
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion recognition using meta-substituted ureidocalix[4]arene receptors†
A.
Surina
a,
J.
Čejka
 b,
K.
Salvadori
cd and
P.
Lhoták
*a
b,
K.
Salvadori
cd and
P.
Lhoták
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague (UCTP), Technicka 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz; Fax: +420-220444288; Tel: +420-220445055
bDepartment of Solid State Chemistry, UCTP, 166 28 Prague 6, Czech Republic
cInstitute of Chemical Process Fundamentals of Czech Academy of Sciences v.v.i., Rozvojová 135, Prague 6, 16502, Czech Republic
dJ. Heyrovský Institute of Physical Chemistry of Czech Academy of Sciences v.v.i., Dolejškova 2155/3, 182 23 Prague 8, Czech Republic
First published on 2nd October 2024
Abstract
Calix[4]arenes bearing urea units at the meta position(s) of the upper rim of the macrocyclic skeleton were prepared by the reaction of the corresponding amines with aryl isocyanates. As shown by the 1H NMR and UV/vis titration experiments, these systems are capable of effectively complexing selected anions even in a highly competitive environment (such as DMSO-d6). While the monoureido derivatives showed approximately the same complexation ability irrespective of the substitution (para vs. meta isomers), the bisureas at the upper rim demonstrated interesting differences in complexation. The meta,meta and para,para isomers were shown to prefer 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes (anion:receptor) regardless of the anion tested, while the analogous meta,para isomer formed 1:1 complexes with strongly coordinated anions (e.g. H2PO4−) based on synchronous complexation by both ureido groups. This suggests that the regioselective introduction of urea units into the upper rim of calix[4]arene brings with it the possibility of “tuning” the complexation properties depending on the substitution pattern of the functional groups.
:1 complexes (anion:receptor) regardless of the anion tested, while the analogous meta,para isomer formed 1:1 complexes with strongly coordinated anions (e.g. H2PO4−) based on synchronous complexation by both ureido groups. This suggests that the regioselective introduction of urea units into the upper rim of calix[4]arene brings with it the possibility of “tuning” the complexation properties depending on the substitution pattern of the functional groups.
Introduction
Recognition of anions, their targeted complexation and transport have become one of the cornerstones of modern supramolecular chemistry.1 Selective complexation of anions is a topic with enormous application potential, ranging from the removal of toxic anions from the environment, through materials chemistry and catalysis to targeted complexation/decomplexation of anionic species in medicine. It is therefore not surprising that the research and development of new receptors for anions is becoming increasingly important, as evidenced by the huge number of recent articles and reviews2 dealing with this topic.Anion complexation uses many different basic strategies and approaches, but generally all receptors can be divided into charged and neutral systems. The charged ones usually rely on electrostatic interactions, which, however, are in principle not very directional. These positively charged molecules can be exemplified by quaternary ammonium cages, protonated or alkylated azacrowns, cryptands and azamacrocycles, amidinium- and guanidinium-based receptors, etc.2,3
The opposite of these substances are neutral receptors, based on highly directional interactions, such as hydrogen bonds (HBs) or halogen bonding.2d Examples include receptors using amides/thioamides,2f,4 sulfonamides, ureas/thioureas,5 pyrroles,6 triazoles2a or molecules with acidic CH bonds (e.g. bambusurils).7
The effectiveness of these highly directional interactions, particularly HBs, can be further enhanced by the thoughtful design of receptors, using several preorganized functional groups. Although there are a number of such molecules, a completely irreplaceable role is played by calix[n]arene derivatives, especially systems based on calix[4]arene.8 The possibility of tuning their 3D structures by simple alkylations of the lower rim of the macrocycle (phenolic hydroxyls) makes these molecules the preferred choice in the design and synthesis of new receptors.9 The existence of four basic conformations/atropisomers (cone, partial cone, 1,2-alternate, and 1,3-alternate) together with well-established derivatization procedures makes calix[4]arene an ideal molecular scaffold for the design of more complex supramolecular systems, including anion receptors.10
Anion receptors based on ureido/thioureido-calixarenes are very popular because these compounds are synthetically readily available. In particular, calix[4]arene-based receptors immobilised in the cone and 1,3-alternate conformations have been documented many times in the literature11 and some of these systems show interesting complexation properties towards selected anions. All receptors described so far have one common feature – they use para-substituted phenolic subunits of calixarenes, which is related to the methods of their preparation and the general reactivity of calixarenes. Only recently have direct procedures12 for the meta substitution of the upper rim of calix[4]arene, allowing the introduction of various functional groups, been described.13
In this context, we realized that no anion receptors bearing urea groups at the meta position of the calixarene skeleton have been prepared so far (Fig. 1). It would therefore be interesting to find out whether this type of substitution exhibits the same behaviour as the already described para-isomers or whether the meta isomers lead to some new properties. In this paper, we report the first preparation of meta-ureido calix[4]arenes and the study of their complexation abilities towards selected anions in comparison with their para-substituted analogues.
 | ||
| Fig. 1 Ureido receptors based on calix[4]arene: para- vs. meta-regioisomers. | ||
Results and discussion
The synthesis of the corresponding meta-substituted aminocalix[4]arene 2c was accomplished according to published procedures. Thus, direct monomercuration12a (1 equiv. Hg(TFA)2 in CHCl3) of the starting tetrapropoxycalixarene 1 immobilized in the cone conformation and the subsequent reaction with aq. HCl provided 2a in 67% yield. The subsequent transformation (isopentyl nitrite/HCl) of this organomercurial intermediate into nitroso derivative 2b and the final reduction with RANEY® nickel and hydrazine afforded amine 2c in a good overall yield14 (Scheme 1). Target receptors were obtained by the reaction of 2c with appropriate commercially available aromatic isocyanates (p-X-C6H4-NCS, X = NO2, n-Bu, OMe, CF3) in dichloromethane at room temperature. Purification by preparative thin layer chromatography on silica gel provided 3a–d in 33–60% isolated yields. | ||
| Scheme 1 Synthesis of meta substituted ureidocalix[4]arenes: (i) (1) Hg(TFA)2/CHCl3, (2) HCl; (ii) i-amyl nitrite/HCl; (iii) NH2NH2·H2O/Ni(R); and (iv) CH2Cl2, RT. | ||
The structures of receptors 3a–d were confirmed by the combination of HRMS and NMR techniques. Thus, the HRMS ESI+ of 3a showed a peak at m/z = 973.4101, which is in good agreement with the [M + Na]+ (973.4106) ion predicted for the product. The multiplicity and splitting pattern of signals in the 1H NMR spectrum (400 MHz, DMSO-d6, 298 K) are fully consistent with the expected C1 symmetry. Thus, the presence of four different signals for the terminal methyl groups (0.89, 0.90, 1.08 and 1.09 ppm) refers to the inherently chiral structure of the entire system. Also, the presence of two sets of doublets (2 × 4) for the axial and equatorial protons (with typical geminal coupling constants ∼13–15 Hz) of the CH2 bridges is consistent with the predicted structure. The singlets at 9.56 and 8.44 ppm (ureido N–H protons) and two doublets in the aromatic region at 8.19 and 7.70 ppm reflect the presence of the p-nitrophenylureido moiety within the molecule.
The structure of receptor 3a was further confirmed by single crystal X-ray studies. Compound 3a crystallized in the triclinic system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; the unit cell contained two molecules of MeOH (crystallization solvent) that formed the 3a·2MeOH complex. As can be seen in Fig. 2a, the molecule adopts a typical pinched cone conformation, where two aromatic subunits are slightly tilted towards the interior of the cavity, while the other two opposing units are directed outwards. If we define the main plane of the molecule using the carbon atoms of the CH2 bridging groups, the individual interplanar angles Φ are 125.75, 77.52, 125.63 and 82.54° starting clockwise from the subunit bearing the ureido group (see Fig. 2a). The urea function is located on the phenolic subunit pointing out of the cavity (Φ = 125.75), and the planar urea group itself is sharply twisted from the plane of the phenol at an angle of 67.47°.
; the unit cell contained two molecules of MeOH (crystallization solvent) that formed the 3a·2MeOH complex. As can be seen in Fig. 2a, the molecule adopts a typical pinched cone conformation, where two aromatic subunits are slightly tilted towards the interior of the cavity, while the other two opposing units are directed outwards. If we define the main plane of the molecule using the carbon atoms of the CH2 bridging groups, the individual interplanar angles Φ are 125.75, 77.52, 125.63 and 82.54° starting clockwise from the subunit bearing the ureido group (see Fig. 2a). The urea function is located on the phenolic subunit pointing out of the cavity (Φ = 125.75), and the planar urea group itself is sharply twisted from the plane of the phenol at an angle of 67.47°.
 | ||
| Fig. 2 X-ray structure of 3a: (a) top view into the cavity with the corresponding interplanar angles and (b) hydrogen bonding motif of ureido functions. | ||
An interesting binding motif was found within the crystal packing of substance 3a (Fig. 2b). Two methanol molecules form a dimer by short hydrogen bonding (O–H⋯O distance = 2.101 Å) outside the calixarene cavity. The urea unit is connected by HB interactions of NH bonds to the first methanol; the N–H⋯O distances are 2.182 and 2.150 Å. At the same time, the second methanol molecule is attached to another urea through HB interaction with the carbonyl (O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C distance is 2.057 Å) and by the interaction between the ortho CH bond of the adjacent nitro-substituted aromatic unit and the CH bond from the methanol (Car–H⋯H–C = 2.373 Å). The result is an endless belt of interconnected ureidocalixarenes, which have the same stereochemistry (identical enantiomers). Since the inherently chiral compound 3a crystallizes as a racemic mixture, the same arrangement of the opposite enantiomer is found within the crystal (50:50 ratio). Finally, the opposite homochiral bands point towards each other through the ureido functions and are interconnected by the π–π interactions between the nitrophenyl units.
C distance is 2.057 Å) and by the interaction between the ortho CH bond of the adjacent nitro-substituted aromatic unit and the CH bond from the methanol (Car–H⋯H–C = 2.373 Å). The result is an endless belt of interconnected ureidocalixarenes, which have the same stereochemistry (identical enantiomers). Since the inherently chiral compound 3a crystallizes as a racemic mixture, the same arrangement of the opposite enantiomer is found within the crystal (50:50 ratio). Finally, the opposite homochiral bands point towards each other through the ureido functions and are interconnected by the π–π interactions between the nitrophenyl units.
After evaluating interactions in the solid state, the properties of the systems in solution were subsequently monitored. Therefore, the ureido derivatives were studied by dilution experiments in order to confirm or disprove the possible self-aggregation behaviour of receptors.15 Here, the insufficient solubility did not allow us to reach concentrations higher than 10 mM even in DMSO (e.g. compound 3a did not dissolve in concentrations exceeding 5 mM). This observation was general for all mono-meta-substituted receptors. Therefore, the complexation abilities of receptors 3a–d towards selected anions were studied using standard 1H NMR and/or UV/vis titration experiments in a HB competitive environment. The anion solution was gradually added to the solution of 3a in DMSO-d6 to obtain various receptor:anion ratios (specific for each measurement, see the ESI†). In all cases, upon the addition of the anion, a significant down-field shift of ureido NH signals was observed, indicating complexation under fast exchange conditions (for example, see Fig. 3). When the formation of the host–guest complex was monitored by UV/vis, a redshift of receptor absorbance with a distinctive isosbestic point was observed.
 | ||
| Fig. 3 1H NMR titration of receptor 3c with TBA+ H2PO4− (DMSO-d6, 400 MHz, 298 K), inset: the titration curve for the same system. | ||
The calculation of the corresponding complexation constants was based on the analysis of binding isotherms obtained from the urea NH protons, and/or the parts of the absorption curves where the changes in absorbance were the most significant. The non-linear curve fitting of experimental data was performed using Bindfit software.16 The stoichiometry of the complexes was determined by Job plot analysis17 or it was based on the Bindfit output, where the 1:1 model provided the best fit among all the stoichiometries (1:1, 1:2, and 2:1) tested.18
The complexation properties of receptor 3a are summarized in Table 1, where they are also compared with the corresponding para isomer 4 (obtained as previously described19). The receptors showed no changes in the 1H NMR spectra upon dilution of their DMSO-d6 solutions (see Fig. S37 and S38†), which excludes receptor self-aggregation15 under the measurement conditions. As can be seen, the complexation constants K for benzoate, chloride and hydrogen sulfate anions are almost identical for both receptors, indicating no influence of the regioisomers (meta vs. para). The only distinct difference can be found for acetate and especially for dihydrogen phosphate, where the constant for the para isomer 4 is twice that of the meta analogue 3a (compare KH2PO4(4) = 2020 vs. KH2PO4(3a) = 1060).
As several tested species can be considered as basic anions (H2PO4−, benzoate, acetate), the interaction of 3a with tetrabutylammonium hydroxide was also studied. As shown in Fig. S42,† deprotonation of the NH bond of receptor 3a leads to a dramatic shift of the absorbance maximum, corresponding to a wavelength around 480 nm, which we never observed during the common titration experiments with any of the above-discussed anions (compare the record for acetate in Fig. S42†).
In order to demonstrate the effect of the substitution of the aryl residue on the complexation of the anions, a series of meta substituted receptors were further expanded to include 3b–d and their binding abilities towards the dihydrogen phosphate anion were examined. As shown in Table 2, the strength of complexation is proportional to the electronic effect of the chosen substituent. Thus, the nitro group (the strongest EWG) exhibits the highest complexation constant KH2PO4(3a) = 1060, while the weakest binding was found for the methoxy derivative (EDG) KH2PO4(3d) = 300.
It is interesting that the chemical shift of the urea NH signal of the receptor (Table 2) can be correlated with the strength of the complex – the higher the shift, the higher the complexation constant. By plotting the complexation constants K against the chemical shift δ of the attached hydrogen, we even observed a linear dependence (see Fig. 4). A simple prediction of the binding constant for receptors of the same structural type, based on the chemical shift of urea NH signals, can be advantageous, e.g. in determining the correct choice of titration conditions for a new receptor.
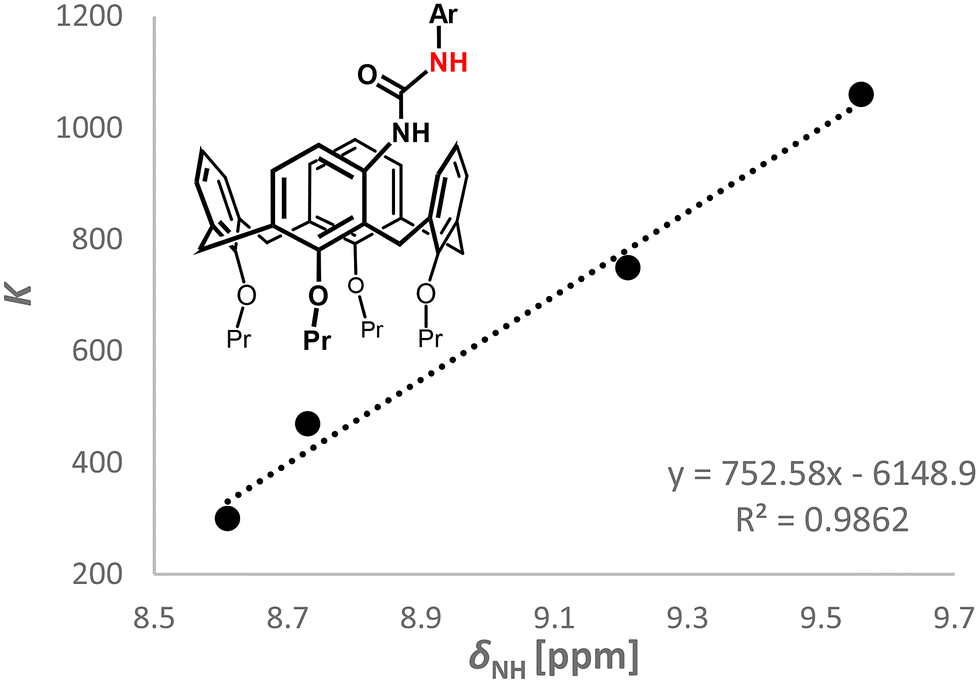 | ||
| Fig. 4 Dependence of the K values on the chemical shifts of NH (compounds 3a–d). | ||
In order to study the effect of system preorganization on the complexation behaviour, we attempted to introduce two urea functionalities into the upper rim of calix[4]arene using an organomercury protocol described by our group.12b Briefly, the starting tetrapropoxycalix[4]arene 1 was reacted with two equivalents of Hg(TFA)2 in chloroform to provide a mixture of two distally dimercurated calixarenes: meta,meta5a and meta,para6a isomers in an approx. 3:2 ratio (Scheme 2). Without any isolation, the crude reaction product was reacted with isoamyl nitrite/aq. HCl to yield a mixture of nitroso derivatives 5b and 6b, which was smoothly separated by column chromatography on silica gel.20 The resulting amines 5c and 6c were obtained by reduction with hydrazine hydrate in the presence of RANEY®-nickel. Since the corresponding nitro derivative 3a showed the highest complexation constants, the reaction with p-nitrophenyl isocyanate was used in both cases. The respective distally substituted diureido isomers 7a and 8a were isolated in 64 and 50% yields, respectively.
 | ||
| Scheme 2 Synthesis of meta substituted diureidocalix[4]arenes 7a and 8a: (i) (1) Hg(TFA)2/CDCl3, (2) HCl; (ii) isoamyl nitrite/HCl; (iii) NH2NH2·H2O/Ni(R); and (iv) CH2Cl2, RT. | ||
The 1H NMR spectra of compounds 7a and 8a (400 MHz, DMSO-d6, 298 K) reflect the expected symmetry of the molecules well. Thus, receptor 7a shows 2 × 2 doublets for the CH2 bridging groups (J ∼ 13 Hz) together with two NH signals (9.59 and 8.54 ppm) of urea groups, suggesting the C2 symmetry of 7a. On the other hand, the splitting pattern and number of signals in the 1H NMR spectrum of 8a (e.g. 2 × 4 doublets for equatorial and axial CH bonds of the bridging CH2 units, four different signals for NH bonds (9.55, 9.36, 8.65 and 8.42 ppm)) are consistent with the predicted C1 symmetry.
Once again, solubility issues play an important role, limiting the range of values of potential binding constants under correct conditions (for the corresponding dilution experiments in DMSO-d6, see Fig. S39–S41†). For receptor 7a, the clear solution was not obtained until the concentration decreased below 2 mM; therefore only strongly interacting anions were studied. The 1H NMR titrations of compound 7a (meta,meta) and the Job plot analysis revealed that 2:1 complexes (anion:calix) are formed for all anions investigated (H2PO4−, BzO− and AcO−). It is therefore obvious that the individual ureido groups function independently and there is no mutual cooperation between them. Thus, each urea grabs its own anionic species, leading to the aforementioned 2:1 stoichiometry. As shown in Table 3, the corresponding binding constants K (1:1) and the overall binding constants β (2:1) were calculated using a non-cooperative model as the most appropriate choice.
:1) and the overall binding constants β (2:1) of receptor 7a towards selected anionsa (1H NMR titration, 400 MHz, DMSO-d6, 298 K)
The corresponding para,para isomer 919 was used for direct comparison of the complexation properties of the receptors. As shown in Table 3, this isomer behaves very similarly to the preceding one. Although dihydrogen phosphate is captured somewhat more strongly, even in this case, a 2:1 complex is formed with independent complexation of both urea moieties.
Finally, the complexation study of receptor 8a (meta,para) yielded unexpected results. With basic anions, this isomer has been shown to form complexes with a 1:1 stoichiometry (Table 4). This suggests that in this case there is a cooperative binding of one anion by both urea units. This leads to the fact that the complexation constant towards dihydrogen phosphate KH2PO4(8a) = 13600 is by far the highest among all measured receptors – essentially an order of magnitude higher than that of the corresponding monoureido receptor 3a (KH2PO4(3a) = 1060). The same trend can be seen for the acetate anion although the effect was not so pronounced. The results for BzO− were somewhat in the middle (between cooperating and independent action of urea's sites). However, as no mathematical tool was able to combine these actions in a single fitting process, we decided to fit the raw data by a simple 1:1 stoichiometry. On the other hand, anions that cannot be captured synchronously from both functional groups (chloride and hydrogen sulphate, Table 4) again clearly form complexes with a 2:1 stoichiometry (Fig. 5).
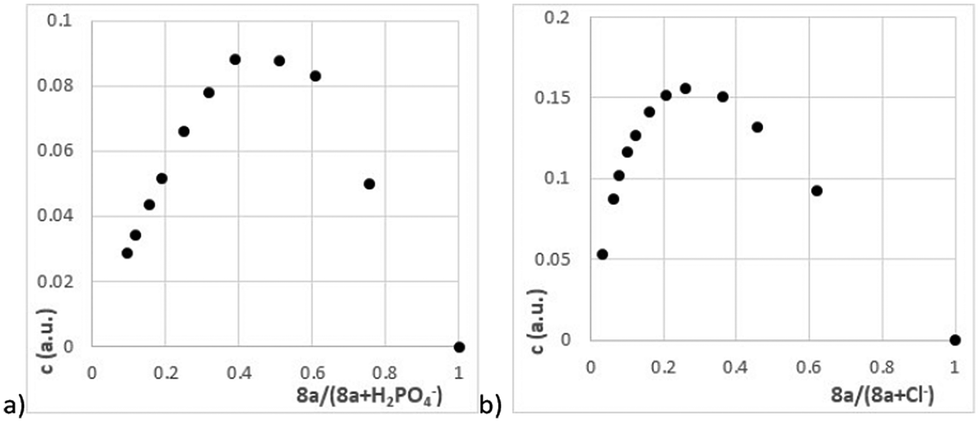 | ||
| Fig. 5 Job plot (1H NMR, 400 MHz, DMSO-d6, 298 K) recorded for receptor 8a (a) with H2PO4− and (b) with Cl− anions. | ||
:1) and the overall binding constants β (2:1) of receptor 8a towards selected anionsa (1H NMR titration, 400 MHz, DMSO-d6, 298 K)
All meta-substituted receptors represent inherently chiral systems that could be useful for chiral anion recognition. To test this hypothesis, we attempted to resolve racemic mixtures of compounds 3a, 7a, and 8a using chiral phase chromatography. Unfortunately, all our efforts did not lead to the goal and it was not possible to separate the urea derivatives into pure enantiomers.
Therefore, we focused on other properties of these receptors. Since our compounds carry the –NO2 group in their structure, we tried to evaluate their electrochemical reduction. The combination of steady state (RDE) and dynamic methods (CV) provided sufficient information about the reduction transformation. Receptors 3a and 4 were shown to be reduced in three well-separated steps (Fig. 6a and S53†), regardless of the substitution (meta vs. para).
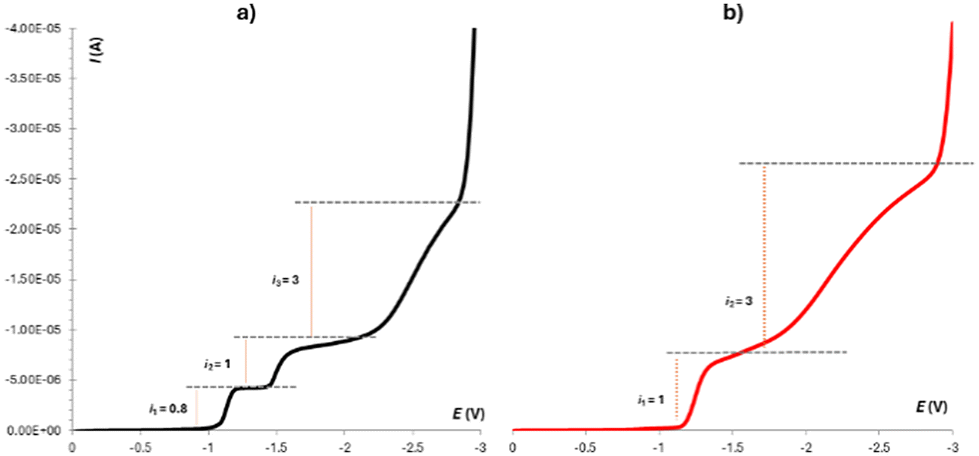 | ||
| Fig. 6 Linear sweep voltammetry of 3a (9 × 10−4 M) in DMSO (0.1 M TBAPF6) (W = RDE = glassy carbon – ∅ 1 mm, Ref = SCE, Aux = Pt) recorded with a scan rate of 10 mV s−1 using a rotating rate of 100 s−1: (a) in the absence of any anion and (b) in the presence of H2PO4−. | ||
This behaviour has been already reported for various compounds bearing acidic hydrogens together with a nitro group within the same structure. The mechanism describing this transformation is known in the literature as the self-protonation mechanism.21 Interestingly, we found out that the urea's hydrogens might be “protected” upon the addition of an anion (H2PO4−), when a strong complex is formed by means of hydrogen bonds from the urea unit (NH). This suppresses the possibility of self-protonation, which results in the common two-step reduction (Fig. 6b).22 This property could therefore be used for electrochemical sensing of phosphates.
Conclusions
In conclusion, calix[4]arenes bearing ureido units at the meta position(s) of the upper rim of the calixarene skeleton were prepared. The 1H NMR and UV/vis titrations proved that these systems are capable of effectively complexing selected anions even in a highly competitive environment (such as DMSO-d6). The monoureido derivatives showed approximately the same complexation ability irrespective of the substitution (para vs. meta isomers). On the other hand, the bisureas at the upper rim demonstrated interesting differences in complexation. While the meta,meta and para,para isomers preferred 2:1 complexes (e.g. with the dihydrogen phosphate anion), a similar meta,para isomer formed a 1:1 complex based on the synchronous complexation of the anion by both ureido groups. This suggests that the regioselective introduction of a urea moiety into the upper rim of calix[4]arene brings with it the possibility of “tuning” the complexation properties depending on the mutual arrangement of the functional groups (substitution pattern).
Experimental
General experimental procedures
All chemicals were obtained from commercial sources and used as received without further purification. Solvents were dried and distilled using conventional methods. TLC was performed on Merck foil sheets with silica gel 60 F254 (Merck). Column chromatography was performed on silica gel 60 with particle size 0.063–0.200 mm (Merck). Preparative thin-layer chromatography was performed on self-prepared glass plates (25 × 25 cm) covered by silica gel 60 GF254 containing CaSO4 (Merck). 1H and 13C NMR spectra were obtained using Agilent 400-MR DDR2, JEOL-ECZL400G (1H: 400 MHz, 13C: 100 MHz) and Bruker Avance DRX 500 (1H: 500 MHz, 13C: 125 MHz) spectrometers at 298 K. Deuterated solvents used are indicated in each case. Chemical shifts are reported as δ values in parts per million (ppm) and were referenced to the residual peak of the solvent or TMS as an internal standard; coupling constants (J) are expressed in Hz. NMR data were processed and displayed using MestReNova and TopSpin software. Melting points were measured on a Heiztisch Mikroskop – Polytherm A (Wagner & Munz, Germany) and are not corrected. The mass spectra analyses were performed on a Q-TOF (Micromass) spectrometer, using ESI ionisation in positive mode. Infrared spectra were measured on an FT–IR spectrometer Nicolet iS50 (Thermo-Nicolet, USA) connected with a heatable Golden Gate Diamante ATR–Unit GladiATR (PIKE, USA) in KBr. 64 Scans for one spectrum were co-added at a spectral resolution of 4 cm−1. The spectra were processed using Omnic 9 (Thermo-Nicolet Instruments Co., USA) with baseline correction.Synthetic procedures
4-{N′-(4-Nitrophenyl)ureido}-25,26,27,28-tetrapropoxycalix[4]arene (cone) (3a). Calixarene 3a was prepared according to the general procedure for ureido derivatives by reacting calixarene 2c (0.141 g, 0.230 mmol) and 4-nitrophenylisocyanate (0.041 g, 0.253 mmol) in 10 mL dry DCM. The product was purified by preparative TLC (eluent acetone
:cyclohexane 10:90) to give the title compound 3a as a yellow solid (0.102 g, 58%), m.p. 143–146 °C. 1H-NMR (500 MHz, DMSO-d6): δ 9.54 (s, 1H, –NH–), 8.43 (s, 1H, –NH–), 8.18 (d, J = 9.10 Hz, 2H, Ar–H), 7.68 (d, J = 9.10 Hz, 2H, Ar–H), 7.20 (d, J = 8.2 Hz, 1H, Ar–H), 7.07 (td, J = 7.0 Hz, J = 1.5 Hz, 2H, Ar–H), 7.06 (d, J = 8.0 Hz, 1H, Ar–H), 6.85 (t, J = 7.5 Hz, 1H, Ar–H), 6.23 (t, J = 7.5 Hz, 1H, Ar–H), 6.18 (t, J = 7.5 Hz, 1H, Ar–H), 6.16 (d, J = 7.5 Hz, 1H, Ar–H), 6.11 (dd, J = 7.5 Hz, J = 1 Hz, 1H, Ar–H), 6.08 (dd, J = 7.5 Hz, J = 1 Hz, 1H, Ar–H), 6.04 (d, J = 7.5 Hz, 1H, Ar–H), 4.34 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 4.33 (d, J = 13.0 Hz, 1H, Ar–CH2–Ar), 4.31 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 4.15 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 4.00–3.87 (m, 4H, –O–CH2–), 3.65–3.59 (m, overlap, 4H, –O–CH2–), 3.49 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 3.16 (d, 1H + 2H, Ar–CH2–Ar), 2.02–1.88 (m, 4H, –O–CH2–CH2–), 1.83 (sextet, J = 7.2 Hz, 4H, –O–CH2–CH2–), 1.08 (t, J = 7.3 Hz, 3H, –O–(CH2)2–CH3), 1.07 (t, J = 7.3 Hz, 3H, –O–(CH2)2–CH3), 0.90 (t, J = 7.5 Hz, 3H, –O–(CH2)2–CH3), 0.88 (t, J = 7.5 Hz, 3H, –O–(CH2)2–CH3). 13C{1H} NMR (125 MHz, DMSO-d6): δ 157.7, 157.3, 154.9, 154.8, 152.7, 146.7, 136.3, 136.2, 134.7, 132.9, 132.83, 132.80, 132.6, 132.5, 130.3, 128.7, 128.6, 127.9, 127.3, 127.2 (2×), 126.7, 125.2 (2×), 121.9, 121.64, 121.59, 118.3, 117.2 (2×), 76.6, 76.4, 76.1, 75.9, 30.2, 30.1, 29.9, 24.0, 23.1, 23.0, 22.6, 22.5, 10.69, 10.67, 9.77, 9.76. HRMS ESI+: (C47H53N3O7) m/z calcd 794.3776 [M + Na]+, found 794.3783 [M + Na]+. IR (KBr) ν 2961, 2931, 2874, 1717, 1667, 1594, 1504, 1455, 1329, 1300, 1245, 1207, 1175, 1111, 1087, 1037, 1005, 964, 850, 753, 691 cm−1.
4-{N′-(4-Trifluoromethylphenyl)ureido}-25,26,27,28-tetrapropoxycalix[4]arene (cone) (3b). Calixarene 3b was prepared according to the general procedure for ureido derivatives by reacting calixarene 2c (0.116 g, 0.190 mmol) and 4-trifluoromethylphenylisocyanate (0.03 mL, 0.21 mmol) in 7 mL dry DCM. The product was purified by preparative TLC (eluent DCM
:cyclohexane 90:10) and then once again (eluent acetone:cyclohexane 15:85) to give the title compound 3b as a white solid (0.09 g, 60%), m.p. 170–173 °C. 1H-NMR (500 MHz, DMSO-d6): δ 9.22 (s, 1H, –NH–), 8.32 (s, 1H, –NH–), 7.65 (d, J = 7.8 Hz, 2H, Ar–H), 7.61 (d, J = 7.8 Hz, 2H, Ar–H), 7.21 (d, J = 8.2 Hz, 1H, Ar–H), 7.07 (td, J = 7.7 Hz, J = 1.1 Hz, 2H, Ar–H), 7.06 (d, J = 8.2 Hz, 1H, Ar–H), 6.85 (t, J = 7.5 Hz, 1H, Ar–H), 6.22 (t, J = 7.5 Hz, 1H, Ar–H), 6.17 (t, J = 8.5 Hz, 1H, Ar–H), 6.16 (d, J = 7.6 Hz, 1H, Ar–H), 6.10 (d, J = 7.6 Hz, 1H, Ar–H), 6.08 (d, J = 7.6 Hz, 1H, Ar–H), 6.03 (d, J = 7.6 Hz, 1H, Ar–H), 4.34 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 4.33 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 4.31 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 4.14 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 4.03–3.86 (m, 4H, –O–CH2–), 3.63 (t, J = 6.3 Hz, 2H, –O–CH2–), 3.62 (t, J = 6.3 Hz, 2H, –O–CH2–), 3.50 (d, J = 13.5 Hz, 1H, Ar–CH2–Ar), 3.16 (∼d, overlap, 1H + 2H, Ar–CH2–Ar), 2.01–1.88 (m, 4H, –O–CH2–CH2–), 1.82 (sextet, J = 7.2 Hz, 4H, –O–CH2–CH2–), 1.08 (t, J = 7.2 Hz, 3H, –O–(CH2)2–CH3), 1.07 (t, J = 7.2 Hz, 3H, –O–(CH2)2–CH3), 0.89 (t, J = 7.6 Hz, 3H, –O–(CH2)2–CH3), 0.88 (t, J = 7.6 Hz, 3H, –O–(CH2)2–CH3). 13C{1H} NMR (125 MHz, DMSO-d6): δ 157.7, 157.3, 154.9, 154.8, 153.0, 143.8, 136.3, 136.2, 135.0, 132.9, 132.83, 132.79, 132.2, 130.1, 128.7, 128.6, 127.8, 127.2, 127.1 (J = 4 Hz), 126.7, 126.09, 126.07, 124.6 (J = 272 Hz), 121.9, 121.64, 121.59, 121.4 (J = 32 Hz), 118.1, 117.5, 76.6, 76.4, 76.1, 75.9, 30.2, 30.1, 29.9, 23.9, 23.1, 23.0, 22.6, 22.5, 10.69, 10.67, 9.76. HRMS-ESI+: (C48H53F3N2O5) m/z calcd 817.3799 [M + Na]+, found 817.3803 [M + Na]+. IR (KBr) ν 2963, 2933, 2875, 1659, 1606, 1550, 1457, 1324, 1208, 1118, 1069, 1008, 965, 840, 760 cm−1.
4-{N′-(4-Butylphenyl)ureido}-25,26,27,28-tetrapropoxycalix[4]arene (cone) (3c). Calixarene 3c was prepared according to the general procedure for ureido derivatives by reacting calixarene 2c (0.112 g, 0.185 mmol) and 4-butylphenylisocyanate (0.036 g, 0.203 mmol) in 6 mL dry DCM. The product was purified by preparative TLC (eluent acetone
:cyclohexane 10:90) to give the title compound 3c as a yellow solid (0.048 g, 33%), m.p. 230–232 °C. 1H-NMR (500 MHz, DMSO-d6): 8.69 (s, 1H, –NH–), 8.15 (s, 1H, –NH–), 7.34 (d, J = 8.0 Hz, 2H, Ar–H), 7.25 (d, J = 8.2 Hz, 1H, Ar–H), 7.12–7.00 (m, overlap, 5H, Ar–H), 6.86 (t, J = 7.50 Hz, 1H, Ar–H), 6.26–6.12 (m, 3H, Ar–H), 6.09 (d, J = 7.50 Hz, 1H, Ar–H), 6.07 (d, J = 7.50 Hz, 1H, Ar–H), 6.03 (d, J = 7.50 Hz, 1H, Ar–H), 4.34 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 4.30 (d, J = 13.3 Hz, 1H, Ar–CH2–Ar), 4.03–3.85 (m, 4H, –O–CH2), 3.66–3.58 (m, 4H, –O–CH2), 3.50 (d, J = 13.3 Hz, 1H, Ar–CH2–Ar), 3.15 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 3.14 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 2.01–1.88 (m, 4H, –O–CH2–CH2–), 1.87–1.77 (m, 4H, –CH2–), 1.55–1.46 (m, 2H, –CH2–), 1.34–1.24 (m, 2H, –CH2–), 1.08 (t, J = 7.50 Hz, 6H, –CH3), 1.08 (overlap, 1H, –CH2–), 0.92–0.83 (m, 9H, –CH3 + 1H, –CH2–). 13C{1H} NMR (125 MHz, DMSO-d6): δ 157.7, 157.3, 154.8, 154.7, 153.2, 137.7, 136.34, 136.26, 135.6, 135.4, 132.9, 132.8, 132.7 (2C), 131.6, 129.5, 128.71, 128.65, 128.5 (2C), 127.8, 127.2, 127.13, 127.09, 126.7, 121.9, 121.64, 121.58, 118.0 (2C), 117.7, 76.6, 76.5, 76.1, 75.9, 34.1, 33.3, 30.2, 30.1, 29.9, 23.7, 23.1, 23.0, 22.6, 22.5, 21.7, 13.8, 10.7 (2C), 9.8 (2C). HRMS-ESI+: (C51H62N2O5) m/z calcd 805.4551 [M + Na]+, found 805.4539 [M + Na]+. IR (KBr) 2960, 2927, 2873, 1655, 1605, 1550, 1456, 1206, 1088, 967, 759 cm−1.
4-{N′-(4-Methoxyphenyl)ureido}-25,26,27,28-tetrapropoxycalix[4]arene (cone) (3d). Calixarene 3d was prepared according to the general procedure for ureido derivatives by reacting calixarene 2c (0.097 g, 0.159 mmol) and 4-methoxyphenylisocyanate (0.02 mL, 0.176 mmol) in 5 mL dry DCM. The product was purified by preparative TLC (eluent acetone
:cyclohexane 20:80) to give the title compound 3d as a white solid (0.051 g, 42%), m.p. 198–200 °C. 1H-NMR (500 MHz, DMSO-d6): 8.64 (s, 1H, –NH–), 8.14 (s, 1H, –NH–), 7.34 (d, J = 9.0 Hz, 2H, Ar–H), 7.23 (d, J = 8.2 Hz, 1H, Ar–H), 7.11–7.00 (m, overlap, 3H, Ar–H), 6.86 (t, J = 7.5 Hz, 1H, Ar–H), 6.85 (d, J = 9.0 Hz, 2H, Ar–H), 6.24–6.11 (m, 3H, Ar–H), 6.08 (td, J = 7.8 Hz, J = 1.2 Hz, 2H, Ar–H), 6.02 (dd, J = 7.7 Hz, J = 1.2 Hz, 1H, Ar–H), 4.33 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 4.30 (d, J = 13.3 Hz, 1H, Ar–CH2–Ar), 4.12 (d, J = 13.3 Hz, 1H, Ar–CH2–Ar), 4.04–3.84 (m, 4H, –O–CH2), 3.70 (s, 3H, –Ar–CH3), 3.66–3.57 (m, 4H, –O–CH2), 3.50 (d, J = 13.3 Hz, 1H, Ar–CH2–Ar), 3.16 (d, J = 13.0 Hz, 2H, Ar–CH2–Ar), 3.13 (d, J = 13.0 Hz, 2H, Ar–CH2–Ar), 2.04–1.87 (m, 4H, –CH2–), 1.87–1.76 (m, 4H, –CH2–), 1.08 (t, J = 7.5 Hz, 6H, –CH3), 0.88 (t, J = 7.5 Hz, 6H, –CH3), 0.87 (t, J = 7.5 Hz, 6H, –CH3). 13C NMR (101 MHz, DMSO-d6) δ 157.7, 157.3, 154.8, 154.8, 154.2, 153.4, 136.3, 136.2, 135.7, 133.1, 132.9, 132.8, 132.74, 132.70, 131.5, 129.5, 128.7, 128.6, 127.7, 127.2, 127.1, 126.7, 121.8, 121.6, 121.6, 119.6, 117.6, 114.0, 76.6, 76.4, 76.1, 75.9, 55.1, 30.2, 30.1, 29.9, 23.7, 23.1, 23.0, 22.6, 22.5, 10.7, 9.8. HRMS-ESI+: (C48H56N2O6) m/z calcd 779.4030 [M + Na]+, found 779.4026 [M + Na]+. IR (KBr) 2961, 2933, 2874, 1647, 1510, 1481, 1455, 1229, 1207, 1089, 1036, 1006, 966, 826, 759 cm−1.
4,16-Bis{N′-(4-nitrophenyl)ureido}-25,26,27,28-tetrapropoxycalix[4]arene (cone) 7a. Calixarene 7a was prepared according to the general procedure for ureido derivatives by reacting calixarene 5c (0.0535 g, 0.0851 mmol) and 4-nitrophenylisocyanate (2.1 eq. 0.029 g, 0.178 mmol) in 7 mL dry DCM. The product was precipitated from acetonitrile to give the title compound 7a as a yellow solid (0.052 g, 64%), m.p. 255–260 °C. 1H-NMR (500 MHz, DMSO-d6): δ 9.57 (s, 1H, –NH–), 8.53 (s, 1H, –NH–), 8.19 (d, J = 9.20 Hz, 4H, Ar–H), 7.69 (d, J = 9.20 Hz, 4H, Ar–H), 7.26 (d, J = 7.75 Hz, 2H, Ar–H), 7.13 (d, J = 7.75 Hz, 2H, Ar–H), 6.17 (t, J = 7.66 Hz, 2H, Ar–H), 6.06 (d, J = 7.45 Hz, 2H, Ar–H), 5.94 (d, J = 7.45 Hz, 2H, Ar–H), 4.30 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 4.13 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 4.05–3.98 (m, 2H, –O–CH2), 3.96–3.88 (m, 2H, –O–CH2), 3.61 (t, J = 6.50 Hz, 4H, –O–CH2), 3.50 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 3.17 (d, J = 13.3 Hz, 2H, Ar–CH2–Ar), 1.97 (sextet, J = 7.61 Hz, 4H, –O–CH2–CH2–), 1.83 (sextet, J = 7.61 Hz, 2H, –O–CH2–CH2–), 1.09 (t, J = 7.3 Hz, 6H, –O–(CH2)2–CH3), 0.88 (t, J = 7.3 Hz, 6H, –O–(CH2)2–CH3). 13C{1H} NMR (125 MHz, DMSO-d6, 333 K): δ 157.7, 154.5, 152.5, 146.4, 140.8, 134.7, 132.5, 132.3, 132.3, 130.2, 127.6, 126.9, 126.3, 124.8, 121.5, 117.8, 117.1, 76.2, 75.7, 29.8, 23.6, 22.8, 22.2, 10.4, 9.4. HRMS-ESI+: (C54H58N6O10) m/z calcd 973.4106 [M + Na]+; found 973.4101 [M + Na]+. IR (KBr) 2965, 2930, 2874, 1664, 1555, 1510, 1482, 1454, 1331, 1218, 1176, 1111, 1085, 965, 851, 752 cm−1.
4,17-Bis{N′-(4-nitrophenyl)ureido}-25,26,27,28-tetrapropoxycalix[4]arene 8a. Calixarene 8a was prepared according to the general procedure for ureido derivatives by reacting calixarene 6c (0.122 g, 0.193 mmol) and 4-nitrophenylisocyanate (2.1 eq. 0.066 g, 0.405 mmol) in 12 mL dry DCM. The product was purified by preparative TLC (eluent acetone
:cyclohexane 30:70) to give the title compound 8a as a yellow solid (0.09 g, 50%), m.p. 220–225 °C. 1H-NMR (500 MHz, DMSO-d6): δ 9.55 (s, 1H, –NH–), 9.36 (s, 1H, –NH–), 8.65 (s, 1H, –NH–), 8.41 (s, 1H, –NH–), 8.18 (d, J = 9.10 Hz, 2H, Ar–H), 8.16 (d, J = 9.10 Hz, 2H, Ar–H), 7.70 (d, J = 9.10 Hz, 2H, Ar–H), 7.68 (d, J = 9.10 Hz, 2H, Ar–H), 7.19 (d, J = 8.2 Hz, 1H, Ar–H), 7.13 (m, 2H, Ar–H), 7.03 (d, J = 8.2 Hz, 1H, Ar–H), 6.28 (t, J = 7.4 Hz, 1H, Ar–H), 6.23–6.18 (m, overlap, 4H, Ar–H), 6.10 (dd, J = 7.0 Hz, J = 1 Hz, 1H, Ar–H), 4.33 (d, J = 13.0 Hz, 1H, Ar–CH2–Ar), 4.32 (d, J = 13.0 Hz, 1H, Ar–CH2–Ar), 4.31 (d, J = 13.0 Hz, 1H, Ar–CH2–Ar), 4.15 (d, J = 13.0 Hz, 1H, Ar–CH2–Ar), 4.02–3.94 (m, 1H, –O–CH2–), 3.94–3.96 (m, 3H, –O–CH2–), 3.68–3.59 (m, 4H, –O–CH2–), 3.49 (d, J = 13.0 Hz, 1H, Ar–CH2–Ar), 3.16 (d, J = 13.0 Hz, 1H, Ar–CH2–Ar), 3.13 (d, J = 13.0 Hz, 2H, Ar–CH2–Ar), 2.01–1.88 (m, 4H, –O–CH2–CH2–), 1.83 (sextet, J = 7.0 Hz, 2H, –O–CH2–CH2–), 1.82 (sextet, J = 7.0 Hz, 2H, –O–CH2–CH2–), 1.07 (t, J = 7.4 Hz, 3H, –O–(CH2)2–CH3), 1.06 (t, J = 7.4 Hz, 3H, –O–(CH2)2–CH3), 0.90 (t, J = 7.4 Hz, 3H, –O–(CH2)2–CH3), 0.89 (t, J = 7.4 Hz, 3H, –O–(CH2)2–CH3). 13C{1H} NMR (125 MHz, DMSO-d6): δ 157.7, 155.0, 154.9, 152.8, 152.7, 152.0, 146.71, 146.66, 140.8, 136.3, 136.2, 134.7, 133.0, 132.8, 132.74, 132.69 (2×), 132.5, 130.2, 127.8, 127.3, 127.2, 127.1, 126.9, 125.2 (2×), 121.7, 119.63, 119.55, 118.3, 117.3, 117.2, 76.7, 76.5, 76.1, 76.0, 30.4, 30.3, 29.9, 24.1, 23.1, 23.0, 22.6, 22.5, 10.7, 10.6, 9.84, 9.79. HRMS-ESI+: (C54H58N6O10): m/z calcd for 973.4106 [M + Na]+, found 973.4110 [M + Na]+. IR (KBr) 2962, 2935, 2874, 1658, 1596, 1552, 1508, 1461, 1328, 1298, 1211, 1178, 1110, 1002, 846, 748, 687 cm−1.
Titration experiments
Dilution experiments preceded titrations to confirm or exclude the self-aggregation processes in the solution. Depending on the solubility of the corresponding urea-based receptors, the studies were performed in DMSO-d6 in the concentration range from 26 mM to 0.6 mM. The appropriate data are shown in the ESI.†All titrations were performed at a constant receptor concentration, specific for individual measurements. Anions were gradually added to the solution of receptors in the form of their TBA salt. The 1H NMR titrations were performed in DMSO-d6 using a Bruker Avance 400 spectrometer (Bruker Biospin, Rheinstetten, Germany). The UV/vis titrations were performed in DMSO (HPLC grade 99.9%, Merck) using a double beam UV-1800 spectrophotometer (Shimadzu). All UV/vis spectra were recorded in the wavelength region from 270 to 800 nm, with steps of 1 nm, in cuvettes with pathlengths of 2 or 1 mm (depending on the absorbance of receptors). The association constants of the resulting complexes were evaluated using the freeware program Bindfit16 using the most pronounced shifts in spectra (1H NMR) or the whole parts of the absorption curves, where the changes in absorbance were the most significant (UV/vis).
The complexation ability of receptors was expressed by association constant K and/or overall association constant β. In the cases of 7a, 8a and 9, the binding ability was evaluated using the overall association constant β by testing four different models for data fitting. The right choice of the model (non-cooperative model) was justified by the accuracy of the fit of experimental points and the fulfilment of the additional fitting conditions, as reported by Thordarson.18
Electrochemistry
A combination of steady-state (RDE) and dynamic methods (CV) was applied to study the electrochemical properties of some prepared molecules. The measurements were performed in DMSO solution (for DNA and peptide synthesis, containing max 0.025% H2O, Merck) using 0.1 M TBAPF6 (from TCI > 98%) as the supporting electrolyte, with the concentration of the corresponding urea derivative as specified in the ESI.† Due to low conductivity, the three-electrode systems were applied in all cases. As the reference electrode, a saturated calomel electrode (SCE) separated from the investigated sample by a bridge filled by the blank (DMSO-electrolyte solution) was applied. As the auxiliary electrode, Pt sheet was chosen. The material of the working electrode was dependent on the particular experiment. For linear sweep voltammetry on RDE (glassy carbon – diameter 1 mm) at a scan rate of 10 mV s−1, several rotation rates (100, 250, 500 and 1000 s−1) were used. For cyclic voltammetry (scan rates 100, 200, and 500 mV s−1), HMDE, glassy carbon electrode (diameter 1 mm) or Pt disk electrode (diameter 1 mm) were used. All these experiments were carried out in an undivided 20 mL cell (using 10 mL of the appropriate solution), and before analyses, the solutions were deaerated using argon (99.998%, Messer). The experiments were carried out using the computer-driven digital potentiostat PGSTAT101 (Autolab-Metrohm) controlled by the software NOVA 2.1.3.X-ray measurements
, a = 9.3111(4) Å, b = 15.9194(7) Å, c = 16.2178(7) Å, α = 98.430(2)°, β = 106.068(2)°, γ = 95.002(2)°, Z = 2, V = 2264.31(17) Å3, Dc = 1.226 g cm−3, μ(Cu-Kα) = 0.68 mm−1, crystal dimensions of 0.202 mm × 0.209 mm × 0.295 mm. Data were collected at 180(2) K on a D8 Venture diffractometer equipped with a Photon II detector and an Incoatec microfocus sealed tube with Cu-Kα radiation. The structure was solved by direct methods23 and anisotropically refined by full matrix least squares on F2 using the CRYSTALS suite of programs24 to a final value of R = 0.0552 and wR = 0.1465, using 8282 independent reflections (θmax = 68.550°), 560 parameters and 0 restrains. The hydrogen atoms bonded to carbon atoms were placed in calculated positions, and all hydrogen atoms were refined with riding constrains. The disordered solvent positions were found in difference electron density maps and refined with no restrictions. MCE25 was used for visualization of electron density maps. The occupancies of disordered functional groups were constrained to full. The structure was deposited in the Cambridge Structural Database under number CCDC 2381330.†
Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for compound 3a have been deposited at the CCDC under CCDC 2381330 and can be obtained from https://www.ccdc.cam.ac.uk.†
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the Czech Science Foundation (Grant 23-07154S and 21-05926X). Financial support from Specific University Research (Grant No. A2 FCHT 2023 051) is also acknowledged.References
- (a) J. L. Sessler, P. Gale and W.-S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, Cambridge, 2006 RSC; (b) Recognition of Anions, R. Vilar, 2008, Springer Berlin, Heidelberg Search PubMed; (c) Anion recognition in supramolecular chemistry, ed. P. Gale and W. Dehaen, 2011, Springer, Berlin, Heidelberg Search PubMed; (d) Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. García-España, 2012, Wiley–VCH Verlag GmbH & Co. KGaA Search PubMed; (e) Anion-Binding Catalysis, ed. O. G. Mancheno, 2022, Wiley–VCH GmbH Search PubMed.
- (a) P. Molina, F. Zapata and A. Caballero, Anion Recognition Strategies Based on Combined Noncovalent Interactions, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed; (b) J. Zhao, D. Yang, X.-J. Yang and B. Wu, Anion coordination chemistry: From recognition to supramolecular assembly, Coord. Chem. Rev., 2019, 378, 415–444 CrossRef CAS; (c) Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Macrocycles as Ion Pair Receptors, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed; (d) M. S. Taylor, Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding, Coord. Chem. Rev., 2020, 413, 213270 CrossRef CAS; (e) R. Hein, P. D. Beer and J. J. Davis, Electrochemical anion sensing: supramolecular approaches, Chem. Rev., 2020, 120, 1888–1935 CrossRef CAS PubMed; (f) U. Manna and G. Das, An overview of anion coordination by hydroxyl, amine and amide based rigid and symmetric neutral dipodal receptors, Coord. Chem. Rev., 2021, 427, 213547 CrossRef CAS; (g) S. C. Patrick, P. D. Beer and J. J. Davis, Solvent effects in anion recognition, Nat. Rev. Chem., 2024, 8, 256–276 CrossRef PubMed.
- S. La Cognata and V. Amendola, Recent applications of organic cages in sensing and separation processes in solution, Chem. Commun., 2023, 59, 13668–13678 RSC.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, The squaramide versus urea contest for anion recognition, Chem. – Eur. J., 2010, 16, 4368–4380 CrossRef CAS PubMed.
- (a) V. Amendola, L. Fabbrizzi and L. Mosca, Anion recognition by hydrogen bonding: urea-based receptors, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC; (b) A.-F. Li, J.-H. Wang, F. Wang and Y.-B. Jiang, Anion complexation and sensing using modified urea and thiourea-based receptors, Chem. Soc. Rev., 2010, 39, 3729–3745 RSC; (c) V. B. Bregović and N. Basarić, Anion binding with urea and thiourea derivatives, Coord. Chem. Rev., 2015, 295, 80–124 CrossRef; (d) C. Jia, W. Zuo, D. Zhang, X.-J. Yang and B. Wu, Anion recognition by oligo-(thio) urea-based receptors, Chem. Commun., 2016, 52, 9614–9627 RSC.
- J. Cai and J. L. Sessler, Neutral CH and cationic CH donor groups as anion receptors, Chem. Soc. Rev., 2014, 43, 6198–6213 RSC.
- L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, The road to aryl CH⋯anion binding was paved with good intentions: fundamental studies, host design, and historical perspectives in CH hydrogen bonding, Chem. Commun., 2019, 55, 5195–5206 RSC.
- (a) P. Neri, J. L. Sessler and M. X. Wang, Calixarenes and Beyond, Springer, Cham, Switzerland, 2016 CrossRef; (b) C. D. Gutsche, Calixarenes: An Introduction, RSC Publishing, Cambridge, U. K., 2nd edn, 2008 Search PubMed; (c) L. Mandolini and R. Ungaro, Calixarenes In Action, Imperial College Press, London, 2000 CrossRef.
- V. M. Mirsky and A. Yatsimirsky, Artificial Receptors for Chemical Sensors, Wiley, Weinheim, 2010 Search PubMed.
- (a) S. A. Wagay, L. Khan and R. Ali, Recent Advancements in Ion-Pair Receptors, Chem. – Asian J., 2023, 18, e202201080 CrossRef CAS PubMed; (b) S. E. Matthews and P. D. Beer, Calixarene-based Anion Receptors, Supramol. Chem., 2005, 17, 411–435 CrossRef CAS; (c) P. Lhoták, in Anion Sensing: -/-, ed. I. Stibor, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 65–95, DOI:10.1007/b101162; (d) N. Y. Edwards and A. L. Possanza, Calixarene-based anionic receptors: highlights from 2011, Supramol. Chem., 2013, 25, 446–463 CrossRef CAS; (e) R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Revisiting Fluorescent Calixarenes: From Molecular Sensors to Smart Materials, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- (a) T. Horackova, J. Budka, V. Eigner, W.-S. Chung, P. Curinova and P. Lhotak, Chiral anion recognition using calix[4]arene-based ureido receptors in a 1,3-alternate conformation, Beilstein J. Org. Chem., 2020, 16, 2999–3007 CrossRef CAS PubMed; (b) M. Rezankova, J. Budka, J. Miksatko, V. Eigner, I. Cisarova, P. Curinova and P. Lhotak, Anion receptors based on intramolecularly bridged calix[4]arenes bearing ureido functions, Tetrahedron, 2017, 73, 742–749 CrossRef CAS; (c) T. Klejch, J. Slavicek, O. Hudecek, V. Eigner, N. A. Gutierrez, P. Curinova and P. Lhotak, Calix[4]arenes containing a ureido functionality on the lower rim as highly efficient receptors for anion recognition, New J. Chem., 2016, 40, 7935–7942 RSC.
- (a) P. Slavik, M. Dudic, K. Flidrova, J. Sykora, I. Cisarova, S. Bohm and P. Lhotak, Unprecedented Meta-Substitution of Calixarenes: Direct Way to Inherently Chiral Derivatives, Org. Lett., 2012, 14, 3628–3631 CrossRef CAS PubMed; (b) K. Flidrova, S. Bohm, H. Dvorakova, V. Eigner and P. Lhotak, Dimercuration of Calix[4]arenes: Novel Substitution Pattern in Calixarene Chemistry, Org. Lett., 2014, 16, 138–141 CrossRef CAS PubMed.
- P. Lhoták, Direct meta substitution of calix[4]arenes, Org. Biomol. Chem., 2022, 20, 7377–7390 RSC.
- M. Tlusty, P. Slavik, M. Kohout, V. Eigner and P. Lhotak, Inherently Chiral Upper-Rim-Bridged Calix[4]arenes Possessing a Seven Membered Ring, Org. Lett., 2017, 19, 2933–2936 CrossRef CAS PubMed.
- C. A. Hunter and H. L. Anderson, What is Cooperativity?, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed.
- Online tools for supramolecular chemistry research and analysis, https://supramolecular.org.
- (a) Z. D. Hill and P. MacCarthy, Novel approach to Job's method: An undergraduate experiment, J. Chem. Educ., 1986, 63, 162 CrossRef CAS; (b) K. Hirose, A Practical Guide for the Determination of Binding Constants, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193–209 CrossRef CAS; (c) D. B. Hibbert and P. Thordarson, The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis, Chem. Commun., 2016, 52, 12792–12805 RSC.
- P. Thordarson, Determining association constants from titration experiments in supramolecular chemistry, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- K. Lang, P. Curinova, M. Dudic, P. Proskova, I. Stibor, V. St'astny and P. Lhotak, Unusual stoichiometry of urea-derivatized calix[4]arenes induced by anion complexation, Tetrahedron Lett., 2005, 46, 4469–4472 CrossRef CAS.
- M. Tlusty, V. Eigner, M. Babor, M. Kohout and P. Lhotak, Synthesis of upper rim-double-bridged calix[4]arenes bearing seven membered rings and related compounds, RSC Adv., 2019, 9, 22017–22030 RSC.
- (a) C. Amatore, G. Capobianco, G. Farnia, G. Sandona, J. M. Saveant, M. G. Severin and E. Vianello, Kinetics and mechanism of self-protonation reactions in organic electrochemical processes, J. Am. Chem. Soc., 1985, 107, 1815–1824 CrossRef CAS; (b) E. Brillas, G. Farnia, M. G. Severin and E. Vianello, Self-protonation effects in the electrochemical reduction mechanism of p-nitrobenzoic acid, Electrochim. Acta, 1986, 31, 759–766 CrossRef CAS; (c) J. A. Morales-Morales, C. Frontana, M. Aguilar-Martínez, J. A. Bautista-Martínez, F. J. González and I. González, Analysis of the Substituent Effect on the Reactivity Modulation during Self-Protonation Processes in 2-Nitrophenols, J. Phys. Chem. A, 2007, 111, 8993–9002 CrossRef CAS PubMed.
- K. Salvadori, J. Ludvík, L. Šimková, P. Matějka and P. Cuřínová, Nitro group as a redox switch in urea-based receptors of anions, J. Electroanal. Chem., 2021, 902, 115816 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, SIRPOW.92 - a program for automatic solution of crystal structures by direct methods optimized for powder data, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- P. Betteridge, J. Carruthers, R. Cooper, K. Prout and D. Watkin, CRYSTALS version 12: software for guided crystal structure analysis, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- J. Rohlicek and M. Husak, MCE2005 - a new version of a program for fast interactive visualization of electron and similar density maps optimized for small molecules, J. Appl. Crystallogr., 2007, 40, 600–601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral characterization of new compounds, measuring of association constants, dilution experiments, Job plot analyses, titration experiments and electrochemical data. CCDC 2381330. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01441c |
| This journal is © The Royal Society of Chemistry 2024 |