °C, as well as the mass spectra, GPC measurements, and SAXS measurements of the samples annealed with SnOct2, show significant differences from those obtained by annealing a PLA ethyl ester of similar molecular masses.
Conclusions
The study shows that PLA with trifluoro ethyl ester end groups can undergo intense transesterification reactions in the solid state, including alcoholytic condensation reactions and ester–ester exchange reactions. These reactions involve the formation and reorganization of loops on the surface of the crystallites and have three significant consequences. The low molecular mass PLA esters undergo polycondensation reactions, resulting in a chain extension of up to a factor of 21(±1). Subsequently, cyclization reactions take place, leading to the formation of extended ring crystals in the mass range below m/z 20000. Within the observed time of 42 days, approximately 90% of the starting material was converted to cyclic or longer linear polylactides. The third significant finding is the separate crystallization of linear and cyclic PLAs, a phenomenon that has been observed in PLAs with other ester end groups. In summary, the chemical structure, average molar masses, and molecular weight distribution of PLAs can be varied over a broad range, which affects their chemical and physical properties. Although studies in this direction are in their infancy, it is important to explore to what extent other polyesters can be modified by transesterification in the solid state. For instance, the study shows that controlling the DP of polyglycolide through the monomer/initiator ratio of alcohol-initiated ROPs is not possible due to rapid polycondensation and cyclization in the solid state interfering with the ROP. Furthermore, recent results shed new light on the mobility of the disordered layer on the surface of crystallites. Studies of the mobility of metal compounds across the surface of crystallites and in the neighbouring amorphous phase may attract the interest of physicochemists.
PLA is particularly well-suited for such studies for two reasons. First, MALDI TOF mass spectrometry allows for differentiation between crystals consisting of cycles and crystals composed of linear chains. Secondly, the Tm is high enough to enable efficient transesterification reactions in the solid state, unlike low-melting polylactones. However, it is not too high, so that thermal decomposition in long term experiments can be avoided.
Author contributions
Steffen M. Weidner: methodology, investigation, visualization, writing – review & editing. Andreas Meyer: investigation, visualization. Hans R. Kricheldorf: conceptualization, investigation, methodology, writing – original draft, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank A. Myxa (BAM, Berlin) for the GPC measurements and Stefan Bleck (Univ. of Hamburg) for the DSC data.References
- M. L. Di Lorenzo and R. Androsch, in Advances in Polymer Science, 279, ed. M. L. Di Lorenzo and R. Androsch, Springer International Publishing, Cham, 2017 Search PubMed.
- L.-T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. López-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS.
- R. A. Auras, L.-T. Lim, S. E. Selke and H. Tsuji, Poly (lactic acid): synthesis, structures, properties, processing, and applications, John Wiley & Sons, 2011 Search PubMed.
- M. L. Di Lorenzo and R. Androsch, in Advances in Polymer Science, 282, ed. M. L. Di Lorenzo and R. Androsch, Springer International Publishing, Cham, 2018 Search PubMed.
- H. R. Kricheldorf and S. M. Weidner, Polym. Chem., 2022, 13, 1618–1647 RSC.
- S. Corneillie and M. Smet, Polym. Chem., 2015, 6, 850–867 RSC.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 7359–7370 CrossRef CAS.
- M. Jalabert, C. Fraschini and R. E. Prud'Homme, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1944–1955 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Macromol. Chem. Phys., 2022, 223, 2100464 CrossRef CAS.
- S. M. Weidner and H. R. Kricheldorf, J. Polym. Sci., 2022, 60, 785–793 CrossRef CAS.
- S. M. Weidner, A. Meyer, J. Falkenhagen and H. R. Kricheldorf, Polym. Chem., 2024, 15, 71–82 RSC.
- H. R. Kricheldorf and S. M. Weidner, Macromol. Rapid Commun., 2020, 41, e2000152 CrossRef PubMed.
- K. Wasanasuk and K. Tashiro, Polymer, 2011, 52, 6097–6109 CrossRef CAS.
- G. Shin, J. H. Kim, S. H. Kim and Y. H. Kim, Korea Polym. J., 1997, 5, 19–25 CAS.
- S.-I. Moon, C.-W. Lee, I. Taniguchi, M. Miyamoto and Y. Kimura, Polymer, 2001, 42, 5059–5062 CrossRef CAS.
- S.-I. Moon, I. Taniguchi, M. Miyamoto, Y. Kimura and C.-W. Lee, High Perform. Polym., 2001, 13, S189–S196 CrossRef CAS.
- V. Katiyar and H. Nanavati, Polym. Eng. Sci., 2011, 51, 2078–2084 CrossRef CAS.
- H. R. Kricheldorf and S. M. Weidner, Eur. Polym. J., 2018, 109, 360–366 CrossRef CAS.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli and P. Müller-Buschbaum, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- S. M. Weidner, A. Meyer, J. Falkenhagen and H. R. Kricheldorf, Polym. Chem., 2024, 15(2), 71–82 RSC.
- D. M. Bigg , presented in part at the Annual Technical Conference of the Society of Plastic Engineers, 1996.
- H. R. Kricheldorf and S. M. Weidner, Polymer, 2023, 276, 125946 CrossRef CAS.
- S. M. Weidner, A. Meyer and H. R. Kricheldorf, Polymer, 2023, 285, 126355 CrossRef CAS.
- S. M. Weidner, A. Meyer and H. Kricheldorf, Polymer, 2022, 255, 125142 CrossRef CAS.
- H. R. Kricheldorf, S. M. Weidner and A. Meyer, Polymer, 2022, 263, 125516 CrossRef CAS.
- H. R. Kricheldorf, F. Scheliga and S. M. Weidner, Macromol. Chem. Phys., 2023, 224, 2300070 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 689–695 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01370g |
| This journal is © The Royal Society of Chemistry 2024 |