
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePillar[5]arene-based dually crosslinked supramolecular gel as a sensor for the detection of adiponitrile†
Maksim
Rodin
 ,
David
Helle
and
Dirk
Kuckling
*
,
David
Helle
and
Dirk
Kuckling
*
Department of Chemistry, Paderborn University, Warburger Str. 100, 33098 Paderborn, Germany. E-mail: dirk.kuckling@uni-paderborn.de
First published on 11th January 2024
Abstract
Host–guest inclusion complex formation between pillar[5]arene (P5A) and various guest molecules (amine- and heterocycle-substituted N-Boc-aminohexanes) in CDCl3 was investigated using NMR titration as well as 2D NMR (NOESY and DOSY) techniques. Supramolecular complexes with methylimidazolium (MIHA) and pyridinium substituted guests were demonstrated to have the highest binding affinity towards P5A (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka = 3.32 ± 0.10, 3.66 ± 0.09, respectively). Dimethyl acrylamide-2-vinyl-4,4-dimethyl azlactone copolymer was modified with mono-substituted P5A and MIHA as well as a photo-crosslinker based on 3,4-dimethyl maleimide to obtain host and guest polymers, which were combined and spin-coated onto a gold-coated quartz substrate to fabricate a dually crosslinked supramolecular gel (DCSG) containing reversible and irreversible crosslinks in a single network. Such DCSG chip was utilized as a sensor for the detection of adiponitrile (AN) in organic solutions based on a competitive complex formation with P5A. Macroscopic changes in the gel (e.g., in swelling degree or in refractive index) upon presence of AN (1 μM–1 mM) in chloroform were monitored with surface plasmon resonance (SPR) spectroscopy. The limit of detection of AN using our platform was determined to be as low as 25 μM.
Ka = 3.32 ± 0.10, 3.66 ± 0.09, respectively). Dimethyl acrylamide-2-vinyl-4,4-dimethyl azlactone copolymer was modified with mono-substituted P5A and MIHA as well as a photo-crosslinker based on 3,4-dimethyl maleimide to obtain host and guest polymers, which were combined and spin-coated onto a gold-coated quartz substrate to fabricate a dually crosslinked supramolecular gel (DCSG) containing reversible and irreversible crosslinks in a single network. Such DCSG chip was utilized as a sensor for the detection of adiponitrile (AN) in organic solutions based on a competitive complex formation with P5A. Macroscopic changes in the gel (e.g., in swelling degree or in refractive index) upon presence of AN (1 μM–1 mM) in chloroform were monitored with surface plasmon resonance (SPR) spectroscopy. The limit of detection of AN using our platform was determined to be as low as 25 μM.
Introduction
Gels, i.e., polymer chains interconnected via covalent crosslinking moieties into three-dimensional networks, represent one of the most important classes of synthetic materials.1,2 Despite their versatility for numerous applications, such networks exhibit often insufficient mechanical properties and poor reversibility, which drastically limits the applicability of the gels. However, their fusion with supramolecular chemistry paved the way for numerous opportunities to enhance the properties of the gels, or even to endow the gels with unprecedented ones.3,4 The introduction of supramolecular interactions (such as ionic interactions,5–7 metal/ligand complexes,8 hydrogen bonding9,10 or host–guest inclusion complexes11–13) into the network results in the reversible crosslinks between the polymer chains. This, in turn, bestows the systems with improved mechanical properties14 and susceptibility towards the changes in the medium and thus with stimuli-responsiveness. This feature is the key for the fabrication of smart materials that can be utilized in diverse areas ranging from tissue engineering,15–18 bioelectronics,19,20 and drug delivery21–24 to self-healing materials,25,26 actuators,27–29 and sensors.12,30,31Lately, the focus of various research groups has been shifted onto polymeric networks that combine multiple types of crosslinks in one system.32,33 Utilization of dual reversible crosslinking, for instance, was shown to endow the networks with multi-stimuli-responsiveness as well as shape memory behavior.34–37 Furthermore, incorporation of a covalent and a non-covalent linkage types in a single gel was demonstrated to be highly promising since it receives the advantages of both types:38,39 the former provides an integrity of structure to the system, whereas the latter is responsible for the reversibility and responsiveness. Such combination proved to be beneficial for the fabrication of reusable devices, which is especially important in sensing applications. Recently, dually cross-linked hydrogel sensors were developed in our working group for the detection of small molecules40 and ovarian cancer biomarker41 – lysophosphatidic acid (LPA). Reversible crosslinks in a form of a host–guest complex with β-cyclodextrin were disrupted in the presence of an analyte – a compound with a higher binding affinity to cyclodextrin – which led to a decrease in the crosslink density and, therefore, to an increased swelling. The swelling behavior was monitored by SPR spectroscopy,42 the sensitivity of which offered a limit of detection of the LPA as low as 0.122 μM.
Nitriles comprise a chemical substance family which is of high importance in various areas such as chemical industry (where they are extensively used as precursors for plastics, fibers, etc.43,44), or medicine (e.g., as biomarkers of chemotherapy-related kidney damage in cancer patients45). In the field of chemical industry adiponitrile is one of the key members of the nitrile family. It is used in the synthesis of nylon-6,6, which is the reason for a very high demand for it (annual production over 1.5 million tons44). Despite being crucial for market, nitriles can exhibit high toxicity,46–48 and adiponitrile can pose a risk for human health (hence being listed in the List of Extremely Hazardous Substances by the US Environmental Protection Agency), especially at high concentrations.49–51 The studies conducted on fish revealed the lethal concentration of AN LC50 between 670 and 2140 mg l−1 (ref. 50) (corresponding to 6.2 to 19.8 mM; the values are dependent on the species and the exposure duration), therefore, making it crucial to introduce a reliable method of monitoring adiponitrile levels in the environment.
Pillar[n]arenes are a comparatively new and highly versatile class of macrocycles constructed by hydroquinone unites para-bridged with methylene groups.52,53 The smallest and the most extensively investigated macrocycle among pillar[n]arenes is pillar[5]arene, which is characterized by pronounced affinity towards various types of molecules, including positively charged and neutral species54–57 such as triazoles, imidazoles,58,59 and pyridine60 derivatives. An especially high affinity is demonstrated in the complexation with various nitriles.51,61 Due to its unique complexation properties and a vast variety of possible property tuning ways via rim group modification62,63 pillar[5]arene has found broad application as adsorbent,51 molecular sensor64–68 and as a building block for the construction of supramolecular responsive materials.69–74
In the current work, we aimed at designing a supramolecular sensor chip for the detection of adiponitrile. Benefiting from a stable complex formation between pillar[5]arenes and adiponitrile, a gel-based sensor was developed bearing two types of crosslinks: chemical (irreversible) and supramolecular (labile), the latter based on a host–guest complex with pillar[5]arene. Upon the presence of the analyte in the medium around a pre-swollen gel, the competitive complexation takes place with the analyte displacing the original guest species, which leads to disruption of the crosslinks and, thus, to the decrease in the crosslink density. This change can be measured by change in the swelling using a sensitive SPR technique (Fig. 1).
 | ||
| Fig. 1 The concept of a pillar[5]arene-based dually crosslinked gel sensor for the detection of adiponitrile. | ||
Results and discussion
Synthesis and general considerations
One of the features of pillar[n]arenes that make them highly versatile host species is the possibility of their functionalization via rim-group substitution.53,63 Hence, various strategies were developed which paved the way for selective substitution of one, two or more alkyl rim groups as well as for achieving regioselectivity thereof.63,90,91 For a fabrication of host polymers with pendant P5A groups a selective mono-substitution is required, which can be implemented by two major approaches: (1) by co-cyclooligomerization (the functionality is introduced directly during the macrocycle formation) and (2) by forming “homo”-P5A followed by mono-demethylation and subsequent etherification. We chose the second pathway (Fig. 2A) for several reasons. P5A in the first step can be produced in a relatively high yield (>70%) and on a gram scale without complex purification. For the selective mono-demethylation using BBr3 it is crucial to find the perfect conditions, however, the unreacted P5A can be recovered from the mixture, which minimizes losses and makes this procedure effective considering the overall yield. P5AOH containing a single hydroxyl group can be easily converted into a mono-functionalized P5A, for instance, by Williamson etherification. In the current work, we sought to improve the versatility of this method by introducing a propargyl function as a side chain. This allows utilizing azide–alkyne [2 + 3]-cycloaddition for further functionalization. As a result, P5A with a single triazole-containing amine-terminated side chain (HT) was synthesized with a yield of 17% over 5 steps. The characterization of the obtained products can be found in the ESI (Fig. S1–S4 and S9–S18†).
 | ||
| Fig. 2 Synthetic routes towards (A) host moiety (HT) and (B) guest moieties (MIHA, PHA-Boc, THA-Boc, IHA-Boc and TAHA-Boc). For the detailed synthetic procedures see experimental part and ESI.† | ||
:1 host:guest ratio is reached, and upon further addition of the guest species the complete disappearance of the free host peaks was observed (Fig. S33†). All these facts speak in favor of a highly stable binding between both species.
Further details of the complexation equilibrium can be provided by 2D NOESY NMR (Fig. S34†). Notably, the strong negative cross-peaks demonstrate that the peaks of both free and “threaded” AN belong to the same molecule, which implies that although the pseudorotaxane exhibits slow equilibrium, the guest molecules still exchange in a cavity within the time of an experiment. This is further proved by NOE cross-peaks (marked green) which appear for all AN protons.
To have an insight into the complexation strength the titration curves were simulated using HySS 2009 (Version 4.0.31) software96 for a two-component system of 1:1 stoichiometry and two different binding constants. The resulting curves (Fig. S35†; solid lines: logKa = 5.0; dashed lines: logKa = 4.0) resemble the evolution of free host [P5A], free guest [AN] and complex [AN@P5A] concentrations depending on the total guest concentration [AN]0. As can be seen from the figure, for both Ka values the curves align almost perfectly with the experimental data (the discrepancies taking place because of the AN and AN@P5A peak broadening, which results in an addition integration error) and with each other, therefore, demonstrating that logKa (AN@P5A) is exceeding 4.0 but cannot be precisely determined using NMR titration.
Ka = 3.5. It is known that pillar[5]arenes can form stable pseudorataxanes with various heterocycle-capped axles.58 Noticeably high binding constant was reported for butanes capped with imidazol-1-yl (logKa = 4.3), 1,2,3-triazol-1-yl (logKa = 4.2) substituents for per-O-ethyl-P5A. For P5A the only reported value considers 1,4-bis(1,2,4-triazol-1-yl)butane (logKa = 3.1) as a guest species. Besides, due to the electron-rich cavity of pillar[n]arenes and, therefore, possibility of exhibiting Coulomb interactions positively charged species can benefit from the complex formation compared to neutral molecules. D'Anna et al.59 studied the effect of solvent and counterion on the stability of complexes of P5A with anthracenyl substituted alkylimidazolium species by UV/Vis spectroscopy. According to their report, the Ka varied between 103–104 M−1 for all the investigated solvents (2300 M−1 in case of chloroform) except DMF where the stability was significantly lower. Besides, the complex with Cl− anion is shown to exhibit especially high stability compared to complexes with other counterions (Ka = 4700 M−1). Bis(imidazolium) substituted butanes were investigated for their interaction with per-hydroxy-P5A97 in DMSO, acetone, their mixtures and a mixture of acetone and chloroform (1:1). It was found that the binding affinity increases with increasing acetone:DMSO ratio (from logKa = 1.74 for pure DMSO up to 2.66 for acetone) and became even stronger in acetone:chloroform mixture (logKa = 3.49). Similar trend was observed for bis(methylimidazolium)butane with logKa reaching 3.00 for acetone:DMSO (3:2). In a solvent mixture with chloroform the complex was insoluble. The same group studied complexation between the same macrocycle and a broad range of paraquats and bis(pyridinium) derivatives in DMSO.60 It was found that the bis(pyridinium)alkanes have a higher binding affinity (up to logKa = 2.65 for bis(pyridinium)butane) towards per-hydroxy-P5A in DMSO than paraquats. Furthermore, methylimidazolium98 and pyridinium70,71,99 derivatives have been successfully used by various working groups as guest moieties for the preparation of pillar[n]arene-based gels. Final condition, since the host and guest moieties are designed as pendant groups on the polymer chains, the desired axles should only have one side substituted.
In this work, with regard to the polymer post-modification arrangement, 6-aminohexan-1-ol was chosen as a basic building block for the guest moieties. After substitution of OH group with a bromine by refluxing in a concentrated HBr and subsequent Boc-protection, it was reacted with various nitrogen-containing species to obtain corresponding guests (Fig. 2B, S5–S8 and S19–S30†). We chose the following axle substituents as potential candidates for the role of the guest in the gel sensor: imidazol-1-yl, (3-methyl)imidazol-1-yl, 1,2,4-triazol-1-yl, pyridinium and trimethylaminium moieties.
NMR investigations of host–guest interactions
 | ||
| Fig. 3 (A) NMR spectra of (from the bottom) P5A, MIHA-Boc@P5A complex (at [G]0 = 3.3 mM), MIHA-Boc. The change in peak shifts of the guest molecule is presented by the arrows. (B) Fragments of the NMR titration (MIHA-Boc@P5A), with shifts of only P5A peaks shown. (C) Job plot of the P5A-MIHA-Boc titration. The maximum of all three peaks of P5A lie around 0.5, which proves a 1:1 stoichiometry of the complex (the lines serve as guides for the eye). (D) Titration curves corresponding to the P5A protons. The fit curves are obtained using a non-linear least square method with Ka as a common parameter for all dependencies. | ||
The results of the NMR titration for the system P5A-MIHA-Boc is shown in Fig. 3 and S36.† In the spectra the shift of the peak positions can be traced. Firstly, the protons of the MIHA-Boc alkyl chain undergo an upfield shift, which is induced by the inclusion of the guest molecule into the macrocycle. It can be noticed that the closer a methylene group is located to the imidazole fragment, the more pronounced the shielding effect is (Fig. 3A): whereas for the proton Ha (for [H]0:[G]0 = 1:0.83) the Δδ = −2.26 ppm and a strong broadening is observed, for the proton He Δδ = −0.14 ppm, and for Hf even a slight deshielding takes place: Δδ = +0.04 ppm with almost no broadening of the signal. Similar trend (upfield shift and broadening of the alkyl chain signals) is observed for alkyl chains of other guest molecules: PHA-Boc, IHA-Boc and TAHA-Boc (Fig. S39, S40, S50, S51, and S54, S55†); in case of THA-Boc the formation of THA-Boc@P5A can be demonstrated100 by broadening but no significant shift of the protons of the guest moiety is observed (Fig. S45 and S46†).
The protons of the functional end group, however, exhibit different trends. For MIHA-Boc, Hg which is normally the most deshielded due to the positive charge of the MIHA-Boc shows a very strong upfield shift and signal broadening in the presence of P5A (Δδ = −2.47 ppm). The proton Hh, however, demonstrates an opposing trend with a downfield shift: Δδ = +0.40 ppm.
As for P5A signals (Fig. 3B, S41, and S47†), a clear trend can be noticed that deshielding takes place for methoxy groups and aromatic protons, which is in accordance with earlier literature reports for P5A.101 Interestingly, the protons of methylene bridges exhibit slight shielding in all fast exchange systems.
Further proof of pseudorotaxane formation can be delivered by 2D NOESY NMR. The presence of NOE cross-peaks between host and guest protons was observed in spectra of PHA-Boc@P5A and THA-Boc@P5A complexes (Fig. S43 and S49†). Surprisingly, the intensity of the NOE signals in case of MIHA-Boc@P5A was too weak (Fig. S37†).
The host peaks can be used to determine the stoichiometry of the Guest@P5A complexes and their binding constants. Job plots obtained from the shifts of each of the protons (Fig. 3C, S42A, and S48A†) indicate a 1:1 stoichiometry of the investigated pseudorataxanes MIHA-Boc@P5A, PHA-Boc@P5A and THA-Boc@P5A due to the maximum of all the curves lying at approx. 0.5. Using the non-linear regression method for the 1:1 complexation model (Fig. 3D, S42B, and S48B†), the constant Ka as a global parameter and the chemical shift at full complexation Δδmax for each of the peaks were obtained (Table S1† and Fig. 4). It can be observed that from the systems with fast exchange on the NMR timescale the charged species (MIHA-Boc and PHA-Boc) have a significantly (by one order of magnitude) higher binding affinity (and therefore, more stable complexes) than neutral THA-Boc: logKa = 3.32 ± 0.10, 3.66 ± 0.09 and 2.27 ± 0.04, respectively. The absence of THA-Boc signal shifts can be hence explained by too weak interactions between the guest and P5A in CDCl3.
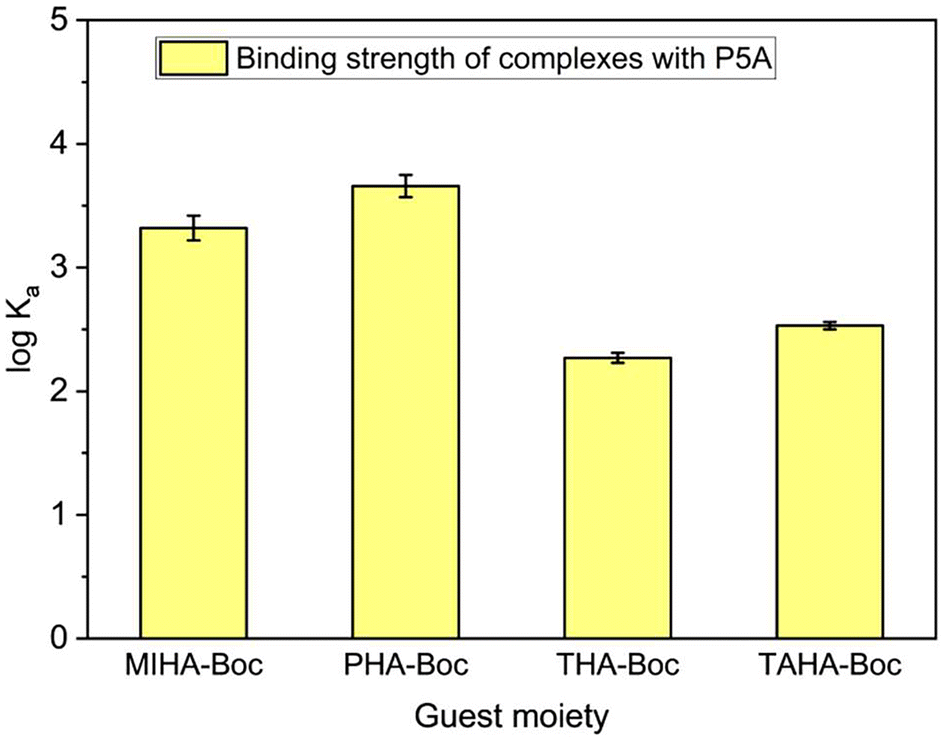 | ||
| Fig. 4 Comparison of the binding affinities of P5A with different guests, as obtained in this work. | ||
As mentioned before, the results of this work suggest that the complexes IHA-Boc and TAHA-Boc exhibit slow exchange on the NMR timescale. For the IHA-Boc@P5A this finding correlates with an extensive studies of P5A complexes with imidazole-substituted alkanes performed by Li et al.101 The protons of the end groups are in both complexes shifted upfield and broadened, and their proximity to the macrocycle protons is additionally confirmed by 2D NOESY for TAHA-Boc@P5A (Fig. S58†). Unfortunately, it was not possible to determine the same for the IHA-Boc@P5A complex (Fig. S53†) because of strong overlapping of signals.
The stoichiometry of the TAHA-Boc@P5A pseudorotaxane was determined by the peak integration to be 1:1 (host:guest), and the binding constant was calculated from the 1:1 mixture to be logKa = 2.53 ± 0.03 (Fig. 4). The titration curves simulated for a system with logKa = 2.53 align well with the experimental data for the beginning of the titration, as shown in Fig. S57.†
Peak integration of bound P5A and IHA-Boc interestingly revealed a 1:2 (P5A:IHA-Boc) geometry for the whole range of titration ratios. The complexation constant Ka was calculated using the following equation from the spectrum of 1:2 (host:guest) mixture:
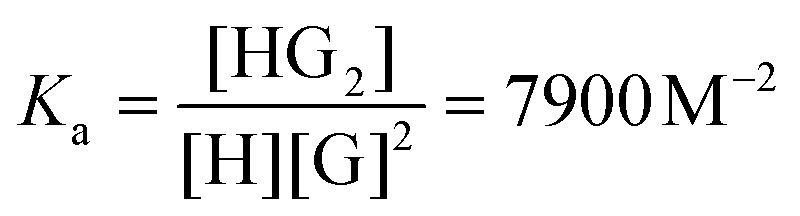
The detailed discussion of the NMR-titration of P5A with PHA-Boc, THA-Boc, IHA-Boc and TAHA-Boc can be found in ESI.†
 | ||
| Fig. 5 Diffusion coefficients D for P5A and the discussed guest molecules alone and as parts of the host–guest inclusion complexes. In case of the complexes with a slow exchange, peaks of free and complexed species were evaluated separately. The dashed line represents a D value for P5A (D = 6.4 × 10−10 m2 s−1). | ||
Being the smallest molecule among the investigated species, AN has by far the highest diffusion coefficient (DAN = 1.48 × 10−9 m2 s−1). Upon presence of P5A the DAN value drops down to 1.02 × 10−9 m2 s−1 for the peaks of the “threaded” AN and to 1.20 × 10−9 m2 s−1 if calculated for the peaks of “free” AN. This highlights the finding made with NOESY NMR that although the interaction is considered “slow” on the NMR timescale, the two states are not fully separated. Therefore, the diffusion coefficients of both free and complexed AN in solution are average values between the isolated and fully threaded AN. It is worth noting that due to the strength of the binding affinity of AN@P5A these two states are distinguishable, which is not the case for other guest species as will be shown further. Moreover, AN@P5A is the only pseudorotaxane by which the mobility of P5A slightly increases up to 6.9 × 10−10 m2 s−1.
Other guest molecules are larger in size and, therefore, possess smaller D values than AN. The diffusion coefficients of neutral THA-Boc (DTHA-Boc = 8.8 × 10−10 m2 s−1) and IHA-Boc (DIHA-Boc = 9.1 × 10−10 m2 s−1) decrease upon presence of P5A in the system. The value of DTHA-Boc@P5A = 7.4 × 10−10 m2 s−1 as expected for a fast exchange complex, lies as an average between pure guest and pure host. P5A diffusion is noticeably hindered as well, which implies a size increase of the macrocycle upon threading with THA-Boc. The same behavior is observed in case of the pseudorotaxane IHA-Boc@P5A, where the differences in mobility between “free” and complexed guest molecules is within the error and, therefore, insignificant (DIHA-Boc@P5A ≈ 7.4 × 10−10 m2 s−1). The diffusion coefficients of the isolated charged guests are comparable to (DMIHA-Boc = 6.5 × 10−10 m2 s−1) or lower than that of P5A (DPHA-Boc = 4.3 × 10−10 m2 s−1, DTAHA-Boc = 5.1 × 10−10 m2 s−1). As was with THA-Boc@P5A, for the complexes exhibiting fast exchange the D value resembles an average between D values for host and guest (for MIHA-Boc the values are within an error from each other). In case of TAHA-Boc@P5A because the diffusion coefficient of the isolated guest (DTAHA-Boc = 5.1 × 10−10 m2 s−1) is close to the D of the complexed P5A, the values of the complexed TAHA-Boc do not change much upon presence of the host species.
Hence, using DOSY NMR it could be demonstrated that the formation of the host–guest assemblies studied in the current work is affecting their mobility and size. Furthermore, alteration of the mobility upon complexation was found to follow the pattern of fast exchange complexes, i.e., where the resulting diffusion coefficient of the guest was an average value rather than completely reduced to the mobility of P5A, which is in agreement with the previously discussed NOESY results.
Polymer synthesis and modification
VDMA as “click”-modifiable monomer was synthesized in two steps form 2-methyl alanine by a slightly modified procedure from ref. 87. DMIEA was obtained according to a known literature procedure.40 Since DMIEA contains a 3,4-maleimide fragment, as mentioned above, it is capable of UV-light-induced [2 + 2]-dimerization for the subsequent gelation of the polymer blends.VDMA was copolymerized with dimethyl acrylamide (DMAAm; DMAAm to VDMA ratio 80:20) using RAFT polymerization with DMP as chain transfer agent and AIBN as initiator (Fig. 6 and S59†). Controlled radical polymerization was selected for achieving a regular distribution of monomers in the copolymer (however, VDMA is known to have a higher copolymerization rate than DMAAm79). The molecular weight (Mn) of p(DMAAm-co-VDMA) copolymer (P0) is presented in the Table 1 and Fig. S63.† Because of the susceptibility of VDMA groups towards hydrolysis, the fresh copolymer was stored under inert atmosphere in a tightly closed vial in a freezer.
 | ||
| Fig. 6 RAFT polymerization of VDMA and DMAAm to a copolymer P0 and polymer modification using host and guest moieties as well as photo-crosslinker. For the detailed synthetic procedures see experimental part and ESI.† | ||
| Polymer | Functionality | Host or guest group,a % | Photo-cross-linker,a % |
M
nb |
Đ |
|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. Per cent of total number of monomer units. b Determined by size-exclusion chromatography. | |||||
| P0 | — | — | — | 53400 |
1.90 |
| PHTP | HT, DMIEA | 11% | 2.8% | 103200 |
2.21 |
| PMIHAP | MIHA, DMIEA | 12% | 2.8% | 68900 |
2.57 |
| PPC | DMIEA | — | 16% | 92700 |
4.45 |
P0 was used for further post-modification to obtain a host polymer (PHTP, containing HT and DMIEA as pendant groups, Fig. S60†), a guest polymer (PMIHAP, containing MIHA and DMIEA, Fig. S61†), and a photo-crosslinker-modified polymer (PPC, containing only DMIEA groups, Fig. S62†). All moieties (HT, MIHA and DMIEA) were attached to the polymer chains by mixing them with P0 in DMF in the presence of DBU. Because VDMA undergoes the ring opening with nucleophiles, DBU was used to deprotonate the amine hydrochlorides thus increasing the nucleophilicity. By devising the modified polymer composition, the amount of the photo-crosslinker DMIEA was aimed at 3 mol% since it was demonstrated earlier in our working group that the DMIEA monomer ratio of as low as 2.3% allows a stable gel formation.40 Furthermore, preliminary studies showed that at the host and guest modifier ratio of 5–6% the responsivity and sensitivity of the system towards AN is insufficient, thus, their content was increased up to 11% and 12% for HT and MIHA, respectively. It is worth mentioning that all polymers including PHTP were water-soluble, therefore, it was possible to purify them by dialysis in water.
The effectiveness of the polymer modification, i.e., the functional groups content in the resulting polymers was calculated (Table 1) by integration of the corresponding NMR signals. GPC results (Table 1 and Fig. S63†) demonstrate that upon modification the increase in Mn of the polymer is observed, which is almost double as high in the case of PHTP compared to P0. This is expected due to a high molecular mass of the attached pendant groups (HT). Noticeably, the Mn of PPC is also significantly higher than that of the unmodified polymer. This can be explained by DMIEA moieties dimerizing during the polymer work up or storing leading to the crosslink formation between chains. This possibility is supported by a remarkably increased polydispersity of the sample.
SPR studies: adiponitrile responsiveness
Although it would be beneficial for the environmental monitoring to be able to determine AN concentration directly from wastewater samples, we consider water to be a poor solvent for the sensing platform in its current design because of, on the one hand, its high polarity which leads to a highly energetic solvation of the charged MIHA pendant groups, thus, hampering the initial host–guest complex formation. On the other hand, considering an exceptional hydrophobicity of P5A groups, water as a solvent promotes their aggregation, which not only hinders the possibility of MIHA@P5A formation even further, but also it decreases the number of P5A units available for binding with AN. As discussed above, CHCl3 was selected for the detection studies due to a good solubility of unmixed host and guest polymers as well as proven high binding affinities of the participating species, which implies a high sensitivity of the designed sensor.The sensor chips prepared by dip-coating in an adhesion promoter solution,102 followed by spin-coating, drying in vacuo, photo-crosslinking using UV irradiation and equilibration in CHCl3 were investigated by surface plasmon resonance spectroscopy (Fig. 7). Here, the gel swelling behavior was studied in CHCl3 and in the presence of the analyte – AN. The scan measurement of a golden layer and a dry gel are demonstrated on Fig. S64.† The plasmon resonance minimum is strongly shifted to higher angles (θPR ≈ 90°) indicating a high refractive index and, accordingly, high gel density. The parameters of the gel layer, obtained by a fit simulation are given in Table S2.† The fit is not matching the experimental SPR curve perfectly because of the inhomogeneities on the gel surface, however, it is possible to draw conclusions about the parameters of the gel by fitting the area around the angle of total internal reflection (20°–25°) and the plasmon resonance minimum.
 | ||
| Fig. 7 SPR sample preparation. Gold-coated N-LaSF9 wafer is immersed in an adhesion promoter solution overnight. Then, a polymer (mixture) solution is spin-coated onto the wafer, followed by drying in vacuo and crosslinking by UV-irradiation. After equilibrating in CHCl3 overnight the sensor chip can be measured. | ||
Through the flow cell 1 ml CHCl3 was pumped using a syringe and the gel was equilibrated for 2 h. Then, the gel layer was treated with AN solutions of increasing concentrations (in each case, 1 ml of the corresponding solution was injected into the flow cell and the gel was equilibrated for 10 min): 1 μM, 5 μM, 10 μM, 20 μM, 50 μM, 100 μM, 200 μM, 500 μM and 1 mM. Between every two injections the flow cell was purged with 1.5 ml CHCl3. The gel behavior was investigated using kinetic measurement at 76°. As can be seen from Fig. 8(A), the system PHTP + PMIHAP exhibits almost no responsiveness up to CAN = 20 μM, followed by a decrease of intensity at a constant angle with an increasing concentration resulting in a linear dependence from logCAN. It is worth noting that the system exhibits almost complete recovery of the intensity upon rinsing with CHCl3, which proves the reusability of the constructed sensor chip.
 | ||
| Fig. 8 SPR measurement of the PHTP + PMIHAP sensor chip with different concentrations of AN in CHCl3. (A) Kinetic studies at 76°. Between swelling in AN solutions the gel was rinsed with CHCl3, which led to the gel returning to its initial state. (B) SPR scan measurements of the gel swollen in AN solutions. (C) Dependence of swelling degree and refractive index with increasing AN concentration (dashed lines serve as guides for the eye). Inset: linear fit of the first section of the graph. | ||
To determine the sensitivity of the chip, scan measurements were performed after each AN solution injection and equilibration. The angular spectra of the swollen gels are presented on Fig. 8(B). In the spectra two features are clearly recognizable and the changes in the system affect these regions the most: the plasmon resonance around 82° and the waveguide (56°–58°). For layers of high thickness (>300 nm) in a one-layer model the former is affected by the refractive index nd, whereas the latter by both the refractive index and the layer thickness.103 The shift of the plasmon minimum to the lower angles observed in Fig. 8(B) implies a decrease in nd is and, therefore, evidence of the decreasing gel density upon the addition of AN, which is expected due to the crosslinks being broken because of the competitive complex formation. Simultaneously, the change in the waveguide region can be noted, serving as a hint about gel thickness changes. For the determination of the gel layer thickness the curve simulation based on Fresnel equations is required. Unfortunately, for the obtained spectra, the one-layer model failed to deliver a precise curve fit. Thus, a two-model simulation was utilized, which considers the anisotropic character of the gel swelling.102,103 In this model, the parameters of the inner layer affect the plasmon minimum region the most, and the waveguide region can be precisely fitted by optimizing the parameters of the outer layer.
From the Fig. 8(C) and Table S2† it is clear that the sensor chip developed in this work shows responsiveness towards adiponitrile. With an increasing concentration of the analyte the gel thickness d (and, therefore, swelling degree Q) increases and the refractive index decreases, indicative of a reduced density likely caused by breaking of the supramolecular host–guest crosslinks through a competitive complex formation. It is worth noting that according to the obtained parameters the inner layer does not undergo significant changes upon increasing AN concentration with the layer thickness din staying between 262–273 nm, whereas the outer layer thickness dout increases from 483 up to 558 nm with a significant decrease in refractive index.
It can also be noticed in linear coordinates that saturation of the supramolecular system is occurring with increasing AN concentration. Thus, a linear section of the swelling degree dependence (Fig. 8, inset of C), was chosen for the determination of the limit of detection for the fabricated sensor chip (Table S4†):104 LoD = 25 μM.
The achieved limit of detection is significantly lower than the concentration limit in the toxicity evaluation against fish as discussed earlier.50 This proves that the sensitivity of the devised sensor chip is sufficient for the detection of AN concentrations that can pose a threat to the environment. Nevertheless, the value can be improved further, in our view, by optimizing the designed sensing platform on chemical and/or methodological levels for further fabrication of sensing devices.
To prove that the responsiveness of the gel sensor is based on the built-in host–guest interactions and to separately evaluate the influence of the other polymer components (DMIEA and DMAAm) on the interaction with AN, an SPR measurement was conducted using a wafer coated with PPC, i.e., not containing any host or guest groups (Fig. S65†). The gel was measured in a dry state as well as swollen in CHCl3 and in 1 mM AN solution (therefore, exposing it to a high concentration). Fig. S74† shows that no difference is noticeable between the two curves, and Table S3† reveals that upon presence of the analyte the PPC gel even exhibits a very slight shrinking, thus confirming the expected mechanism of AN interaction with PHTP + PMIHAP gel sensor.
Experimental section
Materials
N,N-Dimethyl acrylamide (TCI) was purified by vacuum distillation before use. The solvents: acetone, chloroform, dichloromethane (DCM), diethyl ether, ethyl acetate (EtOAc), iso-hexane and methanol (MeOH) were received from Stockmeier and purified by distillation before use. Deuterated solvents: CDCl3 (99.8% D, with Ag), DMSO-d6 (99.8% D), CD3CN (99.0% D) and D2O (99.9% D) were purchased from Deutero GmbH and used as received. All the other chemicals were purchased from the corresponding supplier as listed below and used without further purification.1,2,4-1H-Triazole (98%, Janssen Chimica), 1,2-dichloroethane (DCE) (99.8%, Riedel-de Haën), 1,4-dimethoxybenzene (99+%, Acros Organics), 1,4-dioxane (99.5%, Grüssing GmbH), 1,8-diazabicyclo[5.4.0]undec-7-en (DBU) (98%, Sigma Aldrich), 1-methyl-1H-imidazole (99%, Sigma Aldrich), 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DMP) (98%, Sigma Aldrich), 2,3-dimethylmaleic anhydride (97%, Acros Organics), 2,6-di-tert-butyl-4-methylphenol (BHT) (99%, Fluka), 2-methyl alanine (98%, TCI), 3-bromopropylamine hydrobromide (98%, Sigma Aldrich), 6-aminohexan-1-ol (97%, Sigma Aldrich), acetonitrile (MeCN) (99%, Fisher Chemical), acryloyl chloride (97%, Sigma Aldrich), adiponitrile (AN) (98%, TCI), allyl amine (98+%, Alfa Aesar), azobis(isobutyronitril) (AIBN) (recrystallized from ethanol, Sigma Aldrich), boron tribromide (1 M in dichloromethane, Sigma Aldrich), copper iodide (99.999%, Sigma Aldrich), di-tert-butyl dicarbonate (Boc2O) (97%, Thermo Fisher), ethanol (EtOH) (99.5%, Grüssing GmbH), ethanolamine (99%, Acros Organics), ethyl chloroformate (97%, Sigma Aldrich), ethylene diamine (EDA) (99%, Acros Organics), 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) (99%, Carbolution), hydrogen bromide (48% in H2O, Sigma Aldrich) hydrogen chloride (4 M solution in 1,4-dioxane, Thermo Fisher), hydrogen chloride (37% in H2O, Stockmeier), imidazole (99%, Merck), magnesium sulfate (anhydrous, VWR), N,N-dimethyl formamide (anhydrous, 99.8%, Thermo Fisher), paraformaldehyde (95+%, Sigma Aldrich), potassium trifluoroacetate (CF3COOK) (98%, Sigma Aldrich), propargyl bromide (80% in toluene, Alfa Aesar), pyridine (99.5%, Acros Organics), sodium azide (99%, Merck), sodium carbonate (anhydrous, VWR), sodium chloride (anhydrous, Stockmeier), sodium hydride (60% in mineral oil, Acros Organics), sodium hydroxide (anhydrous, Normapur), sodium sulfate (anhydrous, VWR), tetrahydrofuran (THF) (99.5%, Acros Organics), thioacetic acid (98%, Acros Organics), toluene (technical grade, Stockmeier), triethylamine (TEA) (99.5%, Sigma Aldrich), trifluoroacetic acid (TFA) (99%, Thermo Fisher), trimethylamine (33% in ethanol, Fluka).
Methods
NMR titration was conducted in CDCl3 as follows.105 Into an NMR tube with 0.5 ml host solution (0.004 M) aliquots of host–guest solution (0.004 M host and 0.02 M guest) were successively added. The tube was gently shaken and sonicated for 5 min prior to each NMR measurement. Determination of binding constants Ka for complexes exhibiting a slow exchange proceeded using a single point method.58,61,97,101,106–109 The stoichiometry was determined by the integration of NMR signals, and for 1:1 (host:guest) complexes the binding constant was calculated from the equilibrium:
 :1 complex using HySS 2009 (Version 4.0.31) software.96 For the simulation the Ka value obtained experimentally was used.
:1 complex using HySS 2009 (Version 4.0.31) software.96 For the simulation the Ka value obtained experimentally was used.
In case of the fast exchange complexes for the determination of the stoichiometry of a complex a Job plot was used. The shift of host peaks was monitored, and the titration curves were fitted using the equation:93,94

Capillary voltage: 2.62 kV
Sample peak voltage: 10 kV
Extraction peak voltage: 3.2 kV
Kinetic measurements were performed at the fixed angle corresponding to approx. 30% signal intensity on the left slope of the plasmon minimum. The angular spectra were recorded as reflectivity versus angle R(θ) with θ ranging from 18° to 90°. The obtained spectra were fitted in Winspall software (MPI for polymer research, Mainz) using Fresnel equations to determine layer thicknesses (d) as well as refractive indexes (nd) using a 2-layer model as follows:
The entire spectrum (45°–90°) was simulated and fitted iteratively by optimization of the x-minimum, i.e., the plasmon resonance; only the parameters of the inner layer were varied during the iterative process. Further, d and εreal of the inner layer were fixed for the second iterative process where parameters of the outer gel layer were varied. This was performed using the full curve optimization. The resulting layer thickness d was calculated as a sum of the thickness of both simulated layers, and the average refractive index nav was obtained according to the following equation:
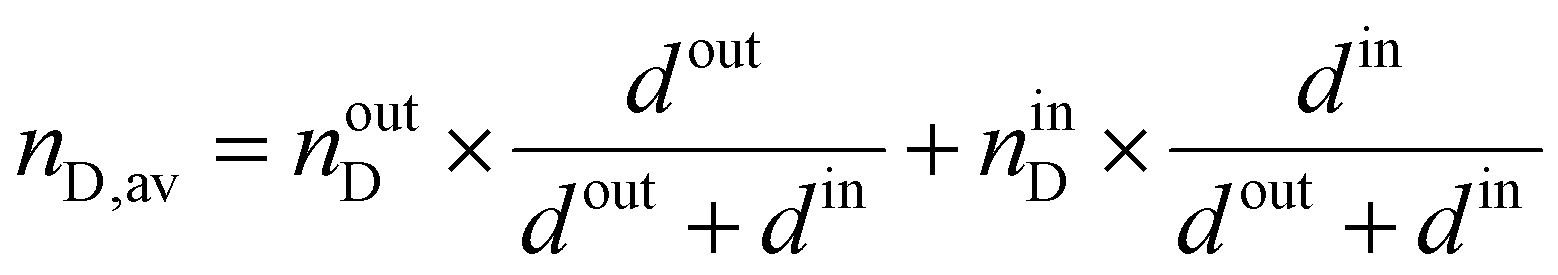
N-LaSF9 (nd = 1.85025) glass wafers (25 × 25 × 1.5 mm) were purchased from ADVANCED OPTICAL COMPONENTS GmbH and coated with gold using PVD. Subsequently, the wafers were immersed in the adhesion promotor solution (1.2 mg ml−1 in EtOH) over night, rinsed with abs. EtOH and dried using a compressed nitrogen flow prior to applying a polymer layer (Fig. S57†).
Host and guest polymer solutions were prepared at a concentration of 4 wt% in DMF separately. After stirring both solutions for 1 h they were mixed at a 1:1 ratio of host and guest groups. The mixture was stirred overnight in a dark place followed by filtering through a Chromafil® PTFE syringe filter (0.45 μm) prior to spin-coating.
Spin-coating was carried out using a G3P-8 spin coater by Specialty Coating Systems. The polymer solution (ca. 80 μl) was applied onto the wafer by static dispense method and coated with a two-step program:
1200 rpm (20 s ramp) for 180 s;
1500 rpm (10 s ramp) for 60 s.
After the completion of the coating process the sample was dried in vacuo overnight in dark to remove the rest of the solvent.
Further, the polymer layer on the wafer was photo-crosslinked by UV-irradiation using an Omnicure S1500 UV lamp (250 mW cm−2) for 300 s. The crosslinked gel layer was measured by SPR (dry layer thickness) after equilibrating in an anhydrous CHCl3 overnight.
For the determination of the swollen gel layer thickness the chip was swollen in CHCl3 in the spectrometer by pumping ca. 1 ml solvent through the cell and equilibrating for ca. 120 min before recording an angular spectrum. Then, for each swelling in AN solution, 1 ml was pumped through the cell and the system was equilibrated for 10 min before a scan measurement. Afterwards, the cell was purged with ca. 1.5 ml CHCl3 the system was equilibrated for 10 min prior to injecting a further AN sample.
The limit of detection (LoD) was calculated by linear fit of the swelling degree dependence as a function of AN concentration according to the definition from:104

where Qi–Qr,i is a difference between experimental and fitted value of swelling degree; n is the amount of data points; A is a slope of the linear fit.
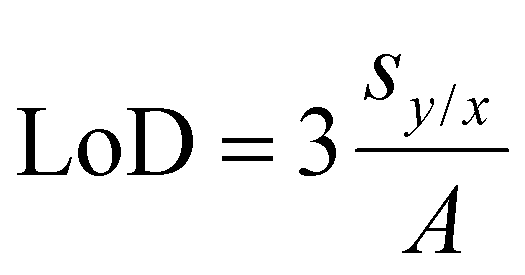
Synthetic procedures
Into a 500 ml 3-necked round-bottom flask 5.528 g (40 mmol) 1,4-dimethoxybenzene and 1.20 g (40 mmol) paraformaldehyde were suspended in 180 ml 1,2-dichloroethane followed by the addition of 20 ml trifluoroacetic acid. The reaction mixture was refluxed for 2 h. After cooling down the mixture was poured into 800 ml MeOH and the formed precipitate was separated using filter paper. Further, the crude product was resolved in 200 ml DCM and filtered through silica. After solvent evaporation the product was stirred in CHCl3 for 30 min to remove the DCM molecules from the cavity. P5A (4.25 g; 5.7 mmol; 71%) was obtained as a white crystalline solid.
T mp = 248.5–249.5 °C (Lit: 248.8 °C;111 248.1–249 °C112).
1H NMR (700 MHz, CDCl3) δ (ppm) = 6.83 (s, 10H, CH), 3.77 (s, 10H, CH2), 3.70 (s, 30H, CH3).
13C NMR (176 MHz, CDCl3) δ (ppm) = 150.8 (CAr–O), 128.4 (CAr–CH2), 113.9 (CAr–H), 55.8 (CH3), 29.6 (CH2).
ESI-MS: cacld for [M]+m/z = 750.3404, found m/z = 750.3396.
:MeOH 100:0 to 100:1, Rf = 0.11). Since the monohydroxy- and dihydroxy-derivatives have similar retention factors,115 for the isolation of the product the mixture was precipitated from 10 ml DCM solution into MeOH (40 ml) twice. Monohydroxy-pillar[5]arene (PA-OH) was obtained as white to slightly pink solid (0.8623 g; 1.17 mmol; 28%).
T mp = 179 °C (Lit.: 203 °C116).
1H NMR (700 MHz, CDCl3) δ (ppm) = 6.88, 6.74, 6.71, 6.70, 6.66, 6.66 (s, 6H, CH); 6.63 (br s, 1H, OH); 6.62, 6.61, 6.59 (s, 3H, CH); 3.81 (s, 3H, CH3); 3.78, 3.78, 3.78, 3.76 (s, 8H, CH2); 3.74 (m, 5H, CH3 and CH2); 3.70, 3.63, 3.61, 3.61, 3.60, 3.57, 3.51 (s, 21H, CH3).
13C NMR (176 MHz, CDCl3) δ (ppm) = 152.1, 151.3, 151.2, 151.2, 151.1, 151.1, 151.1, 151.0, 148.8, 147.8 (CAr–O); 130.2, 129.5, 128.9, 128.8, 128.5, 128.5, 128.3, 127.9, 127.0, 125.2 (CAr–CH2); 119.1, 114.8, 114.8, 114.7, 114.5, 114.3, 114.3, 114.1, 113.2, 113.1 (CAr–H); 56.6, 56.6, 56.3, 56.3, 56.2, 56.1, 56.1, 56.0, 56.0 (CH3); 31.2, 30.3, 30.1, 29.8, 29.1 (CH2).
ESI-MS: cacld for [M + Na]+m/z = 759.3145, found m/z = 759.3137.
T mp = 156.5–158.5 °C.
1H NMR (700 MHz, CDCl3) δ (ppm) = 6.79, 6.78, 6.78, 6.77, 6.76, 6.75, 6.75, 6.75, 6.74, 6.71 (s, 10H, CArH); 4.40 (d, 4JHH = 2.36 Hz, 2H, O–CH2); 3.79–3.76 (m, 10H, CAr–CH2); 3.71, 3.67, 3.67, 3.66, 3.66, 3.6, 3.65, 3.63, 3.61 (s, 27H, CH3); 1.82 (t, 4JHH = 2.02 Hz, 1H, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH).
CH).
13C NMR (176 MHz, CDCl3) δ (ppm) = 151.5, 151.0, 151.0, 151.0, 151.0, 151.0, 151.0, 151.0, 150.9, 149.1 (CAr–O); 129.1, 128.7, 128.7, 128.6, 128.6, 128.5, 128.5, 128.4, 128.3, 128.1 (CAr–CH2); 115.8, 114.6, 114.5, 114.4, 114.4, 114.3, 114.2, 114.2, 114.2, 114.1 (CAr–H); 78.9 (CH2–CCH); 74.8 (CCH); 56.3 (O–CH2); 56.1, 56.1, 56.1, 56.0, 56.0, 56.0, 56.0, 55.9, 55.9 (CH3); 30.7, 30.0, 29.9, 29.9, 29.2 (C–CH2–C).
ESI-MS: cacld for [M + H]+m/z = 775.3482, found m/z = 775.3486.
:MeOH 100:0 to 100:1). HT-Boc was obtained as colorless film (0.5448 g; 0.56 mmol; 76%).
T mp = 159–171 °C.
1H NMR‡ (700 MHz, MeCN-d3:CDCl3 3:1) δ (ppm) = 7.89 (br s, 1H, triazole CH); 7.02, 6.92, 6.91, 6.90, 6.89, 6.89, 6.88, 6.87, 6.86, 6.80 (s, 10H, CAr–H); 5.33 (br s, 1H, NH); 5.03 (s, 2H, O–CH2–C(–N)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH); 4.38 (br tr, 3JHH = 6.2 Hz; 2H, NN–N–CH2); 3.76–3.66, 3.56 (m, 37H, O–CH3 and CAr–CH2); 3.07 (m, 2H, NH–CH2); 2.03 (m, 2H, NH–CH2–CH2); 1.41 (s, 9H, C–CH3).
CH); 4.38 (br tr, 3JHH = 6.2 Hz; 2H, NN–N–CH2); 3.76–3.66, 3.56 (m, 37H, O–CH3 and CAr–CH2); 3.07 (m, 2H, NH–CH2); 2.03 (m, 2H, NH–CH2–CH2); 1.41 (s, 9H, C–CH3).
13C NMR‡ (176 MHz, MeCN-d3:CDCl3 3:1) δ (ppm) = 156.9 (CO); 151.5, 151.0, 151.0, 150.9, 149.7 (CAr–O); 145.0 (Ctriaz–CH2); 130.0, 129.3, 129.3, 129.2 (CAr–CH2); 124.3 (Ctriaz–H); 115.5, 114.1, 114.0, 113.9, 113.9, 113.9, 113.9, 113.8, 113.8 (CAr–H); 79.3 (C(CH3)3); 63.4 (O–CH2); 56.3, 56.3, 56.3, 56.2, 56.1, 56.1 (O–CH3); 48.4 (triazol–CH2); 38.1 (NH–CH2); 31.4 (NH–CH2–CH2); 29.8, 29.8, 29.7 (CAr–CH2); 28.7 (C(CH3)3).
ESI-MS: cacld for [M + H]+m/z = 975.4750, found m/z = 975.4777. Cacld for [M + Na]+m/z = 997.4575, found m/z = 997.4599.
1H NMR§ (700 MHz, MeCN-d3:CDCl3 3:1) δ (ppm) = 8.04 (s, 1H, Ctriaz–H); 7.90 (br s, 3H, NH3+); 7.03, 6.91, 6.90, 6.89, 6.89, 6.88, 6.88, 6.87, 6.86, 6.81 (s, 10H, CAr–H); 5.03 (s, 2H, O–CH2); 4.57 (tr, 3JHH = 6.4 Hz; 2H, triazol–CH2); 3.77–3.65, 3.57 (m, 37H, O–CH3 and CAr–CH2); 3.03 (m, 2H, NH3+–CH2); 2.39 (quint, 3JHH = 6.1 Hz; 2H, NH3+–CH2–CH2).
13C NMR§ (176 MHz, MeCN-d3:CDCl3 3:1) δ (ppm) = 151.5, 151.0, 151.0, 150.9, 149.6 (CAr–O); 145.5 (Ctriaz–CH2); 130.0, 129.3, 129.3, 129.3, 129.3, 129.2, 129.1 (CAr–CH2); 125.0 (Ctriaz–H); 115.5, 114.1, 113.9, 113.9, 113.9, 113.9, 113.8, 113.7 (CAr–H); 63.2 (O–CH2); 56.4, 56.3, 56.3, 56.3, 56.2, 56.1, 56.0 (CH3); 48.0 (triazol–CH2); 38.1 (NH3+–CH2); 29.9, 29.8, 29.7, 29.7 (CAr–CH2); 28.5 (NH3+–CH2–CH2).
ESI-MS: cacld for [M + H]+m/z = 875.4226, found m/z = 875.4221.
1H NMR (700 MHz, CDCl3) δ (ppm) = 10.45 (s, 1H, NCH–N), 7.40 (m, 1H, CH3–N–CHCH), 7.38 (m, 1H, CH3–N–CHCH), 4.71 (s, 1H, NH), 4.33 (t, 3JHH = 7.3 Hz, 2H, imidazolyl–CH2), 4.11 (s, 3H, N−CH3), 3.07 (t, 3JHH = 6.3 Hz, 2H, NH–CH2), 1.92 (m, 2H, imidazolyl–CH2–CH2), 1.47 (m, 2H, NH–CH2–CH2), 1.41 (s, 9H, C(CH3)3), 1.36 (m, 4H, imidazolyl–CH2–CH2–CH2 and NH–CH2–CH2–CH2).
13C NMR (176 MHz, CDCl3) δ (ppm) = 156.3 (CO), 138.1 (NCH–N), 123.4 (CH3–N–CHCH), 122.0 (CH3–N–CHCH), 79.3 (C(CH3)3), 50.2 (imidazolyl–CH2), 40.3 (NH–CH2), 37.0 (N–CH3), 30.2 (imidazolyl–CH2–CH2), 29.9 (NH–CH2–CH2), 28.6 (C(CH3)3), 26.0 (imidazolyl–CH2–CH2–CH2), 25.8 (NH–CH2–CH2–CH2).
ESI-MS: cacld for [M]+m/z = 282.2176, found m/z = 282.2178.
1H NMR (700 MHz, D2O) δ (ppm) = 8.69 (m, 1H, NCH–N); 7.46 (m, 1H, CH3–N–CHCH); 7.41 (m, 1H, CH3–N–CHCH); 4.18 (t, 3JHH = 7.2 Hz, 2H, imidazolyl–CH2); 3.87 (s, 3H, N–CH3); 2.97 (t, 3JHH = 7.6 Hz, 2H, NH3+–CH2); 1.87 (tt, 3JHH = 7.5 Hz, 2H, imidazolyl–CH2–CH2); 1.64 (tt, 3JHH = 7.7 Hz, 2H, NH3+–CH2–CH2); 1.40 (tt, 3JHH = 7.4 Hz, 2H, NH3+–CH2–CH2–CH2); 1.33 (tt, 3JHH = 7.4 Hz, 2H, triazolyl–CH2–CH2–CH2).
13C NMR (176 MHz, D2O) δ (ppm) = 135.8 (NCH–N); 123.5 (CH3–N–CHCH); 122.1 (CH3–N–CHCH); 49.3 (imidazolyl–CH2); 39.3 (NH3+–CH2); 35.6 (N–CH3); 29.0 (imidazolyl–CH2–CH2); 26.5 (NH3+–CH2–CH2); 25.0 (NH3+–CH2–CH2–CH2); 24.9 (imidazolyl–CH2–CH2–CH2).
ESI-MS: cacld for [M]+m/z = 182.1652, found m/z = 182.1631.
1H NMR (700 MHz, CDCl3) δ (ppm) = 9.55 (m, 2H, CHortho); 8.52 (dd, 3JHH = 7.8 Hz, 1H, CHpara); 8.13 (dd, 3JHH = 6.9 Hz; 2H, CHmetha); 4.99 (t, 3JHH = 7.2 Hz, 2H, Py–CH2); 4.79 (s, 1H, NH); 3.03 (m, 2H, NH–CH2); 2.05 (m, 2H, Py–CH2–CH2); 1.44 (m, 2H, NH–CH2–CH2); 1.39 (s, 9H, C(CH3)3); 1.35 (m, 4H, Py–CH2–CH2–CH2 und NH–CH2–CH2–CH2).
13C NMR (176 MHz, CDCl3) δ (ppm) = 156.3 (CO); 145.4 (CHpara); 145.4 (CHortho); 128.6 (CHmeta); 79.2 (C(CH3)3); 61.9 (Py–CH2); 40.3 (NH–CH2); 32.0 (Py–CH2–CH2); 29.8 (NH–CH2–CH2); 28.6 (C(CH3)3); 26.1 (NH–CH2–CH2–CH2); 25.6 (Py–CH2–CH2–CH2).
ESI-MS: cacld for [M]+m/z = 279.2067, found m/z = 279.2082.
T mp = 61 °C.
1H NMR (700 MHz, CDCl3) δ (ppm) = 8.06 (s, 1H, H5triazole), 7.93 (s, 1H, H3triazole), 4.53 (br s, 1H, NH), 4.15 (t, 3JHH = 7.1 Hz; 2H, triazolyl–CH2), 3.09 (m, 2H, NH–CH2), 1.88 (quint, 3JHH = 7.3 Hz; 2H, triazolyl–CH2–CH2), 1.45 (m, 2H, NH–CH2–CH2), 1.43 (s, 9H, CH3), 1.37–1.27 (m, 4H, NH–CH2–CH2–CH2–CH2).
13C NMR (176 MHz, CDCl3) δ (ppm) = 156.2 (CO), 152.0 (C3triazole), 143.0 (C5triazole), 79.3 (C(CH3)3), 49.8 (triazolyl–CH2), 40.5 (NH–CH2), 30.1 (NH–CH2–CH2), 29.9 (triazolyl–CH2–CH2), 28.6 (CH3), 26.3, 26.3 (NH–CH2–CH2–CH2–CH2).
ESI-MS: cacld for [M + Na]+m/z = 291.1797, found m/z = 291.1784.
1H NMR (700 MHz, CDCl3) δ (ppm) = 7.49 (s, 1H, NCH–N); 7.06 (s, 1H, CH2–N–CHCH); 6.90 (s, 1H, CH2–N–CHCH); 4.51 (s, 1H, NH); 3.92 (t, 3JHH = 7.1 Hz, 2H, imidazolyl–CH2); 3.09 (m, 2H, NH–CH2); 1.78 (tt, 3JHH = 7.2 Hz, 2H, imidazolyl–CH2–CH2); 1.46 (m, 2H, NH–CH2–CH2); 1.43 (m, 9H, C(CH3)3); 1.32 (m, 4H, imidazolyl–CH2–CH2–CH2 and NH–CH2–CH2–CH2).
13C NMR (176 MHz, CDCl3) δ (ppm) = 156.2 (CO); 137.2 (NCH–N); 129.4 (CH2–N–CHCH); 119.0 (CH2–N–CHCH); 79.4 (C(CH3)3); 47.2 (imidazolyl–CH2); 40.6 (NH–CH2); 31.2 (imidazolyl–CH2–CH2); 30.2 (NH–CH2–CH2); 28.6 (C(CH3)3); 26.4 (imidazolyl–CH2–CH2–CH2 and NH–CH2–CH2–CH2).
ESI-MS: cacld for [M + H]+m/z = 268.2020, found m/z = 268.2011.
1H NMR (700 MHz, DMSO-d6) δ (ppm) = 6.78 (t, 3JHH = 5.5 Hz, 1H, NH), 3.27 (m, 2H, Me3N+–CH2), 3.05 (s, 9H, N+(CH3)3), 2.90 (dt, 2H, NH–CH2), 1.65 (m, 2H, Me3N+–CH2–CH2), 1.43–1.32 (m, 11H, NH–CH2–CH2 and C(CH3)3), 1.32–1.21 (m, 4H, Me3N+–CH2–CH2–CH2 and NH–CH2–CH2–CH2).
13C NMR (176 MHz, DMSO-d6) δ (ppm) = 155.5 (CO), 77.3 (C(CH3)3), 65.2 (Me3N+–CH2), 52.1 (N+(CH3)3), 39.6 (NH–CH2), 29.2 (NH–CH2–CH2), 28.3 (CH3), 25.7, 25.4 (Me3N+–CH2–CH2–CH2 and NH–CH2–CH2–CH2), 22.0 (Me3N+–CH2–CH2).
ESI-MS: cacld for [M]+m/z = 259.2386, found m/z = 259.2363.
1H NMR (500 MHz, CDCl3) δ (ppm) = 3.62–2.71 (m, 2850H, N(CH3)2); 2.71–2.14 (m, 540H, backbone–CH); 2.14–0.90 (m, 1720H, backbone–CH2 and VDMA–CH3); 0.87 (t, 3JHH = 7.0 Hz; 3H, S-C11H22-CH3).
SEC: Mn = 53400, Đ = 1.90.
1H NMR (500 MHz, CDCl3) δ (ppm) = 8.00–7.56 (br, 1H, triazole CH); 6.89, 6.82–6.62 (br s, 10H, P5A CHAr), 5.00 (s, 2H, O–CH2–C(–N)CH); 4.41 (br, 2H, NN–N–CH2); 3.82–3.70 (br s, 10H, P5A CAr–CH2); 3.70–3.53, 3.48 (br s, 27H, P5A O–CH3); 3.23–2.74 (m, 52H, N(CH3)2); 2.74–2.27 (m, 10H, backbone CH); 2.27–0.92 (m, 48H, backbone CH2, C(O)–NH–C(CH3)2, C(CH3)C(CH3), C(O)–NH–CH2–CH2).
SEC: Mn = 103200, Đ = 2.21.
1H NMR (500 MHz, D2O) δ (ppm) = 7.51 (s, 1H, CH3–N–CHCH–N); 7.47 (s, 1H, CH3–N–CHCH–N); 4.22 (t, 3JHH = 6.8 Hz; 2H, CH3–C3H3N2+–CH2); 3.92 (s, 3H, CH3–C3H3N2+); 3.30–2.84 (m, 48H, N(CH3)2 and C3H3N2+–C5H10–CH2); 2.84–2.35 (m, 8H, backbone CH); 1.96 (s, 1H, C(CH2)C(CH2)); 1.88 (br, 2H, C3H3N2+–CH2–CH2); 1.85–1.14 (m, 35H, backbone CH2, C(O)–NH–C(CH3)2 and C3H3N2+–CH2–CH2–CH2–CH2–CH2).
SEC: Mn = 68900, Đ = 2.57.
Conclusions
In this work, we constructed a sensor chip for the detection of adiponitrile based on a dually crosslinked gel. Chemical crosslinks formed by dimerization of DMIEA endowed the network with stability, whereas supramolecular host–guest complexes with P5A acted as reversible crosslinks disrupted upon presence of AN, due to the competitive complexation between the moieties. For the design of the gel MIHA was selected as a guest moiety from various neutral and positively charged amine- and heterocycle-substituted aminohexanes. The selection was conducted by the evaluation of the binding affinity of Boc-protected axles with P5A. NMR titration revealed that Ka of MIHA-Boc and PHA-Boc with the macrocycle are by one order of magnitude higher than those of THA-Boc and TAHA-Boc complexes, while still being lower than Ka of the AN@P5A complex. IHA-Boc was found to form complex of 1:2 (H:G) stoichiometry with P5A. Additionally, it was demonstrated using 2D DOSY NMR that for neither fast nor slow exchange complexes the mobility of the bound species is fully reduced to the mobility of P5A. The diffusion coefficient, therefore, stays an average between free and “completely bound” states. HT as a host moiety was synthesized by a mono-demethylation of P5A and further etherification of the phenolic OH-group.
Host and guest polymers were synthesized by modification of a P0 copolymer containing VDMA as a comonomer with HT, MIHA (which was our guest moiety of choice) and a photo-crosslinker. By spin-coating and subsequent curing under UV-irradiation, a thin gel layer was fabricated, which was treated with CHCl3 and AN solution of different concentrations (1 μM up to 1 mM). By measuring the change in swelling degree and refractive index using SPR technique, a limit of detection was estimated as a sensitivity parameter of the sensor chip (LoD = 25 μM). Thus, the designed polymeric gel system can serve as a starting point for the development of a sensor for adiponitrile as well as other small molecules capable of forming stable complexes with pillar[n]arenes. We believe that further tuning of the polymer parameters as well as methodology will help improve the performance of the sensing platform for the fabrication of sensors in a form of devices.
Author contributions
Maksim Rodin: conceptualization, investigation, formal analysis, writing – original draft. David Helle: investigation, formal analysis. Dirk Kuckling: conceptualization, writing – review & editing, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG, 501220359).References
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- C. Creton, Macromolecules, 2017, 50, 8297–8316 CrossRef CAS.
- X. Huang, R. Li, Z. Duan, F. Xu and H. Li, Soft Matter, 2022, 18, 3828–3844 RSC.
- A. D. O'Donnell, S. Salimi, L. R. Hart, T. S. Babra, B. W. Greenland and W. Hayes, React. Funct. Polym., 2022, 172, 105209 CrossRef.
- S. L. Craig, Angew. Chem., Int. Ed., 2009, 48, 2645–2647 CrossRef CAS PubMed.
- M. A. Aboudzadeh, M. E. Muñoz, A. Santamaría, R. Marcilla and D. Mecerreyes, Macromol. Rapid Commun., 2012, 33, 314–318 CrossRef CAS PubMed.
- Y. Sun, Y.-Y. Ren, Q. Li, R.-W. Shi, Y. Hu, J.-N. Guo, Z. Sun and F. Yan, Chin. J. Polym. Sci., 2019, 37, 1053–1059 CrossRef CAS.
- J. Sautaux, F. Marx, I. Gunkel, C. Weder and S. Schrettl, Nat. Commun., 2022, 13, 356 CrossRef CAS PubMed.
- M. Ahmadi, A. Jangizehi and S. Seiffert, Macromolecules, 2022, 55, 5514–5526 CrossRef CAS.
- Z. Jing, X. Huang, X. Liu, M. Liao and Y. Li, Polym. Int., 2023, 72, 39–53 CrossRef CAS.
- X. Yang, R. Bai, Z. Zhang, Y. Liu and X. Yan, J. Polym. Sci., 2023, 61, 1105–1110 CrossRef CAS.
- J. Park, Y. Sasaki, Y. Ishii, S. Murayama, K. Ohshiro, K. Nishiura, R. Ikura, H. Yamaguchi, A. Harada, G. Matsuba, H. Washizu, T. Minami and Y. Takashima, ACS Appl. Mater. Interfaces, 2023, 15, 39777–39785 CrossRef CAS PubMed.
- K. Belal, F. Stoffelbach, D. Hourdet, A. Marcellan, J. Lyskawa, L. de Smet, A. Vebr, J. Potier, G. Cooke, R. Hoogenboom and P. Woisel, Macromolecules, 2021, 54, 1926–1933 CrossRef CAS.
- Y. Guo, Y. Liu, X. Zhao, J. Zhao, Y. Wang, X. Zhang, Z. Guo and X. Yan, ACS Appl. Mater. Interfaces, 2023, 15, 25161–25172 CrossRef CAS PubMed.
- Y. Zhao, Z. Li, Q. Li, L. Yang, H. Liu, R. Yan, L. Xiao, H. Liu, J. Wang, B. Yang and Q. Lin, Macromol. Rapid Commun., 2020, 41, e2000441 CrossRef PubMed.
- P. Chakraborty, H. Oved, D. Bychenko, Y. Yao, Y. Tang, S. Zilberzwige-Tal, G. Wei, T. Dvir and E. Gazit, Adv. Mater., 2021, 33, e2008715 CrossRef PubMed.
- Y. Liang, Z. Li, Y. Huang, R. Yu and B. Guo, ACS Nano, 2021, 15, 7078–7093 CrossRef CAS PubMed.
- Y. Zhao, S. Song, X. Ren, J. Zhang, Q. Lin and Y. Zhao, Chem. Rev., 2022, 122, 5604–5640 CrossRef CAS PubMed.
- S. J. K. O'Neill, Z. Huang, M. H. Ahmed, A. J. Boys, S. Velasco-Bosom, J. Li, R. M. Owens, J. A. McCune, G. G. Malliaras and O. A. Scherman, Adv. Mater., 2023, 35, e2207634 CrossRef PubMed.
- G. Su, S. Yin, Y. Guo, F. Zhao, Q. Guo, X. Zhang, T. Zhou and G. Yu, Mater. Horiz., 2021, 8, 1795–1804 RSC.
- C. B. Rodell, R. J. Wade, B. P. Purcell, N. N. Dusaj and J. A. Burdick, ACS Biomater. Sci. Eng., 2015, 1, 277–286 CrossRef CAS PubMed.
- M. Tao, K. Xu, S. He, H. Li, L. Zhang, X. Luo and W. Zhong, Chem. Commun., 2018, 54, 4673–4676 RSC.
- S. Bernhard and M. W. Tibbitt, Adv. Drug Delivery Rev., 2021, 171, 240–256 CrossRef CAS PubMed.
- G. Zhang, J. Li, W. Cai, S. Li, Y. Kong and Z.-Z. Yin, Microchem. J., 2023, 193, 109160 CrossRef CAS.
- M. I. Rial-Hermida, D. C. S. Costa, L. Jiang, J. M. M. Rodrigues, K. Ito and J. F. Mano, Gels, 2023, 9(2), 85 CrossRef CAS PubMed.
- R. Ikura, J. Park, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, NPG Asia Mater., 2022, 14, 10 CrossRef CAS.
- K. Belal, F. Stoffelbach, J. Lyskawa, M. Fumagalli, D. Hourdet, A. Marcellan, L. de Smet, V. R. de La Rosa, G. Cooke, R. Hoogenboom and P. Woisel, Angew. Chem., Int. Ed., 2016, 55, 13974–13978 CrossRef CAS PubMed.
- M. Mauro, J. Mater. Chem. B, 2019, 7, 4234–4242 RSC.
- D. Tian, W. Ma, L. Zheng, K. Jiang, H. He and R. Sun, ACS Appl. Polym. Mater., 2023, 5, 8641–8649 CrossRef CAS.
- Z. Huang, X. Chen, S. J. K. O'Neill, G. Wu, D. J. Whitaker, J. Li, J. A. McCune and O. A. Scherman, Nat. Mater., 2022, 21, 103–109 CrossRef CAS PubMed.
- X. Ji, Y. Yao, J. Li, X. Yan and F. Huang, J. Am. Chem. Soc., 2013, 135, 74–77 CrossRef CAS PubMed.
- M. Rodin, J. Li and D. Kuckling, Chem. Soc. Rev., 2021, 50, 8147–8177 RSC.
- L. Hammer, N. J. van Zee and R. Nicolaÿ, Polymers, 2021, 13(3), 396 CrossRef CAS PubMed.
- J. Chen, Q. Su, R. Guo, J. Zhang, A. Dong, C. Lin and J. Zhang, Macromol. Chem. Phys., 2017, 218, 1700166 CrossRef.
- G. Davidson-Rozenfeld, L. Stricker, J. Simke, M. Fadeev, M. Vázquez-González, B. J. Ravoo and I. Willner, Polym. Chem., 2019, 10, 4106–4115 RSC.
- C. Qian, T.-A. Asoh and H. Uyama, Macromol. Rapid Commun., 2020, 41, e2000406 CrossRef PubMed.
- Y. Liu, L. Wang, H. Lu and Z. Huang, Chem. Eng. J., 2022, 431, 133338 CrossRef CAS.
- J. Deng, R. Bai, J. Zhao, G. Liu, Z. Zhang, W. You, W. Yu and X. Yan, Angew. Chem., Int. Ed., 2023, 62, e202309058 CrossRef CAS PubMed.
- N. Liubimtsev, Z. Zagradska-Paromova, D. Appelhans, J. Gaitzsch and B. Voit, Macromol. Chem. Phys., 2023, 224(3), 2200372 CrossRef CAS.
- J. Li, C. Ji, X. Yu, M. Yin and D. Kuckling, Macromol. Rapid Commun., 2019, 40, e1900189 CrossRef PubMed.
- J. Li, C. Ji, B. Lü, M. Rodin, J. Paradies, M. Yin and D. Kuckling, ACS Appl. Mater. Interfaces, 2020, 12, 36873–36881 CrossRef CAS PubMed.
- A. Azzouz, L. Hejji, K.-H. Kim, D. Kukkar, B. Souhail, N. Bhardwaj, R. J. C. Brown and W. Zhang, Biosens. Bioelectron., 2022, 197, 113767 CrossRef CAS PubMed.
- J. Kümper, J. Meyers, R. Sebers, N. Kurig and R. Palkovits, Green Chem., 2023, 25, 6231–6237 RSC.
- D. E. Blanco, A. Z. Dookhith and M. A. Modestino, React. Chem. Eng., 2019, 4, 8–16 RSC.
- V. Vaks, V. Anfertev, M. Chernyaeva, E. Domracheva, A. Yablokov, A. Maslennikova, A. Zhelesnyak, A. Baranov, Y. Schevchenko and M. F. Pereira, Sci. Rep., 2022, 12, 18117 CrossRef CAS PubMed.
- R. A. Woutersen, Scand. J. Work, Environ. Health, 1998, 24, 5–9 CAS.
- D. Coggon and P. Cole, Scand. J. Work, Environ. Health, 1998, 24, 81–82 CrossRef CAS PubMed.
- S. Caito, Y. Yu and M. Aschner, NeuroToxicology, 2013, 37, 93–99 CrossRef CAS PubMed.
- R. D. Short, W. V. Roloff, L. D. Kier and W. E. Ribelin, J. Toxicol. Environ. Health, 1990, 30, 199–207 CrossRef CAS PubMed.
- G. L. Kennedy, Drug Chem. Toxicol., 2004, 27, 123–131 CrossRef CAS PubMed.
- B. Shi, L. Shangguan, H. Wang, H. Zhu, H. Xing, P. Liu, Y. Liu, J. Liu and F. Huang, ACS Mater. Lett., 2019, 1, 111–115 CrossRef CAS.
- T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- X. Shu, J. Fan, J. Li, X. Wang, W. Chen, X. Jia and C. Li, Org. Biomol. Chem., 2012, 10, 3393–3397 RSC.
- Y. Wang, G. Ping and C. Li, Chem. Commun., 2016, 52, 9858–9872 RSC.
- X. Lou, H. Chen, X. Jia and C. Li, Chin. J. Chem., 2015, 33, 335–338 CrossRef CAS.
- N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed.
- K. Han, Y. Zhang, J. Li, Y. Yu, X. Jia and C. Li, Eur. J. Org. Chem., 2013, 2057–2060 CrossRef CAS.
- F. D'Anna, C. Rizzo, P. Vitale, S. Marullo and F. Ferrante, New J. Chem., 2017, 41, 12490–12505 RSC.
- C. Li, Q. Xu, J. Li, F. Yao and X. Jia, Org. Biomol. Chem., 2010, 8, 1568–1576 RSC.
- X. Shu, S. Chen, J. Li, Z. Chen, L. Weng, X. Jia and C. Li, Chem. Commun., 2012, 48, 2967–2969 RSC.
- P. Demay-Drouhard, K. Du, K. Samanta, X. Wan, W. Yang, R. Srinivasan, A. C.-H. Sue and H. Zuilhof, Org. Lett., 2019, 21, 3976–3980 CrossRef CAS PubMed.
- N. L. Strutt, H. Zhang, S. T. Schneebeli and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2631–2642 CrossRef CAS PubMed.
- S. Sun, X.-Y. Hu, D. Chen, J. Shi, Y. Dong, C. Lin, Y. Pan and L. Wang, Polym. Chem., 2013, 4, 2224 RSC.
- Q. Lin, Y.-Q. Fan, G.-F. Gong, P.-P. Mao, J. Wang, X.-W. Guan, J. Liu, Y.-M. Zhang, H. Yao and T.-B. Wei, ACS Sustainable Chem. Eng., 2018, 6, 8775–8781 CrossRef CAS.
- Y. Mei, Q.-W. Zhang, Q. Gu, Z. Liu, X. He and Y. Tian, J. Am. Chem. Soc., 2022, 144, 2351–2359 CrossRef CAS PubMed.
- R. R. Kothur, B. A. Patel and P. J. Cragg, Chem. Commun., 2017, 53, 9078–9080 RSC.
- X. Tan, X. Liu, W. Zeng, Z. Zhang, T. Huang, L. Yu and G. Zhao, Spectrochim. Acta, Part A, 2019, 221, 117176 CrossRef CAS PubMed.
- N. Laggoune, F. Delattre, J. Lyskawa, F. Stoffelbach, J. M. Guigner, S. Ruellan, G. Cooke and P. Woisel, Polym. Chem., 2015, 6, 7389–7394 RSC.
- J. Chang, Q. Zhao, Le Kang, H. Li, M. Xie and X. Liao, Macromolecules, 2016, 49, 2814–2820 CrossRef CAS.
- C. Huang, H. Zhang, Z. Hu, Y. Zhang and X. Ji, Gels, 2022, 8(8), 475 CrossRef CAS PubMed.
- Q. Zhang, L.-L. Fan, T.-J. Yue, Z.-G. Hu, N. Li, J. Li, Y.-Q. Jiang, K.-Q. Li and H.-M. Guo, Macromol. Chem. Phys., 2022, 2200253 CrossRef CAS.
- Y. Cheng, X. Lv, B. Liang, X. Wei, P. Wang and D. Xia, Polym. Chem., 2023, 14, 191–200 RSC.
- L. Sheng, H. Liu, Z. Hu and X. Ji, Chemistry, 2023, 29, e202300990 CrossRef CAS PubMed.
- C. D. Vo, D. Kuckling, H.-J. P. Adler and M. Schönhoff, Colloid Polym. Sci., 2002, 280, 400–409 CrossRef CAS.
- D. Roy and B. S. Sumerlin, Macromol. Rapid Commun., 2014, 35, 174–179 CrossRef CAS PubMed.
- M. Seuss, W. Schmolke, A. Drechsler, A. Fery and S. Seiffert, ACS Appl. Mater. Interfaces, 2016, 8, 16317–16327 CrossRef CAS PubMed.
- W. Schmolke, M. Ahmadi and S. Seiffert, Phys. Chem. Chem. Phys., 2019, 21, 19623–19638 RSC.
- J. Li, X. Yu, A. Herberg and D. Kuckling, Macromol. Rapid Commun., 2019, 40, e1800674 CrossRef PubMed.
- L. D. Taylor, H. S. Kolesinski, A. C. Mehta, L. Locatell and P. S. Larson, Makromol. Chem., Rapid Commun., 1982, 3, 779–782 CrossRef CAS.
- M. W. Jones, S.-J. Richards, D. M. Haddleton and M. I. Gibson, Polym. Chem., 2013, 4, 717–723 RSC.
- A. Guyomard, D. Fournier, S. Pascual, L. Fontaine and J.-F. Bardeau, Eur. Polym. J., 2004, 40, 2343–2348 CrossRef CAS.
- H. T. Ho, M. E. Levere, D. Fournier, V. Montembault, S. Pascual and L. Fontaine, Aust. J. Chem., 2012, 65, 970 CrossRef CAS.
- C. M. Gardner, C. E. Brown and H. D. H. Stöver, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4674–4685 CrossRef CAS.
- F. François, C. Nicolas, G. Forcher, L. Fontaine and V. Montembault, Eur. Polym. J., 2020, 141, 110081 CrossRef.
- L. Jia, S. M. Kilbey and X. Wang, Langmuir, 2020, 36, 10200–10209 CrossRef CAS PubMed.
- M. E. Levere, H. T. Ho, S. Pascual and L. Fontaine, Polym. Chem., 2011, 2, 2878 RSC.
- S. Pascual, T. Blin, P. J. Saikia, M. Thomas, P. Gosselin and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5053–5062 CrossRef CAS.
- X. Yu, A. Herberg and D. Kuckling, Eur. Polym. J., 2019, 120, 109207 CrossRef CAS.
- G. Wang, H. Qiang, Y.-Z. Guo, J. Yang, K. Wen and W.-B. Hu, Org. Biomol. Chem., 2019, 17, 4600–4604 RSC.
- L. Wu, C. Han, X. Jing and Y. Yao, Chin. Chem. Lett., 2021, 32, 3322–3330 CrossRef CAS.
- C. Schönbeck, H. Li, B.-H. Han and B. W. Laursen, J. Phys. Chem. B, 2015, 119, 6711–6720 CrossRef PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- P. Thordarson, in Supramolecular chemistry, ed. J. W. Steed and P. A. Gale, Wiley, Chichester, 2012 Search PubMed.
- K. Hirose, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193–209 CrossRef CAS.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- C. Li, L. Zhao, J. Li, X. Ding, S. Chen, Q. Zhang, Y. Yu and X. Jia, Chem. Commun., 2010, 46, 9016–9018 RSC.
- M. Boominathan and M. Arunachalam, ACS Appl. Polym. Mater., 2020, 2, 4368–4372 CrossRef CAS.
- M. Boominathan, J. Kiruthika and M. Arunachalam, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1508–1515 CrossRef CAS.
- M. P. Foster, C. A. McElroy and C. D. Amero, Biochemistry, 2007, 46, 331–340 CrossRef CAS PubMed.
- C. Li, S. Chen, J. Li, K. Han, M. Xu, B. Hu, Y. Yu and X. Jia, Chem. Commun., 2011, 47, 11294–11296 RSC.
- M. E. Harmon, D. Kuckling, P. Pareek and C. W. Frank, Langmuir, 2003, 19, 10947–10956 CrossRef CAS.
- M. E. Harmon, D. Kuckling and C. W. Frank, Macromolecules, 2003, 36, 162–172 CrossRef CAS.
- D. J. Anderson, Clin. Chem., 1989, 35, 2152–2153 CrossRef CAS.
- E. G. Sheetz, D. van Craen and A. H. Flood, in Anion-binding catalysis, ed. O. García Mancheño, Wiley–VCH GmbH, 2022, pp. 79–109 Search PubMed.
- D. A. Stauffer, R. E. Barrans and D. A. Dougherty, J. Org. Chem., 1990, 55, 2762–2767 CrossRef CAS.
- J. C. Adrian and C. S. Wilcox, J. Am. Chem. Soc., 1991, 113, 678–680 CrossRef CAS.
- A. B. Braunschweig, C. M. Ronconi, J.-Y. Han, F. Aricó, S. J. Cantrill, J. F. Stoddart, S. I. Khan, A. J. P. White and D. J. Williams, Eur. J. Org. Chem., 2006, 1857–1866 CrossRef CAS.
- L. Li and G. J. Clarkson, Org. Lett., 2007, 9, 497–500 CrossRef CAS PubMed.
- T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419–9422 CrossRef CAS.
- Z. Zhang, B. Xia, C. Han, Y. Yu and F. Huang, Org. Lett., 2010, 12, 3285–3287 CrossRef CAS PubMed.
- Y. Ma, Z. Zhang, X. Ji, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Eur. J. Org. Chem., 2011, 5331–5335 CrossRef CAS.
- T. Ogoshi, K. Demachi, K. Kitajima and T. Yamagishi, Chem. Commun., 2011, 47, 7164–7166 RSC.
- Y. Chen, M. He, B. Li, L. Wang, H. Meier and D. Cao, RSC Adv., 2013, 3, 21405 RSC.
- J. Han, X. Hou, C. Ke, H. Zhang, N. L. Strutt, C. L. Stern and J. F. Stoddart, Org. Lett., 2015, 17, 3260–3263 CrossRef CAS PubMed.
- D. N. Shurpik and I. I. Stoikov, Russ. J. Gen. Chem., 2016, 86, 752–755 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01354e |
| ‡ The compound HT-Boc is inclined to formation of inter- or even intramolecular inclusion complexes in CDCl3, where the side chain acts as a guest moiety for the macrocycle. For this reason, CD3CN was used as a co-solvent for the analysis. |
| § The compound HT is inclined to formation of inter- or even intramolecular inclusion complexes in CDCl3, where the side chain acts as a guest moiety for the macrocycle. For this reason, CD3CN was used as a co-solvent for the analysis. |
| This journal is © The Royal Society of Chemistry 2024 |