
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-based, random terpolymers with defined functionality based on poly(limonene carbonate-ran-menth-1-ene carbonate)†
Marcel
Höferth
,
Holger
Schmalz
 and
Andreas
Greiner
*
and
Andreas
Greiner
*
University of Bayreuth, Macromolecular Chemistry and Bavarian Polymer Institute, Universitätsstraße 30, 95440 Bayreuth, Germany. E-mail: andreas.greiner@uni-bayreuth.de
First published on 1st March 2024
Abstract
The increasing shortage in fossil resources creates a strong demand for the development of bio-based polymers with tailored properties, not competing with food resources. Polycarbonates, produced by ring-opening copolymerization (ROCOP) of epoxides and CO2, are one promising material class to solve this issue. Poly(limonene carbonate) (PLimC) is a bio-based and non-food based polycarbonate made from limonene oxide (LimO) and CO2. It features an exocyclic double bond at each repeating unit, which can be utilized for further functionalization reactions. However, the degree of functionalization is hardly controllable. Here, we demonstrate that random terpolymerization of LimO with its hydrogenated analogue menth-1-ene oxide (Men1O) and CO2 gives access to polycarbonates with a defined number and homogeneous distribution of functional groups within the polymer chain. The reactivity ratios, determined by the Fineman-Ross and non-linear least square (NLLS) methods, are close to 1.0 for both LimO and Men1O, proving the random nature of the terpolymerization. The versatility of this synthetic platform is shown by thiol–ene click functionalization with 2-mercaptoethanol, yielding terpolymers with a defined number of pendant hydroxy groups. These are exemplarily used for fluorescence labelling of solution cast films with 5-fluorescein isothiocyanate (FITC), revealing a high accessibility of the pendant hydroxy groups for further reactions, even in heterogeneous systems.
Introduction
In view of environmental challenges, the demand for non-fossil-based polymers is increasing. Polymers based on renewable bio-based resources contribute to a possible solution.1 However, it is not only the environmental challenge but also new structure–property relationships that could make bio-based polymers of interest both for research and industrial applications. With the increasing shortage of food resources, the use of non-food relevant bio-based polymers should be preferred, ideally based on biowaste.Polycarbonates that can be prepared either by polycondensation or ring-opening copolymerization (ROCOP) from epoxides and CO2 have attracted much attention among bio-based polymers and have been addressed in several reviews.2,3 Bio-based polycarbonates made with polycondensation are mostly based on sugars and therefore compete with the food market. Besides, the source for epoxides for ROCOP can vary from fatty acids and terpenes to essential oils, and the use of CO2 as a renewable C1 building block is also highly beneficial.4 Poly(cyclohexene carbonate) (PCHC) is a common example for an aliphatic polycarbonate produced by ROCOP of an epoxide (cyclohexene oxide) and CO2. Cyclohexene oxide could be obtained as a waste product in the self-metathesis of plant oils, but the brittleness of PCHC makes it impractical for industrial applications.3,5,6 Another excellent example for a bio- and non-food-based polymer is poly(limonene carbonate) (PLimC). PLimC is based on R-(+)-limonene, which is extracted from orange peel and subsequently oxidized to trans-limonene oxide (LimO) and copolymerized with CO2 using a β-diiminate zinc catalyst.7,8 PLimC is highly transparent, harder than bisphenol A (BPA)-based aromatic polycarbonates and its surprisingly high gas permeation and good selectivity towards CO2 make it suitable for “breathing glass” applications.9 The possibility for chemical recycling to recover LimO and the promising life cycle assessment underline its potential for future applications in a circular economy.10 An additional advantage of PLimC is the possibility for post-modification at the exocyclic double bond.11,12 However, functionalization reactions with the exocyclic double bond have only been performed on PLimC homopolymers yet and, thus, the degree of functionalization was hardly controllable (Scheme 1).
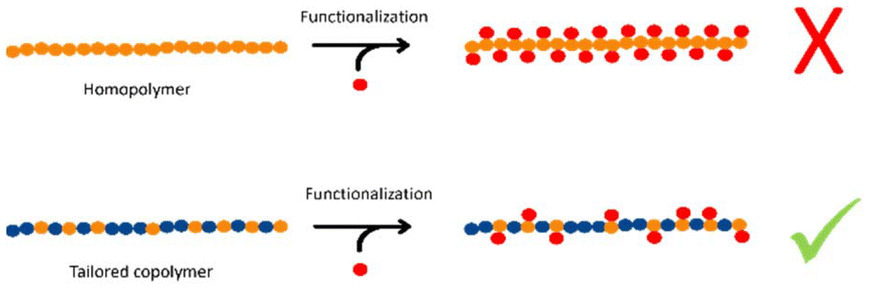 | ||
| Scheme 1 A PLimC homopolymer can be functionalized at the exocyclic double bond of every single repeating unit. In order to control the number of functional groups it is, however, necessary to produce a copolymer with the desired number of LimC repeating units. | ||
The number of functional groups can only be tuned by adjusting the conversion during the functionalization reaction, which is commonly not possible in a reproducible way. In addition, steric effects might produce an irregular distribution of functional groups along the polymer backbone. It is therefore indispensable to construct random copolymers with a tailored number of double bonds, being equally distributed within the polymer chain. Darensbourg et al. reported several reactivity ratios of CO2 based terpolycarbonates, e.g. for poly(1,3-cyclohexadiene carbonate-co-propylene carbonate) the reactivity ratios were determined to be 0.846 and 0.553,13 and for poly(cyclohexene carbonate-co-vinylcyclohexene carbonate) 1.032 and 0.847.14 Additionally, they showed for the latter system that at a reaction temperature of 40 °C a more equal distribution of the comonomers could be achieved compared to that at 25 °C. Frey et al. demonstrated the terpolymerization of propylene oxide with 1,2-epoxy-5-hexene and CO2, Zhang et al. produced random copolycarbonates and Williams et al. produced poly(carbonate-ran-ester)s at low CO2 pressure, however, reactivity ratios were not determined in these studies.15
LimO has been copolymerized with other monomers. However, either sequential monomer addition was employed yielding block copolymers,5 or the difference in monomer reactivity resulted in copolymers with a block-like structure.16 The only exception is a statistical terpolymer of PLimC and poly(propylene carbonate), which was realized by adding LimO over the course of the reaction.17
An elegant way to produce LimO-based polycarbonates with a random distribution of exocyclic double bonds would be the terpolymerization of CO2 with LimO and its hydrogenated analogue trans-menth-1-ene oxide (Men1O, Fig. 1a). Men1O can be obtained by stereoselective epoxidation of R-(+)-menth-1-ene via its bromohydrin (NBS method), and R-(+)-menth-1-ene is readily available from R-(+)-limonene by catalytic hydrogenation of the exocyclic double bond.20 As both comonomers, LimO and Men1O, have very similar structures the formation of random terpolymers should be feasible.
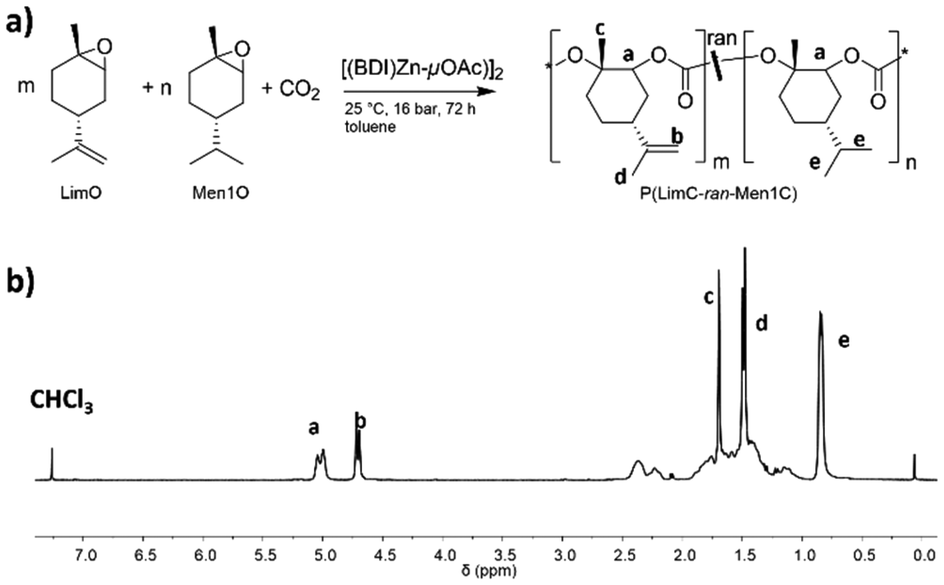 | ||
| Fig. 1 (a) Terpolymerization of LimO, Men1O and CO2 in the presence of the β-diiminate zinc catalyst [(BDI)Zn-μOAc]. (b) 1H NMR spectrum of P(LimC-ran-Men1C) with 43 mol% LimC in CDCl3. | ||
The aim of this work is to produce bio-based polycarbonates with a defined degree of functionalization (pendant exocyclic double bonds) by terpolymerization of LimO and Men1O in various feed ratios with CO2. The Fineman-Ross and non-linear least square (NLLS) methods were applied to determine the respective reactivity ratios and to probe for the random character of the terpolymerization to achieve an even distribution of the LimC units in the polymer chain. Subsequently, the terpolymers were functionalized on the exocyclic double bond of the LimC repeating units with hydroxyl groups by employing the highly efficient thiol–ene click reaction with 2-mercaptoethanol. To prove the accessibility of the pendant hydroxyl groups to further reactions and to visually confirm the functionalization, the terpolymers were labelled with fluorescein isothiocyanate (FITC).
Experimental
Materials
The monomers LimO and Men1O as well as the β-diiminate zinc catalyst [(BDI)Zn-μOAc] were synthesized according to literature procedures.8,11,18 LimO and Men1O had a purity of 84% and 92% trans-conformation, respectively, according to gas chromatography (GC) measurements. R-(+)-Limonene (96%) and 2-mercaptoethanol (99%) were purchased from Carl Roth. Azobisisobutyronitrile (AIBN) was purchased from Sigma-Aldrich and purified by recrystallization from methanol. 5 wt% platinum on charcoal (Pt/C; 99%) was purchased from Acros. FITC was purchased from Fisher Scientific. Toluene (technical grade) was refluxed over 1,1-diphenyl-3-methylpentyllithium, prepared in situ by the reaction of sec-butyllithium (sec-BuLi, 1.3 M in cyclohexane/hexane (92/8), Thermo Scientific, AcroSeal) with 1,1-diphenylethylene (>98.0%, TCI), under nitrogen atmosphere, distilled and freshly condensed from 1,1-diphenyl-3-methylpentyllithium on the day of polymerization. CO2 (>99.995%, Rießner Gase) was dried by passing over a column filled with molecular sieves.Methods
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Avance-300 NMR spectrometer operating at 300 MHz, using deuterated chloroform (CDCl3) as a solvent. Chemical shifts, δ, are indicated in parts per million (ppm) with respect to tetramethylsilane (TMS) as reference.Differential scanning calorimetry (DSC) was performed on a Netzsch 204 F1 Phoenix system using a scanning rate of 40 K min−1 under N2 atmosphere.
Thermogravimetric analysis (TGA) was conducted on a Netzsch TG 209 F1 Libra at a scanning rate of 10 K min−1 under N2 atmosphere.
Size exclusion chromatography (SEC) analyses were carried out on an Agilent 1200 system equipped with a SDV precolumn (particle size 5 μm; PSS Mainz), a SDV linear XL column (particle size 5 μm, PSS Mainz) and a refractive index detector (G1362A, Agilent Technologies). Chloroform (high-performance liquid chromatography (HPLC) grade) was used as a solvent at a flowrate of 0.5 mL min−1 at room temperature. The calibration was done with narrowly distributed polystyrene standards (PSS calibration kit) and toluene (HPLC grade) was used as the internal standard.
Infrared (IR) spectra were recorded with a PerkinElmer Spectrum Two Fourier-transform (FT)-IR spectrometer.
Monomer syntheses
500 mL R-(+)-limonene were added to a stainless-steel hydrogenation reactor (Büchi AG) with 6 g of Pt/C (5 wt%), pressurized with 5 bar of hydrogen gas and stirred for 6 h at 25 °C. The product was filtered over alumina to remove the Pt/C catalyst and the purity of the obtained R-(+)-menth-1-ene was checked with GC to be 96%. For stereoselective epoxidation 500 mL of R-(+)-menth-1-ene were added to a 4 L three-neck round bottom flask and stirred with acetone and water at 0 °C, while 400 g of N-bromo succinimide (NBS) were added slowly. The acetone was removed with a rotary evaporator and 1 L of an aqueous 4 M NaOH solution were added and stirred at 60 °C for 2 h. Subsequently, 1 L of ethyl acetate was added, and the aqueous phase was removed. Finally, the organic solvent was distilled off by rotary evaporation and the purity of the isolated Men1O was checked with GC. LimO was prepared in an analogous manner by stereoselective epoxidation of R-(+)-limonene.Synthesis of P(LimC-ran-Men1C)
The catalyst [(BDI)Zn-μOAc] (80 mg; 0.162 mmol) was added into a previously dried Schlenk flask with a magnetic stirring bar in a glove box. LimO (8.3 mL; 50.6 mmol) and Men1O (8.3 mL; 50.0 mmol) were dried over n-butyllithium (0.8 mL 1.6 M in hexanes; Acros), which was added to the monomer at −78 °C (acetone-dry ice-bath) under dry nitrogen atmosphere followed by slow heating to room temperature and stirring over night. The purified monomers were subsequently distilled and added via a nitrogen flushed syringe through a rubber septum to the Schlenk flask with the catalyst followed by stirring to completely dissolve the catalyst. The monomer/catalyst solution and dry toluene (42 mL) were then added in nitrogen counterflow to a 200 mL oven-dried stainless-steel autoclave (Büchi AG), equipped with a sampling tube and a magnetic stirring bar. The autoclave was then pressurized with dry CO2 (20 bar) and stirred at 25 °C for 72 h. To the resulting mixture 20 wt% of acetic anhydride were added for end-group functionalization to increase thermal stability and stirred for 2 h before precipitating the mixture in MeOH. After drying in rotary vane pump vacuum at 80 °C, the resulting polymers were analyzed by 1H NMR spectroscopy and SEC.Determination of reactivity ratios by Fineman-Ross and NLLS methods
For the Fineman-Ross method terpolymerizations were typically conducted to low total monomer conversions of 5–10%. To figure out the time until 10% monomer conversion was reached, a kinetic study was undertaken. Samples of around 0.5 mL were taken at various times (0 h, 2 h, 4 h, 6 h, 8 h, 24 h) during the terpolymerization with equimolar amounts of LimO and Men1O. Deuterated dichloromethane (CD2Cl2) was added, and the samples were analyzed with 1H NMR spectroscopy. After 6 h a conversion of 10% was reached (Fig. S1 and S2†).Samples of around 1 mL were then withdrawn from the terpolymerizations with various monomer feed ratios (20 mol%, 40 mol%, 60 mol%, 80 mol% LimO) via the sampling tube after 6 h, precipitated in MeOH and dried at 80 °C in vacuo. Then, the composition of the terpolymers was analyzed with 1H NMR spectroscopy and the reactivity ratios were determined according to the Fineman-Ross method. The obtained reactivity ratios were further verified employing the NLLS method (for details please see ESI†).
Functionalization of P(LimC-ran-Men1C) with 2-mercaptoethanol
The procedure was adapted from literature and is exemplarily described for a random terpolymer with 10 mol% LimC repeating units.19 P(LimC-ran-Men1C) (15.0 g, 10 mol% (7.58 mmol) LimC repeating units) was dissolved in 250 mL chloroform. 373 mg AIBN (2.27 mmol, 0.3 eq.) and 16.0 mL 2-mercaptoethanol (227.4 mmol, 30 eq.) were added and heated under reflux for 24 h. The polymer was then precipitated in methanol and dried under reduced pressure at 80 °C.Solvent casting and functionalization with FITC
The functionalized polymers were cast into films by dissolving 1.3 g in 20 mL of dichloromethane and pouring the solutions into glass Petri dishes (Ø = 8 cm), followed by slowly drying at 25 °C for 96 h and subsequently with a rotary vane vacuum pump for 24 h.For FITC functionalization the films were cut into rectangles (1 cm × 2 cm; m = 40 mg) and added to a FITC solution in EtOH/H2O = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) (c = 5 g L−1, V = 10 mL) for 24 h at RT. Afterwards, the films were washed with a large excess of deionized water for 16 h and dried at RT.
:1 (v/v) (c = 5 g L−1, V = 10 mL) for 24 h at RT. Afterwards, the films were washed with a large excess of deionized water for 16 h and dried at RT.
Results and discussion
Terpolymerization of LimO, Men1O and CO2
The monomers LimO and Men1O, as well as the β-diiminate zinc catalyst [(BDI)Zn-μOAc] were synthesized and purified according to previously reported procedures.8,11,18 Terpolymerizations were performed by adding both purified monomers and the catalyst to a stainless-steel autoclave, applying pressurized CO2 and stirring at room temperature for 3 days. For the determination of reactivity ratios by the Fineman-Ross method samples were withdrawn after 6 h, i.e., after a conversion of ca. 10% was reached (Fig. S1 and S2†). The polymers were then purified by precipitation of the reaction mixture in methanol. A detailed description of the syntheses is provided in the Experimental section.The structure as well as the composition of the terpolymers were studied by 1H NMR (Fig. 1, S3†). The signals at 5.00 ppm (a) and 4.70 ppm (b) confirm the successful terpolymerization and can be used to calculate the content of LimC repeating units and accordingly the content of exocyclic C–C double bonds. No ether linkages or cyclic carbonates were detected by 1H NMR spectroscopy for the polymerization with the [(BDI)Zn-μOAc] catalyst. All terpolymers show narrow molar mass distributions and apparent number-averaged molecular weights in the range of ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n,app ≈ 60–120 kg mol−1, as determined by SEC (Fig. 2). The weak shoulder towards higher molecular weights might arise from the presence of a certain fraction of catalytic sites with a different reactivity, or an incomplete deactivation of the β-diiminate zinc complex, as a dimeric active center with two growing chains attached is involved in the polymerization.8 The molecular characteristics of the synthesized P(LimC-ran-Men1C) are summarized in Table 1.
n,app ≈ 60–120 kg mol−1, as determined by SEC (Fig. 2). The weak shoulder towards higher molecular weights might arise from the presence of a certain fraction of catalytic sites with a different reactivity, or an incomplete deactivation of the β-diiminate zinc complex, as a dimeric active center with two growing chains attached is involved in the polymerization.8 The molecular characteristics of the synthesized P(LimC-ran-Men1C) are summarized in Table 1.
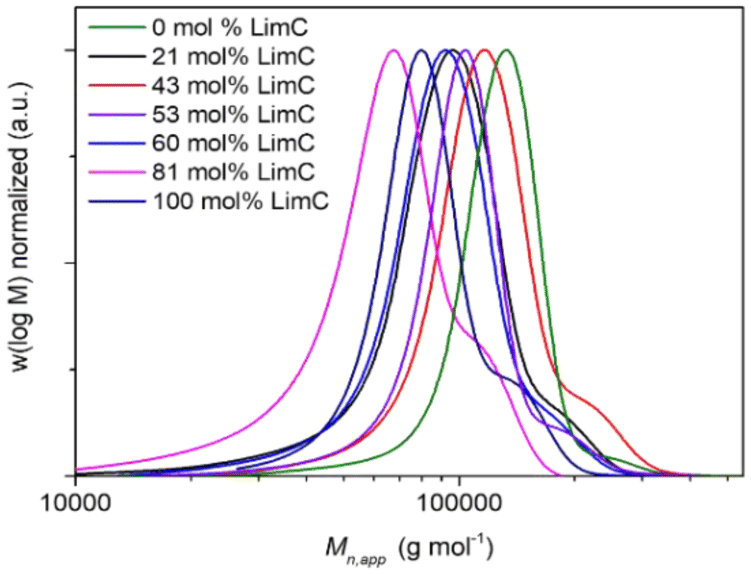 | ||
| Fig. 2 Apparent molar mass distributions of P(LimC-ran-Men1C) with varying LimC content as indicated (CHCl3-SEC, PS calibration). | ||
| Entry | LimO in feed (mol%) | LimO incorp.a (mol%) |
n,theor.
(kg mol−1) |
n,app
(kg mol−1) |
Đ | T g (°C) | T 5% (°C) | OH-groupsa,f (mol%) |
|---|---|---|---|---|---|---|---|---|
|
a Calculated from 1H NMR spectroscopy (300 MHz, CDCl3).
b Calculated from the molar ratio of repeating units resulting from added monomers and CO2 to catalyst.
c Apparent number-averaged molecular weight (n,app) and dispersity (Đ) determined by CHCl3-SEC (PS calibration).
d Glass transition temperature (Tg) determined from DSC (scanning rate 40 K min−1).
e Temperature at 5% weight loss (T5%) determined by TGA (heating rate 10 K min−1, N2 atmosphere).
f The content of OH-groups after thiol–ene click functionalization is given in mol% with respect to PLimC and PMen1C repeating units.
|
||||||||
| 1 | 0 | 0.0 | 100 | 119 | 1.09 | 133 | 232 | 0 |
| 2 | 20 | 21.0 | 100 | 80 | 1.23 | 131 | 230 | 19 |
| 3 | 40 | 43.0 | 100 | 107 | 1.15 | 133 | 234 | 36 |
| 4 | 50 | 52.5 | 100 | 93 | 1.13 | 130 | 246 | 50 |
| 5 | 60 | 60.2 | 100 | 81 | 1.17 | 133 | 229 | 60 |
| 6 | 80 | 80.6 | 100 | 54 | 1.26 | 132 | 246 | 80 |
| 7 | 100 | 100.0 | 100 | 73 | 1.16 | 136 | 248 | 99 |
The measured n,app of 54 kg mol−1 for the terpolymer with 81 mol% LimC is quite low compared to the other terpolymers and with respect to the theoretical n of 100 kg mol−1. This deviation might be caused by sudden changes in the conditions like pressure when the sample for the Fineman-Ross evaluation was taken through the sampling tube of the reactor. The composition of the terpolymers is in each case close to the monomer feed composition, which already points to a random terpolymerization.
This will be later on confirmed by the determined reactivity ratios. The thermal properties of the terpolymers as determined by TGA (Fig. S4†) and DSC (Fig. S5†) are comparable to that of neat PLimC (T5% = 240 °C; Tg = 130 °C) and PMen1C (T5% = 240 °C; Tg = 130 °C) as both homopolymers already behave quite similarly (Table 1).19 After multiple precipitations in methanol or a mixture of ethanol and isopropanol all terpolymers show a Tg at 130–133 °C (Fig. S5)†.
Determination of reactivity ratios
To prove the assumption that terpolymerization proceeds in a random manner, the reactivity ratios were determined with the Fineman-Ross20 and the non-linear least square (NLLS) method,21 both being commonly employed for polycarbonate terpolymers.14,22 As discussed above, terpolymerizations with varying monomer feed ratios were conducted and samples were taken after 6 h reaction time (conversion of ca. 10%, Fig. S2†) and analyzed by 1H NMR (Fig. 1b and S3†). The reactivity ratios were then determined from the molar ratios of the comonomers in the feed (F) and the molar ratio of PLimC and PMen1C repeating units in the obtained terpolymers (f), respectively. The values were plotted according to eqn (1) and the reactivity ratios r1(LimO) and r2(Men1O) were obtained from the slope and intercept of the corresponding linear fit as depicted in Fig. 3.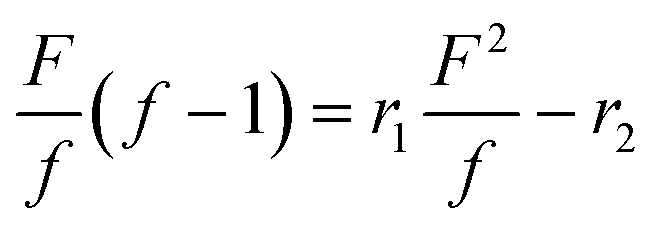 | (1) |
 | ||
| Fig. 3 Fineman-Ross plot for the terpolymerization of LimO (M1) and Men1O (M2) with CO2. | ||
with 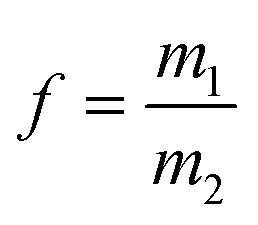 and
and 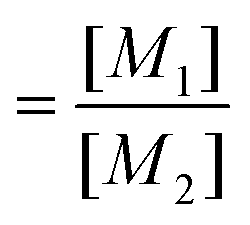 ; [M1] and [M2] are the monomer concentrations in solution and m1/m2 is the molar ratio of the monomer units in the copolymer.
; [M1] and [M2] are the monomer concentrations in solution and m1/m2 is the molar ratio of the monomer units in the copolymer.
The obtained reactivity ratios are r1 = 1.025 for LimO and r2 = 0.956 for Men1O. For comparison, the reactivity ratios were also determined employing the NLLS method, resulting in similar values of r1 = 1.052 for LimO and r2 = 0.924 (Fig. S6†). As both reactivity ratios are close to 1.0 a random character can be deduced for the terpolymerization, which is attributed to the almost identical structure of LimO and Men1O. This allows the production of truly random terpolymers with an even distribution of functional groups (pendant exocyclic double bonds) that can be used for further polymer analogous reactions. In contrast, the reported reactivity ratios for CO2 based terpolycarbonates in literature commonly deviate significantly from 1.0, which leads to terpolymers with a gradient in composition and, thus, a certain inhomogeneity in the distribution of functional groups along the chain.13,14
Functionalization via thiol–ene click reaction
Following the successful synthesis of random P(LimC-ran-Men1C) terpolymers with varying compositions, the exocyclic double bonds at the LimC repeating units were functionalized with 2-mercaptoethanol by thiol–ene click reaction to introduce pendant hydroxy groups (Scheme 2).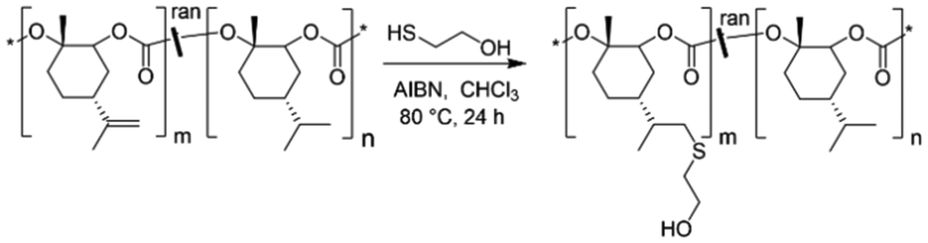 | ||
| Scheme 2 Functionalization of bio-based P(LimC-ran-Men1C) with 2-mercaptoethanol. | ||
Successful functionalization was confirmed for all samples via1H NMR spectroscopy (Fig. 4, Table 1) by the absence of the characteristic signal of the isopropenyl protons (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) of the PLimC repeating units, which would be expected at 4.71 ppm. Consequently, the mole fraction of repeating units with pendant OH-groups matches well with the mole fraction of PLimC units in the employed terpolymers (Table 1). Accordingly, the integral of the methylene protons (b) from the functionalized PMen1C-OH units at 3.71 ppm increases with the degree of functionalization. Successful functionalization can also be probed by FT-IR spectroscopy by the appearance of the characteristic O–H stretching vibration at ca. 3400 cm−1 (Fig. S7†).
CH2) of the PLimC repeating units, which would be expected at 4.71 ppm. Consequently, the mole fraction of repeating units with pendant OH-groups matches well with the mole fraction of PLimC units in the employed terpolymers (Table 1). Accordingly, the integral of the methylene protons (b) from the functionalized PMen1C-OH units at 3.71 ppm increases with the degree of functionalization. Successful functionalization can also be probed by FT-IR spectroscopy by the appearance of the characteristic O–H stretching vibration at ca. 3400 cm−1 (Fig. S7†).
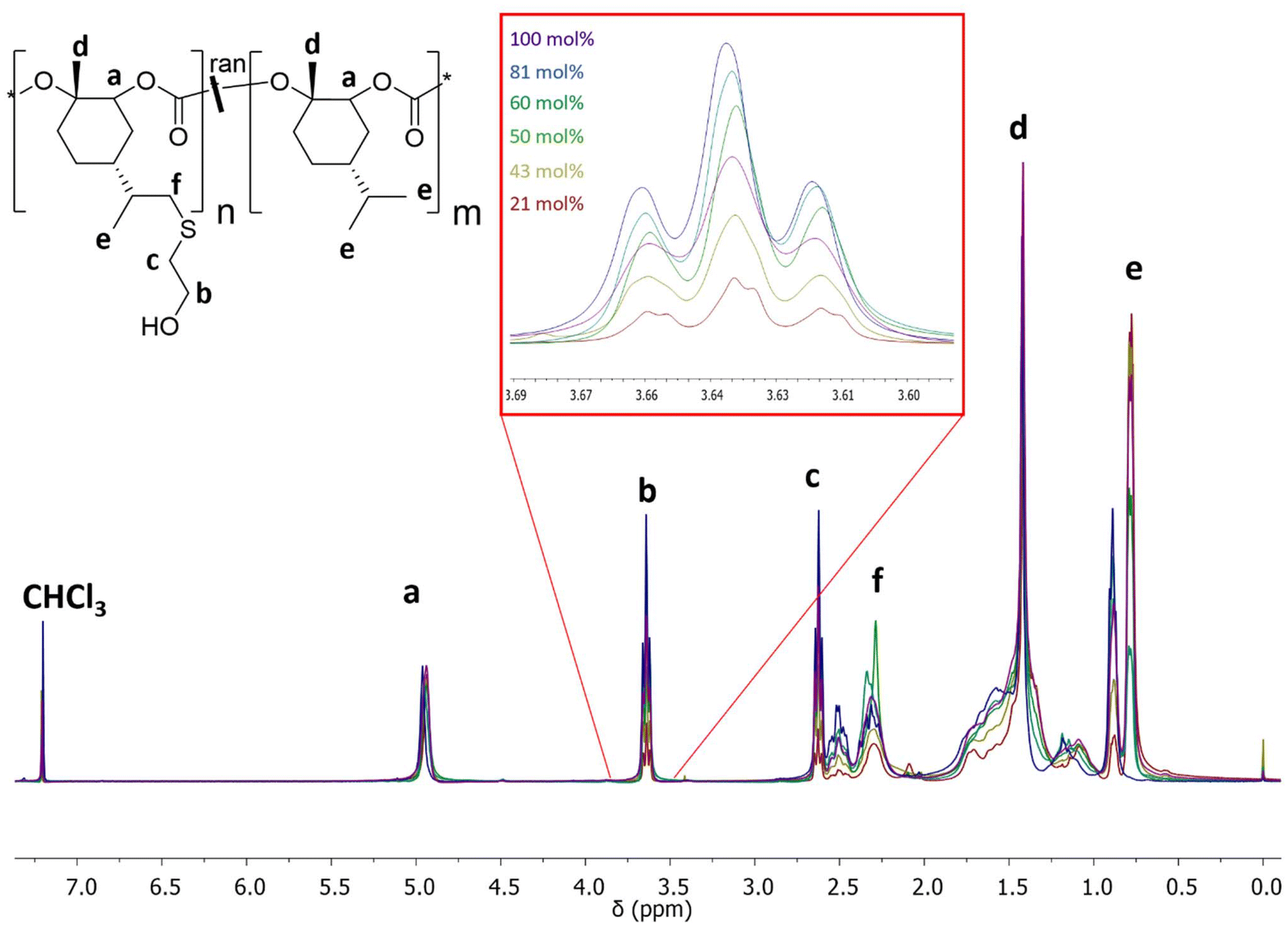 | ||
| Fig. 4 1H NMR spectra of P(Men1C-ran-Men1C-OH) with varying degree of functionalization. | ||
Next to a successful functionalization it is important that undesired crosslinking reactions are efficiently suppressed by the employed excess of 2-mercaptoethanol (30-fold excess with respect to pendant double bonds). This is exemplarily proven by SEC for the terpolymer with 81 mol% PLimC repeating units, revealing a comparable molar mass distribution after functionalization without a noticeable increase in high molar mass fractions (Fig. 5). The slight broadening and shift of the distribution toward lower molecular weights after functionalization can be attributed to decreased hydrodynamic radii of the polymer coil caused by the interaction of the pendant OH-groups with the “poorer” solvent.23
 | ||
| Fig. 5 Apparent molar mass distributions of P(LimC-ran-Men1C) with 81 mol% PLimC repeating units and the respective OH-functionalized P(Men1C-ran-Men1C-OH) (80 mol% OH groups) after thiol–ene reaction (CHCl3-SEC, PS calibration). | ||
Fluorescence labeling
To test the availability of the pendant OH-groups in the terpolymers for subsequent reactions, fluorescein 5-isothiocyanate (FITC, Fig. 6a) was used as a fluorescent marker.24 The reaction was performed on bulk films, including PMen1C (unfunctionalized) as negative control (for details see Experimental section). The success of the reaction was probed by the appearance of a characteristic yellow-orange fluorescence upon illumination with monochromatic light (λ = 366 nm, Fig. 6b). The fluorescence intensity of the FITC treated films increases with the content of pendant OH-groups in the terpolymers (from 36 mol% to 80 mol%). Due to the lack of pendant OH-groups the negative control PMen1C shows no fluorescence, proving the absence of any unspecific reactions with FITC or adsorption effects. It is noted that FITC functionalization was conducted on bulk films in ethanol/water mixtures where only FITC is soluble. This shows that the pendant OH-groups are highly accessible for further functionalization reactions even in heterogenous systems. | ||
| Fig. 6 (a) Reaction scheme for the functionalization of pendant hydroxy groups in P(Men1C-ran-Men1C-OH) with FITC; (b) Films of FITC-functionalized P(Men1C-ran-Men1C-OH) with varying Men1C-OH contents as indicated. Unfunctionalized PMen1C homopolymer was added as a negative control. | ||
Conclusions
We have shown that fully bio-based P(LimC-ran-Men1C) terpolycarbonates with defined functionality are accessible by the terpolymerization of limonene oxide (LimO) and menth-1-ene oxide (Men1O) with CO2, employing the β-diiminate zinc catalyst [(BDI)Zn-μOAc]. The random nature of the terpolymerization was proven by determining the reactivity ratios with the Fineman-Ross and NLLS methods, revealing reactivity ratios close to 1.0 for both monomers. This gives access to random terpolymers with a defined number and uniform distribution of pendant exocyclic double bonds arising from the PLimC repeating units, representing a versatile platform for further functionalization reactions. Based on the highly efficient thiol–ene click reaction a series of hydroxy functionalized terpolymers were prepared and subsequently marked with the common fluorescence dye FITC, underlining the versatility of these truly random terpolymers.This concept could be harnessed to construct bio-based polycarbonates with tailored functionality utilizing thiol–ene click chemistry to introduce a variety of functional groups, like ionic groups, alkyl or oligo(ethylene glycol) segments, etc. to tune hydrophilicity, solubility, as well as thermal and mechanical properties. In a next step, the P(LimC-ran-Men1C)-OH terpolymers with defined number of pendant evenly distributed OH-groups can be used for the preparation of graft copolymers employing ring-opening polymerization of lactones and lactides or using reversible deactivation radical polymerization (RDRP) techniques after proper functionalization with the respective initiating sites, which will be the focus of future studies.
Author contributions
All experiments were conducted by Marcel Höferth. Andreas Greiner and Holger Schmalz created the concept and supervised the work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by German Research Foundation (DFG, 438886960). The authors would like to thank Rika Schneider for the SEC measurements and the keylab “Synthesis and Molecular Characterization” of the Bavarian Polymer Institute (BPI) for support.References
- (a) R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef; (b) J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed; (c) V. Siracusa and I. Blanco, Polymers, 2020, 12, 1641–1657 CrossRef CAS; (d) F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed; (e) R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y.-X. Chen, Nat. Rev. Mater., 2022, 7, 83–103 CrossRef CAS.
- (a) H. R. Kricheldorf, J. Macromol. Sci., Polym. Rev., 1997, 37, 599–631 CrossRef; (b) W. T. Diment, W. Lindeboom, F. Fiorentini, A. C. Deacy and C. K. Williams, Acc. Chem. Res., 2022, 55, 1997–2010 CrossRef CAS PubMed; (c) Y. Liu and X.-B. Lu, J. Polym. Sci., 2022, 60, 3256–3268 CrossRef CAS; (d) D. Yu, J. Zhong, Z. Pu, H. Hou, X. Li, R. Zhu, X. Wang, F. Wu, P. Zheng and J. Liu, J. Polym. Res., 2023, 30, 204 CrossRef CAS; (e) M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef; (f) M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 41141 CrossRef; (g) S. Cui, J. Borgemenke, Z. Liu and Y. Li, J. CO2 Util., 2019, 34, 40–52 CrossRef CAS; (h) S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC; (i) F. Siragusa, C. Detrembleur and B. Grignard, Polym. Chem., 2023, 14, 1164–1183 RSC; (j) O. Santoro, L. Izzo and F. Della Monica, Sustainable Chem., 2022, 3, 259–285 CrossRef CAS.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- (a) M. Sengoden, G. A. Bhat and D. J. Darensbourg, Macromolecules, 2023, 56, 2362–2369 CrossRef CAS; (b) S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC; (c) V. C. Eze, A. Rehman, M. Patel, S. Ahmad and A. P. Harvey, RSC Adv., 2022, 12, 17454–17465 RSC.
- J. Bailer, S. Feth, F. Bretschneider, S. Rosenfeldt, M. Drechsler, V. Abetz, H. Schmalz and A. Greiner, Green Chem., 2019, 21, 2266–2272 RSC.
- (a) M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196 RSC; (b) J. G. Kim, C. D. Cowman, A. M. LaPointe, U. Wiesner and G. W. Coates, Macromolecules, 2011, 44, 1110–1113 CrossRef CAS; (c) Y. Song, K. Yin, Y. Chen, B. Zhao, Y. Zhang, X. Zhu, D. Yuan and Y. Yao, Chin. J. Chem., 2023, 41, 805–813 CrossRef CAS.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- O. Hauenstein, M. M. Rahman, M. Elsayed, R. Krause-Rehberg, S. Agarwal, V. Abetz and A. Greiner, Adv. Mater. Technol., 2017, 2, 1700026 CrossRef.
- (a) A. Durkin, I. Taptygin, Q. Kong, M. F. M. Gunam Resul, A. Rehman, A. M. L. Fernández, A. P. Harvey, N. Shah and M. Guo, ChemistryOpen, 2019, 8, 668–688 CrossRef CAS PubMed; (b) D. H. Lamparelli, A. Villar-Yánez, L. Dittrich, J. Rintjema, F. Bravo, C. Bo and A. W. Kleij, Angew. Chem., Int. Ed. Engl., 2023, 62, e202314659 CrossRef CAS; (c) C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS PubMed; (d) D. Zhang, E. A. Del Rio-Chanona, J. L. Wagner and N. Shah, Sustain. Prod. Consum., 2018, 14, 152–160 CrossRef; (e) F. Parrino, A. Fidalgo, L. Palmisano, L. M. Ilharco, M. Pagliaro and R. Ciriminna, ACS Omega, 2018, 3, 4884–4890 CrossRef CAS PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- (a) C. Li, S. van Berkel, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2016, 85, 466–477 CrossRef CAS; (b) N. Kindermann, À. Cristòfol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS; (c) D. Ghosh and S. Agarwal, Polym. Chem., 2023, 14, 4626–4635 RSC; (d) C. Li, T. Veldhuis, B. Reuvers, R. J. Sablong and C. E. Koning, Polym. Int., 2020, 69, 24–30 CrossRef CAS.
- D. J. Darensbourg, W.-C. Chung, A. D. Yeung and M. Luna, Macromolecules, 2015, 48, 1679–1687 CrossRef CAS.
- D. J. Darensbourg, R. R. Poland and A. L. Strickland, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 127–133 CrossRef CAS.
- (a) G. Rosetto, A. C. Deacy and C. K. Williams, Chem. Sci., 2021, 12, 12315–12325 RSC; (b) H. Zhang, B. Liu, H. Ding, J. Chen and Z. Duan, Polymer, 2017, 129, 5–11 CrossRef CAS; (c) J. Geschwind, F. Wurm and H. Frey, Macromol. Chem. Phys., 2013, 214, 892–901 CrossRef CAS.
- (a) S. Neumann, S. B. Däbritz, S. E. Fritze, L.-C. Leitner, A. Anand, A. Greiner and S. Agarwal, Polym. Chem., 2021, 12, 903–910 RSC; (b) S. Kernbichl and B. Rieger, Polymer, 2020, 205, 122667 CrossRef CAS.
- A. Denk, E. Fulajtar, C. Troll and B. Rieger, Macromol. Chem. Phys., 2023, 308, 2300097 CAS.
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Organomet. Chem., 2003, 683, 137–148 CrossRef CAS.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- M. Fineman and S. D. Ross, J. Appl. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
- A. M. van Herk and T. Dröge, Macromol. Theory Simul., 1997, 6, 1263–1276 CrossRef CAS.
- (a) D. J. Darensbourg and W.-C. Chung, Polyhedron, 2013, 58, 139–143 CrossRef CAS; (b) D. J. Darensbourg and Y. Wang, Polym. Chem., 2015, 6, 1768–1776 RSC; (c) L.-L. Mei, G.-P. Yan, X.-H. Yu, S.-X. Cheng and J.-Y. Wu, J. Appl. Polym. Sci., 2008, 108, 93–98 CrossRef CAS; (d) Z.-M. Li, G.-P. Yan, C.-W. Ai, Q. Zhang, L. Li, F. Liu, X.-H. Yu and B. Zhao, J. Appl. Polym. Sci., 2012, 124, 3704–3713 CrossRef CAS.
- A. M. Caltabiano, J. P. Foley and H. G. Barth, J. Chromatogr. A, 2016, 1437, 74–87 CrossRef CAS PubMed.
- K. Neumann, S. Jain, J. Geng and M. Bradley, Chem. Commun., 2016, 52, 11223–11226 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01331f |
| This journal is © The Royal Society of Chemistry 2024 |