
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceP-Stereodefined morpholino dinucleoside 3′,5′-phosphorothioates†
Katarzyna
Jastrzębska
 *,
Patrycja
Antończyk
and
Rafał
Dolot
*,
Patrycja
Antończyk
and
Rafał
Dolot
Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Department of Bioorganic Chemistry, Sienkiewicza 112, 90-363 Łódź, Poland. E-mail: katarzyna.jastrzebska@cbmm.lodz.pl
First published on 11th October 2024
Abstract
Here, we present for the first time the synthesis of P-stereodefined morpholino phosphorothioate analogs by using a modified 1,3,2-oxathiaphospholane method (OTP method) and provide valuable structural insights into their stereochemistry. N-(2-Thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) derivatives of morpholino-type nucleosides (mU-OTPs) were synthesized, separated into pure P-diastereomers and used to prepare P-stereodefined morpholino dinucleoside 3′,5′-phosphorothioates.
Introduction
Recently, Caruthers’ lab has successfully developed a strategy for the chemical synthesis and preparation of a new class of synthetic molecules called thiophosphoramidate morpholinos (TMOs) using phosphoramidite chemistry.1 These TMOs contain two moieties, namely a phosphorothioate (PS) and a morpholine ring, and have been tested in the oligotherapeutics arena.2 Dumbović et al. and Le et al. have described how these P-stereorandom TMO constructs exhibit interesting biological properties with considerable therapeutic potential.3,4Recent reports have shown that stereoisomerically pure PS oligos have better in vitro and in vivo efficacy than their diastereomeric mixture.5 Therefore, from the racemic mixtures of thousands of diastereomeric species, only a few molecules may represent functional and hopefully non-harmful species, of which only some are active. Identifying the most active stereoisomers requires the development of stereodefined synthesis of oligonucleotide drugs, which allows lower doses with more potent compounds. Many stereopure PS oligonucleotides are currently in clinical development at Wave Life Sciences.6 Wave is a leading genetic drug company focused on exploiting the potential of stereopure oligonucleotides to treat diseases of the central nervous system,7 liver8 and eyes.9 Therefore, the stereoselective synthesis of potential drug candidates is an important goal to achieve.
The increased interest in the stereocontrolled chemical synthesis of phosphorothioate analogs has prompted us to adapt the 1,3,2-oxathiaphospholane (OTP) method for the synthesis of P-stereodefined morpholino phosphorothioate analogs (Fig. 1). The OTP method, as developed by Stec et al.,10,11 has already been successfully used for the stereocontrolled synthesis of PS-DNA, PS-LNA (Locked Nucleic Acids),12,13 PS-GNA (Glycol Nucleic Acids),14 PS-RNA and PS-(2′-OMe) RNA phosphorothioate analogs.15 Recently, we extended the OTP method and applied TBD and Verkade bases, which can be successfully used instead of DBU.16
 | ||
| Fig. 1 Schematic representation of P-stereodefined morpholino dinucleoside 3′,5′-phosphorothioates. | ||
Herein, we describe the synthesis and HPLC separation of mU-OTPs (2) into pure P-diastereomers (2afast- and 2bslow-eluting), as well as the successful crystallographic analyses of the above-mentioned monomers.
These P-diastereomerically pure monomers 2a and 2b were then used for the synthesis of P-stereodefined dinucleotides 4a and 4b (Scheme 1) and their enzymatic stability were tested. X-ray analysis of 5b showed the SP absolute configuration of the phosphorus atom.
 | ||
| Scheme 1 The synthesis of morpholino OTP and mUPST dinucleotides. 2a: (fast); 2b: (slow); 3a: (from 2a); 3b: (from 2b); 4a: (from 2a); 4b: (from 2b); 5a: (from 4a); and 5b: (from 4b). Conditions: (i) cat. NaHSO4/SiO2 in CH3CN; (ii) 1.5 eq. of Verkade base and 2 eq. of 3′-O-acetyl-2′-deoxythymidine in CH3CN; (iii) 1.5% dichloroacetic acid (DCA) in CH2Cl2, NH4OH, and rt. | ||
Results and discussion
Preparation of P-diastereomerically pure oxathiaphospholane derivatives of morpholino nucleosides (mU-OTPs)
The morpholino nucleoside of uridine (mU, 1) was synthesized according to published literature protocols.1 Briefly, 5′-O-dimethoxytrityl ribonucleoside (Scheme 2) was oxidatively converted into an acyclic dialdehyde derivative, followed by reductive amination.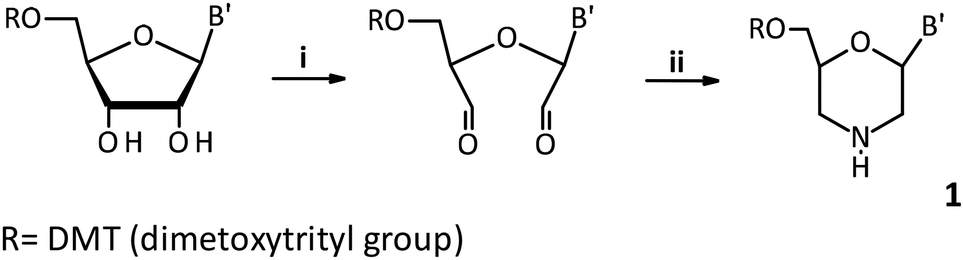 | ||
| Scheme 2 (i) NaIO4 (1.2 equiv.), anhydrous methanol, and 3 h; (ii) (NH4)2B2O7 (1.2 equiv.), NaCNBH3 (2.0 equiv.), CH3COOH (2.0 equiv.), and 16 h. | ||
Then, the morpholino oxathiaphospholane monomer (mU-OTP, 2) was synthesized according to a recently published protocol.16 The oxathiaphospholane monomers mU-OTPs (2) were synthesized according to a general procedure published for the synthesis of standard OTP monomers. Briefly, the overnight dried morpholino nucleoside was phosphitylated with 2-chloro-“spiro”-4,4-pentamethylene-1,3,2-oxathiaphospholane, followed by sulfurization with elemental sulfur. The monomer 2 (as a mixture of P-diastereomers) was obtained with a reasonable yield (∼80%). The P-diastereomers were formed in nearly equimolar amounts: ca. 55% of fast-(2a) and 45% of slow-eluting (2b) P-diastereomers, as indicated by 31P NMR, respectively (Fig. S2 and S3, ESI†). The P-diastereomers were separated by preparative HPLC (Fig. S4, ESI†) using a silica gel column and their diastereomeric purity was confirmed by 31P NMR, 1H NMR, 13C NMR (Fig. S5–S10, ESI†) and HRMS (Fig. S11 and S12, ESI†).
Additionally, 31P NMR analysis showed that the fast-eluting isomer had higher chemical shifts than its slow-eluting counterpart, which is opposite to the OTP derivatives of 2′-deoxyribonucleosides and LNA-OTP analogs (Table 1). Interestingly, the correlation is identical to that observed for the OTP derivatives of 2′-OMe-ribonucleosides and 3′-amino-2′,3′-dideoxyribonucleosides.17
| For mU-OTP | Fast | Slow |
|---|---|---|
a Yield of the isolated mixture of P-diastereomers.
b CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (9:1).
c Separation of P-diastereomers at an AcOEt:hexane ratio (v/v), isocratically.
d Pursuit XRs silica gel column (10 μm, 250 × 21.2 mm, flow rate 10 mL min−1, and isocratically).
e Recorded with a SYNAPT G2-Si high definition mass spectrometer.
f In anhydrous CD3CN. :MeOH (9:1).
c Separation of P-diastereomers at an AcOEt:hexane ratio (v/v), isocratically.
d Pursuit XRs silica gel column (10 μm, 250 × 21.2 mm, flow rate 10 mL min−1, and isocratically).
e Recorded with a SYNAPT G2-Si high definition mass spectrometer.
f In anhydrous CD3CN.
|
||
| R t (min)d | 21 | 30 |
| HR MSe (m/z)e | 734.2134 | 734.2133 |
| δ 31P NMRf (ppm) | 100.29 | 100.22 |
X-ray crystallography analysis of the 6′-OH oxathiaphospholane monomer mU-OTPs
In order to gain structural insights into the stereochemistry of P-stereodefined morpholino analogs, we studied the crystalized structures of 3a/3b and the morpholino dinucleoside 3′,5′-phosphorothioates. Here, we present for the first time the X-ray crystal structures of separated P-diastereomers with their determined absolute configuration. Crystallization attempts with DMT-labeled OTP derivatives were however unsuccessful. Based on previous experience with the crystallization of oxathiaphospholane derivatives in the DNA11 and LNA series,12 we decided to remove the DMT group in mU-OTPs to obtain mU derivatives. Detritylation was carried out in anhydrous acetonitrile using a sodium hydrogen sulfate suspension deposited on silica gel.18 Crystals suitable for X-ray analysis were obtained from the 6′-OH deprotected fast- and slow-eluting monomers, respectively. Crystals suitable for diffraction experiments were obtained by slow evaporation of the solvent. Single colorless transparent prism-shaped crystals of 3a were obtained by recrystallization from a mixture of acetonitrile and methanol (2:1 v/v), while 3b was recrystallized from a mixture of chloroform and methanol (1:1 v/v), yielding colorless, needle-shaped crystals (Fig. S13 and S14, ESI†). Suitable crystals of 3a and 3b with dimensions of 0.57 × 0.57 × 0.28 mm and 0.67 × 0.15 × 0.10 mm, respectively, were selected and mounted on a suitable support on an XtaLAB Synergy Dualflex HyPix diffractometer. The crystals were maintained at a steady T = 293.88(10) K during data collection. The structures were solved using the ShelXT19 structure solution program and the intrinsic phasing solution method and by using Olex220 as the graphical interface. The models were refined with version 2018/3 of ShelXL21 using least squares minimization. The crystal data and refinement parameters for 3a and 3b are collected in Tables S1–S6 (ESI†).
Crystallographic analysis showed that fast- (Fig. 2a) and slow-eluting (Fig. 2b) isomers of mU-OTP have the P-atom in the RP and SP absolute configurations (Fig. 2c), respectively (according to the Cahn–Ingold–Prelog rules, the endocyclic sulfur atom has a higher priority than the exocyclic one).
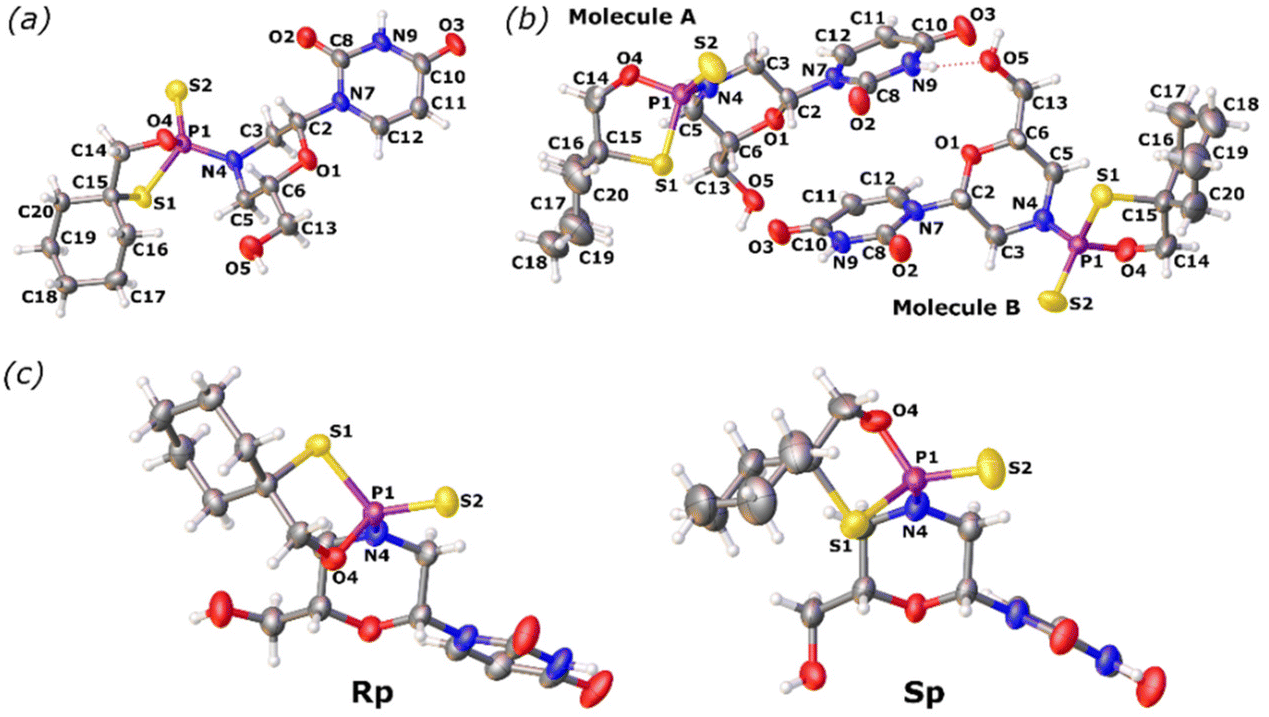 | ||
| Fig. 2 X-ray ORTEP diagrams of (a) the 6′-OH fast-eluting P-diastereomer of 3a and (b) the 6′-OH slow-eluting P-diastereomer of 3b, showing (c) the RP and SP absolute configurations, respectively. Colors: red: oxygen, blue: nitrogen, yellow: sulfur, and purple: phosphorus. CCDC: (a) 2079450 and (b) 2079449.† | ||
Stereocontrolled synthesis of morpholino dinucleoside phosphorothioate mUPST through the oxathiaphospholane approach
To study and optimize the coupling reaction (Scheme 1(ii)), we investigated the reactivity of morpholino oxathiaphospholane monomer 2 (as a mixture of P-diastereomers) towards 3′-O-acetyl-2′-deoxythymidine in the presence of various activators by performing 31P NMR spectroscopy. Here, it should be emphasized that mixtures of P-diastereomers and non-deuterium acetonitrile were used to optimize the synthetic protocol. The reactions were carried out at room temperature with 1 equivalent of monomer 2 and 1.5 equivalents of 3′-O-acetyl-2′-deoxythymidine in dry acetonitrile in the presence of various activators. First, the optimization was carried out using the standard conditions of the oxathiaphospholane method, namely with 1.2 DBU equivalents. After 15 minutes, roughly 90% of the reaction mixture consisted of unreacted starting material 2 (δ: 100.79, 100.08 ppm; Fig. S15, ESI†). After 60 minutes, the conversion reaction was not complete (Fig. S17, ESI†). Attempts to increase the yield by extension of the coupling time or increasing the DBU concentration were unsuccessful. Since longer reaction times led to other by-products, we decided to utilize TBD22 instead of DBU. With 1.2 eq. of TBD, we observed the formation of a major product 4, which showed two resonance signals at around 61 ppm in the 31P NMR spectrum and was accompanied by significant amounts of unreacted substrate and a phosphate moiety, which can be seen as a signal at 45 ppm (9%) (Fig. S18, ESI†). After 45 minutes, the substrate disappeared completely with the formation of the major product 4, accompanied by a considerable amount of a minor product (20%, 45 ppm, Fig. S19, ESI†). When a reaction was performed with 5 eq. of TBD, the same result was observed within 15 minutes. Further research demonstrated that the best activator for synthesizing the P-stereodefined phosphorothioate of morpholino analogs (4) using the OTP method proved to be the Verkade base (Vb). Using 1.5 equivalents of Vb, the reaction proceeded very rapidly (15 minutes) and 31P NMR showed the presence of only one major product 4 (Fig. S20, ESI†). These results suggest that the formation of an internucleotide phosphorothioate linkage involving the morpholino nucleoside via the oxathiaphospholane route is highly efficient and stereocontrolled.The synthesis of P-stereodefined phosphorothioate dinucleotides mUPST was next carried out on a semi-preparative scale. Pure P-diastereomers fast- (3a) and slow-eluting (3b) were reacted with 3′-O-Ac-Thy in the presence of Vb to yield the corresponding protected dinucleotides mUPST (4a and 4b, Scheme 1), which were isolated using silica gel of a Reveleris Flash Chromatography system and analyzed by 31P NMR and HRMS (yield around 70%, Fig. S21 and S22, ESI†).
X-ray analysis of the 6′-OH dinucleotide mUPST
Since X-ray analysis worked best when using detritylated P-diastereomers of fast- (3a) and slow-eluting (3b), further analyses were performed with 5a and 5b in order to complete the stereochemical assignments of the P-diastereomeric diesters. The DMT moiety was removed with 1.5% aqueous dichloroacetic acid (DCA) in dichloromethane over 10 minutes at room temperature. The resultant detritylated dinucleotides mUPST 5a and 5b (Fig. S23, ESI†) were isolated in 66% and 61% yields, respectively. The structures of the dinucleotides mUPST were confirmed by HRMS (Fig. S26 and S28, ESI†). Numerous crystallization attempts of the DMT-protected (4a and 4b) and unprotected (5a and 5b, Scheme 1) dimers were not successful. Based on the literature, we decided to explore the possibility of crystal formation after complexation with selected proteins.23 Initial attempts of 5a and 5b complex formation with human serum albumin (HSA) and histidine triad nucleotide-binding protein 1 (HINT1) were unsuccessful. However, successful crystal formation was achieved by soaking the dinucleotides in bovine pancreatic RNase A as the RNase A/dimer complex was obtained. Briefly, native RNase A crystals were grown by vapor diffusion with hanging droplets. The droplets consisted of 2 μL of protein at a concentration of 10 mg mL−1 and 2 μL of well solution containing 22–25% (w/v) PEG 4000 and 20 mM sodium citrate (pH 5.5). The prepared solution was incubated at 8 °C and RNase A crystals formed after 3–5 days. Crystals of the complex were obtained by soaking preformed RNase A crystals in mother liquor containing 25 mM of 5b for 10–30 minutes. The soaked crystals prepared for analysis did not need to be cryopreserved, but were grown on a very thin film of crystallization buffer. The excess liquid was removed by gently touching the embedding loop on the plate surface. Then the prepared crystals for analysis were cooled directly in a stream of nitrogen.The crystal data and refinement parameters for 5b are collected in Tables S7 and S8 (ESI†). The analysis of the crystallographic data showed that the mUPST dimer (5b), which was obtained from slow-eluting (3b), has the P-atom with the SP absolute configuration (according to the Cahn–Ingold–Prelog rules, the endocyclic sulfur atom has higher priority than the exocyclic one) (Fig. 3).
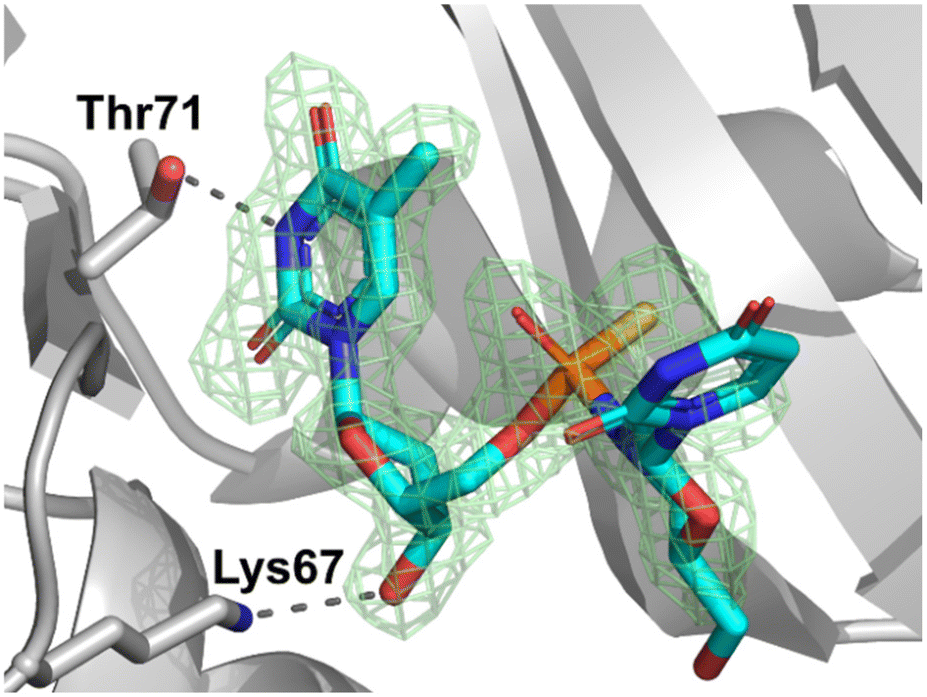 | ||
| Fig. 3 The electron density map (Fo–Fc omit map) at 1.80 Å resolution from the crystal structure of SP-dimer 5b with RNase A. The map is contoured at the 3.0 level. Lys67 and Thr71 residues are involved in direct dimer binding. Hydrogen bonds are indicated by dashed lines. (PDB ID: 8R5V). | ||
Conclusions
In summary, the results presented in this work provide evidence that a modified oxathiaphospholane method can be successfully applied for the stereocontrolled synthesis of novel P-stereodefined phosphorothioate morpholino analogs. Oxathiaphospholane derivatives of morpholino nucleosides (mU-OTPs) were synthesized with a good yield and effectively separated into pure P-diastereomers. The analysis of the crystallographic data showed that fast- and slow-eluting mU-OTP have the P atom in the RP and SP absolute configurations, respectively, according to the Cahn–Ingold–Prelog rules, where the endocyclic sulfur atom has a higher priority than the exocyclic one. The synthesis of P-stereodefined dinucleotide phosphorothioates mUPST (5) was also performed with a relatively high yield. X-ray crystallographic analysis revealed that the phosphorus atom in 5b, which was obtained from the slow-eluting diastereomer 3b, had the SP absolute configuration. Here, we have presented the first-time determination of stereochemistry at the stereogenic phosphorus atom in morpholino analogs. Further research will focus on preparing stereodefined morpholino phosphorothioate oligonucleotides having all four bases and determining their biochemical and biological properties.Author contributions
K. J. supervised the project and prepared and edited the manuscript. K. J. and P. A. performed all the chemical synthesis and data analysis. R. D. performed crystal preparation, diffraction data collection and data analysis. All authors approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Centre of Science, Poland, grant 2021/43/D/ST4/02433 (to K. J.). An Avance Neo 400 NMR spectrometer and Rigaku XtaLAB Synergy-S X-Ray diffractometer systems were purchased using funds provided by the EU Regional Operational Program of the Lodz Region RPLD.01.01.00-10-0008/18. The authors are grateful to Professor Arkadiusz Chworos (CMMS PAS) for constructive suggestions and discussions. Katarzyna Jastrzębska expresses gratitude to Professor Marvin H. Caruthers (University of Colorado Boulder) for scientific inspiration, continuous support and helpful suggestions.References
- H. Langner, K. Jastrzebska and M. Caruthers, Synthesis and Characterization of Thiophosphoramidate Morpholino Oligonucleotides and Chimeras, J. Am. Chem. Soc., 2020, 142, 16240–16253 CrossRef CAS PubMed.
- S. Paul and M. Caruthers, Synthesis of Backbone-Modified Morpholino Oligonucleotides Using Phosphoramidite Chemistry, Molecules, 2023, 28, 5380 CrossRef CAS PubMed.
- (a) G. Dumbović, U. Braunschweig, H. K. Langner, M. Smallegan, J. Biayna, E. P. Hass, K. Jastrzebska, B. Blencowe, T. R. Cech, M. H. Caruthers and J. L. Rinn, Nuclear compartmentalization of TERT mRNA and TUG1 lncRNA is driven by intron retention, Nat. Commun., 2021, 12, 3308–3326 CrossRef; (b) G. Dumbović, H. Krishna, K. Jastrzebska, M. Caruthers and J. L. Rinn, Method for retaining splicing RNAs in the nucleus based on chemically modified antisense oligonucleotides, Patent US 2021/015673, 2021.
- (a) B. T. Le, S. Paul, K. Jastrzebska, H. Langer, M. H. Caruthers and R. N. Veedu, Thiomorpholino oligonucleotides as a robust class of next generation platforms for alternate mRNA splicing, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 1–8 CrossRef; (b) M. Caruthers, S. Paul, R. N. Veedu, K. Jastrzebska and H. Krishna, Thiomorpholino oligonucleotides for the treatment of Duchene muscular dystrophy, Patent US 2023/0193266-A1, 2023.
- H. Jahns, M. Roos, J. Imig, F. Baumann, Y. Wang, R. Gilmour and J. Hall, Stereochemical bias introduced during RNA synthesis modulates the activity of phosphorothioate siRNAs, Nat. Commun., 2015, 6, 6317 CrossRef CAS PubMed.
- W. Liu, N. Iwamoto, S. Marappan, K. Luu, S. Tripathi, E. Purcell-Estabrook, J. D. Shelke, H. Shah, A. Lamattina, Q. Pan, B. Schrand, F. Favaloro, M. Bedekar, A. Chatterjee, J. Desai, T. Kawamoto, G. Lu, J. Metterville, M. Samaraweera, P. S. Prakasha, H. Yang, Y. Yin, H. Yu, P. H. Giangrande, M. Byrne, P. Kandasamy and C. Vargeese, Impact of stereopure chimeric backbone chemistries on the potency and durability of gene silencing by RNA interference, Nucleic Acids Res., 2023, 51, 4126–4147 CrossRef CAS PubMed.
- P. Kandasamy, Y. Liu, V. Aduda, V. S. Akare, R. Alam, A. Andreucci, D. Boulay, K. Bowman, M. Byrne, M. Cannon, O. Chivatakarn, J. D. Shelke, N. Iwamoto, T. Kawamoto, J. Kumarasamy, S. Lamore, M. Lemaitre, X. Lin, K. Longo, R. Looby, S. Marappan, J. Metterville, S. Mohapatra, B. Newman, I. H. Paik, S. Patil, E. Purcell-Estabrook, M. Shimizu, P. Shum, S. Standley, K. Taborn, S. Tripathi, H. Yang, Y. Yin, X. Zhao, E. Dale and C. Vargeese, Impact of guanidine-containing backbone linkages on stereopure antisense oligonucleotides in the CNS, Nucleic Acids Res., 2022, 50, 5401–5423 CrossRef CAS PubMed.
- P. Monian, C. Shivalila, G. Lu, M. Shimizu, D. Boulay, K. Bussow, M. Byrne, A. Bezigian, A. Chatterjee, D. Chew, J. Desai, F. Favaloro, J. Godfrey, A. Hoss, N. Iwamoto, T. Kawamoto, J. Kumarasamy, A. Lamattina, A. Lindsey, F. Liu, R. Looby, S. Marappan, J. Metterville, R. Murphy, J. Rossi, T. Pu, B. Bhattarai, S. Standley, S. Tripathi, H. Yang, Y. Yin, H. Yu, C. Zhou, L. H. Apponi, P. Kandasamy and C. Vargeese, Endogenous ADAR-mediated RNA editing in non-human primates using stereopure chemically modified oligonucleotides, Nat. Biotechnol., 2022, 40, 1093–1102 CrossRef CAS PubMed.
- M. Byrne, V. Vathipadiekal, L. Apponi, N. Iwamoto, P. Kandasamy, K. Longo, F. Liu, R. Looby, L. Norwood, A. Shah, J. D. Shelke, C. Shivalila, H. Yang, Y. Yin, L. Guo, K. Bowman and C. Vargeese, Stereochemistry Enhances Potency, Efficacy, and Durability of Malat1 Antisense Oligonucleotides In Vitro and In Vivo in Multiple Species, Transl. Vis. Sci. Technol., 2021, 10, 1–10 Search PubMed.
- W. J. Stec, A. Grajkowski, M. Koziołkiewicz and B. Uznański, Novel route to oligo(deoxyribonucleoside phosphorothioates). Stereocontrolled synthesis of P-chiral oligo(deoxyribonucleoside phosphorothioates), Nucleic Acids Res., 1991, 19, 5883–5888 CrossRef CAS PubMed.
- W. J. Stec, B. Karwowski, M. Boczkowska, P. Guga, M. Koziołkiewicz, M. Sochacki, M. W. Wieczorek and J. Błaszczyk, Deoxyribonucleoside 3′-O-(2-Thio- and 2-Oxo-“spiro”-4,4-pentamethylene-1,3,2-oxathiaphospholane)s: Monomers for Stereocontrolled Synthesis of Oligo(deoxyribonucleoside phosphorothioate)s and Chimeric PS/PO Oligonucleotide, J. Am. Chem. Soc., 1998, 120, 7156–7167 CrossRef CAS.
- K. Jastrzębska, A. Maciaszek, R. Dolot, G. Bujacz and P. Guga, Thermal Stability and Conformation of Antiparallel Duplexes Formed by P-Stereodefined Phosphorothioate DNA/LNA Chimeric Oligomers with DNA and RNA Matrices, Org. Biomol. Chem., 2015, 13, 10032–10040 RSC.
- K. Jastrzębska, B. Mikołajczyk and P. Guga, LNA units present in [RP-PS]-(DNA#LNA) chimeras enhance the thermal stability of parallel duplexes and triplexes formed with (2′-OMe)-RNA strands, RSC Adv., 2020, 10, 22370–22376 RSC.
- A. Tomaszewska-Antczak, K. Jastrzębska, A. Maciaszek, B. Mikołajczyk and P. Guga, P-Stereodefined, phosphorothioate analogs of glycol nucleic acids – synthesis and structural properties, RSC Adv., 2018, 8, 24942–24952 RSC.
- K. Jastrzębska, A. Maciaszek, R. Dolot, A. Tomaszewska-Antczak, B. Mikołajczyk and P. Guga, Synthesis and hybridizing properties of P-stereodefined chimeric [PS]-{DNA:RNA} and [PS]-{DNA:(2′-OMe)-RNA} oligomers, RSC Adv., 2022, 12, 26815–26824 RSC.
- K. Jastrzębska, An efficient alternative to DBU in the oxathiaphospholane (OTP) method for the solid phase synthesis of P-stereodefined phosphorothioate analogs, RSC Adv., 2024, 14, 21174–21179 RSC.
- E. Radzikowska, R. Kaczmarek, D. Korczyński, A. Krakowiak, B. Mikołajczyk, J. Baraniak, P. Guga, K. A. Wheeler, T. Pawlak and B. Nawrot, P-stereocontrolled synthesis of oligo(nucleoside N3′→O5′ phosphoramidothioate)s – opportunities and limitations, RSC Adv., 2020, 10, 35185–35197 RSC.
- R. K. Kannasani, V. V. S. Peruri and S. R. Battula, NaHSO4-SiO2 as an efficient and chemoselective catalyst, for the synthesis of acylal from aldehydes under, solvent-free conditions, Chem. Cent. J., 2012, 6, 136 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment - Olex2 dissected, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- E. Fritz-Langhals, Unique Superbase TBD (1,5,7-Triazabicyclo[4.4.0]dec-5-ene): From Catalytic Activity and One-Pot Synthesis to Broader Application in Industrial Chemistry, Org. Process Res. Dev., 2022, 26(11), 3015–3023 CrossRef CAS.
- R. Dolot, R. Kaczmarek, A. Sęda, A. Krakowiak, J. Baraniak and B. Nawrot, Crystallographic studies of the complex of human HINT1 protein with a non-hydrolyzable analog of Ap4A, Int. J. Biol. Macromol., 2016, 87, 62–69 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and associated data. CCDC: 2079450 and 2079449; PDB: 8R5V. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01437e |
| This journal is © The Royal Society of Chemistry 2024 |