
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of novel hybrid compounds bearing pyrazine and 1,2,4-triazole analogues as potent antitubercular agents†
Shivakumar
Naik
 a,
Dinesha
Puttachari
a,
Vanishree
A. L.
a,
Udayakumar
D.
*a,
Varsha Prakash
Shetty
b,
Chaitra
Prabhu
b and
Vijaya Kumar
Deekshit
b
a,
Dinesha
Puttachari
a,
Vanishree
A. L.
a,
Udayakumar
D.
*a,
Varsha Prakash
Shetty
b,
Chaitra
Prabhu
b and
Vijaya Kumar
Deekshit
b
aOrganic and Medicinal Chemistry Laboratory, Department of Chemistry, National Institute of Technology Karnataka, Surathkal-575025, Mangalore, Karnataka, India. E-mail: udayakumar@nitk.edu.in; udayaravi80@gmail.com
bDivision of Infectious Diseases, NITTE University Center for Science Education and Research, NITTE (Deemed to be University), Deralakatte-575018, Mangalore, Karnataka, India
First published on 13th March 2024
Abstract
In this study, we elucidate the conceptualization and synthesis of hybrid compounds (T1–T18) amalgamating pyrazine and 1,2,4-triazole scaffolds. A total of eighteen compounds were screened in vitro for their efficacy against the Mycobacterium tuberculosis H37Rv strain via the MABA assay. The results revealed that eight compounds (T4, T5, T6, T11, T14, T15, T16, and T18) manifested noteworthy activity against Mtb, with minimum inhibitory concentration (MIC) values of ≤21.25 μM. Furthermore, we also examined these compounds for their antibacterial and antifungal properties against various strains. Compounds T4, T9, T10, T16, and T18 displayed significant antibacterial activity, while compounds T12 and T14 demonstrated significant antifungal activity. Subsequently, the most potent compounds were evaluated for their potential cytotoxicity to the Vero cell line via the MTT assay, revealing IC50 values surpassing 375 μM, indicative of minimal cytotoxicity. Additionally, we conducted in silico studies on these target molecules to better understand their action mechanisms. The in silico investigations suggest that the target enzyme involved in the action of the compounds may be DprE1. However, further experimental validation is necessary to ascertain the target responsible for the whole cell activity. All the target compounds are docked within the active site of the DprE1 enzyme, demonstrating favorable binding interactions. Furthermore, we predicted the ADME properties, physicochemical characteristics, and drug-like qualities of the target compounds using in silico methods. We also performed DFT studies to examine their electronic properties. These findings collectively indicate that the active compounds hold substantial promise as prospective contenders for the development of novel antitubercular agents.
Introduction
Tuberculosis (TB), an infectious disease caused by Mycobacterium tuberculosis, persists as a formidable global health challenge. With millions of new cases and deaths recorded annually, it endures as a predominant source of morbidity and mortality worldwide.1 The advent of drug-resistant variants, notably multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB), has intensified the exigency for the formulation of groundbreaking and more efficacious antitubercular agents.1 The current treatment regimens for TB are often lengthy, costly, and associated with adverse side effects, underscoring the critical need for innovative therapies.2 Over the preceding decades, notable advancements have transpired in the exploration of novel antimicrobial agents characterized by heightened efficacy and diminished toxicity. In this context, hybrid compounds have emerged as a compelling strategy to address the growing threat of drug-resistant TB.3,4 Hybrid compounds are designed by combining distinct pharmacophores in a single molecule, leveraging the unique properties of each component to enhance biological activity. Pyrazine and 1,2,4-triazole analogues, well-known for their diverse pharmacological properties, have garnered substantial attention as potential building blocks for such hybrid compounds.5,6Pyrazinamide serves as a primary pharmacotherapeutic agent in the treatment of tuberculosis, commonly employed in conjunction with other anti-TB medications. Its robust anti-TB efficacy plays a pivotal role in abbreviating the duration of tuberculosis therapy.7 Various modified versions of pyrazinamide have been explored as effective antitubercular agents. For instance, Reddyrajula et al. investigated bioisosteric modifications of pyrazinamide derivatives, resulting in the development of potent antitubercular compounds.8 Srinivasarao and team have focused on N-(6-(4-(pyrazine-2-carbonyl)piperazine/homopiperazine-1-yl)pyridin-3-yl)benzamide derivatives as antitubercular agents.9 Additionally, Panda et al. reported pyrazolopyridones as a novel class of noncovalent DprE1 inhibitors with strong anti-mycobacterial activity.10 Zhou and his team have reported pyrazine-2-carboxamide derivatives that possess antitubercular properties.11 Kumar et al. reported a series of pyrazine analogues as promising inhibitors of DprE1 for the treatment of tuberculosis.12 In parallel, 1,2,4-triazoles, a subclass of five-membered heterocyclic compounds, have a well-established history of pharmacological importance. In recent research, Karczmarzyk and team have explored derivatives of 1,2,4-triazoles combined with pyridine for their antitubercular activity.13 Oh et al. reported a series of 1,2,4-triazole derivatives with antitubercular properties.14 Karabanovich et al. reported a series of 3,5-dinitrophenyl-containing 1,2,4-triazoles and their trifluoromethyl analogues as potent inhibitors of DprE1 for tuberculosis treatment.15 Combining these two structural elements within a single hybrid molecule offers a unique opportunity to leverage their combined strengths, thereby potentially culminating in the conception of more potent and versatile antitubercular pharmaceutical agents. In light of the information provided, this study aims to illustrate the synthesis of an array of novel hybrid molecules by linking the pyrazine ring with a biologically active 1,2,4-triazole moiety to assess the effectiveness of these compounds in combating tuberculosis, evaluate their safety through cytotoxicity testing, investigate their physicochemical and pharmacokinetic properties, and explicate the plausible mechanism of action.
Materials and methods
The required chemical reagents were procured from diverse commercial suppliers, including Sigma Aldrich, TCI, and Alfa-Aesar. To monitor the progress of the chemical reaction, we employed TLC with alumina plates coated with silica gel (Merck 60 F254) as the stationary phase and a mobile phase comprising a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 amalgamation of ethyl acetate and petroleum ether. Subsequently, we scrutinized the resultant spots under a UV chamber. For the determination of the melting points of the synthesized compounds, we utilized a digital melting point apparatus without any adjustments. Furthermore, for in-depth structural analysis, we conducted spectroscopic analysis on the synthesized compounds, encompassing proton nuclear magnetic resonance (1H-NMR) and carbon nuclear magnetic resonance (13C-NMR) spectroscopy. These spectroscopic assessments were carried out using a Bruker Avance Fourier transform-NMR (FT-NMR) spectrometer operating at 400 MHz for 1H-NMR and 100 MHz for 13C-NMR. CDCl3 or DMSO-d6 was employed as a solvent, with tetramethyl silane (TMS) as an internal standard. Chemical shifts are articulated in parts per million on the δ-scale, while coupling constants are presented in Hertz (Hz). NMR spectral analysis was conducted utilizing Bruker NMR software (TopSpin 4.1.4). Furthermore, we acquired mass spectra of the synthesized compounds employing a Waters Xevo QTOF MS system equipped with an electrospray ionization (ESI) source.
:7 amalgamation of ethyl acetate and petroleum ether. Subsequently, we scrutinized the resultant spots under a UV chamber. For the determination of the melting points of the synthesized compounds, we utilized a digital melting point apparatus without any adjustments. Furthermore, for in-depth structural analysis, we conducted spectroscopic analysis on the synthesized compounds, encompassing proton nuclear magnetic resonance (1H-NMR) and carbon nuclear magnetic resonance (13C-NMR) spectroscopy. These spectroscopic assessments were carried out using a Bruker Avance Fourier transform-NMR (FT-NMR) spectrometer operating at 400 MHz for 1H-NMR and 100 MHz for 13C-NMR. CDCl3 or DMSO-d6 was employed as a solvent, with tetramethyl silane (TMS) as an internal standard. Chemical shifts are articulated in parts per million on the δ-scale, while coupling constants are presented in Hertz (Hz). NMR spectral analysis was conducted utilizing Bruker NMR software (TopSpin 4.1.4). Furthermore, we acquired mass spectra of the synthesized compounds employing a Waters Xevo QTOF MS system equipped with an electrospray ionization (ESI) source.
Chemistry
Procedure for the synthesis of (Z)-pyrazine-2-carbohydrazonamide (2). In a clean and dry 100 mL round bottom flask, pyrazine-2-carbonitrile (1) (10 g, 95.14 mmol) was combined with anhydrous methanol (50 mL). To this mixture, hydrazine hydrate (8.33 mL, 166.50 mmol) was introduced. The reaction mixture was then stirred at room temperature for 24 hours. The progression of the reaction was scrutinized using thin-layer chromatography. Following the reaction, the obtained solid product was isolated by filtration, washed with cold methanol, and subsequently dried. The purification process involved recrystallization in methanol, resulting in the formation of yellow crystals.16 Yield: 12.83 g, 98%; m.p.: 127–128 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 9.24 (s, 1H), 8.48 (d, J = 2.36 Hz, 1H), 8.40 (d, J = 1.28 Hz, 1H), 5.10 (s, 2H), 4.70 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 146.60, 146.44, 144.11, 142.81, 142.40; ESI-MS (m/z) = 106.03 [M + H]+.
Procedure for the synthesis of 5-(pyrazin-2-yl)-4H-1,2,4-triazole-3-thiol (3). Pyrazine-2-carbohydrazonide (2) (10 g, 72 mmol) and KOH (4.5 g, 80 mmol) were taken in anhydrous methanol (100 mL) in a clean 250 mL round bottom flask. Carbon disulfide (4.40 g, 72 mmol) was introduced into the reaction mixture and refluxed for 24 hours at 65 °C. The progress of the reaction was scrutinized using TLC. The reaction mixture was then cooled to room temperature and excess solvent was removed using a rotorvap. The obtained residue was poured into crushed ice and made acidic (pH = 6) using 20% HCl, and the obtained solid was filtered and dried. Recrystallization was performed using methanol.17 Pale yellow solid, yield: 92%, m.p.: 176–177 °C, 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 14.1692 (s, 1H), 13.9546 (s, 1H), 9.1809 (s, 1H), 8.7571 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm):167.9086, 148.3865, 145.8394, 144.3842, 142.3343, 140.5931, 40.1253, 39.9179, 39.7089, 39.5005, 39.2919, 39.0836, 38.8748; ESI-MS (m/z) = 180.03 [M + H]+.
General procedure for the synthesis of 1-phenyl-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one derivatives (T1–T9). A mixture of 5-(pyrazin-2-yl)-4H-1,2,4-triazole-3-thiol (3) (1.0 mmol) and sodium hydroxide (1.1 mmol) was taken in aqueous methanol (80%) (10 mL) in a clean 50 mL round bottom flask and the reaction mixture was stirred for 10 minutes at room temperature. Then substituted phenacyl bromide (1.0 mmol) was added and the reaction mixture was stirred at room temperature for 4 hours. The progression of the reaction was scrutinized using thin-layer chromatography. The reaction mixture was then poured into ice-cold water. The precipitated solid was filtered, washed with ice-cold water, and dried. Recrystallization was performed using methanol.18
1-Phenyl-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T1). White solid, yield: 86%; m.p.: 193–194 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.7680 (s, 1H), 9.4716 (s, 1H), 8.6173 (s, 1H), 8.5612 (s, 1H), 8.0357 (s, 2H), 7.5983 (s, 1H), 7.4901 (d, J = 5.68 Hz, 2H), 5.0396 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 191.91, 166.40, 163.37, 146.63, 144.76, 144.07, 139.41, 134.92, 134.49, 129.15, 128.69, 41.97; ESI-MS (m/z) = 298.11 [M + H]+.
2-((5-(Pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)-1-(p-tolyl)ethan-1-one (T2). White solid, yield: 88%; m.p.: 190–191 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.9680 (s, 1H), 9.4716 (s, 1H), 8.6260 (d, J = 2.16 Hz, 1H), 8.5711 (s, 1H), 7.9356 (d, J = 8.16 Hz, 2H), 7.2870 (d, J = 8.08 Hz, 2H), 5.0317 (s, 2H), 2.4134 (s, 3H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 191.53, 166.57, 163.36, 146.63, 145.66, 144.77, 144.09, 139.45, 132.45, 129.84, 128.83, 42.07, 21.99; ESI-MS (m/z) = 312.09 [M + H]+.
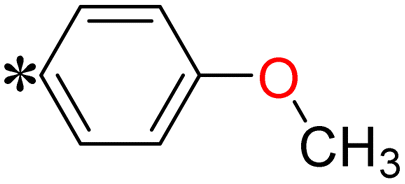
1-(4-Methoxyphenyl)-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T3). White solid, yield: 93%; m.p.: 198–199 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.7783 (s, 1H), 9.4574 (s, 1H), 8.6143 (d, J = 2.44 Hz, 1H), 8.5622 (d, J = 1.44 Hz, 1H), 8.0076 (d, J = 8.8 Hz, 2H), 6.9419 (d, J = 8.84, 2H), 5.0007 (s, 2H), 3.8567 (s, 3H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 190.39, 166.70, 164.64, 163.35, 146.64, 144.79, 144.10, 139.47, 131.15, 127.95, 114.35, 55.81, 41.94; ESI-MS (m/z) = 328.13 [M + H]+.
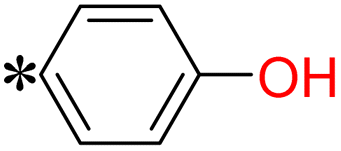
1-(4-Hydroxyphenyl)-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T4). White solid, yield: 87%; m.p.: 250–251 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.9889 (s, 1H), 10.5739 (s, 1H), 9.3073 (s, 1H), 8.8451 (t, J = 5.80 Hz, 2H), 7.9481 (d, J = 8.52 Hz, 2H), 6.9016 (d, J = 8.52 Hz, 2H), 5.1359 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 190.28, 165.39, 162.90, 162.87, 146.95, 145.02, 143.43, 138.69, 131.21, 126.43, 115.48; ESI-MS (m/z) = 314.06 [M + H]+.
1-(4-Nitrophenyl)-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T5). White solid, yield: 81%; m.p.: 229–230 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.6630 (s, 1H), 9.3847 (d, J = 1.20 Hz, 1H), 8.8110 (d, J = 2.48 Hz, 1H), 8.7697 (d, J = 1.52 Hz, 1H), 8.2163 (d, J = 8.68 Hz, 2H), 7.7926 (d, J = 8.64 Hz, 2H), 4.8030 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 191.83, 164.80, 162.96, 150.21, 146.90, 144.92, 143.35, 139.54, 138.53, 129.85, 123.88; ESI-MS (m/z) = 343.08 [M + H]+.
4-(2-((5-(Pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)acetyl)benzonitrile (T6). White solid, yield: 85%; m.p.: 215–216 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.7140 (s, 1H), 9.3132 (d, J = 1.32 Hz, 1H), 8.8724 (d, J = 2.48 Hz, 1H), 8.8494 (t, J = 1.48 Hz, 1H), 8.4048 (d, J = 8.84 Hz, 2H), 8.3055 (d, J = 8.84 Hz, 2H), 5.2958 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 190.28, 165.39, 162.90, 162.87, 146.95, 145.02, 143.43, 138.69, 131.21, 126.43, 121.44, 115.48; ESI-MS (m/z) = 323.10 [M + H]+.
1-(4-Fluorophenyl)-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T7). White solid, yield: 92%; m.p.: 202–203 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.9680 (s, 1H), 9.4573 (s, 1H), 8.6234 (t, J = 1.56 Hz, 2H), 8.0875 (d, J = 5.56 Hz, 1H), 8.0679 (d, J = 5.56 Hz, 1H), 7.2400 (s, 1H), 7.1642 (t, J = 8.28 Hz, 1H), 5.0019 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 190.42, 167.87, 166.30, 165.32, 163.46, 146.70, 144.79, 144.11, 139.40, 131.57, 131.47, 116.56, 116.34, 41.69; ESI-MS (m/z) = 316.09 [M + H]+.
1-(4-Chlorophenyl)-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T8). White solid, yield: 96%; m.p.: 197–198 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.9686 (s, 1H), 9.4573 (s, 1H), 8.6234 (t, J = 1.56 Hz, 2H), 8.0875 (d, J = 5.56 Hz, 1H), 8.0679 (d, J = 5.56 Hz, 1H), 7.2400 (s, 1H), 7.1642 (t, J = 8.28 Hz, 1H), 5.0019 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 190.84, 166.21, 163.48, 146.70, 144.79, 144.11, 141.14, 139.38, 130.11, 129.55, 41.64; ESI-MS (m/z) = 332.04 [M + H]+.
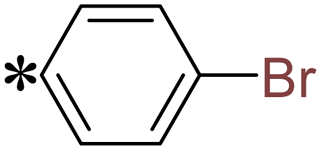
1-(4-Bromophenyl)-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T9). White solid, yield: 94%; m.p.: 203–204 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.8630 (s, 1H), 9.4631 (s, 1H), 8.6313 (s, 1H), 8.5737 (s, 1H), 7.9071 (d, J = 7.00 Hz, 2H), 7.6406 (d, J = 7.08 Hz, 2H), 4.9843 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 191.07, 166.21, 163.50, 146.72, 144.80, 144.13, 139.40, 133.73, 132.57, 130.18, 129.97, 41.61; ESI-MS (m/z) = 375.99 [M + H]+.
General procedure for the synthesis of 2-(5-(benzylthio)-4H-1,2,4-triazol-3-yl)pyrazine derivatives (T10–T18). A mixture of 5-(pyrazin-2-yl)-4H-1,2,4-triazole-3-thiol (3) (1.0 mmol) and potassium carbonate (1.0 mmol) was taken in acetone (10 mL) in a clean 50 mL round bottom flask and the reaction mixture was stirred for 10 minutes at room temperature. Then, substituted benzyl bromide (1.0 mmol) was introduced and the reaction mixture was stirred at room temperature for 2 hours. The progression of the reaction was scrutinized using thin-layer chromatography. The reaction mixture was then poured into ice-cold water. The precipitated solid was filtered, washed with ice-cold water, and dried. Recrystallization was performed using methanol.18
2-(5-(Benzylthio)-4H-1,2,4-triazol-3-yl)pyrazine (T10). Brown solid, yield: 96%; m.p.: 198–199 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.7482 (s, 1H), 9.5144 (d, J = 1.00 Hz, 1H), 8.6320 (d, J = 2.44 Hz, 1H), 8.5723 (d, J = 1.44 Hz, 1H), 7.4541 (d, J = 7.16 Hz, 2H), 7.3169 (m, J = 6.88 Hz, 3H), 4.6244 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 168.19, 162.49, 160.02, 145.89, 144.90, 144.38, 142.46, 131.67, 131.63, 130.20, 130.11, 124.49, 31.46; ESI-MS (m/z) = 270.08 [M + H]+.
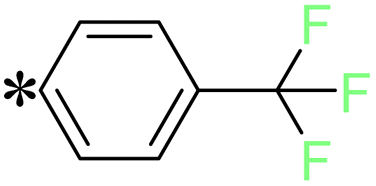
2-(5-((4-(Trifluoromethyl)benzyl)thio)-4H-1,2,4-triazol-3-yl)pyrazine (T11). White solid, yield: 92%; m.p.: 154–155 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.9783 (s, 1H), 9.3917 (d, J = 1.36 Hz, 1H), 8.8125 (d, J = 2.56 Hz, 1H), 8.7720 (t, J = 1.52 Hz, 1H), 7.7360 (s, 2H), 7.6738 (s, 2H), 4.7617 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 168.19, 162.49, 160.02, 145.89, 144.90, 144.36, 142.46, 131.66, 131.63, 130.20, 130.11, 115.63; ESI-MS (m/z) = 338.11 [M + H]+.
2-(5-((4-Fluorobenzyl)thio)-4H-1,2,4-triazol-3-yl)pyrazine (T12). White solid, yield: 86%; m.p.: 150–151 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.8980 (s, 1H), 9.4857 (d, J = 1.12 Hz, 1H), 8.6161 (d, J = 2.44 Hz, 1H), 8.5516 (t, J = 1.52 Hz, 1H), 7.4241 (d, J = 5.40 Hz, 1H), 7.4030 (d, J = 5.36 Hz, 1H), 7.0115 (s, 1H), 6.9791 (d, J = 8.60 Hz, 1H), 4.5731 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 167.88, 163.78, 161.34, 145.90, 144.85, 144.37, 142.45, 131.74, 131.71, 131.17, 131.09, 115.78, 37.44; ESI-MS (m/z) = 288.08 [M + H]+.
2-(5-((2-Fluorobenzyl)thio)-4H-1,2,4-triazol-3-yl)pyrazinepyrazine (T13). Yellow solid, yield: 98%; m.p.: 130–131 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.9637 (s, 1H), 9.5007 (d, 1H), 8.6205 (d, J = 2.52 Hz, 1H), 8.5582 (t, J = 1.56 Hz, 1H), 7.5256 (m, J = 1.56 Hz, 1H), 7.2660 (m, J = 2.04 Hz, 1H), 7.0876 (m, J = 7.60 Hz, 2H), 4.6542 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 168.19, 162.49, 160.02, 145.89, 144.90, 144.38, 142.46, 131.67, 131.63, 130.20, 130.11, 115.73, 31.46; ESI-MS (m/z) = 288.10 [M + H]+.
4-(((5-(Pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)benzonitrile (T14). White solid, yield: 89%; m.p.: 209–210 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.9220 (s, 1H), 9.3871 (d, 1H), 8.8122 (d, J = 2.52 Hz, 1H), 8.7701 (t, J = 1.56 Hz, 1H), 7.8314 (d, J = 8.28 Hz, 2H), 7.7139 (d, J = 8.24 Hz, 2H), 4.7464 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 167.39, 163.89, 159.34, 146.58, 144.95, 142.62, 141.39, 132.49, 130.13, 123.49, 120.13, 110.37; ESI-MS (m/z) = 295.07 [M + H]+.
2-(5-((4-Bromobenzyl)thio)-4H-1,2,4-triazol-3-yl)pyrazine (T15). Brown solid, yield: 92%; m.p.: 155–156 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.9880 (s, 1H), 8.8150 (d, J = 2.52 Hz, 1H), 8.7779 (d, J = 1.52 Hz, 1H), 7.5579 (d, J = 8.40 Hz, 2H), 7.4731 (d, J = 8.44 Hz, 2H), 4.6444 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 168.09, 163.79, 161.34, 146.54, 144.93, 143.72, 141.37, 135.99, 131.50, 131.35, 120.89; ESI-MS (m/z) = 347.98 [M + H]+.
2-(5-((2-Bromobenzyl)thio)-4H-1,2,4-triazol-3-yl)pyrazine (T16). Brown solid, yield: 92%; m.p.: 156–157 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.9889 (s, 1H), 9.5008 (d, J = 1.28 Hz, 1H), 8.6200 (d, J = 2.48 Hz, 1H), 8.5581 (t, J = 1.56 Hz, 1H), 7.6238 (m, J = 6.08 Hz, 1H), 7.5658 (m, J = 7.04 Hz, 1H), 7.2533 (m, J = 6.44 Hz, 1H), 7.1454 (m, J = 6.12 Hz, 1H), 4.7505 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 168.19, 162.49, 160.02, 145.89, 144.90, 144.38, 142.46, 131.67, 131.63, 130.20, 130.11, 115.94, 31.46; ESI-MS (m/z) = 347.97 [M + H]+.
2-(5-((4-Chlorobenzyl)thio)-4H-1,2,4-triazol-3-yl)pyrazine (T17). White solid, yield: 94%; m.p.: 162–163 °C; 1H-NMR (CDCl3, 400 MHz, δ in ppm): 12.9881 (s, 1H), 9.4932 (d, J = 1.20 Hz, 1H), 8.6239 (d, J = 2.48 Hz, 1H), 8.5600 (t, J = 1.60 Hz, 1H), 7.3880 (d, J = 8.40 Hz, 2H), 7.2692 (t, J = 8.40 Hz, 2H), 4.5689 (s, 2H); 13C-NMR (CDCl3, 100 MHz, δ in ppm): 168.09, 163.79, 161.34, 145.90, 144.85, 144.37, 142.45, 131.74, 131.71, 131.17, 131.09, 116.00, 37.44; ESI-MS (m/z) = 304.05 [M + H]+.
2-(5-((4-Nitrobenzyl)thio)-4H-1,2,4-triazol-3-yl)pyrazine (T18). White solid, yield: 97%; m.p.: 194–195 °C; 1H-NMR (DMSO-d6, 400 MHz, δ in ppm): 12.9680 (s, 1H), 9.3846 (d, J = 1.16 Hz, 1H), 8.8109 (d, J = 2.52 Hz, 1H), 8.7696 (d, J = 1.48 Hz, 1H), 8.2163 (d, J = 8.68 Hz, 2H), 7.7925 (d, J = 8.68 Hz, 2H), 4.8030 (s, 2H); 13C-NMR (DMSO-d6, 100 MHz, δ in ppm): 167.83, 167.34, 146.85, 146.57, 144.93, 144.78, 143.67, 141.37, 130.40, 123.67; ESI-MS (m/z) = 315.08 [M + H]+.
Computational studies
Biological studies
:1 ratio was added to the wells. A subsequent 24-hour incubation at 37 °C ensued. Thereafter, a visual examination of the well contents was carried out, with a pink hue signifying bacterial proliferation and a blue tint indicating the suppression of bacterial growth. The minimum inhibitory concentration was delineated as the lowest concentration of a compound required to impede bacterial growth. For comparative analysis, pyrazinamide (PZA), ciprofloxacin (INN), and streptomycin (STM) were employed as benchmark pharmaceuticals.
In vitro antibacterial and antifungal activity (broth microdilution method)
Cytotoxicity studies
The Vero cell line, sourced from the National Center for Cell Sciences (NCCS) in Pune, India, comprises African green monkey kidney cells designated by Catalog number 11965-092. These cellular entities were cultivated in 96-well flat-bottomed microtiter plates utilizing DMEM supplemented with 10% heat-inactivated fetal calf serum (FBS) and 1% antibiotic–antimycotic 100× solution.28 Subsequently, they were housed in an incubator maintained at a temperature of 37 °C, 95% humidity, and 5% CO2 concentration for 24 hours. The cellular entities were then exposed to these distinct drug concentrations and subjected to an additional incubation period of 72 hours. Following this incubation, a thorough washing of cells in each well was executed using a phosphate buffer solution. After this step, a meticulously prepared stock solution of MTT (20 μL, 5 mg mL−1 in sterile phosphate-buffered saline) was instilled into each well, followed by an additional incubation for 4 hours in an environment comprising 5% CO2. Upon removal of the supernatant, 100 μL of DMSO was introduced to dissolve the precipitated crystals. The absorbance levels of the wells housing the cells and the corresponding blanks were quantified at 570 nm using a microplate reader. The determination of the extent of growth inhibition was executed through the application of the following formula: % Growth Inhibition = (mean optical density (OD) of the test compound/mean OD of the negative control) × 100.Results and discussion
Chemistry
Based on the favorable results obtained through in silico studies, we have progressed toward the synthesis of the envisioned compounds by following the synthetic pathways outlined in Scheme 1. The process commenced with the reaction of readily available pyrazine-2-carbonitrile (1) with hydrazine hydrate in the presence of methanol, yielding (Z)-pyrazine-2-carbohydrazonamide (2). Subsequently, compound 2 underwent cyclization when treated with KOH and CS2, resulting in the formation of 5-(pyrazin-2-yl)-4H-1,2,4-triazole-3-thiol (3). Compounds T1–T9 (Table 1) were produced by reacting compound 3 with commercially accessible phenacyl bromides, while compounds T10–T18 (Table 1) were synthesized by reacting compound 3 with commercially available substituted benzyl bromides.| Compound code | R | Compound code | R |
|---|---|---|---|
| The symbol ‘*’ denotes the point of attachment. | |||
| T1, T10 |
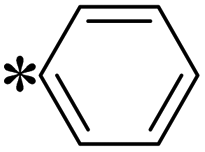
|
T7, T12 |
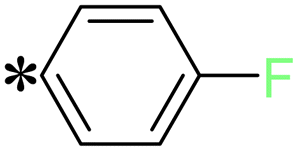
|
| T2 |
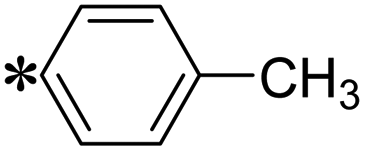
|
T8, T17 |
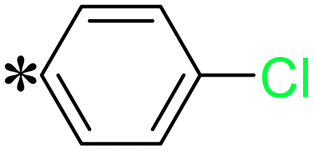
|
| T3 |
|
T9, T15 |
|
| T4 |
|
T11 |
|
| T5, T18 |
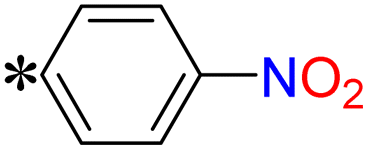
|
T13 |
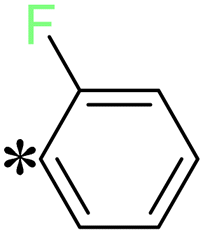
|
| T6, T14 |
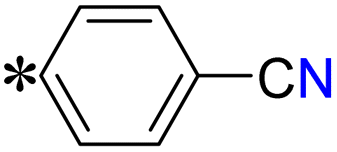
|
T16 |
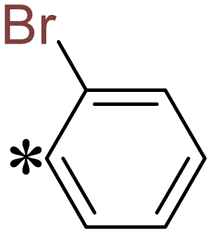
|
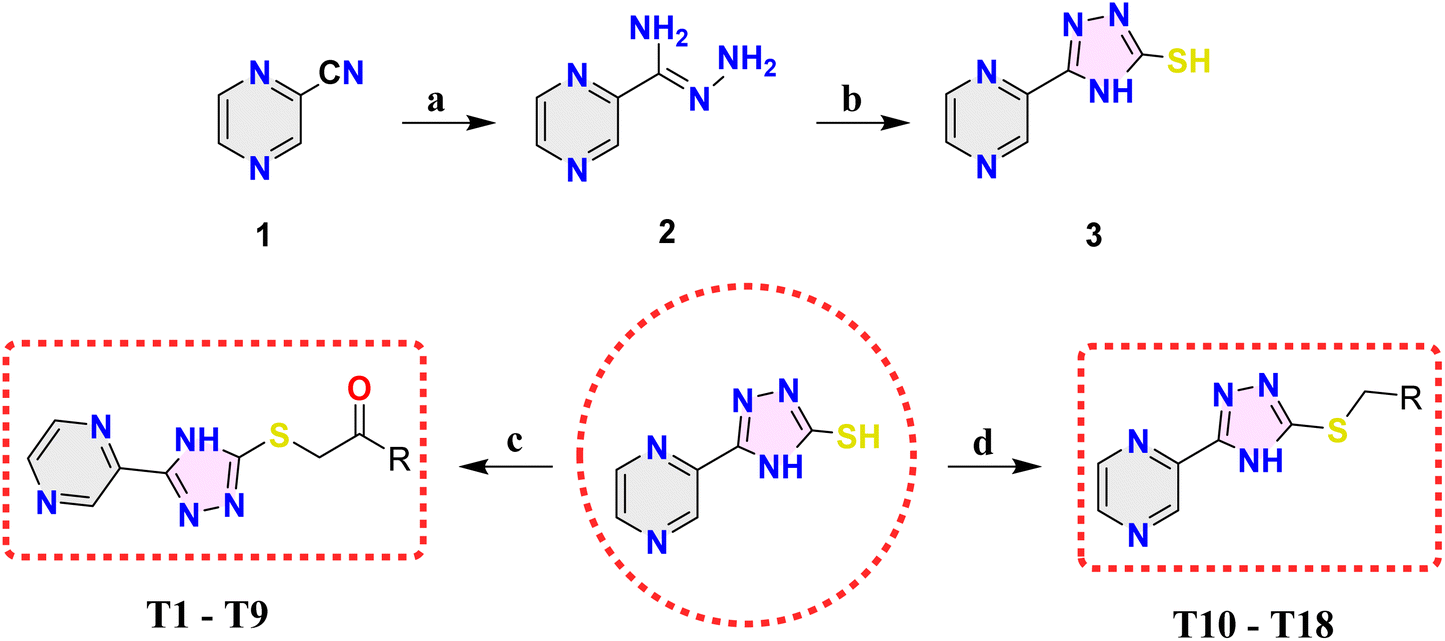 | ||
| Scheme 1 Synthesis protocol for the synthesis of target compounds T1–T18. Reagents and conditions: (a) N2H4·H2O, CH3OH, RT, 24 hours, (b) CS2, KOH, CH3OH, reflux, 24 hours, (c) phenacyl bromide derivatives, 10% NaOH, CH3OH, RT, 6 hours and (d) benzyl bromide derivatives, K2CO3, acetone, reflux, 2 hours. | ||
The validation of the intermediates and target compounds (T1–T18) involved a combination of analytical techniques, including 1H NMR, 13C NMR, and mass spectrometry. The 1H NMR spectrum of compound T4 showed two singlet peaks at δ: 12.98 and 10.57 ppm, corresponding to the –NH proton of 1,2,4-triazole and the –OH proton, respectively. The pyrazine ring's three aromatic protons were observed with two singlet peaks at δ: 9.30 and 8.84 ppm and one doublet peak at δ: 8.85 ppm. Additionally, two doublet peaks at δ: 7.95 and 6.90 ppm indicated the presence of four aromatic protons from the phenyl ring. Finally, a sharp singlet peak at δ: 5.13 ppm was attributed to the –CH2 protons. The 13C NMR spectrum of compound T4 showed characteristic peaks corresponding to its molecular structure. The peaks at δ: 190.28 and 165.39 ppm were assigned to the carbonyl carbon and the carbon attached to the hydroxy group respectively. The two carbons of the 1,2,4-triazole were denoted by peaks at δ 162.90 and 162.87 ppm. The four carbons of the pyrazine ring were represented by peaks at δ: 146.95, 145.02, 143.43, and 138.69 ppm. Five carbons from the phenyl ring were identified by three peaks at δ: 131.21, 126.43, and 115.48 ppm. Furthermore, a peak at δ: 38.88 ppm confirmed the presence of –CH2 carbon. The molecular mass of compound T4 was unambiguously confirmed through the mass spectrum, revealing a molecular ion peak (M + H peak) observed at (m/z) 314.06. Similarly, the 1H NMR spectrum of compound T11 presented a singlet peak at δ: 12.97 ppm, signifying the –NH proton of the 1,2,4-triazole. The three aromatic protons within the pyrazine ring manifested as two doublet peaks at δ: 9.39 and 8.81 ppm, along with one triplet peak at δ: 8.77 ppm. In the region of δ: 7.73–7.67 ppm four aromatic protons originating from the phenyl ring were detected. Additionally, a distinct sharp singlet peak at δ: 4.76 ppm represents the presence of –CH2 protons. In the 13C NMR spectrum of compound T11, characteristic peaks aligned with its molecular structure. Peaks at δ: 168.19 and 162.49 ppm were assigned to two carbons of 1,2,4-triazole. The four carbons of the pyrazine ring were represented by peaks at δ: 160.02, 145.89, 144.90, and 144.36 ppm. Furthermore, the phenyl ring contributed five peaks in the range of δ: 142.46 to 130.11 ppm, reflecting the presence of six carbon atoms. Peaks at δ: 115.63 and 36.59 ppm were attributed to the –CF3 and –CH2 carbons, respectively. The molecular mass of compound T11 was conclusively confirmed through mass spectrometry, revealing the presence of the molecular ion peak (M + H peak) at (m/z) 338.18, thus validating its molecular weight.
Computational studies
logS) (ranging from 54.31 to 1128.37 nm s−1 and −5.25 to −3.38, respectively) further enhance the potential for efficient absorption in the intestines. Significantly, the blood–brain partition coefficient (QPlogBB) values are within an acceptable range (−2.02 to −0.24), indicating that these compounds can cross the blood–brain barrier. Furthermore, the human serum albumin binding coefficient (QPlogKhsa) falls comfortably within the desired range (ranging from −0.50 to 0.20), underscoring the potential of these compounds to effectively bind to human serum albumin, a critical factor in how drugs are distributed in the body. Notably, a majority of these compounds demonstrate a human oral absorption rate exceeding 85%, which is a strong indicator of their outstanding oral bioavailability. In summary, the comprehensive in silico ADME predictions provide strong support for advancing these compounds as promising candidates for further development in drug discovery.
| Comp. | MW (≤500 Da) | HBD (≤5) | HBA (≤10) | QPlogP (o/w) (≤5) |
QPlogS (≤0.5) |
nRB (0–15) | PSA (≤140 Å) | QPPCaco (<25 nm s−1 is low; >500 nm s−1 is high) | QPlogKhsa (−1.5 to 1.5) |
QPlogBB (−3.0 to 1.2) |
%OA (>80% is high: <25% is low) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp.: compound, MW: molecular weight, HBD: number of hydrogen bond donors, HBA: number of hydrogen bond acceptors, QPlogP (o/w): logarithm of the partition coefficient between n-octanol and water, QPlogS: aqua solubility parameter, nRB: number of rotatable bonds, PSA: polar surface area, QPPCaco: caco-2 cell permeability, QPlogKhsa: human serum albumin binding co-efficient, QPlogBB: blood/brain partition co-efficient, and %OA: percentage of oral absorption. |
|||||||||||
| T1 | 297.33 | 1 | 6.5 | 1.84 | −3.49 | 4 | 87.14 | 454.69 | −0.32 | −0.95 | 85.31 |
| T2 | 311.36 | 1 | 6.5 | 2.10 | −4.03 | 4 | 87.16 | 454.22 | −0.17 | −0.99 | 86.83 |
| T3 | 327.36 | 1 | 7.25 | 1.95 | −3.73 | 5 | 95.37 | 453.58 | −0.32 | −1.05 | 85.90 |
| T4 | 313.33 | 2 | 7.25 | 1.21 | −3.38 | 5 | 109.73 | 137.55 | −0.40 | −1.57 | 72.31 |
| T5 | 342.33 | 1 | 7.5 | 1.13 | −3.60 | 5 | 131.86 | 54.32 | −0.39 | −2.03 | 64.61 |
| T6 | 322.34 | 1 | 8 | 1.08 | −4.41 | 5 | 112.94 | 96.13 | −0.51 | −1.78 | 68.78 |
| T7 | 315.32 | 1 | 6.5 | 2.02 | −3.78 | 4 | 87.15 | 454.64 | −0.28 | −0.85 | 86.31 |
| T8 | 331.78 | 1 | 6.5 | 2.35 | −4.23 | 4 | 87.14 | 454.89 | −0.22 | −0.81 | 88.27 |
| T9 | 376.23 | 1 | 6.5 | 2.42 | −4.33 | 4 | 87.14 | 454.87 | −0.20 | −0.80 | 88.70 |
| T10 | 269.32 | 1 | 4.5 | 2.61 | −3.85 | 3 | 60.78 | 1109.17 | −0.05 | −0.48 | 96.73 |
| T11 | 337.32 | 1 | 4.5 | 3.58 | −5.26 | 3 | 60.78 | 1109.04 | 0.20 | −0.24 | 100 |
| T12 | 287.31 | 1 | 4.5 | 2.83 | −4.19 | 3 | 60.78 | 1109.26 | −0.01 | −0.38 | 100 |
| T13 | 287.31 | 1 | 4.5 | 2.78 | −4.08 | 3 | 60.77 | 1109.81 | −0.02 | −0.40 | 100 |
| T14 | 294.33 | 1 | 6 | 1.83 | −4.78 | 4 | 86.58 | 234.64 | −0.22 | −1.29 | 80.10 |
| T15 | 348.22 | 1 | 4.5 | 3.17 | −4.68 | 3 | 60.78 | 1109.31 | 0.09 | −0.32 | 100 |
| T16 | 348.22 | 1 | 4.5 | 3.08 | −4.48 | 3 | 60.70 | 1128.37 | 0.06 | −0.33 | 100 |
| T17 | 303.77 | 1 | 4.5 | 3.09 | −4.57 | 3 | 60.78 | 1109.50 | 0.06 | −0.33 | 100 |
| T18 | 314.32 | 1 | 5.5 | 1.88 | −3.99 | 4 | 105.46 | 132.99 | −0.10 | −1.54 | 75.99 |
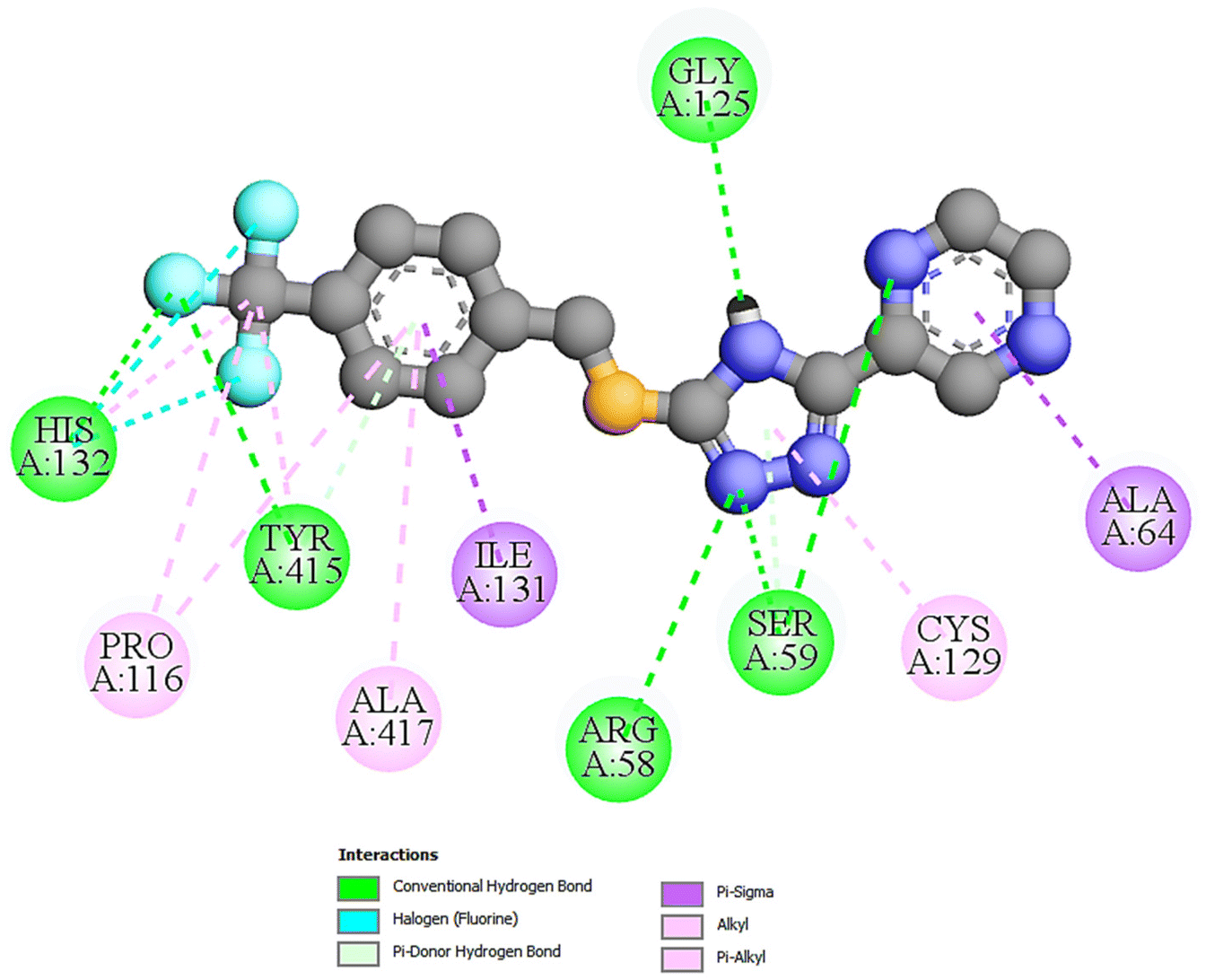 | ||
| Fig. 1 The 2D docking poses of compound T11 with receptor 4P8N. | ||
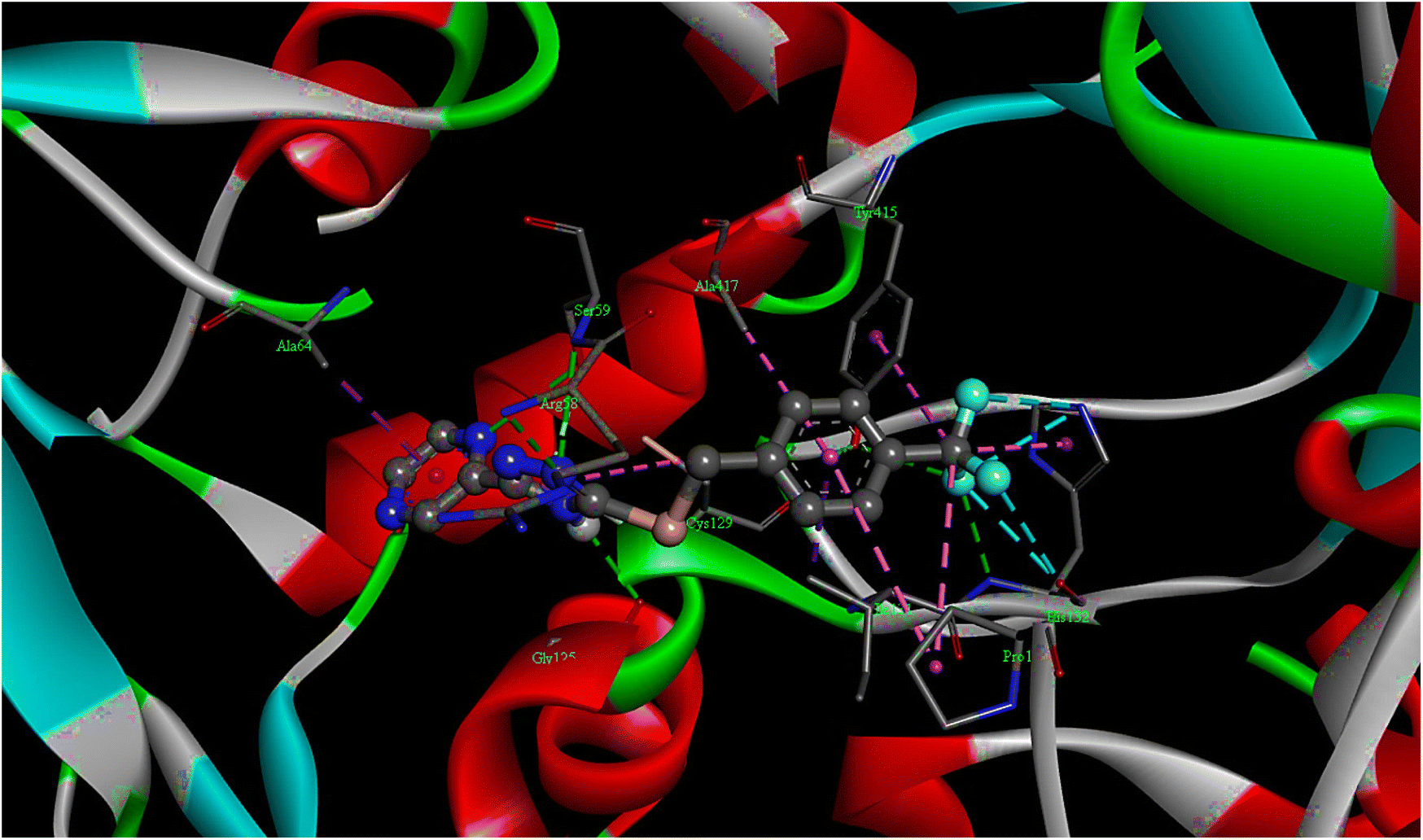 | ||
| Fig. 2 The 3D docking poses of compound T11 with receptor 4P8N. | ||
| Comp. | Binding energy (kcal mol−1) | Inhibition constant (Ki) (nM) | No. of H-bonds | Interacting amino acid residues |
|---|---|---|---|---|
| Comp.: compound, PZA: pyrazinamide, INN: ciprofloxacin, STM: streptomycin. | ||||
| T1 | −8.3 | 393.27 | 03 | GLY57, ARG58, SER59, CYS129 |
| T2 | −9.4 | 125.14 | 04 | GLY55, GLY57, ARG58, SER59, CYS129 |
| T3 | −9.1 | 156.21 | 02 | ARG58, SER59, TYR60, ALA64 |
| T4 | −9.4 | 125.38 | 03 | ALA53, ARG58, SER59, GLY125 |
| T5 | −8.8 | 277.76 | 04 | SER59, GLY117, THR118, LYS134, GLN336 |
| T6 | −9.7 | 79.26 | 04 | ARG58, SER59, CYS129, ASN63 |
| T7 | −9.3 | 149.61 | 03 | ARG58, SER59, CYS129, ASN63 |
| T8 | −8.8 | 277.93 | 02 | ALA53, SER59, GLY124, GLY125 |
| T9 | −8.9 | 281.47 | 02 | ALA53, SER59, GLY124, GLY125, VAL121 |
| T10 | −9.0 | 243.35 | 02 | SER59, ALA64, VAL121, GLY125, CYS129 |
| T11 | −10.1 | 39.47 | 05 | ARG58, SER59, GLY125, HIS132, TYR415 |
| T12 | −9.2 | 151.87 | 02 | SER59, ALA64, GLY125, CYS129 |
| T13 | −9.3 | 149.82 | 02 | SER59, ALA64, GLY125, CYS129 |
| T14 | −9.7 | 74.65 | 04 | SER59, ALA64, GLY117, GLY125, CYS129 |
| T15 | −9.3 | 149.47 | 05 | SER59, GLY117, THR118, LYS134, TYR415 |
| T16 | −9.1 | 156.07 | 02 | SER59, ALA64, GLY125, CYS129, LYS418 |
| T17 | −9.1 | 156.03 | 02 | SER59, ALA64, GLY125, CYS129 |
| T18 | −9.8 | 69.08 | 04 | ARG58, SER59, ALA64, HIS132, TYR415 |
| PZA | −5.3 | 21.74 μM | 04 | GLY57, ARG58, SER59, GLY125, CYS129 |
| INN | −8.2 | 17.93 μM | 04 | ALA53, ARG54, THR122, GLY125, ILE184 |
| STM | −9.1 | 201.09 | 06 | GLY117, HIS132, SER228, GLN336, TYR415 |
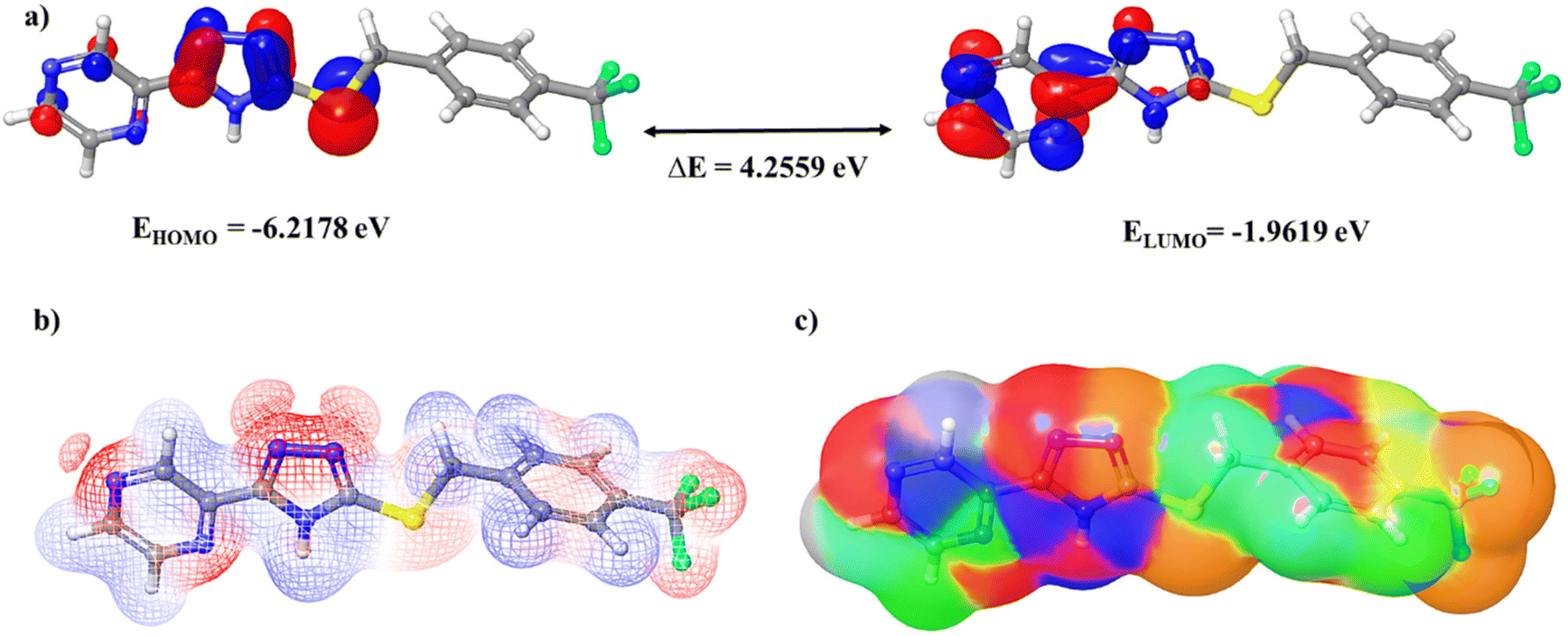 | ||
| Fig. 3 (a) Frontier molecular orbitals, (b) molecular electrostatic potentials, and (c) the electron density surface of compound T11. | ||
| Comp. | E HOMO (eV) | E LUMO (eV) | ΔE (eV) | IP (eV) | EA (eV) | η (eV) | σ (eV) | μ (eV) | ω (eV) | χ (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| Comp.: compound, bandgap (ΔE) = EHOMO − ELUMO, ionization potential (IP) = −EHOMO, electron affinity (EA) = −ELUMO, chemical hardness (η) = (IA − EA)/2, chemical softness (σ) = 1/2η, chemical potential (μ) = −η, electrophilicity index (ω) = η/2, and electronegativity (χ) = (IP + EA)/2. | ||||||||||
| T1 | −6.0954 | −1.8858 | 4.2096 | 6.0954 | 1.8858 | 2.1048 | 0.2376 | −2.1048 | 1.0524 | 3.9906 |
| T2 | −6.0437 | −1.8531 | 4.1906 | 6.0437 | 1.8531 | 2.0953 | 0.2386 | −2.0953 | 1.0476 | 3.9484 |
| T3 | −6.0518 | −1.7905 | 4.2613 | 6.0518 | 1.7905 | 2.1307 | 0.2347 | −2.1307 | 1.0653 | 3.9212 |
| T4 | −6.0300 | −1.8449 | 4.1851 | 6.0300 | 1.8449 | 2.0926 | 0.2389 | −2.0926 | 1.0463 | 3.9375 |
| T5 | −6.3212 | −3.1266 | 3.1946 | 6.3212 | 3.1266 | 1.5973 | 0.3130 | −1.5973 | 0.7987 | 4.7239 |
| T6 | −6.3049 | −2.6694 | 3.6354 | 6.3049 | 2.6694 | 1.8177 | 0.2751 | −1.8177 | 0.9089 | 4.4872 |
| T7 | −6.1362 | −1.9184 | 4.2178 | 6.1362 | 1.9184 | 2.1089 | 0.2371 | −2.1089 | 1.0544 | 4.0273 |
| T8 | −6.1743 | −2.0871 | 4.0872 | 6.1743 | 2.0871 | 2.0436 | 0.2447 | −2.0436 | 1.0218 | 4.1307 |
| T9 | −6.1824 | −2.1034 | 4.0790 | 6.1824 | 2.1034 | 2.0395 | 0.2452 | −2.0395 | 1.0197 | 4.1429 |
| T10 | −6.0437 | −1.8585 | 4.1851 | 6.0437 | 1.8585 | 2.0926 | 0.2389 | −2.0926 | 1.0463 | 3.9511 |
| T11 | −6.2178 | −1.9619 | 4.2559 | 6.2178 | 1.9619 | 2.1279 | 0.2350 | −2.1279 | 1.0640 | 4.0899 |
| T12 | −6.1144 | −1.8994 | 4.2150 | 6.1144 | 1.8994 | 2.1075 | 0.2372 | −2.1075 | 1.0538 | 4.0069 |
| T13 | −6.0573 | −1.8558 | 4.2014 | 6.0573 | 1.8558 | 2.1007 | 0.2380 | −2.1007 | 1.0504 | 3.9565 |
| T14 | −6.3130 | −2.0218 | 4.2912 | 6.3130 | 2.0218 | 2.1456 | 0.2330 | −2.1456 | 1.0728 | 4.1674 |
| T15 | −6.1688 | −1.9347 | 4.2341 | 6.1688 | 1.9347 | 2.1170 | 0.2362 | −2.1170 | 1.0585 | 4.0518 |
| T16 | −6.0709 | −1.8613 | 4.2096 | 6.0709 | 1.8613 | 2.1048 | 0.2376 | −2.1048 | 1.0524 | 3.9661 |
| T17 | −6.1661 | −1.9293 | 4.2368 | 6.1661 | 1.9293 | 2.1184 | 0.2360 | −2.1184 | 1.0592 | 4.0477 |
| T18 | −6.3457 | −2.6096 | 3.7361 | 6.3457 | 2.6096 | 1.8681 | 0.2677 | −1.8681 | 0.9340 | 4.4776 |
| PZA | −6.7838 | −1.9347 | 4.8491 | 6.7838 | 1.9347 | 2.4246 | 0.2062 | −2.4246 | 1.2123 | 4.3592 |
| INN | −5.7007 | −1.2027 | 4.4980 | 5.7007 | 1.2027 | 2.2490 | 0.2223 | −2.2490 | 1.1245 | 3.4517 |
Comprehending overarching chemical reactivity descriptors on a global scale is imperative for establishing intricate correlations among the structural attributes, stability, and reactivity of a molecule.30 These descriptors are ascertained through the meticulous evaluation of the HOMO and LUMO energy levels of the molecules, with precise numerical values delineated in Table 4.31 An elevated chemical softness parameter signifies an augmented proclivity for binding with the receptor, whereas an increased chemical hardness value implies a diminished inclination for binding. Significantly, all scrutinized compounds manifest heightened chemical softness and diminished chemical hardness in contrast to the reference pharmaceutical agents. With the augmentation of the chemical potential, the discernibility of interaction between the ligand and the receptor intensifies, and notably, all target compounds manifest elevated chemical potential values in comparison to the benchmark pharmaceuticals. The electrophilicity index and electronegativity serve as metrics gauging a molecule's capacity to attract electrons from its ambient milieu. Molecules endowed with a heightened electrophilicity index and electronegativity values evince an augmented predisposition for forging interactions with receptors. It is of significance that all target compounds exhibit electronegativity and electrophilicity index values akin to those inherent in the reference pharmaceuticals. Fig. 3c elucidates the electron density surface of compound T11. The ionization potential (IP) and electron affinity (EA) serve as quantitative measures for appraising a molecule's proclivity to either gain or relinquish electrons within its proximate milieu. In the general context, diminished values of IP and EA are correlated with an increased proclivity toward toxicity. It merits emphasis that all investigated compounds align within the stipulated IP and EA value range characteristic of the FDA-approved pharmaceuticals, with EA values spanning the range from −3 to 7 eV and IP values encompassing the interval of 4–15 eV.
To evaluate the reactivity of these compounds, we undertook molecular electrostatic potential (MEP) computations, a pivotal methodology for prognosticating the interaction modalities of these target compounds based on their physicochemical attributes and comprehending their predisposition toward electrophilic and nucleophilic reactions.32 Through the employment of DFT, we ascertained the MEP profiles of the specified compounds. In Fig. 3b, the MEPs of compound T11 are delineated in detail, with the MEP surface meticulously color-coded for optimal elucidation. The blue domains in the figure delineate regions characterized by a positive electrostatic potential, while the red domains signify areas distinguished by a negative electrostatic potential. The white regions denote a neutral electrostatic potential. Across the entirety of these molecules, regions characterized by a negative electrostatic potential are chiefly congregated in the proximity of nitrogen, sulfur, and fluorine atoms. Conversely, zones exhibiting a positive electrostatic potential are predominantly situated in the vicinity of hydrogen atoms and an intricately structured phenyl ring.
Biological studies
| Comp. | M. tuberculosis H37Rv | Comp. | M. tuberculosis H37Rv | ||
|---|---|---|---|---|---|
| MIC (μg mL−1) | MIC (μM) | MIC (μg mL−1) | MIC (μM) | ||
| Comp.: compound, PZA: pyrazinamide, STM: streptomycin, INN: ciprofloxacin. | |||||
| T1 | 12.5 | 42.07 | T12 | 12.5 | 43.54 |
| T2 | 12.5 | 40.18 | T13 | 12.5 | 43.54 |
| T3 | 12.5 | 38.21 | T14 | 6.25 | 21.25 |
| T4 | 6.25 | 19.96 | T15 | 6.25 | 18.01 |
| T5 | 6.25 | 18.27 | T16 | 6.25 | 18.01 |
| T6 | 6.25 | 19.40 | T17 | 12.5 | 41.25 |
| T7 | 12.5 | 39.67 | T18 | 6.25 | 19.90 |
| T8 | 12.5 | 37.76 | PZA | 3.12 | 25.34 |
| T9 | 12.5 | 33.33 | STM | 6.25 | 10.74 |
| T10 | 12.5 | 46.45 | INN | 3.12 | 9.41 |
| T11 | 3.12 | 9.25 | |||
| Comp. | MIC (mg mL−1) | |||
|---|---|---|---|---|
| S. aureus | S. mutans | E. coli | S. typhi | |
| Comp.: compound, STM: streptomycin, CHL: chloramphenicol, control: DMSO. | ||||
| T1 | 2.5 | >5.0 | 5.0 | >5.0 |
| T2 | 2.5 | >5.0 | 5.0 | >5.0 |
| T3 | 5.0 | 5.0 | 5.0 | >5.0 |
| T4 | 2.5 | 1.25 | 0.625 | >5.0 |
| T5 | 1.25 | >5.0 | >5.0 | >5.0 |
| T6 | 5.0 | >5.0 | >5.0 | >5.0 |
| T7 | 2.5 | >5.0 | 5.0 | >5.0 |
| T8 | 5.0 | 5.0 | 5.0 | >5.0 |
| T9 | 0.01953 | 1.25 | >5.0 | 1.25 |
| T10 | 0.625 | 2.5 | 5.0 | >5.0 |
| T11 | 5.0 | 5.0 | 1.26 | 5.0 |
| T12 | 2.5 | 5.0 | 5.0 | 5.0 |
| T13 | 2.5 | 5.0 | 5.0 | 5.0 |
| T14 | 2.5 | >5.0 | >5.0 | >5.0 |
| T15 | 2.5 | 5.0 | 2.5 | >5.0 |
| T16 | 0.01953 | 0.01953 | 0.625 | 2.5 |
| T17 | 2.5 | >5.0 | >5.0 | 5.0 |
| T18 | 0.625 | >5.0 | 1.25 | 5.0 |
| STM | 0.012 | 0.010 | 0.010 | 0.011 |
| CHL | 0.010 | 0.010 | 0.012 | 0.010 |
| Control | 00 | 00 | 00 | 00 |
| Comp. | MIC (mg mL−1) | Comp. | MIC (mg mL−1) |
|---|---|---|---|
| A. niger | A. niger | ||
| Comp.: compound, AmB: amphotericin B, control: DMSO. | |||
| T1 | 2.5 | T11 | 1.25 |
| T2 | 5.0 | T12 | 0.625 |
| T3 | 5.0 | T13 | 5.0 |
| T4 | 5.0 | T14 | 0.625 |
| T5 | 5.0 | T15 | 1.25 |
| T6 | 5.0 | T16 | 2.5 |
| T7 | 5.0 | T17 | 5.0 |
| T8 | 2.5 | T18 | 5.0 |
| T9 | 5.0 | AmB | 0.004 |
| T10 | 5.0 | Control | 00 |
| Compound | % of inhibition | |||
|---|---|---|---|---|
| 25 μM | 50 μM | 75 μM | 100 μM | |
| The values represent the mean ± standard error of the mean obtained from three separate determinations after 72 hours of exposure. | ||||
| T4 | 3.21 ± 0.34 | 6.46 ± 0.21 | 9.61 ± 0.18 | 12.92 ± 0.26 |
| T5 | 3.05 ± 0.23 | 6.38 ± 0.17 | 9.53 ± 0.15 | 12.87 ± 0.19 |
| T6 | 3.14 ± 0.27 | 6.35 ± 0.32 | 9.89 ± 0.23 | 13.05 ± 0.23 |
| T11 | 2.97 ± 0.15 | 6.03 ± 0.22 | 9.08 ± 0.27 | 12.10 ± 0.14 |
| T14 | 3.02 ± 0.18 | 6.24 ± 0.31 | 9.63 ± 0.20 | 13.09 ± 0.17 |
| T15 | 2.99 ± 0.31 | 6.12 ± 0.19 | 9.74 ± 0.38 | 12.64 ± 0.27 |
| T16 | 3.04 ± 0.25 | 6.16 ± 0.27 | 9.37 ± 0.16 | 12.35 ± 0.22 |
| T18 | 3.17 ± 0.23 | 6.38 ± 0.22 | 9.71 ± 0.21 | 12.59 ± 0.25 |
| Compound | IC50 (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| The IC50 values represent the concentration of a substance required to inhibit 50% of cell viability calculated after a 72-hour exposure. | |||
| T4 | 387.53 ± 10.15 | T14 | 379.86 ± 11.47 |
| T5 | 387.29 ± 12.03 | T15 | 389.47 ± 10.73 |
| T6 | 379.88 ± 10.49 | T16 | 402.72 ± 13.48 |
| T11 | 412.32 ± 11.63 | T18 | 393.80 ± 12.44 |
Conclusions
This study entailed the synthesis of a new series of 1-phenyl-2-((5-(pyrazin-2-yl)-4H-1,2,4-triazol-3-yl)thio)ethan-1-one (T1–T9) and 2-(5-(benzylthio)-4H-1,2,4-triazol-3-yl)pyrazine (T10–T18) derivatives. The synthesized compounds were characterized using various analytical techniques. The compounds underwent in vitro testing to assess their potential as antimicrobial agents against diverse bacteria, fungi, and the tuberculosis-inducing M. tuberculosis H37Rv strain. Among the tested compounds, eight exhibited notable activity against tuberculosis, with a MIC ≤ 6.25 μg mL−1. Particularly noteworthy was compound T11, showcasing the most potent activity with a MIC of 3.12 μg mL−1. To ascertain their suitability for pharmaceutical development, cytotoxicity evaluations were conducted, revealing that these active compounds did not induce harm to normal cells. Additionally, computational analysis was employed to scrutinize the binding interactions between these compounds and the active site of the DprE1 enzyme through in silico molecular docking. Although these studies indicated strong binding interactions, further experimental validation is necessary to confirm the mechanism of inhibition action. DFT studies provided insights into the electronic properties of these compounds, indicating a narrower HOMO–LUMO energy gap, augmented chemical softness, diminished chemical hardness, lower electron affinity, and higher chemical potential in comparison with reference compounds. These findings collectively imply heightened reactivity and more robust interactions with the target receptor. Beyond their antitubercular attributes, compounds T9, T10, and T18 manifested significant antibacterial activity against S. aureus, whereas compound T16 exhibited antibacterial activity against S. aureus, S. mutans, and E. coli. Compounds T12 and T14 evinced substantial antifungal activity against A. niger. This exhaustive study emphasizes the importance of further delving into the pharmacological characteristics of these compounds, inclusive of in vivo investigations, to comprehensively fathom their potential in combating infectious diseases.Author contributions
Shivakumar: conceptualization, methodology, computational studies, data curation, visualization, biological studies, and writing – original draft. P Dinesha: data curation and visualization. Vanishree A. L.: data curation and visualization. Udayakumar D.: supervision, validation, and writing – review and editing. Varsha Prakash Shetty: bacterial and fungal activity. Vijaya Kumar Deekshit: bacterial and fungal activity.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the National Institute of Technology Karnataka (NITK), Surathkal, India for providing all the necessary infrastructure and resources to carry out this research work and CRF, NITK for the instrumentation facilities.References
- Global tuberculosis report 2023, World Health Organization, 2023, ISBN 978-92-4-008385-1.
- A. Gupta, S. Juneja, S. Sahu, M. Yassin, G. Brigden, E. Wandwalo, S. Rane, F. Mirzayev and M. Zignol, PLOS Glob. Public Health, 2022, 2, e0001287, DOI:10.1371/journal.pgph.0001287.
- V. Singh and K. Chibale, Acc. Chem. Res., 2021, 54, 2361–2376, DOI:10.1021/acs.accounts.0c00878.
- G. Stelitano, J. C. Sammartino and L. R. Chiarelli, Molecules, 2020, 25, 1239, DOI:10.3390/molecules25051239.
- P. Miniyar, P. Murumkar, P. Patil, M. Barmade and K. Bothara, Mini-Rev. Med. Chem., 2013, 13, 1607–1625, DOI:10.2174/1389557511313110007.
- V. Rangaswamy and U. Laddi, Antiinfect. Agents, 2023, 21, e111023222113, DOI:10.2174/0122113525268198230921071016.
- Y. Zhang, W. Shi, W. Zhang and D. Mitchison, Microbiol. Spectr., 2014, 2 DOI:10.1128/microbiolspec.mgm2-0023-2013.
- R. Reddyrajula and U. Dalimba, Bioorg. Med. Chem. Lett., 2020, 30, 126846, DOI:10.1016/j.bmcl.2019.126846.
- S. Srinivasarao, A. Nandikolla, A. Suresh, K. Van Calster, L. De Voogt, D. Cappoen, B. Ghosh, H. Aggarwal, S. Murugesan and K. V. G. Chandra Sekhar, RSC Adv., 2020, 10, 12272–12288, 10.1039/d0ra01348j.
- M. Panda, S. Ramachandran, V. Ramachandran, P. S. Shirude, V. Humnabadkar, K. Nagalapur, S. Sharma, P. Kaur, S. Guptha, A. Narayan, J. Mahadevaswamy, A. Ambady, N. Hegde, S. S. Rudrapatna, V. P. Hosagrahara, V. K. Sambandamurthy and A. Raichurkar, J. Med. Chem., 2014, 57, 4761–4771, DOI:10.1021/jm5002937.
- S. Zhou, S. Yang and G. Huang, J. Enzyme Inhib. Med. Chem., 2017, 32, 1183–1186, DOI:10.1080/14756366.2017.1367774.
- S. Kumar, P. Dinesha, D. Udayakumar, V. P. Shetty and V. K. Deekshit, J. Mol. Struct., 2024, 1304, 137657, DOI:10.1016/j.molstruc.2024.137657.
- Z. Karczmarzyk, M. Swatko-Ossor, W. Wysocki, M. Drozd, G. Ginalska, A. Pachuta-Stec and M. Pitucha, Molecules, 2020, 25, 6033, DOI:10.3390/MOLECULES25246033.
- T. Oh, A. Hayat, E. Yoo, S. N. Cho, Y. Y. Sheen, D. K. Kim and H. Y. P. Choo, Bull. Korean Chem. Soc., 2015, 36, 43–51, DOI:10.1002/bkcs.10009.
- G. Karabanovich, J. Dušek, K. Savková, O. Pavliš, I. Pávková, J. Korábečný, T. Kučera, H. Kočová Vlčková, S. Huszár, Z. Konyariková, K. Konečná, O. Jand'Ourek, J. Stolaříková, J. Korduláková, K. Vávrová, P. Pávek, V. Klimešová, A. Hrabálek, K. Mikušová and J. Roh, J. Med. Chem., 2019, 62, 8115–8139, DOI:10.1021/acs.jmedchem.9b00912.
- F. E. Bradford, L. P. Connor, C. A. Kilner and M. A. Halcrow, Polyhedron, 2004, 23, 2141–2151, DOI:10.1016/j.poly.2004.06.018.
- S. R. Patil, A. Asrondkar, V. Patil, J. N. Sangshetti, F. A. Kalam Khan, M. G. Damale, R. H. Patil, A. S. Bobade and D. B. Shinde, Bioorg. Med. Chem. Lett., 2017, 27, 3845–3850, DOI:10.1016/j.bmcl.2017.06.053.
- L. R. Zhang, Z. J. Liu, H. Zhang, J. Sun, Y. Luo, T. T. Zhao, H. Bin Gong and H. L. Zhu, Bioorg. Med. Chem., 2012, 20, 3615–3621, DOI:10.1016/j.bmc.2012.03.061.
- J. Vrbanac and R. Slauter, in A Comprehensive Guide to Toxicology in Nonclinical Drug Development, Elsevier, 2016, pp. 39–67. DOI:10.1016/B978-0-12-803620-4.00003-7.
- H. Beard, A. Cholleti, D. Pearlman, W. Sherman and K. A. Loving, PLoS One, 2013, 8, e82849, DOI:10.1371/journal.pone.0082849.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898, DOI:10.1021/acs.jcim.1c00203.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455–461, DOI:10.1002/jcc.21334.
- K. R. Cousins, J. Am. Chem. Soc., 2005, 127, 4115–4116, DOI:10.1002/jcc.21334.
- A. Fiser, R. Kinh, G. Do and A. S. Ali, Modeling of loops in protein structures, 2000, https://www.rcsb.org0pdb Search PubMed.
- J. Yang, A. Roy and Y. Zhang, Bioinformatics, 2013, 29, 2588–2595, DOI:10.1093/bioinformatics/btt447.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306, DOI:10.1021/ct700248k.
- S. G. Franzblau, R. S. Witzig, J. C. Mclaughlin, P. Torres, G. Madico, A. Hernandez, M. T. Degnan, M. B. Cook, V. K. Quenzer, R. M. Ferguson and R. H. Gilman, J. Clin. Microbiol., 1998, 36, 362–366, DOI:10.1128/jcm.36.2.362-366.1998.
- M. R. Espíndola, F. de P. Varotti, A. C. C. Aguiar, S. N. Andrade and E. M. M. da Rocha, Braz. J. Pharm. Sci., 2022, 58, e18308, DOI:10.1590/s2175-97902022e18308.
- J. Gawad and C. Bonde, Chem. Cent. J., 2018, 12, 72, DOI:10.1186/s13065-018-0441-2.
- A. Bendjeddou, T. Abbaz, A. Gouasmia and D. Villemin, Acta Chim. Pharm. Indica, 2016, 6, 32–44 CAS.
- P. S. Sen Gupta, H. R. Bhat, S. Biswal and M. K. Rana, J. Mol. Liq., 2020, 320, 114375, DOI:10.1016/j.molliq.2020.114375.
- M. G. Srinivasa, Shivakumar, D. U. Kumar, C. H. Mehta, U. Y. Nayak and B. C. Revanasiddappa, J. Comput. Biophys. Chem., 2024, 23, 117–136, DOI:10.1142/S2737416523500540.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3pm00054k |
| This journal is © The Royal Society of Chemistry 2024 |