
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Syntheses of bottromycin derivatives via Ugi-reactions and Matteson homologations†
Etienne
Bickel
and
Uli
Kazmaier
 *
*
Institute of Organic Chemistry, Saarland University, P.O. Box 151150, 66041 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
First published on 2nd October 2024
Abstract
New bottromycin derivatives have been prepared using flexible Ugi and Matteson reactions. The Ugi reaction allows the fast and direct assembly of sterically hindered peptide fragments, while the Matteson homologation is excellently suited for the stereoselective synthesis of unusual amino acids like β-methylphenylalanine. Some of the new compounds show excellent activity against Streptococcus pneumoniae.
Introduction
The rise of antimicrobial resistance (AMR) causes severe healthcare problems and, based on urgency and the need for new antibiotics, multidrug-resistant pathogens are classified as the highest priority by the World Health-Organisation (WHO).1 Such pathogens can develop resistance towards almost all currently used antibiotics, so the development of new antimicrobial agents, with new modes of action, is highly desired.2In 1957, Waisvisz et al. reported the isolation of a new peptide from the fermentation broth of Streptomyces bottropensis and called it bottromycin.3 Biosynthetically, bottromycins are formed from ribosomally synthesized peptides via post-translationally modifications.4 Mode of action (MoA) studies indicated that bottromycin inhibits protein biosynthesis by binding to the aminoacyl-tRNA binding site (A site) of the 50S ribosome.5 This biological target would avoid the cross-resistance issue but, to date, has not been addressed by any other antibiotic. Therefore, bottromycin is effective against problematic bacterial strains such as vancomycin-resistant Enterococci (VRE) and methicillin-resistant Staphylococcus aureus (MRSA).4b
Although, the bottromycins were discovered and described as early as the late 1950′s, only one total synthesis of bottromycin A2 and analogues by Ōmura and Sunazuka et al. has been reported.6 A reason for this may be several incorrect structures proposed during these early years.7 The latest, and correct, proposal, as confirmed by the first total synthesis, was described by Shipper in 1983 (Fig. 1).8
 | ||
| Fig. 1 Bottromycin A2. | ||
Several research groups,9 including our own,10 have carried out synthetic studies towards the various bottromycin building blocks and proposed structures. Researchers at AiCuris GmbH (Germany) saponified the C-terminal methyl ester of biotechnologically produced bottromycin and converted the acid into several amides.11 Further modifications on this C-terminal unusual amino acid were carried out by Ōmura and Sunazuka based on their previously developed total synthesis.6 Structure–activity relation (SAR) studies on these different derivatives indicate that especially the β-substituted phenylalanine is required for high activity. Obviously, the methyl group on the phenylalanine influences the conformation of the side chain and probably of the whole molecule as indicated by 1H-NMR.6 Interestingly, while the C-terminal thiazolyl amino acid ester is not required for activity, the free amino acid or its complete removal render the derivatives almost inactive. However, replacing the amino acid by a simple benzylamide had no significant effect on the activity. Clearly, only an amide bond is required, and an aromatic substituent has a positive effect on the activity.
In the context of our interest in the total synthesis of natural products with anticancer12 or antibiotic13 activities, we were motivated to develop a flexible synthetic approach towards bottromycin derivatives (Fig. 2) which should allow us to readily modify the core structure of the molecule.
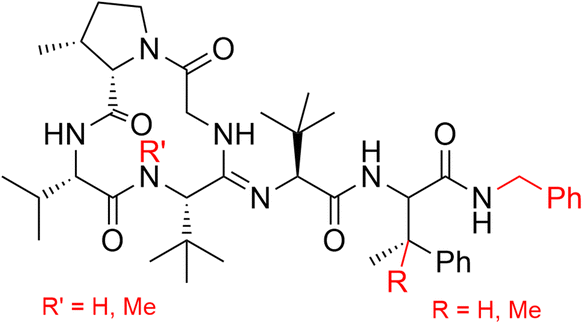 | ||
| Fig. 2 Modification of bottromycin. | ||
Results and discussion
For a flexible synthesis of the side chain, we investigated two different approaches. First, a synthesis of the β-branched phenylalanine should give us the freedom to modify this amino acid. Because β-methylphenylalanine (MePhe) is incorporated not only in bottromycin but also several other natural products,14 various syntheses for this building block already exist, although many are specific for this amino acid. Therefore, we decided to apply the Matteson homologation15 which should give us a versatile and stereoselective means to modify every carbon atom of the amino acid side chain.Starting from the known methyl boronic ester 1,15 addition of lithiated dichloromethane (DCM) (generated in situ by deprotonating DCM with LDA) provided the α-chloroboronic ester 2 in a highly stereoselective fashion (Scheme 1). Addition of phenylmagnesium bromide generated a boronate complex which underwent a 1,2-phenyl shift replacing the chlorine in an SN2 fashion. The desired α-phenyl substituted boronic ester 3 was obtained as a single stereoisomer. Repeating the sequence using vinylmagnesium bromide in the second step gave access to allylboronate 4.
 | ||
| Scheme 1 Synthesis of dipeptide 8via Matteson homologation. Abbreviations: NMM: N-methylmorphiline; HATU: [O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium-hexafluorphosphat]. | ||
According to a protocol described by Morken, 4 was reacted with deprotonated O-methylhydroxylamine resulting in replacement of the boronic ester by an NH2 group,16 which was directly Boc-protected (5). Subsequent ozonolysis and Pinnick oxidation17 gave the N-protected amino acid 6 which was converted into benzylamide 7. Cleavage of the Boc protecting group, peptide coupling, and further N-deprotection gave the desired dipeptide 8. In principle, by using other starting boronic esters or other aryl Grignard reagents, a wide range of derivatives should be accessible.
As a complementary and powerful protocol, we took advantage of the Ugi reaction18 to generate the entire side chain in only one or two steps. In principle, enantiomerically pure 2-phenylpropionaldehyde can be used as the aldehyde component in this 4-component coupling for direct access to the bottromycin side chain. However, we decided to use achiral 2-methyl-2-phenylpropionaldehyde, generating a quaternary β-carbon (Scheme 2). Ugi reaction with Boc–t-leucine (Boc–Tle), ammonia, and benzyl isocyanide in trifluoroethanol generated the desired dipeptide 9 in high yield as a mixture of diastereomers and rotamers. Purification by flash chromatography provided a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture in 60% yield, which was subjected to N-Boc deprotection. The diastereomers of dipeptide 10 with the free amino terminus could be separated by reversed-phase chromatography or crystallisation. The crystals obtained were suitable for X-ray structure analysis which allowed us to determine the absolute configuration of the two diastereomers.
:1 diastereomeric mixture in 60% yield, which was subjected to N-Boc deprotection. The diastereomers of dipeptide 10 with the free amino terminus could be separated by reversed-phase chromatography or crystallisation. The crystals obtained were suitable for X-ray structure analysis which allowed us to determine the absolute configuration of the two diastereomers.
 | ||
| Scheme 2 Synthesis and separation of diastereomers of dipeptide 10. | ||
The Ugi reaction was also used to generate the sterically demanding peptide fragment applying our previously developed thio-Ugi approach (Scheme 3).19 Alloc-protected valine was activated with carbonyldiimidazol (Im2CO) to the corresponding imidazolide which was directly reacted with H2S to afford the corresponding thioacid. Without purification, 11 was reacted with pivalaldehyde, methylamine, and the isocyanide obtained from glycine methyl ester. The reaction proceeded cleanly and the two diastereomers of 12 could be separated by flash chromatography. To determine the configuration of the newly formed stereogenic centre of the central amino acid, we took advantage of a side reaction generally observed later during the formation of the amidine. Hence, stirring the separated diastereomers in the presence of Hg(OTf)2 and lutidine led to desulfuration and the formation of tripeptide 13. The configurations of these tripeptides could be correlated with those obtained by standard peptide coupling reactions.
 | ||
| Scheme 3 Synthesis and separation of diastereomers of thioamides 12 and elucidation of their configuration. Abbreviation: BEP: 2-bromo-1-ethyl pyridinium tetrafluoroborate. | ||
With thiopeptides 12 in hand, we next investigated the crucial amidine formation step (Scheme 4). Both diastereomers of 12 were reacted with dipeptide 8 in the presence of Hg(OTf)2. Although the reaction is relatively fast, the desired amidines 14a and 14b were obtained in only moderate yields. A major side reaction was the desulfuration mentioned above, providing peptide 13. Nevertheless, palladium-catalysed Alloc cleavage and coupling with N-Boc-protected 3-methylproline furnished linear peptides 15a and 15b, which were subjected to peptide cyclisation. The yield in the cyclisation step was significantly higher with the (R)-stereoisomer 16a than the (S)-isomer 16b. This is not surprising, as nearly all naturally occurring cyclotetrapeptides contain at least one (R)-amino acid. Such cyclic peptides generally show a cis–trans–cis–trans configuration of the cyclic peptide backbone.20 This is why we incorporated N-methylated Tle opposite to the Pro in the ring, which should form a cis-amide bond more easily.
 | ||
| Scheme 4 Synthesis of bottromycin derivatives 16. Abbreviation: TPPTS: trinatrium-3,3′,3′′-phosphintriyltribenzolsulfonat. | ||
Based on these positive results, we applied the same reaction conditions to the coupling of 12 with the (S,S)-dipeptide 10. The results obtained in the peptide coupling and cyclisation step to 16c and 16d were comparable. Here too, the (R)-Tle isomer 15c resulted in a better cyclisation yield.
Unfortunately, the Ugi approach could not be applied to the synthesis of N-nonmethylated derivatives. Yields for the thio-Ugi reaction were comparable to those obtained with methylamine but the subsequent amidine formation clearly did not tolerate an NH amide bond between Val and Tle. No amidine formation was observed at all.
Therefore, we used the protocol described by Ōmura and Sunazuka in their bottromycin synthesis (Scheme 5). Coupling of thioamide 17 with our dipeptide 8 and (S,S)-10 provided the desired amidines 18a and 18b in high yield. Cleavage of the phthaloyl protecting group and two subsequent peptide coupling steps gave access to the linear precursors 20a and 20b. The TBDPS ethers were cleaved, and the free primary alcohols were oxidised to the corresponding acids 22a and 22b which were then subjected to cyclisation. The yields obtained in both series were almost identical, including the cyclisation step. The cyclisation yields were unsatisfactory but not unexpected. In these cases, an all (S)-cyclopeptide 23 is sterically unfavourable. In addition, the N-methyl group that facilitates the formation of a cis-amide bond is absent and, in the linear peptide, the amide bond between the two sterically demanding amino acids Val and Tle is certainly trans.
 | ||
| Scheme 5 Synthesis of bottromycin-derivatives 23. | ||
The new bottromycin derivatives were evaluated for their biological activity towards Mycobacterium tuberculosis (Mtb) H37Ra and Mycobacterium smegmatis (Msm) mc2155. Unfortunately, none of the N-methylated derivatives 16 showed any significant activity (MIC >64 μG mL−1), either against the Mycobacterium strains or any other Gram-positive or Gram-negative bacteria or yeasts and fungi. In contrast, some activity was observed for the two derivatives 23 with the unmodified peptide ring (Table 1). Although the activity against Mtb was weaker than that of bottromycin A2, a higher activity was observed against Msm. This led us to investigate the activity against other strains that had not yet been tested. Derivative 23a with the natural β-methylphenylalanine was approximately 10-fold more active than the double β-methylated derivative 23b. The compounds proved to be particularly active against Streptococcus pneumoniae.
| Bacterial strain | MIC (μG mL−1) | ||
|---|---|---|---|
| BotA2 | 23a | 23b | |
| M. tuberculosis H37Ra | 1 | 16 | 32 |
| M. smegmatis mc2155 | 8–16 | 2 | 4 |
| E. faecalis 20478 | 8 | >64 | |
| E. faecium DSM-17050 | 32 | >64 | |
| E. faecium 20477 | 2 | 16 | |
| S. pneumoniae DSM-11865 | <0.03 | 0.5 | |
| S. pneumoniae 20566 | <0.03 | 0.25 | |
Conclusions
In conclusion, we have shown that bottromycins are readily accessible when applying Ugi reactions to generate larger fragments. Unfortunately, in SAR studies, no significant structural changes are tolerated on the peptide ring. N-Methylation facilitates cyclisation but, unfortunately, does not provide active derivatives. Matteson homologation was found to be a suitable tool for the stereoselective synthesis of β-methylated phenylalanine.Data availability
The data supporting this article (copies of 1H, 13C NMR spectra and experimental details) have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Saarland University and the DFG (Ka 880/13-1 and Bruker Neo 500 - 447298507) is gratefully acknowledged.References
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini and the WHO Pathogens Priority List Working Group, Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- (a) B. A. Cunha, Antibiotic resistance: A historical perspective, Semin. Respir. Crit. Care Med., 2000, 21, 3–8 CrossRef PubMed; (b) Z. Lin, T. Yuan, L. Zhou, S. Cheng, X. Qu, P. Lu and Q. Feng, Impact factors of the accumulation, migration and spread of antibiotic resistance in the environment, Environ. Geochem. Health, 2021, 43, 1741–1758 CrossRef CAS PubMed.
- (a) J. M. Waisvisz, M. G. van der Hoeven, J. van Peppen and W. C. M. Zwennis, Bottromycin. I. A new sulfurcontaining antibiotic, J. Am. Chem. Soc., 1957, 79, 4520–4521 CrossRef CAS; (b) J. M. Waisvisz, M. G. van der Hoeven, J. F. Hölscher and B. te Nijenhuis, Bottromycin. II. Preliminary degradation studies, J. Am. Chem. Soc., 1957, 79, 4522–4523 CrossRef CAS; (c) J. M. Waisvisz, M. G. van der Hoeven and B. te Nijenhuis, The structure of the sulfur-containing moiety of bottromycin, J. Am. Chem. Soc., 1957, 79, 4524–4527 CrossRef CAS.
- (a) T. H. Eyles, N. M. Vion and A. W. Truman, Rapid and Robust Yeast-Mediated Pathway Refactoring Generates Multiple New Bottromycin-Related Metabolites, ACS Synth. Biol., 2018, 7, 1211–1218 CrossRef CAS PubMed; (b) L. Franz, U. Kazmaier, A. W. Truman and J. Koehnke, Bottromycins - biosynthesis, synthesis and activity, Nat. Prod. Rep., 2021, 38, 1659–1683 RSC; (c) P. H. Krushnamurthy, K. S. Subramanya, D. Simita, G. Dhananjaya and M. Nilkamal, Recent Advancements in Bottromycin Biosynthesis, Synlett, 2023, 793–806 Search PubMed.
- (a) T. Otaka and A. Kaji, Mode of action of bottromycin A2. Release of aminoacyl or peptidyl tRNA from ribosomes, J. Biol. Chem., 1976, 251, 2299–2306 CrossRef PubMed; (b) T. Otaka and A. Kaji, Mode of action of bottromycin A2: Effect of bottromycin A2 on polysomes, FEBS Lett., 1983, 153, 53–59 CrossRef PubMed; (c) T. Otaka and A. Kaji, Mode of action of bottromycin A2: effect on peptide bond formation, FEBS Lett., 1981, 123, 173–176 CrossRef PubMed.
- (a) H. Shimamura, H. Gouda, K. Nagai, T. Hirose, M. Ichioka, Y. Furuya, Y. Kobayashi, S. Hirono, T. Sunazuka and S. Ōmura, Structure determination and total synthesis of bottromycin A2: a potent antibiotic against MRSA and VRE, Angew. Chem., Int. Ed., 2009, 48, 914–917 CrossRef PubMed; (b) Y. Kobayashi, M. Ichioka, T. Hirose, K. Nagai, A. Matsumoto, H. Matsui, H. Hanaki, R. Masuma, Y. Takahashi, S. Ōmura and T. Sunazuka, Bottromycin derivatives: Efficient chemical modifications of the ester moiety and evaluation of anti-MRSA and anti-VRE activities, Bioorg. Med. Chem. Lett., 2010, 20, 6116–6120 CrossRef PubMed; (c) T. Yamada, M. Yagita, Y. Kobayashi, G. Sennari, H. Shimamura, H. Matsui, Y. Horimatsu, H. Hanaki, T. Hirose, S. Ōmura and T. Sunazuka, Synthesis and evaluation of antibacterial activity of bottromycins, J. Org. Chem., 2018, 83, 7135–7149 CrossRef PubMed.
- (a) S. Nakamura, T. Chikaike, H. Yonehara and H. Umezawa, Structures of bottromycins A and B, J. Antibiot., Ser. A, 1965, 18, 60–61 Search PubMed; (b) S. Nakamura, N. Tanaka and H. Umezawa, Bottromycin A1, A2 and their structures, J. Antibiot., 1966, 19, 10–12 Search PubMed; (c) Y. Takahashi, H. Naganawa, T. Takita, H. Umezawa and S. Nakamura, The revised structure of bottromycin A2, J. Antibiot., 1976, 29, 1120–1123 CrossRef PubMed.
- D. Schipper, The revised structure of Bottromycin-A2, J. Antibiot., 1983, 36, 1076–1077 CrossRef PubMed.
- U. Kazmaier, The long, long way to Bottromycin, Isr. J. Chem., 2021, 61, 308–321 CrossRef.
- S. Ackermann, H.-G. Lerchen, D. Haebich, A. Ullrich and U. Kazmaier, Synthetic studies towards bottromycin, Beilstein J. Org. Chem., 2012, 8, 1652–1656 CrossRef PubMed.
- H.-G. Lerchen, G. Schiffer, H. Broetz-Oesterhelt, A. Mayer-Bartschmid, S. Eckermann, C. Freiberg, R. Endermann, J. Schuhmacher, H. Meier, N. Svenstrup, S. Seip, M. Gehling and D. Haebich, Fermentative and chemical synthesis of bottromycin derivative analogs for use in treatment or prevention of bacterial infections in humans or animals, WO2006103010A1, 2006.
- (a) A. Ullrich, Y. Chai, D. Pistorius, Y. A. Elnakady, J. E. Herrmann, K. J. Weissman, U. Kazmaier and R. Müller, Pretubulysin, a potent and chemically-accessible tubulysin precursor from Angiococcus disciformis”, Angew. Chem., Int. Ed., 2009, 47, 4422–4425 CrossRef PubMed; (b) L. Karmann, K. Schulz, J. Herrmann, R. Müller and U. Kazmaier, Total syntheses and biological evaluation of miuraenamides, Angew. Chem., Int. Ed., 2015, 54, 4502–4507 CrossRef PubMed; (c) J. Gorges and U. Kazmaier, Matteson homologation-based total synthesis of lagunamide A, Org. Lett., 2018, 20, 2033–2036 CrossRef PubMed; (d) O. Andler and U. Kazmaier, Total synthesis of apratoxin A using Matteson's homologation approach, Org. Biomol. Chem., 2021, 19, 4866–4870 RSC.
- (a) P. Barbie and U. Kazmaier, Total synthesis of cyclomarin A, a marine cycloheptapeptide with anti-tuberculosis and anti-malaria activity, Org. Lett., 2016, 18, 204–207 CrossRef PubMed; (b) T. Kinsinger and U. Kazmaier, C-H-Functionalization of N-methylated amino acids and peptides as tool in natural product synthesis – Synthesis of abyssenine A and mucronine E, Org. Lett., 2018, 20, 7726–7730 CrossRef PubMed; (c) J. Greve, A. Mogk and U. Kazmaier, Total synthesis and biological evaluation of modified ilamycin derivatives, Mar. Drugs, 2022, 20, 632 CrossRef PubMed; (d) R. Priester and U. Kazmaier, A straightforward synthesis of emericellamide A using Matteson's homologation approach, Synlett, 2023, 2159–2164 Search PubMed; (e) O. Andler and U. Kazmaier, Matteson homologation-based total synthesis of meliponamycin A, Org. Lett., 2024, 26, 148–152 CrossRef PubMed.
- (a) S. Fuse, H. Koinuma, A. Kimbara, M. Izumikawa, Y. Mifune, H. He, K. Shin-ya, T. Takahashi and T. Doi, Total synthesis and stereochemistry revision of mannopeptimycin aglycone, J. Am. Chem. Soc., 2014, 136, 12011–12017 CrossRef PubMed; (b) C. S. V. Houge-Frydrych, M. L. Gilpin, P. W. Skett and J. W. Tyler, SB-203207 and SB-203208, two novel isoleucyl tRNA synthetase inhibitors from a Streptomyces sp. II. Structure determination, J. Antibiot., 2000, 53, 364–372 CrossRef PubMed.
- (a) D. S. Matteson, Boronic esters in stereodirected synthesis, Tetrahedron, 1989, 7, 1859–1885 CrossRef; (b) D. S. Matteson, Boronic esters in asymmetric synthesis, J. Org. Chem., 2013, 78, 10009–10023 CrossRef PubMed; (c) S. Kirupakaran, H.-S. Kort and C. Hirschhäuser, A complementary toolbox of iterative methods for the stereoselective synthesis of heteroatom-rich motives from C1-building blocks, Synthesis, 2018, 2307–2322 Search PubMed; (d) D. S. Matteson, B. S. L. Collins and V. K. Aggarwal, The Matteson Reaction, Org. React., 2021, 105, 427–860 Search PubMed.
- (a) S. N. Mlynarski, A. S. Karns and J. P. Morken, Direct stereospecific amination of alkyl and aryl pinacol boronates, J. Am. Chem. Soc., 2012, 134, 16449–16451 CrossRef PubMed; (b) E. K. Edelstein, A. C. Grote, M. D. Palkowitz and J. P. Morken, A protocol for direct stereospecific amination of primary, secondary, and tertiary alkylboronic esters, Synlett, 2018, 1749–1752 Search PubMed; (c) P. Xu, M. Zhang, B. Ingoglia, C. Allais, A.-M. R. Dechert-Schmitt, R. A. Singer and J. P. Morken, Construction of azacycles by intramolecular amination of organoboronates and organobis (boronates), Org. Lett., 2021, 23, 3379–3383 CrossRef PubMed.
- (a) B. O. Lindgren, T. Nilsson, S. Husebye, Ø. Mikalsen, K. Leander and C.-G. Swahn, Preparation of carboxylic acids from aldehydes (including hydroxylated benzaldehydes) by oxidation with chlorite, Acta Chem. Scand., 1973, 27, 888–890 CrossRef; (b) B. S. Bal, W. E. Childers and H. W. Pinnick, Oxidation of α,β-unsaturated aldehydes, Tetrahedron, 1981, 37, 2091–2096 CrossRef.
- (a) I. Ugi, Isonitrile Chemistry, Academic press, New York, 1971 Search PubMed; (b) A. Dömling and I. Ugi, Multicomponent reactions with isocyanides, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef; (c) A. Ullrich and U. Kazmaier, Organic Reactions, Vol 112a: A half-century of the Ugi reaction: classic variant, John Wiley & Sons, Hoboken, New Jersey, 2023, pp. 1–1050 Search PubMed; (d) C. Liu, L. G. Voskressensky and E. V. Van der Eycken, Recent Advances in the Synthesis of Peptidomimetics via Ugi Reactions, Chem. – Eur. J., 2024, 30, e202303597 CrossRef PubMed.
- (a) S. Heck and A. Dömling, A versatile multi-component one-pot thiazole synthesis, Synlett, 2000, 424–426 Search PubMed; (b) M. Umkehrer, J. Kolb, C. Burdack and W. Hiller, 2, 4, 5-Trisubstituted thiazole building blocks by a novel multi-component reaction, Synlett, 2005, 79–82 Search PubMed; (c) A. Dömling and K. Illgen, 1-Isocyano-2-dimethylamino-alkenes: Versatile reagents in diversity-oriented organic synthesis, Synthesis, 2005, 662–667 Search PubMed; (d) U. Kazmaier and S. Ackermann, A straightforward approach towards thiazoles and endothiopeptides via Ugi reaction, Org. Biomol. Chem., 2005, 3, 3184–3187 RSC; (e) U. Kazmaier and A. Persch, A straightforward approach towards 5-substituted thiazolylpeptides via thio-Ugi-reaction, Org. Biomol. Chem., 2010, 8, 5442–5447 RSC.
- (a) H. Kessler, R. Gratias, G. Hessler, M. Gurrath and G. Müller, Conformation of cyclic peptides. Principle concepts and the design of selectivity and superactivity in bioactive sequences by ‘spatial screening’, Pure Appl. Chem., 1996, 68, 1201–1205 CrossRef; (b) N. Loiseau, J.-M. Gomis, J. Santolini, M. Delaforge and F. André, Predicting the conformational states of cyclic tetrapeptides, Biopolymers, 2003, 69, 363–385 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization, copies of 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d4ob01373e |
| This journal is © The Royal Society of Chemistry 2024 |