
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoiniferter-RAFT polymerization mediated by bis(trithiocarbonate) disulfides†
Magdalena A.
Beres
 a,
Julia Y.
Rho
b,
Andrew
Kerr
b,
Timothy
Smith
c and
Sébastien
Perrier
*abcd
a,
Julia Y.
Rho
b,
Andrew
Kerr
b,
Timothy
Smith
c and
Sébastien
Perrier
*abcd
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: s.perrier@warwick.ac.uk
bWarwick Medical School, University of Warwick, Coventry, CV4 7AL, UK
cLubrizol Limited, The Knowle, Nether Lane, Hazelwood, Derbyshire DE56 4AN, UK
dFaculty of Pharmacy and Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
First published on 2nd January 2024
Abstract
Photoiniferter reversible addition fragmentation chain transfer (PI-RAFT) polymerization has gained significant attention; however, scalable methodologies are still lacking. Here, we investigate the use of a RAFT agent precursor, butyltrithiocarbonate disulfide (BisTTC), as an iniferter agent for the polymerization of two model monomers – methyl acrylate (MA) and methyl methacrylate (MMA). Different wavelengths of light were screened to optimise polymerization conditions for both monomers. While BisTTC can photodissociate and initiate polymerization, its activation under visible light is slow and competes with photodegradation. As for methyl acrylate, degradation only occurs during the induction period, so efficient initiation minimises potential side reactions, but the poly(methyl methacrylate)trithiocarbonate end group continues to degrade during polymerization due to the increased lifetime and stability of the propagating radical. Higher energy blue light gives poorer control than green light for the polymerization of MMA, due to increased photodegradation, whilst methyl acrylate polymerization is retarded under green light and requires conditions that strongly favour initiation.
Introduction
Light-mediated polymerizations have gained significant attention in the past decade, particularly because they can amalgamate the advantages of reversible-deactivation radical polymerization (RDRP) with spatiotemporal control. Harnessing light can also bring us closer to sustainable manufacturing of materials as photopolymerizations require significantly less energy than typical thermal polymerizations.1 Since their discovery, many photocontrolled RDRPs have been reported, including reversible addition fragmentation chain transfer (RAFT) polymerization.2–6 PhotoRAFT can follow three different mechanisms: (1) photoinitiated RAFT where thermal free-radical initiators are replaced with photoinitiators which generate radicals upon activation with light rather than heat,7 (2) photoinduced electron/energy transfer (PET)-RAFT which requires an exogenous catalyst which upon activation transfers energy/electrons to a chain transfer agent (CTA),8,9 and (3) photoiniferter-RAFT (PI-RAFT) where RAFT agents themselves act as both initiators and CTAs.2,4 Photoinitiated RAFT suffers from the same types of termination reactions as those in conventional thermal RAFT because in both cases, radicals are generated throughout the reaction from the decomposition of the initiators. Furthermore, side reactions can be introduced if the wavelength of the light activating initiator also activates the RAFT agent. PET-RAFT offers advantages such as higher livingness, but it also requires an extra component that needs to be tuned for each system and possibly has to be removed after polymerization. PI-RAFT, on the other hand, has a simpler setup as it does not require the presence of either an initiator or a photoactivator.The first examples of photoiniferter polymerizations were conducted by Otsu and co-workers in 1950s. In this seminal work, dithiocarbamates and their derivatives were used as initiating, chain transfer and terminating agents.10–12 The molecular weight of the resulting polymers increased with conversion, but attempts to synthesise block copolymers led to significant broadening of molecular weight distribution. The relatively poor control could be attributed to a mismatch of the iniferter Z group and monomer reactivity, which became apparent when RAFT was reported in the late 90's.13,14 In the PI-RAFT process, there are three distinct mechanistic steps: fragmentation of the RAFT agent, degenerative chain transfer and reversible termination (Scheme 1).2,7,15–17 First, photolysis of the RAFT agent leads to a β-scission – homolytic cleavage of a C–S bond which releases a carbon-centred radical derived from the RAFT agent R group and a thiyl radical. R-group radicals are capable of initiating polymerization and hence no exogenous initiator is needed for the PI-RAFT process. Thiyl radicals can then act as persistent radicals and reversibly terminate with the propagating radical.17 Under certain conditions, thiyl radicals can also initiate polymerization, as shown previously by the Perrier group and expanded upon herein.18 A macroCTA can be reactivated by light to establish reversible termination equilibrium, as in the original iniferter work developed by Otsu and co-workers.10–12,19 In reversible termination, propagating chains are reversibly deactivated by a thiyl radical such as trithiocarbonate. The exchange between dormant and active species must be faster than propagation to allow good control over polymerization. However, it is unlikely that reversible termination alone could account for the level of control that was observed in PI-RAFT polymerization. Indeed, in PI-RAFT, the growing chain can also participate in degenerative chain transfer, thus establishing a RAFT equilibrium as in conventional RAFT polymerization. Currently, it is widely agreed that both processes take place to different extents, depending on the RAFT agent and monomer pair, and reaction conditions.16,17,20 Easterling et al. utilised the photolysis of polymer chains bearing either a xanthate or dithiocarbamate end group to reverse the sequence of block copolymers which would not have worked well if the reversible termination did not take place; however, the same process using trithiocarbonates was less successful.21 Furthermore, as there is no exogenous source of radicals in PI-RAFT, higher levels of livingness can be achieved. Carmean et al. showed that unprecedented high molecular weight polymers could be prepared by PI-RAFT, as short initiator-derived chains are eliminated and polymer chains are unlikely to terminate due to limited mobility in viscous media.6,22,23
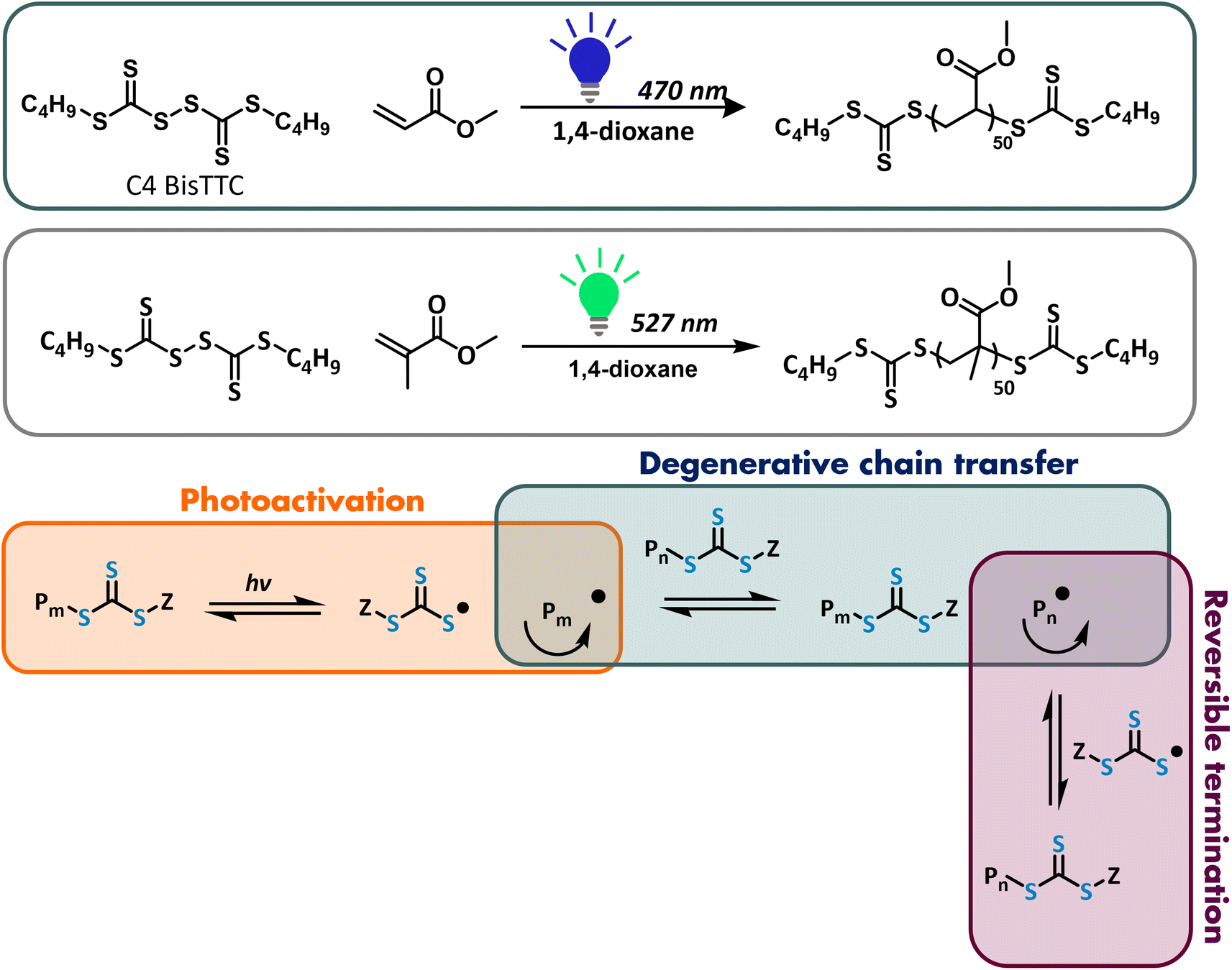 | ||
| Scheme 1 PI-RAFT polymerization of methyl acrylate (MA) and methyl methacrylate (MMA) mediated by C4 BisTTC. | ||
RAFT photopolymerization can be carried out under both visible and UV light, targeting π–π* or n–π* thiocarbonyl transition, respectively. The initial work primarily utilised UV irradiation; however, it was found that UV light could lead to significant degradation of the thiocarbonylthio moiety,7,24 which could be partially suppressed by cutting off short-wavelength UV light.25,26 Subsequently, it was reported independently by Qiao and coworkers and Boyer and coworkers that PI-RAFT could also be carried out under visible light.2,4 Indeed, although the π–π* transition has a higher molar absorption coefficient than the symmetry forbidden n–π* transition, the quantum yield of homolytic bond dissociation is higher for the latter.27 Hughes et al. reported the excitation dependence of photoiniferter kinetics by studying how the reaction rate changes when the π–π* or n–π* transition is targeted.28 They found that for both trithiocarbonates and xanthates, targeting the symmetry-forbidden transition increased the rate of C–S bond photolysis and the rate of initiation. A higher quantum yield, however, does not necessarily directly correlate with the rate of propagation, as the generated radicals can recombine or have poor reactivity.29 Nevertheless, trithiocarbonates are usually used with visible light (mainly blue or green), whereas xanthates, in which the n–π* transition is blue-shifted, are used with UV light.2
The rate of photopolymerizations can be controlled by changing the temperature or light intensity. Increasing the light intensity leads to faster generation of radical/propagating species and can accelerate the reaction.30 Indeed, Konkolewicz and co-workers found that in a model PET-RAFT polymerization, the apparent rate of polymerization scaled with the square root of light intensity.31 However, in photoiniferter polymerization, a higher radical flux will also increase the rate of reversible deactivation, leading to a maximum effective radical concentration, at which point a further increase in light intensity has no effect on the reaction rate.32 Junkers and co-workers reported that for the photoiniferter polymerization of methyl methacrylate, increasing the light intensity increased monomer conversion until light saturation was reached.33 Increased radical concentration could also lead to enhanced bimolecular termination. Johnson and co-workers reported the detrimental effect of increasing light intensity on the polymerization control either due to the increased termination or degradation of CTA, especially when higher degrees of polymerization were targeted.34,35
While thiocarbonylthiyl radicals are not thought to be able to initiate PI polymerization, our group has recently reported successful PI polymerization using bis(trithiocarbonate) disulfide.18 Usually, disulfides are used as precursors of RAFT agents but are rarely used as chain transfer agents themselves.36–38 Disulfides can be reacted with azo initiators to give a RAFT agent with an initiator R group and an Z group from bis(thiocarbonyl) disulfide.39 This reaction can be also carried out in situ40–42 and is triggered by piezoelectrically mediated reduction of alkyl bromides.43 Recently, bisdodecyltrithiocarbonate was used for the polymerization of a range of monomers using a compact fluorescent lamp, to give telechelic polymers that could be cyclised. However, the polymerization only proceeded in the presence of the photoredox catalyst – bismuth oxide.44 Bis(thiocarbonyl) disulfides were also used as chain transfer agents in organo-catalysed photocontrolled radical polymerization with sulfonyl chlorides as initiators.45 The potential of disulfides as agents capable of both initiating and reversibly deactivating polymer chain growth has been recognised as early as the late 1950s by Otsu and co-workers both in photo- and thermal-iniferter systems.19 Using our understanding of both reversible termination and degenerative chain transfer, we aim to study this type of system in depth. Disulfides, which are cheaper and more widely available on a bulk scale, are a promising alternative to conventional thiocarbonyl RAFT agents, whose cost and availability hinder the widespread use of RAFT polymerization in industrial setting.46 Here, we present a detailed study of photopolymerizations mediated by bisbutyltrithiocarbonate in the absence of catalysts and initiators to develop a more scalable and cheaper PI-RAFT methodology. In this approach, disulfide is used as a RAFT agent directly as there is no azo initiator to form a regular chain transfer agent in situ and the resulting polymers are telechelic. The work focuses on two model monomers: methyl acrylate (MA) – fast propagating more activated monomer (MAM) – and methyl methacrylate (MMA) – slow propagating MAM. While many acrylate monomers have been polymerized successfully via photoRAFT, polymerization of methacrylates remains challenging, with high monomer conversion not readily available within a reasonable time scale.4 Furthermore, as methacrylic radicals are more prone to termination by disproportionation, degradation of end groups is observed during the polymerization of MMA. Even in conventional thermal RAFT, the choice of RAFT agents for successful control over the molecular weight and its narrow distribution is limited to dithiobenzoates or trithiocarbonates with a cyano R group. Our previous study showed that bis(trithiocarbonate) disulfides work well with methacrylic monomers in thermal polymerization. Here, we investigate if the control can be further improved in photopolymerization by a systematic study of the impact of wavelengths of light and reactor geometry.
Results and discussion
Choice of the wavelength of light
A range of light wavelengths (UV – 365 nm, blue – 470 nm, cyan – 505 nm, and green – 527 nm) were screened to find the optimal conditions for the polymerization of methyl acrylate and methyl methacrylate (Fig. 1). Light intensity was adjusted to match the photon flux, defined as the number of photons emitted per second, to consider the single parameter of photon energy. Visible and UV light excited RAFT agents to different excited states by targeting separate electronic transitions, which will affect photodissociation and hence different levels of control of polymerization could be expected. Reactions were carried out using a multiwell reaction setup which could fit up to 42 2 ml vials. Polymerization temperature was fixed at 45 °C for all wavelengths of light. While modern LEDs were characterised with high efficiencies, they still generated heat and oftentimes maintaining room temperature, especially across different light wavelengths, was cumbersome. However, the residual heat could be utilised as photopolymerizations were shown to benefit from the increased reaction temperature which resulted in an increased rate of polymerization.47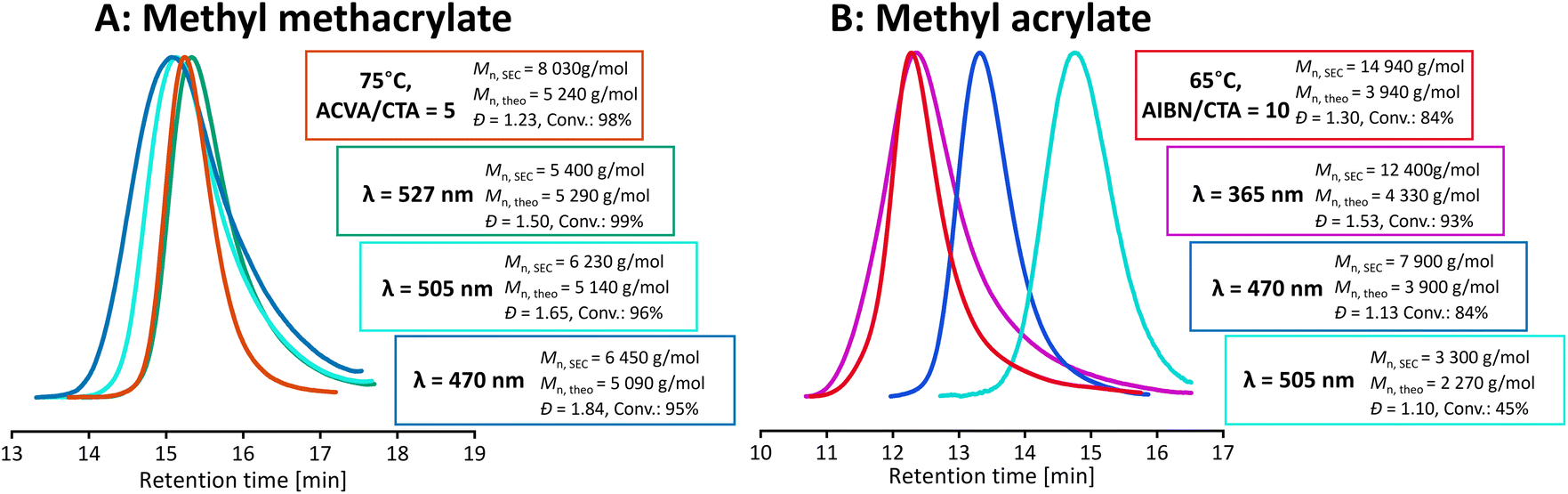 | ||
| Fig. 1 Polymerization of methyl methacrylate (A) and methyl acrylate (B), with the target degree of polymerization (DP) of 50. Comparison of thermal and light conditions at different light wavelengths. All light reactions were carried out in the multiwell setup, at 45 °C. | ||
Our previous study has shown that upon irradiation with UV light, PMMA loses its trithiocarbonate end group.18 Therefore, in this study, we focused our work on using visible light for the polymerization of MMA. Among the wavelengths tested, green light was proved to be most successful albeit the dispersity of PMMA was broad (Đ = 1.50) (Fig. 1). We hypothesise that higher energy blue light is likely to lead to too fast radical generation and loss of control. On the other hand, lower energy cyan light does not provide enough energy for fast fragmentation, thus leading to non-uniform growth of chains. This is investigated further in later sections. For all light mediated reactions, there is a clear low molecular weight tailing present. Since this tailing is absent in the thermal control reaction, we hypothesised that it is not due to poor degenerative chain transfer, but rather due to side reactions related specifically to activation with light. Attempts at polymerizing MA under UV light yield a broad dispersity polymer (Đ > 1.5), whilst polymerization under blue light (470 nm) leads to PMA with narrow and symmetrical molecular weight distribution (Đ < 1.15).
UV light excites trithiocarbonates into a higher energy second excited singlet state – S2, whereas visible light leads to the first singlet or first triplet state (S1 and T1).27 As the intersystem crossing from S1 to T1 is fast for thiocarbonyl compounds, most of the photochemical reactions are related to S2 and T1 states. S2 is relatively long lived as there is a significant energy gap between π–π* and n–π*, which retards radiationless decay to the lower energy triplet state, therefore increasing the chances of undesirable reactions taking place. Indeed, UV light-driven polymerizations often suffer from increased side reactions, including decomposition of the RAFT agent, which leads to loss of control and poor end group fidelity, especially at high conversions.48 We hypothesised that the lower quantum yield of homolytic bond dissociation for π–π* transition compared to n–π* transition could lead to slow initiation and hence non-uniform growth of polymer chains. As thiyl radicals are known to be poor initiators, a Bis-TTC-type RAFT agent needs conditions that strongly favour homolytic bond cleavage. Indeed, when lower energy cyan (505 nm) light was used, although the control was similar to that in blue light polymerization, the reaction was significantly slower, reaching only 45% monomer conversion in 16 h (83% in 10 h for blue light). When cyan light reaction was carried out until similar conversion was reached, the same molecular weight discrepancy was observed (Fig. S9†).
No polymerization was observed under green light (527 nm) under the otherwise same conditions. Although cyan and green light targets the n–π* transition, the LED emission spectra have progressively poorer overlap with the CTA absorption spectrum, which leads to a decreased activation of the RAFT agent. The polymerization of methyl acrylate under green light and using a TTC-based RAFT agent with a R group mimicking a methyl acrylate propagating radical (PMBTC) reached nearly full conversion (>95%) within the same reaction time, and gave well-defined PMA (Đ = 1.13, Mn,GPC = 4100 g mol−1) (see Fig. S10†). As the maximum of the n–π* absorption band of PMBTC is blue-shifted when compared to BisTTC, the poorer overlap of emission–absorption spectra cannot be the sole reason for the lack of polymerization of MA with BisTTC under green light. Polymerization with C4 BisTTC could only proceed to near full conversion and gave PMA of Mn,GPC = 9500 g mol−1 and Đ = 1.20 by increasing the irradiated surface of the sample (by placing the reaction vial above the LED array) and performing the reaction for 64 h. The difference between polymerizations mediated by BisTTC and PMBTC could be due to an increased cage effect for the S–S bond cleavage, or the influence of the α-TTC group on the excitation of TTC leading to the poorer activation of the RAFT agent and lower quantum yield of bond dissociation. This again highlights that bis(trithiocarbonate) disulfides require conditions strongly favouring photodissociation.
Finally, in all polymerizations of MA, including that thermally controlled with AIBN, significant deviation from targeted molecular weights was observed, with experimental molecular weight even as high as 4 times the target. This is in sharp contrast with narrow and symmetrical molecular weight distribution which suggests good control over polymerization.
MMA polymerization kinetics
For MMA DP 50 polymerization with BisTTC under 527 nm, 505 nm, 470 nm and 445 nm light irradiation, kinetic analysis showed varied rates of polymerization (Fig. 2). Indigo light (445 nm) which overlaps most closely with the absorption maximum of C4 BisTTC (symmetry forbidden n–π* transition) led to slower polymerization than blue light (470 nm), which in turn was faster than green and cyan (527 nm and 505 nm) light. This is similar to the findings of Nardi et al., who showed that the action spectrum of the photoactive species (copper(II) catalyst) was red shifted in relation to its absorption spectrum.49 This rate enhancement, however, has a negative effect on the control over polymerization of MMA. While the molecular weight evolution for green light polymerization is almost linear and in line with the theoretical molecular weight, a large discrepancy between theoretical and experimental molecular weight is observed in the early stages of polymerization mediated by blue light. In both polymerizations, dispersity increases over time. Broad dispersity for methacrylate is in sharp contrast with narrow values obtained for acrylate polymerization. Similar observations were performed for conventional trithiocarbonate (TTC) photoiniferter systems. Carmean et al. observed loss of control for methacrylates but not acrylates or acrylamides, and hypothesised that the TTC radical could abstract hydrogen from a propagating chain end.6 As methacrylic radicals are more prone to termination by disproportionation, degradation of end groups is observed during the polymerization of MMA but not MA. In the BisTTC case, it is the low molecular weight tailing which contributes significantly to the broadness of molecular weight distribution. This tailing is present from the early stages of the reaction and shifts to higher molecular weight with increasing conversion. Hence, we hypothesise that this tailing arises from the non-uniform initiation of polymer chains at the beginning of the polymerization.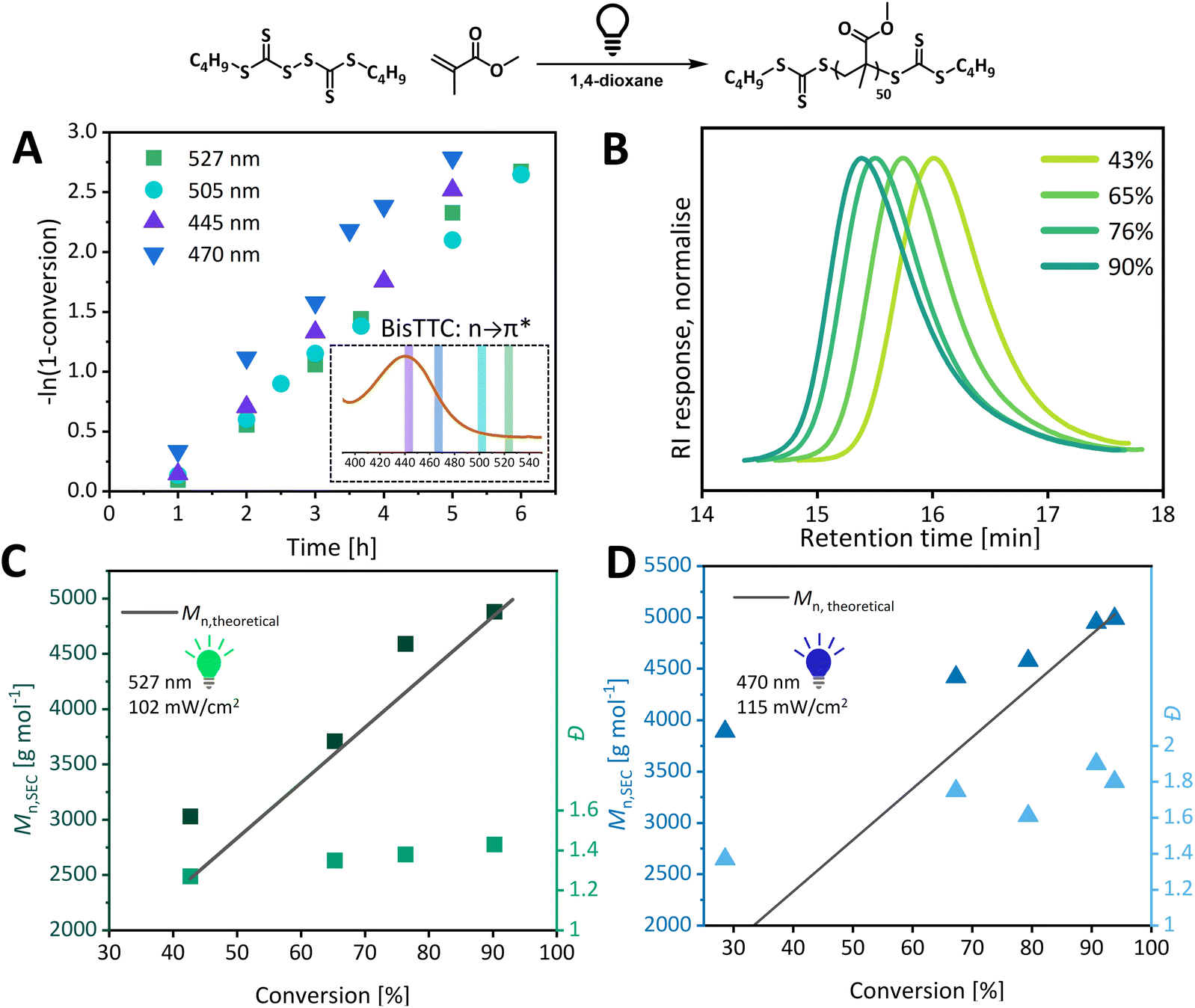 | ||
| Fig. 2 Analysis of photoiniferter DP50 MMA polymerization at different light wavelengths. (A) Pseudo-first order kinetic plot and UV-vis absorption for symmetry forbidden transition of C4 BisTTC annotated with the emission of LEDs used. (B) THF SEC traces of 527 nm, 102 mW cm−2 polymerization. (C) Plot of Mn,SEC and Đ against monomer conversion for 527 nm polymerization. (D) Plot of Mn,SEC and Đ against monomer conversion for 470 nm polymerization. | ||
MA polymerization kinetics
While narrow dispersity PMA was obtained under blue light irradiation, significant deviation from theoretical molecular weight was observed. To elucidate the origin of molecular weight discrepancy, the polymerizations were analyzed by NMR, SEC and HPLC. It was hypothesised that higher molecular weights for acrylates could arise from the partial consumption of the RAFT agent during photopolymerization. Monitoring reaction progress with HPLC showed that BisTTC is fully consumed by the time polymerization reaches 7% monomer conversion, as confirmed by the disappearance of BisTTC at the 1 hour polymerization time point (Fig. 3). At the beginning of the reaction, there is an initialisation period of about 2 hours during which BisTTC is converted into a single monomer unit insertion product (SMUI), retarding the polymerization, as reported in our previous study on bistrithiocarbonates.18 As more monomer units add to the SMUI, several oligomeric species appear on the chromatogram until HPLC can no longer separate higher molecular weight polymers, and only one broad peak is present at the later stages of polymerization (4 h, 44% monomer conversion). Unlike in the previously reported MMA polymerization with C12 BisTTC, the induction period is over once the SMUI is fully formed. The single monomer insertion can significantly affect the reactivity of the RAFT agent47,50,51 and we hypothesise that the MA SMUI has a higher chain transfer constant than the MMA SMUI. Dispersity remains low throughout polymerization and molecular weight increases linearly with conversion. Furthermore, the discrepancy between theoretical and experimental molecular weights is observed from the early stages of the reaction. The polymerization was notably slow as the polymerization required more than 8 hours to reach a conversion above 80%.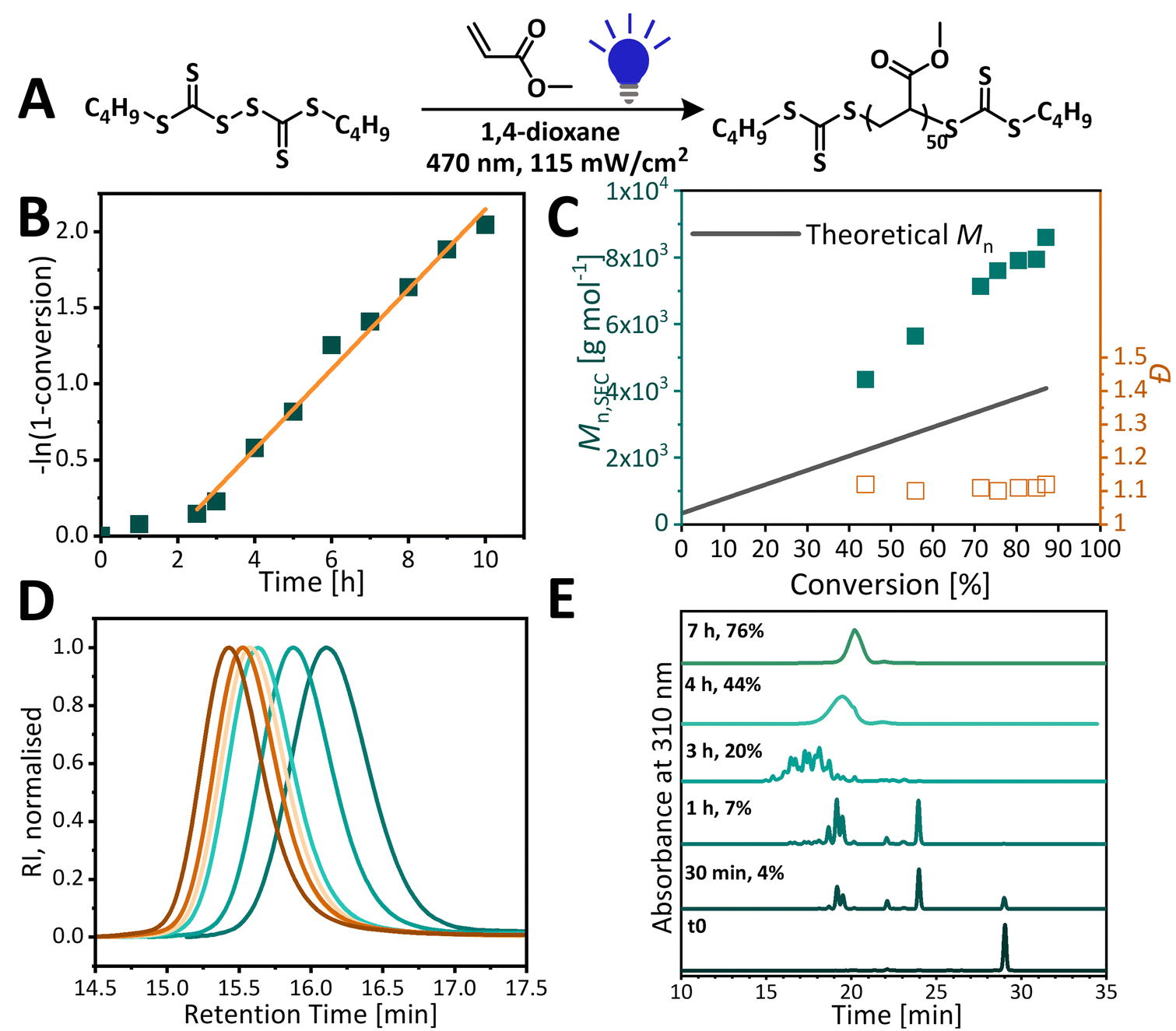 | ||
| Fig. 3 (A) Analysis of DP50 MA polymerization under blue light (470 nm) in the multiwell setup (115 mW cm−2). (B) Pseudo-first order kinetic plot. (C) Evolution of the molecular weight and dispersity over time. (D) Molecular weight distribution over the course of reaction, THF SEC. (E) HPLC kinetics analysis of methyl acrylate polymerization – monitoring RAFT agent consumption. | ||
We were interested to assess how increasing light exposure and light intensity, both of which increase the radical flux, would affect polymerization kinetics. We modified our reaction setup from the multiwell setup, where the vial is irradiated from the bottom, thus leading to poor light penetration, to a vertical setup, where the vial is irradiated from its side, resulting in larger irradiated areas. In this setup, polymerization time to achieve >85% conversion was reduced from 10 h (multiwell setup) to 5 h (Fig. 4), without detrimental effects on the control. The initialisation period was reduced from 2 h to 1 h. As in the multiwell setup polymerization, BisTTC is first converted into SMUI with the simultaneous formation of oligomeric species, but in this case, the SMUI is consumed faster and it disappears by the time polymerization reaches 4% (Fig. 4D). A further increase in the radical flux by increasing light intensity from 120 to 170 mW cm−2 increased the polymerization rate and decreased the initialisation period without negative effects on polymerization control. In both cases, the evolution of molecular weight with monomer conversion is linear albeit it is does not follow the theoretical molecular weight. In addition, good temporal control could be achieved by periodically switching light on and off (Fig. 4E): no polymerization is observed in the dark, indicating that the polymerization is both initiated and controlled with light, and that a continuous source of photons is necessary to sustain the reaction. Furthermore, this experiment demonstrates the livingness of the polymerization since chains are efficiently re-initiated upon switching the light on again.
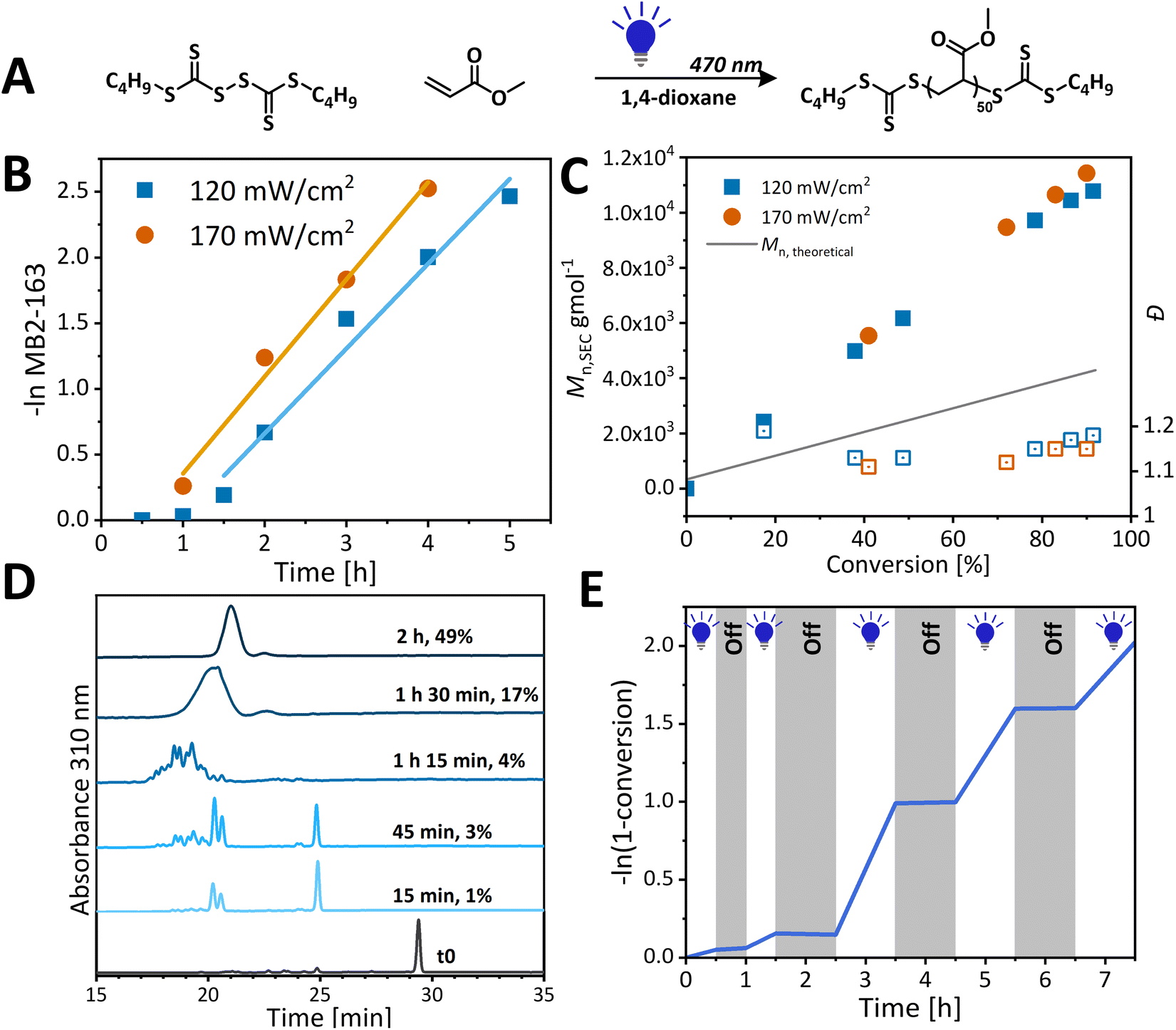 | ||
| Fig. 4 (A) Polymerization of methyl acrylate with the target DP of 50 under blue light (470 nm) in the test-tube setup. (B) Pseudo-first order kinetic plot for polymerization at two different light intensities. (C) Evolution of the molecular weight over the course of polymerization. (D) HPLC kinetics analysis of methyl acrylate polymerization – monitoring RAFT agent consumption. (E) Demonstration of temporal control. | ||
This new scaled-up setup did not work for the polymerization of MMA and this reaction resulted in a large increase in dispersity, from Đ values around 1.5 to Đ = 2.31 (Fig. S11†). Increased photodissociation has a detrimental effect on control over polymerization of a slowly polymerizing MMA monomer. If polymer chains are spending more time in the active form rather than the dormant form, the polymer growth will be uncontrolled. Furthermore, when the polymeric chain is not in its dormant state, TTC radicals can possibly degrade, further affecting polymerization livingness. As PMMA gives a tertiary radical upon cleavage of TTC as compared to a secondary radical from PMA, this effect is exacerbated for PMMA due to the resulting tertiary radical stability and lifetime.
To test if reducing the radical flux could improve control over MMA polymerization, the reaction was studied again in the multivial setup, but with a lower light intensity at the same temperature (82 mW cm−2 compared to previous 102 mW m−2). Reducing the light intensity did not improve reaction control and resulted in an induction period of about 2 h, after which the polymerization proceeded at a similar rate to that of the higher light intensity polymerization (Fig. S12†). The induction period is in agreement with our hypothesis that BisTTC requires conditions which strongly favour its photodissociation due to either the poor quantum yield of bond scission or the significant radical cage effect.29 Similar rates of polymerization for both 82 and 102 mW cm−2 suggest that once RAFT equilibrium is established, the rate-determining step for the BisTTC-mediated polymerization of MMA is propagation and increasing the reaction temperature rather than the light intensity should increase the rate of polymerization without detrimental effects on control. While the light intensities employed in this study are higher those typically reported, including that in our previous work on a BisTTC-type system, we found that such a setup leads to improved kinetics (faster rate of polymerization and reduced induction time) without detrimental effects on the control over polymerization. Furthermore, as in the multivial reactor, only the bottom of the reaction vial is irradiated with light, the light intensity per volume of the reaction vessel is significantly lower.
While low to medium molecular weight PMA polymerizations (2000 to 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) were controlled in both photoreactors, higher molecular weight PMA polymerization was only controlled in the multiwell setup, not in the scaled up setup, giving PMA Đ = 1.23, Mn,SEC = 60700 g mol−1 and Đ = 1.91, Mn,SEC = 59900 g mol−1 respectively (Fig. S13†). The loss of control at higher degrees of polymerization can arise from the photodegradation of the RAFT agent, which is more prevalent for lower concentrations of the RAFT agent. Wang et al. reported that a high concentration of 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid as the RAFT agent was necessary to achieve controlled radical polymerization as the RAFT agent decomposed irreversibly at a low concentration while at high concentrations the photolysis process was reversible.52
000 g mol−1) were controlled in both photoreactors, higher molecular weight PMA polymerization was only controlled in the multiwell setup, not in the scaled up setup, giving PMA Đ = 1.23, Mn,SEC = 60700 g mol−1 and Đ = 1.91, Mn,SEC = 59900 g mol−1 respectively (Fig. S13†). The loss of control at higher degrees of polymerization can arise from the photodegradation of the RAFT agent, which is more prevalent for lower concentrations of the RAFT agent. Wang et al. reported that a high concentration of 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid as the RAFT agent was necessary to achieve controlled radical polymerization as the RAFT agent decomposed irreversibly at a low concentration while at high concentrations the photolysis process was reversible.52
When higher DP is targeted, photoiniferter polymerization is slower as there is a less amount of the RAFT agent per monomer, so effectively there is a less amount of the initiator available and its decomposition will have a detrimental effect on the control over polymerization. We hypothesise that with increased exposure to light in the scaled up setup, there is more degradation of the RAFT agent than that in the multiwell setup.
Chain end group fidelity
Polymers resulting from iniferter polymerization mediated by a bis(trithiocarbonate) disulfide-type RAFT agent are expected to have trithiocarbonate on both α and ω ends. Detailed polymer characterisation is discussed in our previously published work.18 A PMA macroCTA (Đ = 1.13, Mn,SEC = 12220 g mol−1) synthesised via the vertical setup was purified and the chain extended with another aliquot of methyl acrylate (MA). Although there is a clear shift to a higher molecular weight, a significant low molecular weight shoulder corresponding to unreacted first block is still presented (see Fig. 5). This suggests partial loss of the end group, due to termination reactions and/or end group degradation. To ascertain if the unreacted first block is due to the loss of the end group from termination reactions, a new macroCTA was synthesised by targeting higher DP and stopping the polymerization at lower conversions (<50%), leading to a similar final molecular weight (Đ = 1.10, Mn,SEC = 12200 g mol−1). By keeping conversions low, the livingness (ratio of number of living chains/total number of chains) was maintained high in a reversible termination process. This macroCTA was then chain extended and led to a well-defined polymer (Đ = 1.18, Mn,SEC = 19630 g mol−1). Therefore, it is likely that the loss of the end group is due to termination reactions occurring throughout the polymerization.
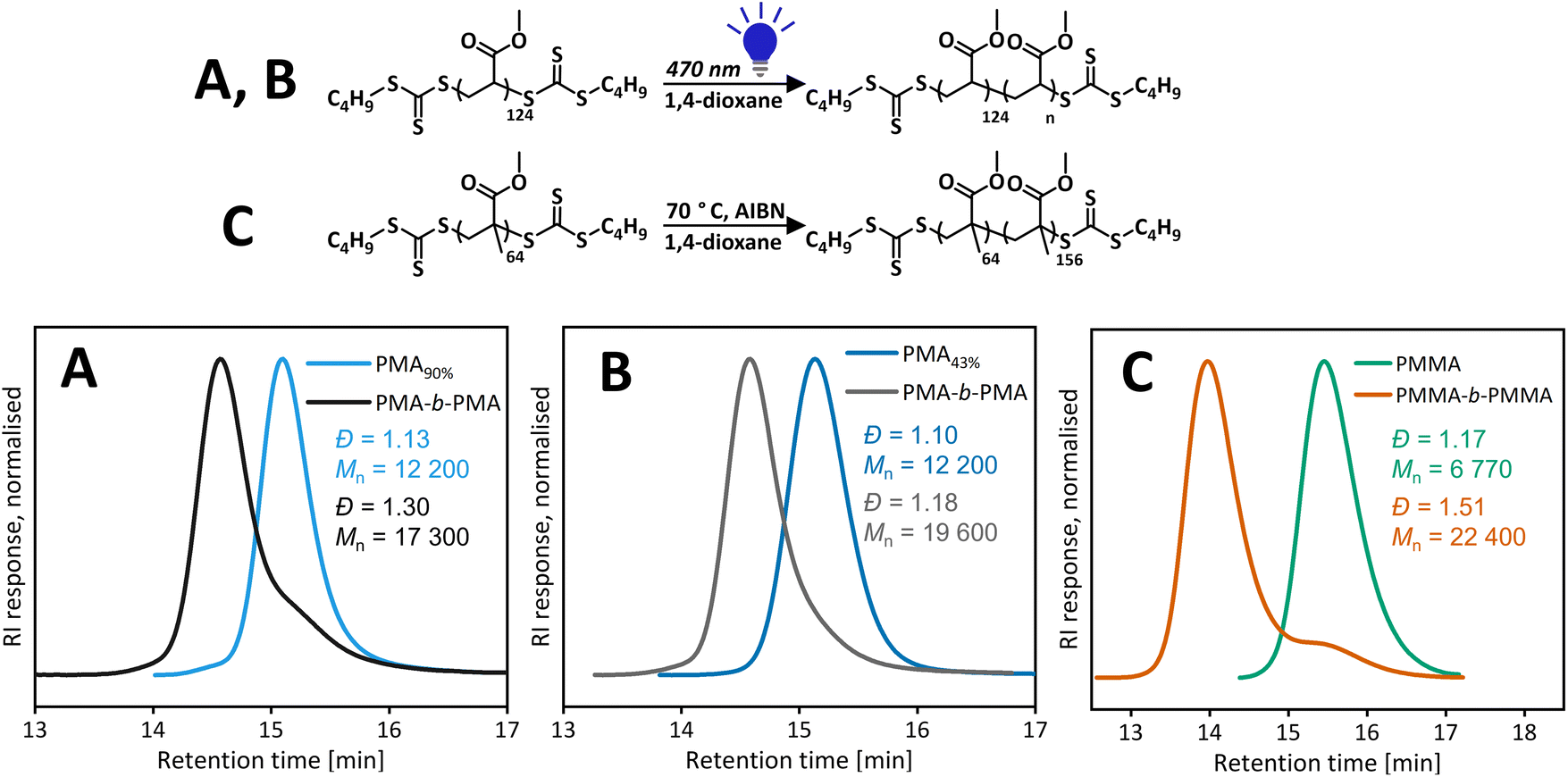 | ||
| Fig. 5 SEC chromatograms for pseudo block copolymers. (A) Blue light chain extension of PMA prepared by photoiniferter polymerization. (B) Blue light chain extension of PMA prepared by photoiniferter polymerization taken to 43% conversion. (C) Thermal chain extension of PMMA prepared using a green light photoiniferter. | ||
A further study of the PMA end group was undertaken using MALDI-TOF spectroscopy. Three distributions were isolated for the PMA sample with Đ = 1.11and Mn,SEC = 2180 g mol−1, all with MA as the repeating unit (m/z = 86) (Fig. S14†), including thioether-capped chains, which suggests photodegradation of the thiocarbonyl group. Indeed, photodegradation of the trithiocarbonate from the RAFT agent or the end group typically led to thiol radicals and the release of carbon disulfide; we believe the thiol radical either reinitiated polymerization, or end capped a propagating polymeric chain, leading to the thioether end group.52,53 The end group for the third PMA distribution could not be assigned.
As the photoiniferter polymerization of MMA under green light was not well controlled, photoiniferter chain extension would not give a good block copolymer and the termination/loss of control would be a combined result from the polymerization of the first block and chain extension. Hence, we performed the thermal chain extension of the purified PMMA sample prepared under green light (Đ = 1.17, Mn,SEC = 6770 g mol−1). After the purification of PMMA by precipitation, the low molecular weight tail was removed; however, this did not affect chain extension and chain extension of a crude PMMA sample gave similar results (Fig. S15†). In both cases, there was a clear shift to the higher molecular weight upon chain extension with an aliquot of MMA, there was also significant leftover of the first block. The same result from purified and crude PMMA confirmed our hypothesis that the low molecular weight tailing was not formed of dead chains formed during the reaction but rather living chains that were initiated later during polymerization.
Photodegradation
To determine if the RAFT agent or thiocarbonyl polymer end group is most likely to photodegrade, the photostabilities of the CTA and polymers were tested independently. A purified PMA (Đ = 1.15, Mn,SEC = 9990 g mol−1) sample was redissolved in 1,4-dioxane, degassed and irradiated with blue light in the test tube setup for 4 h. GPC analysis of 5 mg ml−1 sample before and after light irradiation showed no difference in the shape or intensity of UV and RI signals, thus confirming the stability of the polymeric chain (from RI) and the polymeric end group (used as the UV chromophore in the UV detector), Fig. 6. Similarly, the photostability of the PMMA sample (Đ = 1.14, Mn,SEC = 7310 g mol−1) was tested under green light. Over time, the polymer's 310 nm UV signal decreases and a low molecular weight signal associated with TTC related species appears simultaneously, confirming end group removal by green light. After a sharp decrease in the UV signal at the 8 h time point, only a small further decrease is observed after 24 h of light irradiation, suggesting that most end groups are cleaved by this point. The UV signal does not disappear completely due to the absorbance of the stable α-end TTC group. From the RI signal, it becomes apparent that some chains terminate by combination rather than only disproportionation as shown in our previous study with UV light,18 as evidenced by the appearance of the high molecular weight shoulder. While the end group cleavage kinetics are different in the presence of the monomer, the cleavage will inevitably happen during the polymerization of MMA, leading to accumulation of terminated chains. The released TTC end groups can either participate in chain transfer with the remaining living chains or potentially initiate new chains which would contribute to the low molecular weight tailing. Importantly, this side reaction can take place with conventional RAFT agents, especially when polymerization is taken to high conversion, or the reaction is left under light after all the monomer has been consumed. The difference in the photostability of PMMA and PMA arises from the difference in their radical stability, just like the stability of the RAFT agent is affected by the R group.53 The tertiary methacrylic radical is relatively long lived compared to the secondary acrylic radical and this increased lifetime increases chances of side reactions, alongside with the well-known tendency of methacrylates to terminate by disproportionation.54 The end group cleavage is significantly faster under blue light – in just 2 hours of light irradiation; the polymer UV signal decreases sharply and does not decrease further over 24 h suggesting the complete removal of ω-TTC in 2 h (Fig. S16†). This increased rate of end group cleavage for blue light can explain poorer control over MMA polymerization under blue light than green light.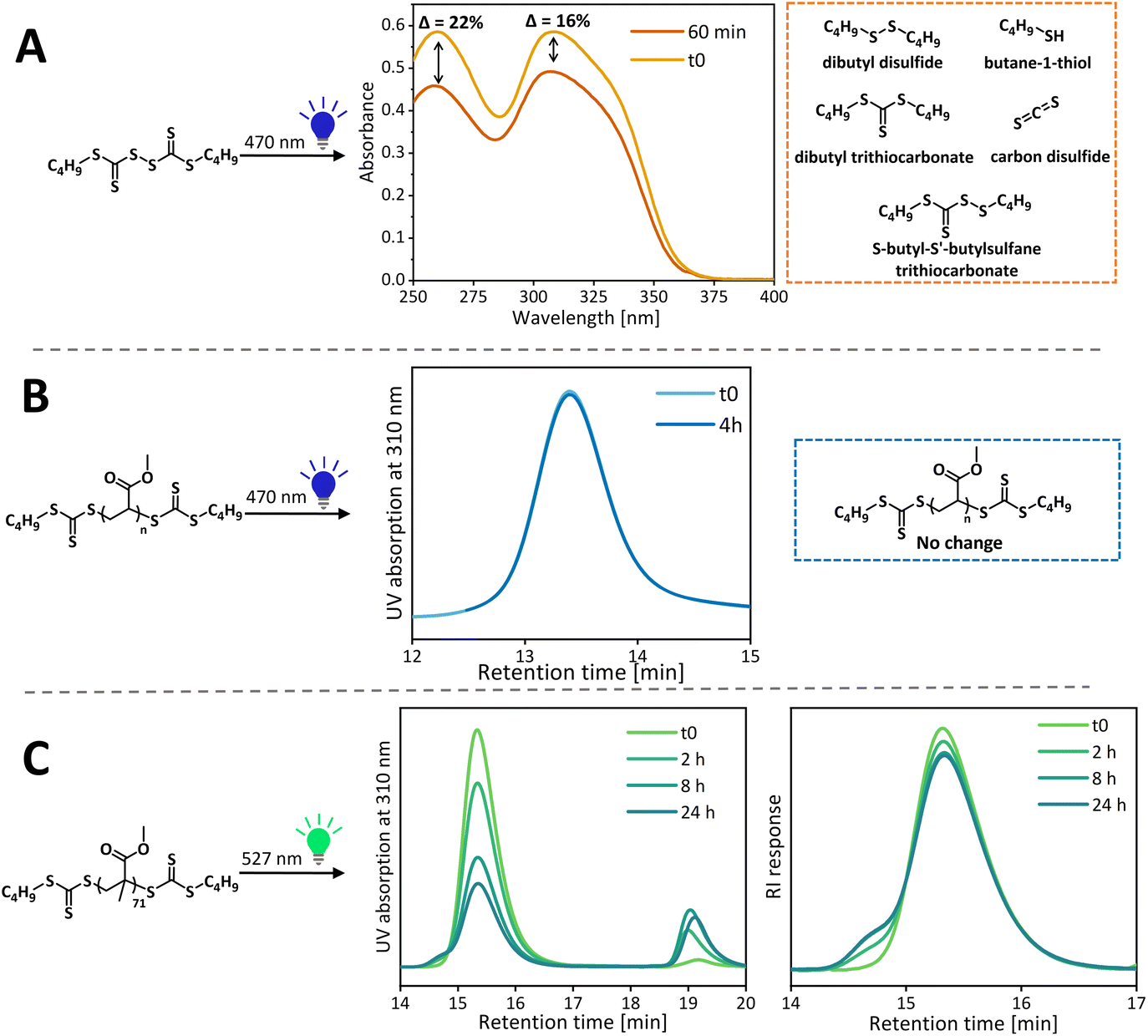 | ||
| Fig. 6 (A) UV-vis spectra before and after blue light treatment of C4 BisTTC and suggested degradation products. (B) Size exclusion chromatograms before and after blue light treatment of the PMA sample. (C) Size exclusion chromatograms before and after green light treatment of the PMMA sample. | ||
Similarly, the photostability of C4 BisTTC alone was tested under conditions mimicking MA polymerization (monomer volume was replaced with an additional solvent to maintain the same molar concentration of CTA, [CTA] = 0.08 M). Degradation of the RAFT agent leads to an increased molecular weight of the resulting polymers due to an increased [CTA]/[monomer] ratio, as well as an increased concentration of propagating radicals and hence the amplified likelihood of termination. Photodegradation was evaluated by UV-Vis spectrometry and the extent of degradation was quantified by measuring the decrease of 310 nm maxima corresponding to the n–π* band of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond, as reported previously.53 For BisTTC, two substituents with markedly different electronic properties (alkyl chain and another trithiocarbonate) on the cross-conjugated thiocarbonyl group gave rise to two absorption maxima for the n–π* band.55 After irradiation for 1 h, a larger decrease in the 260 nm band (22%) compared to the 310 nm band (16%) was observed (Fig. 6); this was even more apparent when higher percentage degradation was observed (Fig. S17†). We postulate that if degradation products are derivatives of trithiocarbonate, they should also possess a maximum absorption at 310 nm, thus artificially increase the absorption at 310 nm and mask the extend of BisTTC degradation. Hence, we assessed the degradation of BisTTC by following the decrease of the 260 nm band.
S bond, as reported previously.53 For BisTTC, two substituents with markedly different electronic properties (alkyl chain and another trithiocarbonate) on the cross-conjugated thiocarbonyl group gave rise to two absorption maxima for the n–π* band.55 After irradiation for 1 h, a larger decrease in the 260 nm band (22%) compared to the 310 nm band (16%) was observed (Fig. 6); this was even more apparent when higher percentage degradation was observed (Fig. S17†). We postulate that if degradation products are derivatives of trithiocarbonate, they should also possess a maximum absorption at 310 nm, thus artificially increase the absorption at 310 nm and mask the extend of BisTTC degradation. Hence, we assessed the degradation of BisTTC by following the decrease of the 260 nm band.
The characterisation of the impurities proved to be very challenging due to the structural similarities between the side products and BisTTC itself, which made isolation of impurities difficult. From the 1H-NMR analysis of a sample taken from a blue light degradation study (Fig. S6†), it became apparent that BisTTC degrades to multiple compounds. Preparative HPLC enabled us to isolate two of the impurities which were identified as dibutyl trithiocarbonate (based on 1H NMR) and S-butyl-S′-butylsulfane trithiocarbonate (based on literature spectra), details are given in the ESI.† Although not isolated due to small quantities released during degradation and volatility of the product, butyl sulfide and dibutyl disulfide were identified as possible degradation products by comparing the shift of the characteristic triplet signal of the CH2 group attached to sulphur to literature data. This is in line with the expected trithiocarbonate degradation pathway, which results in the release of a thiol radical and carbon disulfide.52,53 The thiol radicals can typically abstract a hydrogen to from a free thiol, or more typically combine and form a disulfide. Here, we hypothesise that the thiol radicals can also terminate a propagating thiyl radical and/or take part in the thiol–ene reaction. This was supported by the aforementioned MALDI analysis which showed the PMA chain with a thioester end group (Fig. S14†). Dibutyl trithiocarbonate is expected to be inert during polymerization while thiol and S-butyl-S′-butylsulfane trithiocarbonate prematurely terminate propagating radicals. S-Butyl-S′-butylsulfane trithiocarbonate can also further degrade to a thiol radical and carbon disulfide.
To assess the presence of the products of BisTTC degradation in the polymer product, a PMA sample, synthesised under blue light, was purified by precipitation in hexane, in order to separate the polymer form small molecule side products. The supernatant was analysed and revealed the presence of hexane-soluble PMA oligomers, as well as dibutyl trithiocarbonate and dibutyldisulfide. Thiol trithiocarbonate disulfide was not detected, presumably because it further degraded into the abovementioned impurities or reacted with the propagating polymeric chains.
Qiao and coworkers have shown that a tertiary amine catalyst can prevent the degradation of trithiocarbonates. In this suggested mechanism, the amine acts as a single-electron reductant, leading to the formation of an amine radical cation and a TTC radical anion pair, and avoiding unstable TTC radicals.56 Similar improvements in control over molecular weights and dispersities were reported by several groups.57,58 Unfortunately, no such improvements were observed in our system; in fact, the addition of triethylamine resulted in lower conversions and broader dispersities (Fig. S18†).
Significant differences in the rate of degradation of BisTTC at different wavelengths of light were observed, with blue light degrading 65% of BisTTC in 24 h, followed by cyan light (45%) and green light (29%) (Fig. S17†). The increased degradation of the RAFT agent correlates with the loss of control over polymerization when higher energy is used for the polymerization of MMA. This prompted us to test lower energy light for the polymerization of MMA. Usually, to enable polymerization under longer light wavelengths, catalysts such as zinc(II) porphyrin are necessary to activate the RAFT agent indirectly in the PET-RAFT process.59,60 In the case of BisTTC, we found that it can be activated directly with red light (630 nm). Despite being slow, the polymerization was controlled and gave narrow dispersity PMMA (Đ = 1.31, Mn,SEC = 5410 g mol−1), Fig. 7. We hypothesise that the improved control is a result of the lower radical flux and the slower degradation of BisTTC and the PMMA TTC end group. The long reaction time (56 h) could be potentially shortened by increasing the temperature, which should increase the rate of propagation.
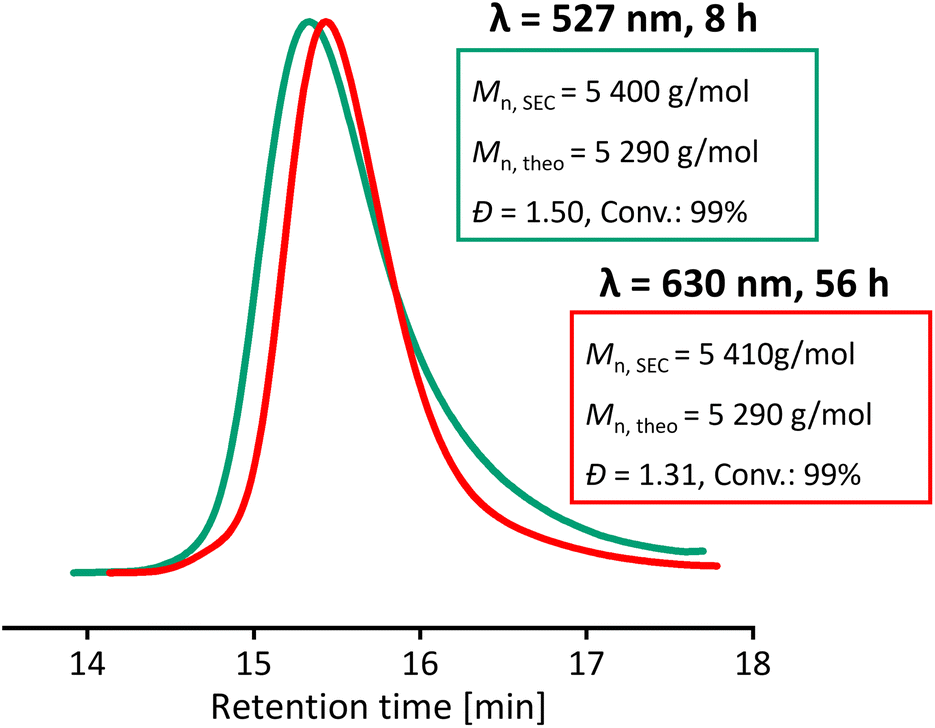 | ||
| Fig. 7 SEC chromatograms of MMA DP50 photoiniferter polymerization – comparison of green (527 nm) and red light (630 nm). | ||
Conclusions
A bis(trithiocarbonate) disulfide RAFT agent can mediate the photoiniferter-RAFT polymerization of more activated monomers, albeit with various levels of control. While BisTTC can photodissociate and initiate polymerization, its activation under visible light is slow but degradation is fast. Polymerization of methyl acrylate is retarded under green light as the addition of a monomer unit to the RAFT agent significantly reduces the stability of the R group, further reducing the activity of the RAFT agent which is already lower than the conventional equivalent. Under blue light, molecular weight evolution for PMA polymerization is linear but there is a big discrepancy from theoretical molecular weight. This is caused by the photodegradation of the RAFT agent during the induction period and interference of the degradation products with polymerization. While in thermal polymerization with BisTTC polymerization of methyl acrylate is less controlled than methyl methacrylate, in the case of photopolymerization this monomer compatibility is reversed. The level of control and kinetics of MMA polymerization are highly dependent on the wavelength of light. Lower energy green light gives the best results, as photodegradation of BisTTC and the polymeric end group is slower than that under blue light. Blue light leads to faster polymerization but worse control. Due to the increased radical stability and lifetime, the PMMA TTC end group can degrade readily even under visible light, leading to the termination of growing chains and initiation of new chains from released TTC radicals. Lowering the light intensity leads to the induction period but no decrease in the apparent rate of propagation, indicating that propagation is the rate-determining step. Too fast photodissociation leads to uncontrolled growth of polymers and degradation of end groups. These side reactions can be suppressed by using an even lower-energy wavelength of light such as red light. While polymerization under red light is slow, optimizing the reaction setup and utilising continuous flow and higher temperatures to aid propagation could significantly reduce the reaction time.Conflicts of interest
There are no conficts to declare.Acknowledgements
M. A. B. acknowledges a scholarship from Lubrizol. We are grateful for the Warwick Polymer Characterisation RTP for allowing the use of the following equipment: THF SEC.References
- M. A. Tehfe, F. Louradour, J. Lalevée and J. P. Fouassier, Appl. Sci., 2013, 3, 490–514 CrossRef CAS.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3(9), 1500394 CrossRef PubMed.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, in Controlled Radical Polymerization: Mechanisms, ed. K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky and J. Chiefari, American Chemical Society, Washington, 1st edn, 2015, pp. 247–267 Search PubMed.
- A.-C. Lehnen, J. Gurke, A. M. Bapolisi, M. Reifarth, M. Bekir and M. Hartlieb, Chem. Sci., 2023, 14, 593–603 RSC.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS PubMed.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- T. Otsu and A. Kuriyama, J. Macromol. Sci., Part A: Pure Appl. Chem., 1984, 21, 961–977 CrossRef.
- T. Otsu and M. Yoshida, Makromol. Chem., Rapid Commun., 1982, 3, 127–132 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- P. Corpart, D. Charmot, T. Biadatti, S. Zard and D. Michelet, Rhodia Chimie, WO, 9858974, 1998 Search PubMed.
- M. Y. Khan, M. S. Cho and Y. J. Kwark, Macromolecules, 2014, 47, 1929–1934 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS.
- M. Hartlieb, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS.
- A. Kerr, G. Moriceau, M. A. Przybyla, T. Smith and S. Perrier, Macromolecules, 2021, 54, 6649–6661 CrossRef CAS.
- T. Otsu, K. Nayatani, I. Muto and M. Imai, Makromol. Chem., 1958, 27, 142–148 CrossRef CAS.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, ACS Macro Lett., 2019, 8, 1461–1466 CrossRef CAS.
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS.
- R. A. Olson, M. E. Lott, J. B. Garrison, C. L. G. Davidson, L. Trachsel, D. I. Pedro, W. G. Sawyer and B. S. Sumerlin, Macromolecules, 2022, 55(19), 8451–8460 CrossRef CAS.
- P. Lambrinos, M. Tardi, A. Polton and P. Sigwalt, Eur. Polym. J., 1990, 26, 1125–1135 CrossRef CAS.
- L. Lu, H. Zhang, N. Yang and Y. Cai, Macromolecules, 2006, 39, 3770–3776 CrossRef CAS.
- H. Zhang, J. Deng, L. Lu and Y. Cai, Macromolecules, 2007, 40, 9252–9261 CrossRef CAS.
- J. D. Coyle, Tetrahedron, 1985, 41, 5393–5425 CrossRef CAS.
- R. W. Hughes, M. E. Lott, J. I. Bowman and B. S. Sumerlin, ACS Macro Lett., 2022, 12, 14–19 CrossRef PubMed.
- B. Cabannes-Boué, Q. Yang, J. Lalevée, F. Morlet-Savary and J. Poly, Polym. Chem., 2017, 8, 1760–1770 RSC.
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., 2019, 131, 5224–5243 CrossRef.
- P. N. Kurek, A. J. Kloster, K. A. Weaver, R. Manahan, M. L. Allegrezza, N. De Alwis Watuthanthrige, C. Boyer, J. A. Reeves and D. Konkolewicz, Ind. Eng. Chem. Res., 2018, 57, 4203–4213 CrossRef CAS.
- T. Eckert and V. Abetz, J. Polym. Sci., 2020, 58, 3050–3060 CrossRef CAS.
- G. D. Ammini, J. P. Hooker, J. Van Herck, A. Kumar and T. Junkers, Polym. Chem., 2023, 14, 2708–2716 RSC.
- H. Zhou and J. A. Johnson, Angew. Chem.,– Int. Ed., 2013, 52, 2235–2238 CrossRef CAS.
- M. Chen and J. A. Johnson, Chem. Commun., 2015, 51, 6742–6745 RSC.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- Q. Yang, Y. Yang, W. Liu, W. Tian, F. Xing and P. Xiao, ChemistrySelect, 2021, 6, 13270–13276 CrossRef CAS.
- J. J. Vosloo, D. de Wet-Roos, M. P. Tonge and R. D. Sanderson, Macromolecules, 2002, 35, 4894–4902 CrossRef CAS.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 2435–2438 CrossRef CAS.
- D. Zhou, Y. Gao, A. Sigen, Q. Xu, Z. Meng, U. Greiser and W. Wang, ACS Macro Lett., 2016, 5, 1266–1272 CrossRef CAS PubMed.
- B. Liu, L. Deng, C. Guo, R. Guo, A. Dong and J. Zhang, J. Appl. Polym. Sci., 2012, 126, 740–748 CrossRef CAS.
- X. Liu, G. Zhang, B. Li, Y. Bai, D. Pan and Y. Li, Eur. Polym. J., 2008, 44, 1200–1208 CrossRef CAS.
- C. Ding, Y. Yan, Y. Peng, D. Wu, H. Shen, J. Zhang, Z. Wang and Z. Zhang, Macromolecules, 2022, 55(10), 4056–4063 CrossRef CAS.
- K. Hakobyan, C. S. P. McErlean and M. Müllner, Macromolecules, 2021, 54, 7732–7742 CrossRef CAS.
- L. Zhang, H. Zhou, Y. Gu and M. Chen, Macromolecules, 2023, 56(15), 6010–6018 CrossRef CAS.
- M. Destarac, Macromol. React. Eng., 2010, 4, 165–179 CrossRef CAS.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
- L. Lu, N. Yang and Y. Cai, Chem. Commun., 2005, 5287–5288 RSC.
- M. Nardi, E. Blasco and C. Barner-Kowollik, J. Am. Chem. Soc., 2022, 144, 1094–1098 CrossRef CAS.
- A. Aerts, R. W. Lewis, Y. Zhou, N. Malic, G. Moad and A. Postma, Macromol. Rapid Commun., 2018, 39, 1–7 CrossRef PubMed.
- L. Zhang, R. Liu, Z. Huang and J. Xu, Polym. Chem., 2021, 12, 581–593 RSC.
- H. Wang, Q. Li, J. Dai, F. Du, H. Zheng and R. Bai, Macromolecules, 2013, 46, 2576–2582 CrossRef CAS.
- T. G. McKenzie, L. P. M. Da Costa, Q. Fu, D. E. Dunstan and G. G. Qiao, Polym. Chem., 2016, 7, 4246–4253 RSC.
- Y. Nakamura and S. Yamago, Macromolecules, 2015, 48, 6450–6456 CrossRef CAS.
- K. Skrabania, A. Miasnikova, A. M. Bivigou-Koumba, D. Zehm and A. Laschewsky, Polym. Chem., 2011, 2, 2074–2083 RSC.
- Q. Fu, T. G. McKenzie, S. Tan, E. Nam and G. G. Qiao, Polym. Chem., 2015, 6, 5362–5368 RSC.
- M. L. Allegrezza, Z. M. Demartini, A. J. Kloster, Z. A. Digby and D. Konkolewicz, Polym. Chem., 2016, 7, 6626–6636 RSC.
- B. Nomeir, O. Fabre and K. Ferji, Macromolecules, 2019, 52, 6898–6903 CrossRef CAS.
- Y. Zhou, Z. Zhang, C. M. Reese, D. L. Patton and J. Xu, Macromol. Rapid Commun., 2020, 1–6 Search PubMed , 1900478.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, Chem. Commun., 2018, 54, 6591–6606 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01307c |
| This journal is © The Royal Society of Chemistry 2024 |