
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow synthesis of the antiviral drug tecovirimat and related sp3-rich scaffolds†
Arlene
Bonner
 and
Marcus
Baumann
*
and
Marcus
Baumann
*
University College Dublin, School of Chemistry, Science Centre South, D04 N2E5 Dublin, Ireland. E-mail: marcus.baumann@ucd.ie
First published on 24th September 2024
Abstract
Herein we report a 2-step continuous flow synthesis of the antiviral drug tecovirimat, which is used for the treatment of monkeypox and smallpox. This work exploits a high-temperature pericyclic cascade process between cycloheptatriene and maleic anhydride generating a key sp3-rich scaffold, which affords the desired API after further condensation with an acyl hydrazide. Additional investigations of the key intermediate in reactions with different hydrazines revealed the accessibility of different heterocyclic chemotypes, depending on the substitution pattern of the hydrazine used. Ultimately, the streamlined and scalable access to these sp3-rich scaffolds enables improved access to tecovirimat and structurally related entities with high drug-like character.
Introduction
Recent years have witnessed an increasing demand for chemical motifs that are rich in sp3-character. The advantages of such scaffolds in modern medicinal chemistry campaigns arise from the potentially improved binding of such motifs in restricted binding pockets, as well as improved solubility. This has led to defining the fraction of sp3 hybridised carbon atoms as a metric for assessing drug likeness.1 Classical three-dimensional scaffolds such as norbornenes,2 adamantanes3 and cubanes4 have thus experienced a revival, with new and improved synthetic routes being of high value (Fig. 1).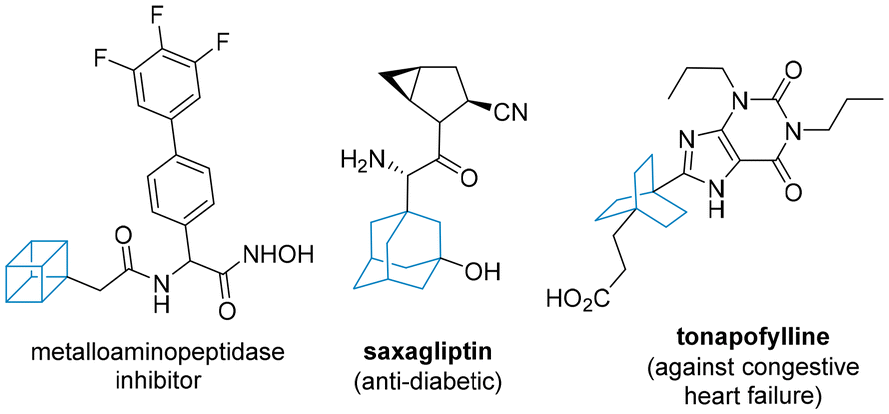 | ||
| Fig. 1 Representative drugs containing 3D-rich scaffolds. | ||
Whilst symmetrical three-dimensional motifs dominate in most reported drugs due to simpler synthetic access and the absence of stereoisomerism, several studies employ scaffolds that are desymmetrised. Crucially, for these scaffolds to be synthetically viable, they must be easily accessible from readily available starting materials and their preparation must be safe and scalable to multi-gram quantities.
A unique polycyclic scaffold can be found in the antiviral drug tecovirimat which is used in the treatment of monkeypox and smallpox.5 The synthesis of the carbocyclic fragment relies on a pericyclic cascade process in which cycloheptatriene and maleic anhydride are reacted at elevated temperature. Detailed studies by Leitich and Sprintschnik have confirmed that an initial 6π-electrocyclisation thereby renders the bicyclic diene, which then engages in a thermal Diels–Alder cycloaddition reaction (Scheme 1).6 Condensation of the resulting anhydride adduct with an acyl hydrazide subsequently affords the desired drug target. Earlier patented routes7 often describe the use of xylenes as high-boiling solvent to facilitate the initial pericyclic cascade, however, more recent studies8 show further improvements in terms of in situ generation of cycloheptatriene, solvent choice and yields en route to kilogram quantities of tecovirimat.
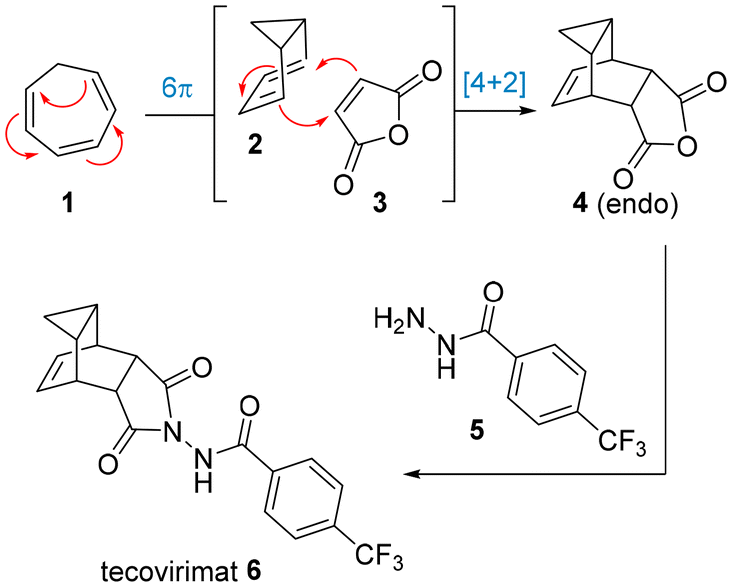 | ||
| Scheme 1 Structure of tecovirimat and its synthesis in batch mode. | ||
Due to our interest in exploiting continuous flow approaches for the intensification of chemical reactions, as well as the generation of strained ring systems that have potential applications in medicinal chemistry programs,9 we wished to develop a fast high-temperature flow process towards tecovirimat. This was expected to deliver multigram quantities of the desired cycloadduct in a safe and streamlined manner which would additionally enable subsequent synthetic studies on this intriguing yet underexplored scaffold. In particular, the use of miniaturised flow set-ups was deemed crucial in achieving improved heat and mass transfer whilst mitigating any safety concerns as only small amounts of material are within the contained flow system at any given time.10 As reported previously, these advantages are crucial in the exploitation of continuous flow technology for the generation of active pharmaceutical ingredients (APIs),11 which oftentimes includes reaction telescoping,12 inline analysis13 and inline purification strategies.14
Results and discussion
We initiated our study towards scaffold 4 using commercially available cycloheptatriene in combination with maleic anhydride. When replicating the reported batch conditions which entailed longer reaction times (3–12 hours)7,8,15 in refluxing xylene (b. p. 139 °C), discolouration of the reaction mixture was observed over time indicating thermal degradation. However, shortening the reaction time to 1 hour afforded scaffold 4 in high yield (83%, entry 1), which is comparable to previous reports.7j Next, we investigated suitable replacements for xylene as maleic anhydride is not sufficiently soluble in this solvent and its evaporation is time consuming. While toluene was initially investigated for the flow process (89%, 145 °C, tRes 30 min, 0.15 M, 4 bar BPR), its non-polar nature restricts solubility of maleic anhydride and consequently, lowers reaction throughput. Thus, acetonitrile (b. p. 83 °C) was favoured as it is more polar and can be superheated above its atmospheric boiling point using a back-pressure regulator (BPR) in flow. A trial reaction in continuous flow mode rendered the desired product in comparable yield (85%, tRes 45 min) under high temperature/high pressure conditions (entry 2, 145 °C, 8 bar BPR). Encouraged by this improvement we wished to evaluate whether an additional temperature increase would accelerate this pericyclic cascade further. As the PFA tubing (perfluoroalkoxy) starts to soften above 150 °C, we opted to use a batch microwave reactor in this instance which pleasingly gave near quantitative conversion in only 15 min reaction time at 169 °C (toluene, entry 3). This result was replicated on 25 mmol scale, giving the desired product in excellent yield with a productivity of 18.6 g h−1 (98 mmol h−1, entry 4). The superiority of the microwave process can be demonstrated by its higher space–time-yield of 0.75 g mL−1 h−1 of scaffold 4, in comparison to those of batch and flow (0.16 g mL−1 h−1 and 0.22 g mL−1 h−1 respectively). Pleasingly, multigram quantities of 4 were found to be bench stable for months at ambient conditions (Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | Reaction mode | Solvent | Temperature and time | Yielda of 4 | Space-time-yield |
| a Isolated yield. b 25 mmol scale. c 10 mmol scale. | |||||
| 1 | Batch | Xylene | 139 °C, 1 h | 83%b | 0.16 g mL−1 h−1 |
| 2 | Flow | MeCN | 145 °C, 45 min | 85%c | 0.22 g mL−1 h−1 |
| 3 | MW | Toluene | 169 °C, 15 min | 98% | 0.75 g mL−1 h−1 |
| 4 | MW | Toluene | 169 °C, 15 min | 98%b | 0.75 g mL−1 h−1 |
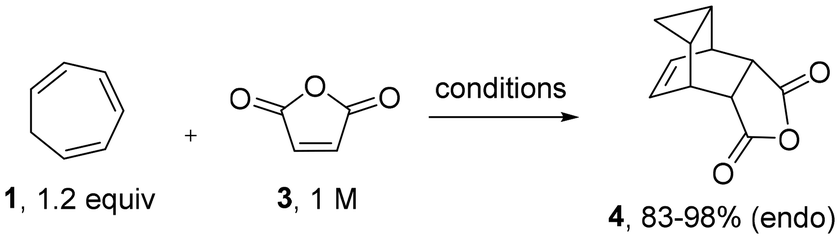
Being able to rapidly generate gram quantities of 4 in both batch (MW) and flow mode, we next investigated the flow synthesis of tecovirimat by condensing the commercially available acyl hydrazide 5 with scaffold 4. For this step ethanol was favoured as a polar, protic solvent and solvent superheating using a BPR was again envisioned to accelerate the desired transformation.
As shown in Table 2, initial trials at 110–130 °C in ethanol in the presence of small amounts of DIPEA gave tecovirimat in up to 85% yield with small amounts of the acyl hydrazide remaining (entries 1–3). Extending the reaction time from 15 to 20 minutes gave a lower yield due to competing side reactions (entry 4). A similar outcome was observed when increasing the temperature to 125 °C (entry 5). Doubling the reaction concentration from 50 mM to 100 mM gave near identical results (entry 6 vs. 2), however, the limited solubility of 4 in ethanol did preclude further increasing the concentration. Lastly, a gram-scale experiment was conducted (entry 7) which afforded tecovirimat in 86% isolated yield after hot recrystallisation from a mixture of ethanol/water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). This equates to a throughput of 3.3 mmol h−1 (ca. 1.3 g h−1) representing a highly efficient continuous synthesis of tecovirimat in a total residence time of 1 hour and an overall yield of 84%. The telescoping of these two steps in an acetonitrile/ethanol mixed solvent system did only give a modest yield of tecovirimat (30% overall yield, unoptimised process).
:1). This equates to a throughput of 3.3 mmol h−1 (ca. 1.3 g h−1) representing a highly efficient continuous synthesis of tecovirimat in a total residence time of 1 hour and an overall yield of 84%. The telescoping of these two steps in an acetonitrile/ethanol mixed solvent system did only give a modest yield of tecovirimat (30% overall yield, unoptimised process).
|
|
|||
|---|---|---|---|
| Entry | Temperature and time | Yielda of 6 | Remaining 5a |
| a Determined by 1H NMR of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. b 0.1 M in ethanol. c 1 g scale. d Isolated yield. | |||
| 1 | 110 °C, 15 min | 77% | 20% |
| 2 | 120 °C, 15 min | 85% | 14% |
| 3 | 130 °C, 15 min | 73% | 7% |
| 4 | 120 °C, 20 min | 63% | 20% |
| 5 | 125 °C, 15 min | 67% | 20% |
| 6b | 120 °C, 15 min | 84% | 14% |
| 7 , | 120 °C, 15 min | 86%d | — |

Having a achieved a robust 2-step flow synthesis for the antiviral API tecovirimat, our next target concerned the study of scaffold 4 as a building block towards related entities. Previous studies by Efremova and coworkers16 had already established that the steric hinderance around the alkene in 4 prevents its reaction in dipolar cycloadditions, which directed our attention to the alternative anhydride functionality. Therefore, we wished to reductively desymmetrise 4via a NaBH4-mediated conversion of the anhydride moiety. As shown in Scheme 2, this transformation gave product 7 as a single diastereoisomer in a high yield of 74% along with the lactone structure 8. The latter can be obtained as the major product (87% yield) when using an excess of the reducing agent. As both products are crystalline materials, single crystal X-ray diffraction was used to unambiguously confirm their structure.17 Pleasingly, gram quantities of 7 could be accessed via this simple protocol which enabled further studies on this versatile scaffold.
 | ||
| Scheme 2 Reductive desymmetrisation of 4 using NaBH4 (X-ray structures are shown at the 50% probability level). | ||
Next, we evaluated the reactivity of compound 7 with different commercially available aryl hydrazine species. This showed that the corresponding condensation products 10 can be obtained in high chemical yields and as single isomers when treating 7 with the desired aryl hydrazine (as HCl salt, 1.5 equiv.) in the presence of triethylamine in refluxing methanol (Scheme 3). As shown below, the desired adducts are obtained in high yields within 1 hour. Amongst the hydrazine partners investigated only 2-fluorophenyl hydrazine showed a slightly different reactivity as a secondary adduct (10d′) was isolated as a minor component under these conditions.
 | ||
| Scheme 3 Derivatisation studies between 7 and aryl hydrazine hydrochlorides. | ||
Interestingly, a different product outcome was observed when subjecting scaffold 7 to analogous condensation conditions with hydrazine hydrate. As depicted in Scheme 4, a 6-membered ring product was obtained in 97% yield whereby two molecules of water were eliminated to give the embedded 4,5-dihydropyridazin-3(2H)-one ring system. These findings differ from the observations of Martin and coworkers18 who reported that both 5- and 6-membered ring products can be formed when condensing hydrazine with simpler 5-hydroxydihydrofuran-2(3H)-ones. Given the different functionalities present, this unexpected outcome opens opportunities for further elaboration of the resulting heterocyclic product (11) to generate libraries of drug-like products.
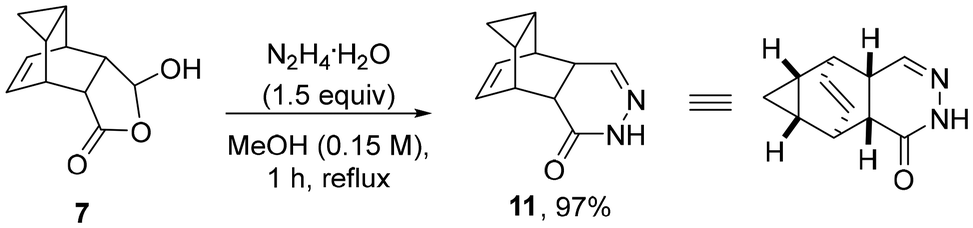 | ||
| Scheme 4 Reaction between scaffold 7 and hydrazine hydrate. | ||
Conclusions
In conclusion, we report an efficient 2-step continuous flow synthesis of the antiviral tecovirimat under high-temperature conditions. Reaction intensification thereby provides access to this API in 1 hour with an overall yield of 84%. Key to this success was the generation of a complex anhydride intermediate (4) with high productivity, which enabled further synthetic studies via a reductive desymmetrisation process. Condensation reactions of the resulting products with different hydrazines afforded additional heterocyclic scaffolds in high chemical yields. These results demonstrate the value of flow-based reaction cascades for the streamlined generation of complex building blocks, that can find applications as medicinally relevant scaffolds.Author contributions
A. B. conducted the experimental work, purified and characterised the reported products, and wrote the initial draft of this paper. M. B. supervised the project and edited the manuscript. A. B. and M. B. secured the funding and designed the research project.Data availability
All data generated during this study are deposited in the ESI.†1H, 19F and 13C-NMR spectra for all reported compounds can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Irish Research Council for providing a postgraduate scholarship (GOIPG/2020/1177, to A.B.). Dr Yannick Ortin and Dr Jimmy Muldoon are thanked for assistance with NMR and MS analysis (UCD). The authors are grateful to Dr Julia Bruno Colmenarez (UCD) for determining the X-ray structures reported in this work. Instrumentation support via the Comprehensive Molecular Analysis Platform (CMAP) initiative under the SFI Research Infrastructure Programme in 2019 (reference 18/RI/5702), and BiOrbic, the SFI Bioeconomy Research Centre (with the support of the School of Chemistry and UCD) and the UCD Equip Funding programme (2020 and 2022) is gratefully acknowledged. We wish to thank Science Foundation Ireland for supporting our research program through a Future Frontiers Project grant (20/FFP-P/8712). We thank Sarah Flynn (CEM Technology (Ireland) Ltd) for the loan of the microwave reactor used in this study.References
- (a) W. Wei, S. Cherukupalli, L. Jing, X. Liu and P. Zhan, Drug Discovery Today, 2020, 25, 1839–1845 CrossRef PubMed; (b) D. C. Combo, K. Tallapragada, R. Jain, J. Chewning, A. A. Mazurov, J. D. Speake, T. A. Hauser and S. Toler, J. Chem. Inf. Model., 2013, 53, 327–342 CrossRef PubMed; (c) J. Meyers, M. Carter, N. Y. Mok and N. Brown, Future Med. Chem., 2016, 8, 1753–1767 CrossRef PubMed.
- G. Calvo-Martín, D. Plano, N. Martínez-Sáez, C. Aydillo, E. Moreno, S. Espuelas and C. Sanmartín, Pharmaceuticals, 2022, 15, 1465 CrossRef PubMed.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef PubMed.
- K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Chem. Rev., 2015, 115, 6719–6745 CrossRef CAS PubMed.
- (a) C. Hanna, Toxicol. Appl. Pharmacol., 1960, 2, 379–391 CrossRef CAS PubMed; (b) Y. Zhang, T. W. Kensler, C. G. Cho, G. H. Posner and P. Talalay, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3147–3150 CrossRef CAS PubMed; (c) D. B. Calne and J. L. Reid, Drugs, 1972, 4, 49–74 CrossRef CAS PubMed; (d) J. D. Parkes, Drugs, 1981, 21, 341–353 CrossRef; (e) R. Jackisch, H. Y. Huang, W. Reimann and N. Limberger, J. Pharmacol. Exp. Ther., 1993, 264, 889–898 Search PubMed; (f) A. Kostelnik, A. Cegan and M. Pohanka, BioMed Res. Int., 2017, 2017, 2532764 Search PubMed; (g) https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-indication-treatment-smallpox (accessed on August 1st, 2024).
- J. Leitich and G. Sprintschnik, Chem. Ber., 1986, 119, 1640–1660 CrossRef.
- (a) R. Jordan, T. R. Bailey, S. R. Rippin and D. Dai, U.S. Pat, US8530509B2, 2013 Search PubMed; (b) R. Jordan, T. R. Bailey, S. R. Rippin and D. Dai, U.S. Pat, US8802714B2, 2014 Search PubMed; (c) S. R. Tyavanagimatt, M. A. C. L. Stone, W. C. Weimers, D. Nelson, T. C. Bolken, D. E. Hruby, M. H. O'Neill, G. Sweetapple and K. A. McCloughan, U.S. Pat, US20110236434A1, 2011 Search PubMed; (d) S. R. Tyavanagimatt, M. A. C. L. S. Anderson, W. C. Weimers, D. Nelson, T. C. Bolken, D. E. Hruby, M. H. O'Neill, G. Sweetapple and K. A. McCloughan, U.S. Pat, US20160107993A1, 2016 Search PubMed; (e) S. R. Tyavanagimatt, M. A. C. L. S. Anderson, W. C. Weimers, D. Nelson, T. C. Bolken, D. E. Hruby, M. H. O'Neill, G. Sweetapple and K. A. McCloughan, U.S. Pat, US20180193308A1, 2018 Search PubMed; (f) S. R. Tyavanagimatt, M. A. C. L. S. Anderson, W. C. Weimers, D. Nelson, T. C. Bolken, D. E. Hruby, M. H. O'Neill, G. Sweetapple and K. A. McCloughan, U.S. Pat, US20180311213A1, 2018 Search PubMed; (g) S. R. Tyavanagimatt, M. A. C. L. S. Anderson, W. C. Weimers, D. Nelson, T. C. Bolken, D. E. Hruby, M. H. O'Neill, G. Sweetapple and K. A. McCloughan, U.S. Pat, US9744154B2, 2017, B2, 2018 Search PubMed; (h) S. R. Tyavanagimatt, M. A. C. L. S. Anderson, W. C. Weimers, D. Nelson, T. C. Bolken, D. E. Hruby, M. H. O'Neill, G. Sweetapple and K. A. McCloughan, U.S. Pat, US10045964B2, 2018 Search PubMed; (i) S. R. Tyavanagimatt, M. A. C. L. Stone, W. C. Weimers, D. Nelson, T. C. Bolken, D. E. Hruby, M. H. O'Neill, G. Sweetapple and K. A. McCloughan, U.S. Pat, US9339466B2, 2016 Search PubMed; (j) M.-X. Dong, J. Zhang, X.-Q. Peng, H. Lu, L.-H. Yun, S. Jiang and Q. Dai, Eur. J. Med. Chem., 2010, 45, 4096–4103 CrossRef PubMed; (k) Q. Dai, M.-X. Dong and J. Hu, Chin. Pat, CN101445478A, 2009 Search PubMed; (l) H. Wang, D. Zhang, Y. Dou, L. Xu, Y. Guo, Y. Sun, Z. Bei, Y. Yang, P. Dong, L. Li and L. Yang, Chin. Pat, CN101906064A, 2010 Search PubMed; (m) R. F. Jordan and S. R. Rippin, WO2004112718A3, 2005; (n) D. L. Hughes, Org. Process Res. Dev., 2019, 23, 1298–1307 CrossRef.
- E. M. Bonku, H. Qin, A. Odilov, F. Yang, X. Xing, X. Wang, S. D. Guma and J. Shen, Org. Process Res. Dev., 2023, 27(11), 1984–1991 CrossRef.
- (a) K. Donnelly and M. Baumann, Chem. Commun., 2021, 57, 2871–2874 RSC; (b) A. Bonner and M. Baumann, Org. Process Res. Dev., 2024, 28, 1567–1575 CrossRef; (c) R. Crawford and M. Baumann, Chem. – Eur. J., 2024, 30, e202401491 CrossRef; (d) M. Smyth, T. S. Moody, S. Wharry and M. Baumann, Synlett, 2024, 285–290 Search PubMed; (e) K. Donnelly and M. Baumann, Chem. – Eur. J., 2024, 30, e202400758 CrossRef CAS PubMed.
- (a) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS; (b) T. Razzaq and C. O. Kappe, Chem. – Asian J., 2010, 5, 1274–1289 CrossRef CAS PubMed; (c) M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC; (d) M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed.
- (a) C. R. Sagandira, S. Nqeketo, K. Mhlana, T. Sonti, S. Gaqa and P. Watts, React. Chem. Eng., 2022, 7, 214–244 RSC; (b) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6729 CrossRef CAS; (c) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS; (d) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS.
- J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- (a) A. M. Kearney, S. G. Collins and A. R. Maguire, React. Chem. Eng., 2024, 9, 990–1013 RSC; (b) J. D. Williams, P. Sagmeister and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2024, 47, 100921 CrossRef.
- (a) J. García-Lacuna and M. Baumann, Beilstein J. Org. Chem., 2022, 18, 1720–1740 CrossRef.
- J. Blümel and F. H. Köhler, Chem. Ber., 1993, 126, 1283–1290 Search PubMed.
- M. M. Efremova, A. P. Molchanov, A. S. Novikov, G. L. Starova, A. A. Muryleva, A. V. Slita and V. V. Zarubaev, Tetrahedron, 2020, 76, 131104 Search PubMed.
- CCDC 2378696 and 2378697† contain the supplementary crystallographic data for this paper (https://www.ccdc.cam.ac.uk/structures).
- (a) F. Delgado, M. R. Martin and M. V. Martin, Tetrahedron, 2001, 57, 4389–4395 Search PubMed; (b) L. Somogyi, Liebigs Ann. Chem., 1985, 1985, 1679–1691 Search PubMed; (c) J. E. Francis, K. J. Doebel, P. M. Schutte, E. F. Bachmann and R. E. Detlefsen, Can. J. Chem., 1982, 60, 1214–1232 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and 1H, 19F and 13C-NMR spectra for all reported compounds. CCDC 2378696 and 2378697. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01379d |
| This journal is © The Royal Society of Chemistry 2024 |