
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs receptor mediated active macrophage targeting of amphotericin B nanoformulations a promising approach?
Saugandha
Das†
a,
Pooja
Todke†
a,
Manisha
Madkaikar
b and
Padma
Devarajan
 *a
*a
aDepartment of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Deemed University, Elite Status and Centre of Excellence (Maharashtra), N.P. Marg, Matunga (E), Mumbai, 400019, Maharashtra, India. E-mail: pv.devarajan@ictmumbai.edu.in; Fax: +91 22 33611020; Tel: +91 22 33612210
bICMR-National Institute of Immunohaematology, KEM Hospital Campus, Parel, Mumbai, 400012, Maharashtra, India
First published on 5th April 2024
Abstract
We present an AmB-LIPOMER anchored with Acemannan (ACEM), a mannose ligand for active macrophage targeting, via mannose receptor mediated endocytosis (RME). The AmB-LIPOMER prepared by modified nanoprecipitation was anchored with ACEM by simple incubation. FITC was added to obtain fluorescent LIPOMERs. The LIPOMERs revealed a spherical morphology, an average size of 400–450 nm and a PDI < 0.3. Reduction in the zeta potential and FTIR confirmed ACEM anchoring. Flow cytometry demonstrated a >13-fold enhancement of the FITC-ACEM LIPOMER in vitro in RAW 264.7 macrophage cells, compared to the FITC-LIPOMER, ascribed to mannose receptor mediated endocytosis. This was confirmed by the decreased uptake of the FITC-ACEM LIPOMER in the mannose receptor blocking study. Nevertheless, we were surprised by an ∼2-fold decrease in the in vitro antileishmanial efficacy despite the augmented uptake of the ACEM LIPOMER. This poor efficacy was explained by the extensive localization of the FITC-ACEM LIPOMER in the lysosomal compartment, established by confocal microscopy, wherein AmB underwent rapid degradation. On the other hand phagocytic uptake and lipid mediated prolonged localization in the less harsh phagosome enabling lower degradation could have facilitated higher efficacy of the AmB-LIPOMER. The pharmacokinetic and biodistribution studies in rats revealed rapid and high reticuloendothelial system uptake. While the AmB-LIPOMER group exhibited no mortality, the mortality of 5 out of 6 animals in the AmB-ACEM LIPOMER group, within 15–30 minutes caused by lung necrosis was disturbing. While we propose an explanation for the toxicity, our study questions the rationale and safety of active targeting AmB using receptor mediated endocytosis.
1. Introduction
Amphotericin B (AmB), which is a potent polyene macrolide antibiotic, is considered a drug of choice for visceral leishmaniasis (VL), a serious macrophage resident infection. Nevertheless, non-selective binding of AmB to host cells following intravenous administration has resulted in severe and near fatal renal toxicity with conventional AmB injection, leading to a need for safer and targeted alternatives.1,2Drug loaded nanocarriers, by mimicking the intracellular pathway of pathogens, have enabled high intracellular delivery of therapeutic payloads. Based on this premise, AmB loaded nanoformulations which rely on passive targeting approaches have been developed to combine the advantages of high efficacy and low toxicity. Among nanoformulations, micellar formulations revealed significant toxicity, while liposomal AmB demonstrated both good efficacy and safety through macrophage targeted delivery, aided by the nanosize.3 Passive macrophage targeting of AmB was also achieved using different nanocarriers like solid lipid nanoparticles (SLNs),4 emulsomes,5,6 polymeric nanoparticles,7,8 nanodisks,9 and nanoemulsions.10 Our group has earlier reported macrophage targeted delivery of AmB-LIPOMERs by passive targeting. In vitro antileishmanial efficacy in amastigotes, was 2-fold higher and the safety index 3-fold higher compared to liposomal AmB.11
Despite high effectiveness in the treatment of both leishmaniasis as well as a range of fungal infections, the dose-dependent kidney toxicity and systemic toxicity of AmB continue to pose serious concerns. Coupling targeted delivery with dose reduction strategies is one approach to tackle this challenge. In one study administration of oral AmB cochleates exhibited a dose-dependent reduction in mortality, a better therapeutic index with a survival rate of 70% and no compromise on efficacy.12 Similarly, in another study, researchers demonstrated that AmB cochleates protected C. albicans infected ICR mice following intraperitoneal administration at a low dose of 0.1 mg kg−1 day−1.13 In a mouse model of systemic candidiasis, AmB lipid nanospheres (1.0 mg kg−1) demonstrated an enhanced survival rate and greater efficacy compared to AmBisome (8.0 mg kg−1) or Fungizone (1.0 mg kg−1).14 This served as the motivation to design an actively targeted AmB-LIPOMER to further augment intracellular uptake, in an attempt to decrease systemic toxicity and enable a possible reduction in dose without altering the efficacy.
Active targeting approaches can facilitate the delivery of significantly enhanced therapeutic cargoes inside the cell and have become an important focus area for the treatment of cancers and infectious diseases.15 Active targeting can improve uptake and thereby augment efficacy. For conditions like VL where the parasite Leishmania donavani is harboured safely inside macrophages, enhanced uptake through active targeting could provide a major advantage of improved efficacy,16,17 as well as the opportunity to decrease the dose, which could be very beneficial for drugs like AmB which exhibit high toxicity. Among various active targeting strategies, receptor mediated endocytosis is the most widely investigated and relies on ligands to target receptors overexpressed on cells.18 One such receptor overexpressed on macrophages where the leishmanial parasites are harboured is the mannose receptor, which exhibits high affinity for mannose and mannose-based macromolecules.19
Mannan conjugated to didanosine gelatin NPs exhibited a five-fold increase in intracellular macrophage uptake with augmented localization in lymph nodes, spleen, and the brain in the treatment of HIV.20 A mannose-anchored thiolated AmB nanocarrier elicited a very high (71-fold) enhancement in intramacrophage drug uptake compared with the native drug.21 Furthermore, paramomycin loaded PLGA NPs coated with mannosylated thiolated chitosan demonstrated enhanced in vitro antileishmanial efficacy with a 36-fold lower IC50 compared with the native drug. Moreover, the in vivo antileishmanial efficacy study indicated a 3.6-fold decreased parasitic burden in the Leishmania donovani infected BALB/c mice model.22 In another study, high intracellular delivery of rifampicin was achieved by surface modification of lipid NPs with chitosan-conjugated Acemannan (ACEM).23
In the above studies the mannose-based ligands were attached by covalent conjugation of mannose to the nanocarrier, a major limitation being the complexity of synthesis.24 Herein, we report for the first time ACEM as a mannose targeting ligand anchored to an AmB-LIPOMER by simple adsorption.
In an earlier study we reported a near two-fold enhancement in the in vitro efficacy of the AmB-LIPOMER in an antileishmanial amastigote model compared to the marketed liposomal formulation.11 Based on the promise of further enhancement by active targeting, we embarked on designing a mannose receptor targeted AmB-LIPOMER using the known mannose ligand ACEM.25–27 The objective of the study was to confirm enhanced macrophage uptake in RAW 264.7 macrophage cells and enhanced efficacy in an in vitro antileishmanial amastigote model. Yet another objective was to evaluate the effect of ACEM anchoring on the pharmacokinetics and biodistribution of the AmB-LIPOMER in a rat model.
2. Materials and methods
2.1 Materials
AmB was a kind gift by Cipla Limited, Mumbai, India. Plurol Stearique WL1009 (polyglyceryl-6-distearate, PGDS) with a molecular weight (MW) of 995.42 g mol−1 was gifted by Gattefosse, France. Poly(methyl vinyl ether-co-maleic anhydride) and Gantrez® AN 119 (MW: 872.78 g mol−1) were provided by Ashland, Mumbai, India, while Trehalose 100 (MW: 342.29 g mol−1) was gifted by Gangwal Chemicals, Mumbai, India. Gift samples of Acemannan (ACEM) with MW ≥ 1 × 106 Daltons and ≥20% polymeric acetylated mannan content were provided by Naturaloe, Costa Rica. Magnesium acetate tetrahydrate pure, hydrochloric acid (35%) and potassium bromide were procured from Merck, Mumbai, India. Absolute ethanol (99.9% AR) was obtained from Changshu Hongsheng Fine Chemical Co., Ltd, China, while tetrahydrofuran (AR), isopropyl alcohol (HPLC grade), acetonitrile, methanol (HPLC grade), phenol and conc. sulphuric acid were obtained from Qualigens Thermo Fisher Scientific, India. Fetal bovine serum (FBS), Dulbecco's modified Eagle's medium (DMEM), antibiotic solution, 4′,6-diamidino-2-phenylindole (DAPI; MW: 350.25 g mol−1), fluorescein isothiocyanate (FITC; MW: 389.38 g mol−1), dimethyl sulfoxide (cell culture grade) and MTT (3-[4,5-dicell methylthiazol-2-yl]2,5-diphenyltetrazolium bromide) with a MW of 414.32 g mol−1 were bought from HiMedia, Mumbai, India. LysoTracker™ Red and CellMask Deep Red were purchased from Thermo Fisher Scientific, India. Other analytical grade chemicals utilized for the study were procured as per use. The mouse monocyte macrophage cell line J-774A.1 was acquired from the National Centre for Cell Science, Pune, India.2.2 Preparation of the ACEM anchored AmB-LIPOMER
Briefly, 20 mg of Gantrez AN119 (GZ) and 20 mg of AmB were solubilized in 6 ml of organic phase comprising an acidified mixture of tetrahydrofuran![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (1:1) and was added under continuous magnetic stirring to a 30 ml aqueous phase consisting of 30% IPA in double distilled water. Furthermore, 40 mg of polyglyceryl-6-distearate (PGDS) solubilized in 2 ml of the above organic mixture was introduced into the aqueous phase which was followed by the addition of 3 ml, 5% w/v magnesium acetate. The AmB-LIPOMER dispersion was continuously stirred overnight for complete evaporation of the organic solvents. The solvent-free dispersion was centrifuged at 16350g for 10 min resulting in separation of the AmB-LIPOMER pellet, while the supernatant was used for evaluation of the % drug entrapped. The pellet obtained was redispersed in filtered, double distilled water (5 ml) and disaggregated using an ultrasonic probe sonicator (DP120, Dakshin, Mumbai, India) operating at 200 V with 10 s pulses for 5 min, to obtain the AmB-LIPOMER dispersion.
:ethanol (1:1) and was added under continuous magnetic stirring to a 30 ml aqueous phase consisting of 30% IPA in double distilled water. Furthermore, 40 mg of polyglyceryl-6-distearate (PGDS) solubilized in 2 ml of the above organic mixture was introduced into the aqueous phase which was followed by the addition of 3 ml, 5% w/v magnesium acetate. The AmB-LIPOMER dispersion was continuously stirred overnight for complete evaporation of the organic solvents. The solvent-free dispersion was centrifuged at 16350g for 10 min resulting in separation of the AmB-LIPOMER pellet, while the supernatant was used for evaluation of the % drug entrapped. The pellet obtained was redispersed in filtered, double distilled water (5 ml) and disaggregated using an ultrasonic probe sonicator (DP120, Dakshin, Mumbai, India) operating at 200 V with 10 s pulses for 5 min, to obtain the AmB-LIPOMER dispersion.
An aqueous solution of ACEM was prepared by dissolving the ligand in filtered, double distilled water at room temperature (28 °C), and vacuum filtered (0.2 μm filter), prior to use. The ACEM ligand solution was added to the resulting AmB-LIPOMER pellet at a GZ/ligand ratio of 1:1 (w/w), cyclo-mixed and allowed to stand for 10 min. The dispersion was probe sonicated for 5 min on an ice bath with 10 s pulses at 200 V, to obtain the AmB-ACEM LIPOMER.
The FITC loaded LIPOMER and ACEM-LIPOMER were prepared by the same method by replacing AmB with FITC (4 mg ml−1).
2.3 Lyophilization study
AmB-LIPOMER and AmB-ACEM LIPOMER dispersions were lyophilized using a mixture of 10% Trehalose and 0.05% w/v Lutrol F68 as a cryoprotectant. The dispersions containing the cryoprotectant were dispensed into amber coloured glass vials. The vials were frozen for 24 h at −72 °C and lyophilized for another 24 h on a LABCONCO freeze dryer (FreeZone 4.5, USA). At the end of the cycle, the vials were tightly sealed with aluminium crimped rubber closures. The freeze-dried cake was reconstituted to an original volume (5 ml) with filtered double distilled water followed by shaking to obtain a homogeneous nanoparticle dispersion. The reconstituted formulations were further evaluated for particle size from which the Sf/Si ratio was calculated.2.4 ACEM anchored AmB-LIPOMER characterization


2.5 Stability study
Stability was evaluated over a duration of 12 months at 4 ± 2 °C and 25 ± 2 °C/60 ± 5% RH according to the ICH guidelines. The lyophilized AmB-LIPOMER and AmB-ACEM LIPOMER were stored in amber coloured glass vials (10 ml) with rubber stoppers and aluminium-crimped tops. Samples were removed from the stability chambers at fixed time intervals of 0, 1, 2, 3, 6 and 12 months and various parameters, like visual examination, PS, and PDI, were evaluated in triplicate as described in section 2.4.3 while the drug content was evaluated as described below.The AmB content was estimated by reverse phase HPLC using a Jasco LC 2000 (Jasco, Japan), fitted with a PDA detector. Sample analysis was performed under isocratic elution conditions, using a C18 Waters column (250 × 4.6 mm, 5 μm PS) at a temperature of 25 °C. The mobile phase comprising 50 mM disodium EDTA:acetonitrile (55:45) was maintained at a flow rate of 1.2 ml min−1. For sample preparation, the freeze-dried AmB-LIPOMER and AmB-ACEM LIPOMER were accurately weighed and transferred into DMSO and allowed to dissolve by cyclo-mixing and bath sonication for 15 min, followed by suitable dilution with methanol. The concentration of AmB was quantified in triplicate at a λmax of 405 nm by RP-HPLC.
2.6 In vitro serum stability of nanoparticles
Briefly, 0.5 ml each of the AmB-LIPOMER and the AmB-ACEM LIPOMER dispersions (∼2 mg ml−1 AmB) were incubated with an equal volume of rat serum maintained at 37 ± 0.5 °C. Aliquots of 0.1 ml were withdrawn at time intervals of 1, 2, 4, and 6 h, diluted 100-fold with filtered, double distilled water and evaluated for alteration in PS in triplicate as detailed in section 2.4.3.2.7 Macrophage cell viability
The RAW 264.7 macrophage cell line was maintained as reported previously. RAW 264.7 cells grown in 10% FBS supplemented DMEM medium were seeded in a 96-well plate, containing 5000 cells per well. The well plates were incubated overnight, at 37 °C with a supply of 5% CO2 for cell adherence. Serial dilutions of the blank LIPOMER, the blank ACEM LIPOMER, the AmB-LIPOMER and the AmB-ACEM LIPOMER were prepared in complete medium to acquire varying AmB concentrations from 1.56 to 50 μg ml−1, which were added to the wells in triplicate. Plain medium treated cells were used as the control. The well plates were incubated for 24 h at 37 °C, after which the test solutions were discarded. This was followed by the addition of 100 μL of MTT solution (500 μg ml−1) in each well and incubation for another 4 h to result in the formation of purple coloured formazan crystals. The MTT solution was aspirated and the formazan crystals were dissolved in DMSO (200 μL). The absorbance of the wells was recorded at 570 nm employing a multi-well microplate reader (BIO-TEK, Plate reader, Germany) and % cell viability was evaluated as described in the equation below:% Macrophage cell viability = (Absorbancesample/Absorbancecontrol) × 100
2.8 Macrophage uptake
2.9 In vitro anti-leishmanial efficacy
Nanoparticle mediated growth inhibition of transgenic Leishmania donovani amastigote infected J774A.1 macrophage cells was evaluated using a previously reported method.11 Varying dilutions of the AmB-LIPOMER and the AmB-ACEM LIPOMER were prepared in DMEM complete medium to obtain AmB concentrations from 2.5 to 50 ng ml−1. The samples were added to the amastigote (2.5 × 105 per well) infected macrophage cells (5 × 104 cells per well) seeded on a 96-well plate and incubated for 72 h at 37 °C. At the end of 72 h, the samples from each well were discarded and replaced with 50 μL each of Steady-Glo reagent and PBS, and gently mixed for 1–2 min. The luciferase activity of each well was measured in terms of the relative luminescence unit (RLU) and parasitic growth inhibition was calculated using the equation: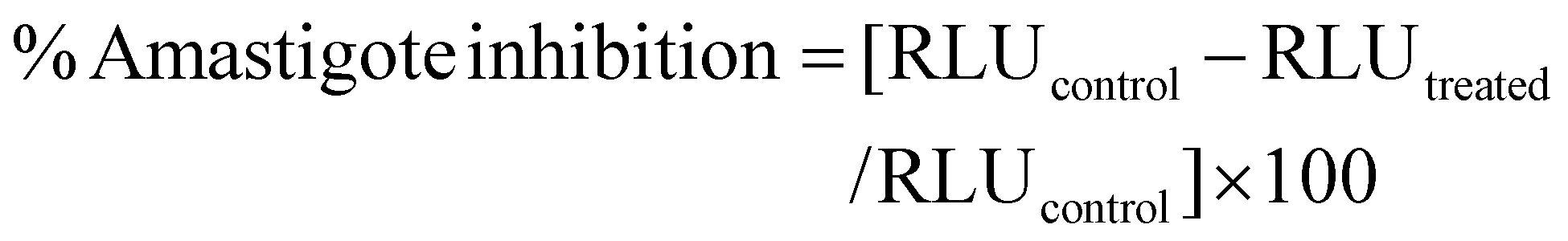
2.10 Safety evaluation
The safety of the AmB-ACEM LIPOMER was evaluated via hemolysis assay. Briefly, varying dilutions of the AmB-LIPOMER, the AmB-ACEM LIPOMER and commercial AmB formulations (Amfocare® and Fungisome™) were prepared in 5% v/v dextrose solution to obtain AmB concentrations from 50 to 1000 μg ml−1. Each sample was added to an erythrocyte suspension of 5% v/v and incubated at 37 °C for 1 h. At the end of 1 h the sample tubes were immersed in ice cold water to halt the process and the intact erythrocytes were separated by centrifuging for 15 min at 1008g. The supernatant was analysed for released hemoglobin by measuring absorbance at 540 nm. Hemolysis observed was compared against the negative control (PBS) and the positive control (deionized water), and percentage hemolysis was calculated employing the equation:
2.11 In vivo pharmacokinetics and biodistribution
The study protocol was approved by the Institutional Animal Ethics Committee (IEAC) of the Institute of Chemical Technology, Mumbai, India, in accordance with the CPCSEA guidelines (protocol no.: ICT/IAEC/2018/P13). Male Sprague–Dawley (SD) rats (250 ± 20 g) were quarantined in the animal house of the Institute of Chemical Technology, Mumbai, India, under controlled temperature and humidity (25 °C ± 2 °C, 50 ± 5% RH) with a 12 h dark/light cycle throughout the study.The overall targeting efficiency of AmB-SLN and the LIPOMER was calculated using the equation:

2.12 Statistical analysis
All studies were carried out in triplicate and the mean ± standard deviation of the three independent experiments was calculated. Statistical analysis was carried out using one-way ANOVA with Tukey's test and a P-value of <0.05 was selected to indicate statistical significance. The IC50 value was calculated by regression of log dose versus drug response obtained through probing analysis.3. Results and discussion
3.1 ACEM anchored AmB-LIPOMER
The AmB-LIPOMER was prepared using a method previously reported by our group.11 The characteristic frequencies of ACEM in the fingerprint region corresponding to the C–O pyranose ring and C–O–C ether groups (<1400 cm−1) remained intact.29,30 However, the sharp bands at 1852.17, 1779.86, and 1725.61 cm−1, characteristic of anhydride lactone of GZ and at 3400 cm−1 and 1740 cm−1 corresponding to the O–H stretching vibrations and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of ACEM, respectively, were absent in the FTIR spectrum of the AmB-ACEM LIPOMER indicating H-bonding interactions between ACEM and GZ (Fig. 1), which aided noncovalent anchoring of ACEM.
O stretching vibrations of ACEM, respectively, were absent in the FTIR spectrum of the AmB-ACEM LIPOMER indicating H-bonding interactions between ACEM and GZ (Fig. 1), which aided noncovalent anchoring of ACEM.
 | ||
| Fig. 1 FTIR spectra of AmB, GZ, PGDS, ACEM and AmB-ACEM LIPOMER. The blue dotted circle represents the characteristic frequencies of ACEM in the fingerprint region that remained intact, thereby confirming anchoring of ACEM. Red dotted circles represent the AmB-ACEM LIPOMER indicating H-bonding between ACEM and GZ. | ||
The AmB-LIPOMER and the AmB-ACEM LIPOMER demonstrated high AmB entrapment efficiencies of 97.76 ± 0.56% and 95.51 ± 0.04% and drug loadings of 21.58 ± 0.39% and 21.16 ± 0.55%. A negative zeta potential (−14.78 ± 0.92 mV and −19.65 ± 0.89 mV), respectively, indicated good colloidal stability. A mean diameter in the range of 300–450 nm with a low PDI (<0.3) is considered apposite for intramacrophage targeting. Lyophilization of the AmB-LIPOMER and the AmB-ACEM LIPOMER to ensure long-term stability revealed Sf/Si ratios of 1.24 ± 0.16 and 1.21 ± 0.02, respectively.
3.2 Imaging by TEM
TEM images of the AmB loaded nanocarriers revealed a nearly spherical morphology with the particle size correlating with those reported by DLS measurements (Fig. 2). | ||
| Fig. 2 Transmission electron microscopy analysis of (A) the AmB-LIPOMER and (B) the AmB-ACEM LIPOMER. | ||
3.3 DSC and XRD analyses
DSC thermograms (Fig. 3A) revealed two broad endothermic peaks at 115.63 °C and 202.64 °C for AmB, which corroborated earlier reports.31,32 The disappearance of these characteristic peaks in the AmB-ACEM LIPOMER suggests decreased crystallinity and partial amorphization of AmB in the NPs. Such a decrease indicates enhanced dissolution.33 | ||
| Fig. 3 (A). DSC thermograms of AmB, GZ, PGDS, ACEM and the AmB-ACEM LIPOMER. (B) XRD spectra of AmB, GZ, PGDS, ACEM, and the AmB-ACEM LIPOMER. | ||
XRD analysis has been used for quantifying the extent of the amorphous or crystalline phase in nanosolids. In agreement with previous reports34,35 AmB demonstrated multiple sharp endothermic peaks at 2θ scattered angles between 10 and 24° indicating a high degree of crystallinity. PGDS also exhibited a sharp peak at a 2θ value of 22° while GZ and ACEM indicated a relatively amorphous state. The absence of the sharp drug and lipid peaks in the AmB-ACEM LIPOMER (Fig. 3B) indicated partial amorphization of the drug in the nanoparticles.
3.4 Long-term stability study
The freeze dried AmB-LIPOMER and AmB-ACEM LIPOMER revealed a drug content of >94% and no significant change in physical appearance or particle size (Fig. 4) over a period of 12 months under refrigerated conditions confirming good stability as per ICH guidelines.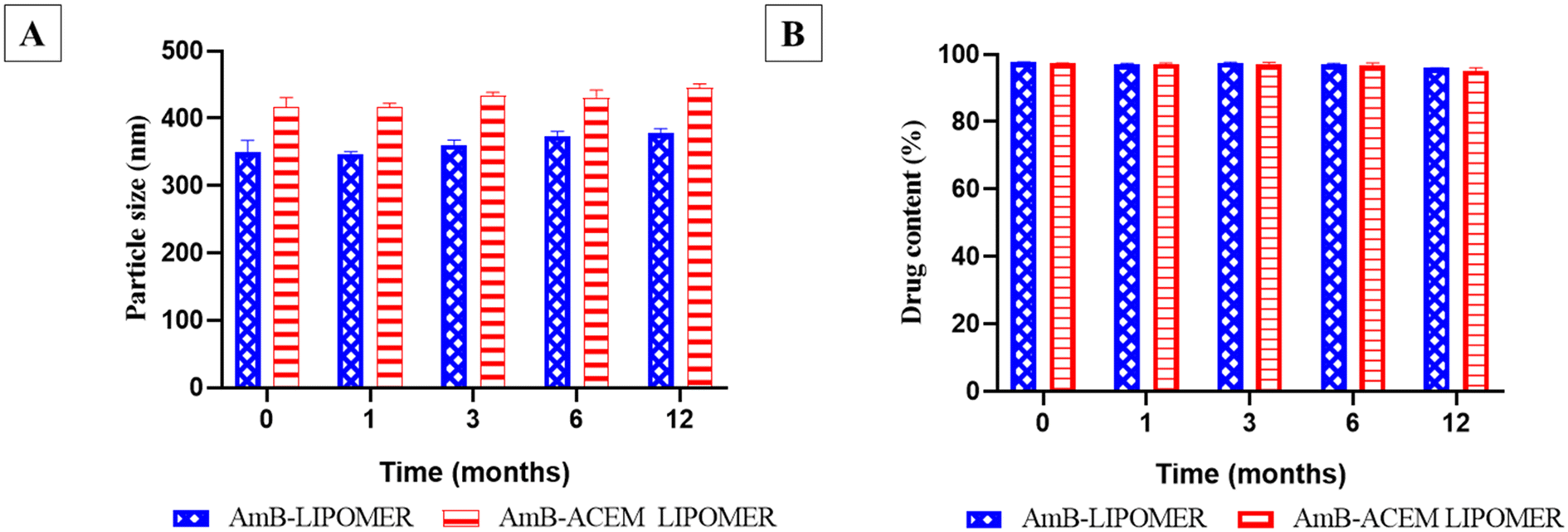 | ||
| Fig. 4 Long term stability study of the AmB-ACEM LIPOMER and the AmB-LIPOMER: (A) particle size (nm) and (B). drug content (%). Data are presented as mean ± SD, n = 3. | ||
3.5 Serum stability
Protein binding has been posited as one of the key factors influencing particle internalization. Serum proteins, particularly opsonins which are adsorbed on the surface of nanocarriers, tag them for recognition and clearance by the macrophages of the RES.36 Such adsorption leads to alterations in the size of nanocarriers. The LIPOMERs were stable up to 6 h revealing no significant increase in the particle size and an average size not exceeding 500 nm, indicating safety for i.v. administration (Fig. 5). The stability of the LIPOMERs is credited to surface hydrophilicity induced steric stabilization imparted by GZ and ACEM.37 | ||
| Fig. 5 Serum stability study of the AmB-ACEM LIPOMER and the AmB-LIPOMER. Data are presented as mean ± SD, n = 3. | ||
3.6 Hemolysis assay
Hemolytic activity is a well-known toxicity associated with AmB. Different marketed formulations served as controls in this study. The conventional micellar AmB, Amphocare®, reported to be toxic, revealed ∼100% hemolysis even at low AmB concentrations (Fig. 6). This is ascribed to the rapid dissociation of AmB from deoxycholate micelles into the aggregated state upon dilution, which is heightened by the hemolytic activity of the surfactant in Amphocare®.38 On the other hand, the AmB-ACEM LIPOMER revealed less than 15% hemolysis even at high AmB concentrations (Fig. 6), comparable with the AmB-LIPOMER and Liposomal AmB (Fungisome™), endorsing safety for i.v. administration.39 | ||
| Fig. 6 Erythrocyte toxicity of the AmB-ACEM LIPOMER and the AmB-LIPOMER, in comparison with Amphocare® and Fungisome™ diluted in 5% dextrose to obtain varying AmB concentrations, after 1 h of incubation. Data are presented as mean ± SD, n = 3, at a significance level of ****p < 0.0001. | ||
3.7 Macrophage cell viability
The AmB-ACEM LIPOMER revealed good safety, as even at a high AmB concentration of 50 μg ml−1, >71% cell viability was achieved (Fig. 7). The blank AmB-LIPOMER and the AmB-LIPOMER revealed near 100% cell viability suggesting good safety while the blank ACEM LIPOMER demonstrated high cell proliferation.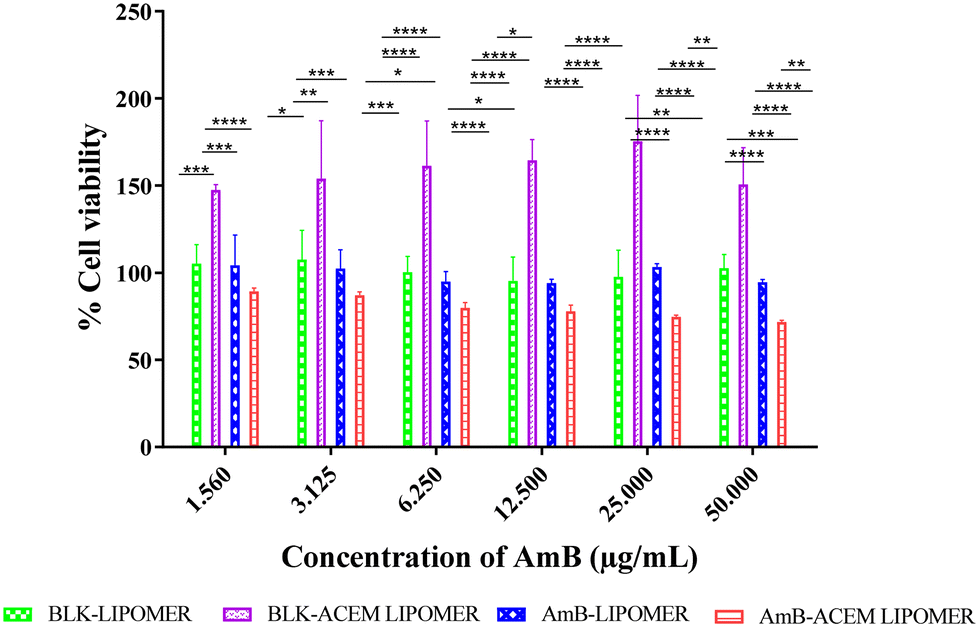 | ||
| Fig. 7 Concentration dependent cytotoxicity of the blank LIPOMER, blank ACEM LIPOMER, AmB-LIPOMER, and AmB-ACEM LIPOMER against the RAW 264.7 cell line after 24 h of incubation. Data are presented as mean ± SD, n = 3, at significance levels of *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. | ||
3.8 Macrophage uptake study
Based on the macrophage viability data, the AmB concentration equivalent to 25 μg ml−1 (∼80% cell viability) was selected for the macrophage uptake study. A 13- to 20-fold enhancement in the fluorescence intensity was observed with the FITC loaded ACEM LIPOMER compared to the LIPOMER at all time points (Fig. 8A and B). The high macrophage uptake confirmed the role of ligand based active targeting in enabling augmented macrophage uptake, and corroborated other reports on carbohydrate enabled mannose receptor mediated uptake.40–42 Nevertheless, we confirmed the same by performing the mannose receptor blocking assay, aided by confocal microscopy. A significant reduction in uptake of the FITC-ACEM LIPOMER in mannose treated macrophages validated the mannose receptor mediated uptake facilitated by ACEM. Furthermore, the yellow fluorescence seen due to integration of the green fluorescence of FITC with the lysotracker red dye also suggested high lysosomal localization of the FITC-ACEM LIPOMER. This could be particularly advantageous in enabling high efficacy in leishmaniasis, wherein the parasite is harboured in the lysosome.43 | ||
| Fig. 8 Macrophage uptake study of the FITC-LIPOMER and the ACEM LIPOMER (A). Uptake by flow cytometry – change in the mean fluorescence intensity with time. (B). Overlay graph of fluorescence intensity. Data are presented as mean ± SD, n = 3, at a significance level of ****p < 0.0001. (C). Uptake by confocal microscopy – fluorescence intensity in macrophages with FITC-ACEM-LIPOMER + D-mannose and the FITC-ACEM-LIPOMER, respectively; scale bar, 10 μm. | ||
3.9 Anti-leishmanial efficacy
The data demonstrate comparable efficacy of the AmB-ACEM LIPOMER with that of liposomal AmB (Fungisome™) which served as the reference (Table 1). Surprisingly the AmB-LIPOMER without ACEM, despite a lower macrophage uptake, revealed a 2-fold lower IC50 value and higher efficacy compared to both liposomal AmB and the AmB-ACEM LIPOMER. The AmB-LIPOMER also demonstrated a significantly higher safety index compared to the AmB-ACEM LIPOMER as well as liposomal AmB.| Formulation | IC50 (ng mL−1) amastigote | CC50 (ng mL−1) | SI |
|---|---|---|---|
| AmB-LIPOMER | 7.55 ± 1.16 | 556.66 ± 15.45 | 73.72 |
| AmB-ACEM LIPOMER | 14.45 ± 0.31 | 198.83 ± 20.67 | 13.75 |
| Fungisome™ (Liposomal AmB) | 13.55 ± 0.91 | 333.83 ± 29.00 | 24.63 |
The lower efficacy is ascribed to the receptor mediated endocytic uptake of the ACEM-LIPOMER and the extensive localization of the AmB-ACEM LIPOMER resulting in the lysosomal compartment as substantiated by the confocal images (Fig. 8C). Rapid degradation of AmB in the harsh and acidic lysosomal pH44,45 resulted in decreased efficacy. The AmB LIPOMER exhibited phagocytic uptake in the absence of the ligand and localisation in the phagosome. Possible delay in phagosomal maturation due to the lipidic nature of the carrier as reported in other studies46–48 resulting in delay in phagolysosomal fusion could have limited the degradation of AmB, thereby enabling higher efficacy.
3.10 Pharmacokinetics and biodistribution study
The plasma profile after intravenous administration of the AmB-LIPOMER and liposomal Fungisome™ demonstrated rapid clearance from the plasma, possibly by the macrophages (Fig. 9). Furthermore, the pharmacokinetic profile of the AmB-LIPOMER and Fungisome™ demonstrated similar areas under the curve (AUC) and extensive tissue distribution was indicated by the high volume of distribution (Vd > 7 L). Although the AmB-LIPOMER exhibited a 1.43-fold lower plasma half-life compared to liposomal Fungisome™, it presented a 1.57-fold higher mean residence time (MRT), respectively, compared to Fungisome™, indicating longer retention in the body (Table 2). | ||
| Fig. 9 Pharmacokinetic profile of AmB after intravenous administration of the AmB-LIPOMER and Fungisome™ at 5 mg kg−1 dose (n = 3). | ||
| Pharmacokinetic parameters | Treatment groups (5 mg kg−1) | |
|---|---|---|
| AmB-LIPOMER | Fungisome™ | |
| C 0 (μg mL−1) | 21.57 ± 4.87 | 56.45 ± 11.42 |
| t 1/2 (h) | 31.89 ± 9.04 | 45.63 ± 5.87 |
| AUC0–t (h μg mL−1) | 12.41 ± 1.06 | 17.96 ± 2.13 |
| V d (L kg−1) | 10.94 ± 2.31 | 7.87 ± 2.11 |
| C l (mL h−1 kg−1) | 0.27 ± 0.05 | 0.20 ± 0.02 |
| MRT0–t (h) | 14.95 ± 1.23 | 9.12 ± 2.32 |
In the case of the AmB-ACEM LIPOMER, rapid plasma clearance was evident following intravenous administration, as at 5 minutes the plasma concentration was very low (0.36 ± 0.09 μg mL−1). This was 25.61-fold and 81-fold lower compared to the AmB-LIPOMER and Fungisome™, respectively, suggesting very rapid distribution into the RES organs. Overall however, while the AmB-ACEM LIPOMER dosed animal revealed an RES targeting efficiency of ∼84%, the AmB-LIPOMER and Fungisome™ demonstrated RES targeting efficiencies of >92%. The AmB-LIPOMER and Fungisome™ revealed high localization in the clinically relevant VL reservoirs, namely the liver and spleen, with significantly lower amounts in the non-target organ, lungs (Fig. 10A). In the case of the AmB-ACEM LIPOMER, within 30 min 5 of the 6 animals exhibited severe respiratory distress, laboured breathing, difficulty in movement, leg twitching and death due to asphyxiation. The AmB-ACEM LIPOMER group revealed enhanced concentration not only in the liver, but also in the lungs, kidneys and heart (Fig. 10A). Furthermore, gross examination of the organs of the deceased animals revealed a significant increase in the size of the lungs, with severe inflammation, congestion and clotting compared to the lungs of AmB-LIPOMER treated animals (Fig. 10B). Neither the AmB-LIPOMER nor Fungisome™ groups showed such adverse effects and mortality.
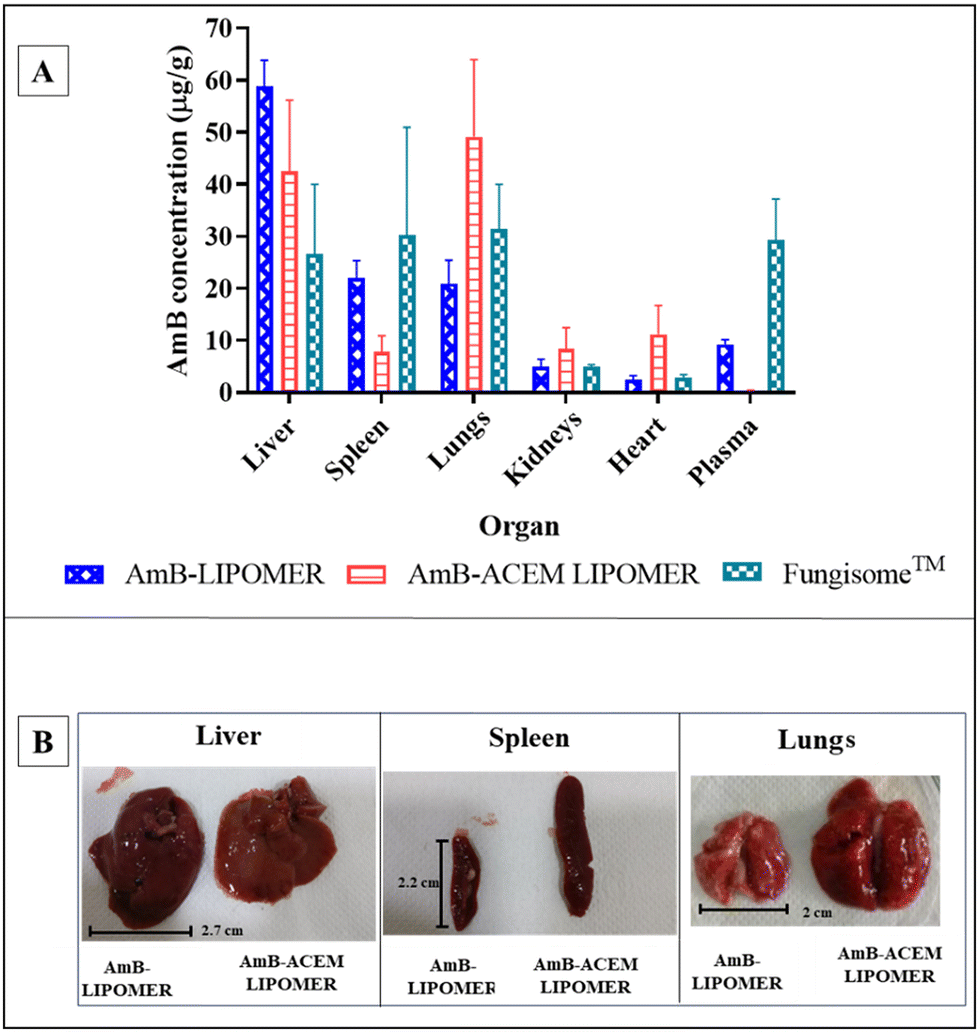 | ||
| Fig. 10 (A). Biodistribution of the AmB-LIPOMER, the AmB-ACEM LIPOMER and Fungisome™, mean ± SD (n = 3); (B). liver, spleen and lungs of the AmB-LIPOMER and the AmB-ACEM LIPOMER dosed rats (<30 minutes) after intravenous administration at a 5 mg kg−1 dose. | ||
Pulmonary accumulation and adverse reactions have been reported in humans, shortly after administration of commercial liposomal AmB, but only in patients with liver damage. The reduced liver clearance of liposomal AmB in such patients consequently enhanced the deposition of liposomes in the lungs, which act as a substitutive clearing organ.49–51
Extrapolating this to our study, the extremely rapid uptake and high concentration of the AmB-ACEM LIPOMER probably resulted in saturation of the liver macrophage receptors supported by the rapid distribution observed in the pharmacokinetic study. This resulted in significantly enhanced deposition in the lungs, the substitutive clearing organ, causing severe inflammation and subsequent mortality (Fig. 10B). Moreover, complex carbohydrates like ACEM act as immune stimulators of both macrophages and T cells which can trigger enhanced macrophage/mononuclear cell infiltration and stimulate the production of proinflammatory agents (cytokines, tumour necrosis factor and nitric oxide) resulting in untoward effects as observed in our study,26 although another report suggests that ACEM could inhibit the cytokine storm in mouse pneumonia.52 Furthermore, pulmonary hypertension and lung damage caused by AmB have also been reported,53 which have been associated with enhanced immune cell aggregation and the production of chemical mediators by macrophages. This could have further aggravated the condition.
This brings to the forefront a serious concern about the strategy of active targeting of AmB, even though the safety of ACEM, a carbohydrate ligand, has been demonstrated. The study also highlights the need for a rational approach to titrating the augmented intracellular drug concentration, particularly for toxic drugs like AmB.
4. Conclusion
Active targeting using a mannose receptor-based ligand is a promising approach for manifold enhancement in the intracellular delivery of drugs. Nevertheless, this study proposes caution in active macrophage targeting of AmB, as the high mannose receptor uptake proved seriously detrimental instead of being advantageous. Our study also demonstrated the AmB-LIPOMER without the ACEM ligand to be comparable with the gold standard liposomal AmB, proposing passive targeting as the safer option.Author contributions
Saugandha Das: conceptualization, methodology, investigation, software, and writing – original draft. Pooja Todke: conceptualization, methodology, investigation, software, and writing – original draft. Manisha Madkaikar: methodology, investigation, and validation. Padma Devarajan: conceptualization, investigation, supervision, validation, funding acquisition, and manuscript – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present research was supported by the Department of Science and Technology (DST) under the INSPIRE fellowship scheme (INSPIRE Fellowship Code No. IF130037), Government of India, New Delhi (India). The authors are thankful to the All India Council for Technical Education (AICTE), Government of India, and TEQIP-III for providing research fellowship to Pooja Todke (Student ID: 1-6431958581).References
- M. Baginski, J. Czub and K. Sternal, Chem. Rec., 2006, 6, 320–332 CrossRef CAS PubMed.
- V. Fanos and L. Cataldi, J. Chemother., 2000, 12, 463–470 CrossRef CAS PubMed.
- B. L. Burgess, Y. He, M. M. Baker, B. Luo, S. F. Carroll, T. M. Forte and M. N. Oda, Int. J. Nanomed., 2013, 4733–4743 Search PubMed.
- S. Gupta, A. Dube and P. Vyas, Pharm. Nanotechnol., 2012, 1, 54–67 CrossRef.
- S. Gupta and S. P. Vyas, J. Drug Targeting, 2007, 15, 206–217 CrossRef CAS PubMed.
- S. Gupta, A. Dube and S. P. Vyas, J. Drug Targeting, 2007, 15, 437–444 CrossRef CAS PubMed.
- M. Nahar and N. K. Jain, Pharm. Res., 2009, 26, 2588–2598 CrossRef CAS PubMed.
- R. Kumar, G. C. Sahoo, K. Pandey, V. N. Das and P. Das, Drug Delivery, 2015, 22, 383–388 CrossRef CAS PubMed.
- K. G. Nelson, J. V. Bishop, R. O. Ryan and R. Titus, Antimicrob. Agents Chemother., 2006, 50, 1238–1244 CrossRef CAS PubMed.
- S. Asthana, A. K. Jaiswal, P. K. Gupta, V. K. Pawar, A. Dube and M. K. Chousaria, Antimicrob. Agents Chemother., 2013, 57, 1714–1722 CrossRef CAS PubMed.
- S. Das and P. V. Devarajan, Mol. Pharm., 2020, 17, 2186–2195 CrossRef CAS PubMed.
- G. Delmas, S. Park, Z. W. Chen, F. Tan, R. Kashiwazaki, L. Zarif and D. S. Perlin, Antimicrob. Agents Chemother., 2002, 46, 2704–2707 CrossRef CAS PubMed.
- L. Zarif, J. R. Graybill, D. Perlin, L. Najvar, R. Bocanegra and R. J. Mannino, Antimicrob. Agents Chemother., 2000, 44, 1463–1469 CrossRef CAS PubMed.
- H. Fukui, T. Koike, T. Nakagawa, A. Saheki, S. Sonoke, Y. Tomii and J. Seki, Int. J. Pharm., 2003, 267, 101–112 CrossRef CAS PubMed.
- A. Jhaveri and V. Torchilin, Expert Opin. Drug Delivery, 2016, 13, 49–70 CrossRef CAS.
- N. K. Jain, V. Mishra and N. K. Mehra, Expert Opin. Drug Delivery, 2013, 10, 353–367 CrossRef CAS PubMed.
- C. P. Thakur, A. K. Pandey, G.P Sinha, S. Roy, K. Behbehani and P. Olliaro, Trans. R. Soc. Trop. Med. Hyg., 1996, 90, 319–322 CrossRef CAS PubMed.
- S. Asthana, P. K. Gupta, A. K. Jaiswal, A. Dube and M. K. Chourasia, Pharm. Res., 2015, 32, 2663–2377 CAS.
- E. Dalle Vedove, G. Costabile and O. M. Merkel, Adv. Healthc. Mater., 2018, 7, 1701398 CrossRef.
- A. Kaur, S. Jain and A. Tiwary, Acta Pharm., 2008, 58, 61–74 CAS.
- G. Shahnaz, B. J. Edagwa, J. McMillan, S. Akhtar, A. Raza, N. A. Qureshi, M. Yasinzai and H. E. Gendelman, Nanomedicine, 2017, 12, 99–115 CrossRef CAS PubMed.
- I. Afzal, H. S. Sarwar, M.F Sohail, S. Varikuti, S. Jahan, S. Akhtar, M. Yasinzai, A. R. Satoskar and G. Shahnaz, Nanomedicine, 2019, 14, 387–406 CrossRef CAS PubMed.
- T. Suciati, P. Rachmawati, E. Soraya, A. B. Mahardhika, R. Hartarti and K. Anggadiredja, J. Appl. Pharm. Sci., 2018, 8, 1–11 CAS.
- J. R. McCombs and S. C. Owen, AAPS J., 2015, 17, 339–351 CrossRef CAS PubMed.
- L. Zhang and I. R. Tizard, Immunopharmacology, 1996, 35, 119–128 CrossRef CAS PubMed.
- L. Ramamoorthy, M. C. Kemp and I. R. Tizard, Mol. Pharmacol., 1996, 50, 878–884 CAS.
- A. Djeraba and P. Quere, Int. J. Immunopharmacol., 2000, 22, 365–372 CrossRef CAS PubMed.
- J. B. Kim, K. Park, J. Ryu, J. J. Lee, M. W. Lee, H. S. Cho, H. S. Nam, O. K. Park, J. W. Song, T. S. Kim and D. J. Oh, Sci. Rep., 2016, 6, 22608 CrossRef CAS PubMed.
- S. Kumar and R. Kumar, Carbohydr. Polym., 2019, 207, 460–470 CrossRef CAS PubMed.
- D. Miramon-Ortíz, W. Argüelles-Monal, E. Carvajal-Millan, Y. L. López-Franco and F. M. Goycoolea, Polymers, 2019, 11, 330 CrossRef PubMed.
- S. Bhatia, V. Kumar, K. Sharma, K. Nagpal and T. Bera, Sci. World J., 2014, 2014, 564573 Search PubMed.
- K. S. Nagarsekar, C. Galdhar, R. Gaikwad, A. Samad and P. V. Devarajan, Drug Delivery Lett., 2014, 4, 208–220 CrossRef CAS.
- M. A. Tawfik, M. I. Tadros and M. I. Mohamed, AAPS PharmSciTech, 2018, 19, 3650–3660 CrossRef CAS.
- C. Shu, T. Li, W. Yang, D. Li, S. Ji and L. Ding, R. Soc. Open Sci., 2018, 5, 171814 CrossRef.
- G. Schwartzman, I. Asher, V. Folen, W. Brannon and J. Taylor, J. Pharm. Sci., 1978, 67, 398–400 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- L. Guerrini, R. Alvarez-Puebla and N. Pazos-Perez, Materials, 2018, 11, 1154 CrossRef PubMed.
- K. Hac-Wydro and P. Dynarowicz-Latka, Biophys. Chem., 2006, 123, 154–161 CrossRef CAS PubMed.
- P. A. Todke and P. V. Devarajan, J. Controlled Release, 2022, 349, 756–764 CrossRef CAS PubMed.
- L. Yang, Y. Sha, Y. Wei, H. Fang, J. Jiang, L. Yin, Z. Zhong and F. Meng, Biomater. Sci., 2023, 11, 2211–2220 RSC.
- E. Maretti, L. Costantino, F. Buttini, C. Rustichelli, E. Leo, E. Truzzi and V. Iannuccelli, Drug Delivery Transl. Res., 2019, 9, 298–310 CrossRef CAS PubMed.
- I. D. Zlotnikov, M. A. Vigovskiy, M. P. Davydova, M. R. Danilov, U. D. Dyachkova, O. A. Grigorieva and E. V. Kudryashova, Int. J. Mol. Sci., 2022, 23, 16144 CrossRef CAS PubMed.
- V. Liévin-Le Moal and P. M. Loiseau, FEBS Lett., 2016, 283, 598–607 CrossRef PubMed.
- J. G. Visser, A. D. P. Van Staden and C. Smith, Front. Pharmacol., 2019, 10, 22 CrossRef CAS PubMed.
- P. Sawangchan, F. A. Júnior, É. N. Alencar, E. S. Egito and L. E. Kirsch, Int. J. Pharm., 2023, 632, 122586 CrossRef CAS PubMed.
- K. C. Nicolaou, T. K. Chakraborty, Y. Ogawa, R. A. Daines, N. S. Simpkins and G. T. Furst, J. Am. Chem. Soc., 1988, 110, 4660–4672 CrossRef CAS.
- S. Axelrod, H. Oschkinat, J. Enders, B. Schlegel, V. Brinkmann, S. H. Kaufmann, A. Haas and U. E. Schaible, Cell. Microbiol., 2008, 10, 1530–1545 CrossRef CAS PubMed.
- A. M. Pauwels, M. Trost, R. Beyaert and E. Hoffmann, Trends Immunol., 2017, 38, 407–422 CrossRef CAS PubMed.
- Y. Kao and R. Juliano, Biochim. Biophys. Acta, 1981, 677, 453–461 CrossRef CAS PubMed.
- R. L. Souhami, H. M. Patel and B. E. Ryman, Biochim. Biophys. Acta, 1981, 674, 354–371 CrossRef CAS PubMed.
- J. Dave and H. M. Patel, BBA, Biochim. Biophys. Acta, Mol. Cell Res., 1986, 888, 184–190 CrossRef CAS PubMed.
- L. Li, W. Xu, Y. Luo, C. Lao, X. Tong, J. Du, B. Huang, D. Li, J. Chen, H. Ye and F. Cong, Carbohydr. Polym., 2022, 297, 120032 CrossRef CAS PubMed.
- T. J. McDonnell, S. W. Chang, J. Y. Westcott and N. F. Voelkel, J. Appl. Physiol., 1988, 65, 2195–2206 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |