
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of lipid A derived from commensal Bacteroides†
Enrico C. J. M.
Verpalen
a,
Anna M.
Ehlers
a,
Aldo C. A.
van Wingaarden
 a,
Arwin J.
Brouwer
a and
Geert-Jan
Boons
*abcd
a,
Arwin J.
Brouwer
a and
Geert-Jan
Boons
*abcd
aDepartment of Chemical Biology and Drug Discovery, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, 3584 CG Utrecht, The Netherlands. E-mail: g.j.p.h.boons@uu.nl
bComplex Carbohydrate Research Center, University of Georgia, Athens, GA 30602, USA. E-mail: gjboons@ccrc.uga.edu
cBijvoet Center for Biomolecular Research, Utrecht University, Utrecht, The Netherlands
dChemistry Department, University of Georgia, Athens, GA 30602, USA
First published on 4th October 2024
Abstract
The inflammation-inducing properties of lipopolysaccharides (LPS) of Gram-negative bacteria reside in their lipid A moiety. Bacillus fragilis, which is a commensal Gram-negative bacterium, biosynthesises lipid A that is structurally distinct from that of E. coli and other enteric bacteria. It is composed of a β1,6-linked glucosamine (GlcN) disaccharide that is only phosphorylated at the anomeric center. The major species of B. fragilis has five fatty acids and the amine of the distal GlcN moiety carries the unusual (R)-3-(13-methyltetradecanoyloxy)-1.5-methylhexadecanoic acid. A recent study indicates that the LPS of B. fragilis has anti-viral activity by selective induction of interferon (IFN)-β and is protective in mouse models of vesicular stomatitis virus (VSV) and influenza A. Heterogeneity in the structures of LPS and lipid A and possible contamination with other inflammatory components make it difficult to unambiguously define the immune-modulatory properties of LPS or lipid A. Therefore, we developed a synthetic approach for the preparation of the unusual major lipid A species derived from B. fragilis, which includes a synthetic approach for (R)-3-(13-methyltetradecanoyloxy)-1.5-methylhexadecanoic acid by the Wittig olefination to install the terminal isopropyl moiety. The proinflammatory and antiviral responses of synthetic B. fragilis lipid A were investigated in several cell lines and primary human monocytes by examining the production of interleukin (IL)-6 and IFN-β. It was found that B. fragilis does not induce the production of IL-6 and IFN-β but can partially antagonize the production of pro-inflammatory cytokines induced by E. coli LPS and lipid A.
Introduction
Members of the genus Bacteroides, such as B. thetaiotaomicron and B. fragilis, are commensal Gram-negative bacteria that are abundant in the human ileum and large intestine.1 They benefit the host by their ability to degrade indigestible plant polysaccharides, thereby providing cross feeding. Furthermore, several cell surface structures of Bacteroidetes, most notably lipopolysaccharide (LPS), exhibit immunomodulatory properties potentially beneficial for the host.2,3LPS extracted from B. fragilis has low endotoxic activity and can antagonize proinflammatory responses induced by enteric LPS.4 Furthermore, LPS from B. vulgatus, which is a commensal bacterium of the murine intestine, can restore intestinal homeostasis in mice in which colitis was induced.5,6 A recent study has indicated that a heterogeneous LPS mixture of B. fragilis has anti-viral activity by selective induction of interferon (IFN)-β and is protective in mouse models of vesicular stomatitis virus (VSV) and influenza A virus.4 Microbial compounds that induce IFN-β have the potential to function as therapeutics for various human diseases, making the LPS of B. fragilis an interesting target to pursue such opportunities.
The inflammation-inducing properties of LPS reside in its lipid A moiety.7 Lipid A can be recognized by a heterodimeric complex of Toll-like receptor (TLR) 4 and myeloid differentiation factor 2 (MD-2) that is found on the surface of myeloid cells.8 The binding of lipid A results in the formation of an m-shaped receptor multimer comprised of two TLR4–MD2–LPS complexes.9 The resulting dimerization of the intracellular domains of two TLR4s creates binding sites for adapter proteins – myeloid differentiation primary response protein (MyD88) and Toll/IL-1R domain containing adaptor-inducing IFN-β (TRIF). MyD88-dependent cellular activation leads to the production of (pro)inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6. On the other hand, the TRIF-dependent pathway induces phosphorylation and dimerization of the transcription factor interferon regulatory factor 3 (IRF-3), which then activates the IFN pathway, resulting in IFN-β production. Thus, it appears that the LPS of B. fragilis B. skews immune cell activation towards the TRIF pathway.4
Bacteroidetes species, such as B. fragilis, biosynthesise lipid As that are structurally distinct from that of E. coli and other enteric bacteria (Fig. 1).10,11 They are composed of a β1,6-linked glucosamine (GlcN) disaccharide that is only phosphorylated at the reducing anomeric center. The major species of B. fragilis has five fatty acids. The amine of the proximal GlcN moiety is modified as (R)-3-hydroxyhexadecanoate and the C-3 hydroxyl as 3-(R)-hydroxylpentadecanoyl ester. The amine of the distal GlcN moiety carries the unusual (R)-3-(13-methyltetradecanoyloxy)-1.5-methylhexadecanoic acid and the C-3′ position is modified by an ester of (R)-3-hydroxyhexadecanoic acid. Lipid As from other Bacteroidetes species, including that of B. thetaiotaomicron, appear to have similar structures.2 On the other hand, lipid A from E. coli is 1,4′-bis-phosphorylated and has (R)-3-hydroxymyristyl residues at the C-2, C-2′, C-3 and C-3′ positions (Fig. 1). The (3)-hydroxyacyl moieties of the distal GlcN moiety are further modified by lauric and myristic acids.
 | ||
| Fig. 1 Structures of B. fragilis-type lipid A and E. coli-type lipid A. | ||
Human gut microbes, including Bacteroidetes, are in general resistant to inflammation-associated antimicrobial peptides.12 Interestingly, a mutant of Bacteroides thetaiotaomicron that cannot remove the C-4′ phosphate from its lipid A was displaced from the microbiota during inflammation caused by infection.13 Thus, it appears that the fine structural features of lipid A of Bacteroides contribute not only to immune-modulatory activity but also to maintenance of a healthy microbiota.
Heterogeneity in the structure of lipid A and possible contaminations with other inflammatory components make it difficult to unambiguously define the immune properties of LPS or lipid A.14,15 Chemical synthesis offers an attractive approach to obtain well-defined lipid A derivatives for structure–activity relationship studies.7,16–18 Therefore, we set out to develop a synthetic approach to prepare the major lipid A derived from B. fragilis. It includes a synthetic approach for the unusual (R)-3-(13-methyltetradecanoyloxy)-1.5-methylhexadecanoic acid by the Wittig olefination to install the terminal isopropyl moiety.19 The resulting synthetic lipid A was examined for its ability to induce the production of pro-inflammatory (IL-6) and anti-viral (IFN-β) cytokines and the results were compared with similar responses induced by LPS, lipid A and monophosphoryl lipid A from E. coli. In addition, the antagonistic properties of lipid A from B. fragilis to reduce proinflammatory responses induced by LPS and lipid A were investigated. It was found that lipid A from B. fragilis does not induce the production of IL-6 and IFN-β but can partially antagonize the production of pro-inflammatory cytokines induced by E. coli LPS and lipid A.
Results and discussion
Chemical synthesis
It was envisaged that lipid A from B. fragilis (1) could be synthesised from glycosyl donor 2 and acceptor 3 and lipids 4, 5, and 6 (Fig. 2). 2-Methylnaphthyl ethers (Nap) was selected as permanent protecting group because it can be more readily removed by hydrogenation20,21 compared to the conventionally employed benzyl ether. The C-3 hydroxyl groups of glycosyl donor 2 and acceptor 3 were modified by (R)-3-(2-naphthylmethoxy)hexadecanoic ester and (R)-3-(2-naphthylmethoxy)pentadecanoic ester, respectively, to minimize the number of synthetic steps that need to be performed at the disaccharide stage. The amine of the donor was protected as 2,2,2-trichloroethoxycarbonyl (Troc) carbamate because it can perform neighboring group participation during glycosylation, resulting in the selective formation of 1,2-trans-glycosides. It can be selectively cleaved by zinc in acetic acid to give a free amine that can be acylated with 6. The amine of acceptor 3 was protected as 9-fluorenylmethyloxycarbonyl (Fmoc) carbonate because it can be removed under mild basic conditions without influencing other functionalities, allowing the introduction of 4. The anomeric phosphate was installed at a late stage using diallyl diisopropylphosphoramidite and can be readily deprotected by PdCl2.22 | ||
| Fig. 2 B. fragilis lipid A (1) and building blocks (2–6) for chemical synthesis. | ||
The preparation of glycosyl donor 2 started from compound 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 having an amino masking azido functionality at the C-2 position. The C-4 and C-6 hydroxyls of this compound can be readily protected by Nap ethers by Williamson ether synthesis using 2-methylnaphthyl bromide (NapBr) in the presence of NaH in DMF to give compound 8. We also attempted to install Nap ethers on a similar compound having Troc at the C-2 position under acid conditions using O-(2-naphthylmethyl) trichloroacetamidate; however, these efforts were unsuccessful.24 Next, the azido group of 8 was reduced using Zn in a mixture of THF and acetic acid and the resulting amine was immediately protected as Troc carbamate by reaction with 2,2,2-trichloroethoxycarbonyl chloride (TrocCl) in the presence of N,N-diisopropylethylamine (DIPEA). After silica gel column chromatography, 9 was obtained in a yield of 59% over two steps. The allyl ether at the C-3 position of 9 was removed using PdCl2 in a mixture of CH2Cl2 and MeOH to give 10. In the next step, the C-3 hydroxyl group of 10 was modified by the Steglich acylation using lipid 4 in the presence of N,N-dicyclohexylcarbodiimide (DCC) and a catalytic amount of 4-(dimethylamino)pyridine (DMAP) to give 11. The anomeric thexyldimethylsilyl (TDS) group of 11 was removed by HF in pyridine and the resulting 12 could be isolated by precipitation from water (Scheme 1). Finally, conversion of the anomeric hydroxyl group of 12 into N-phenyl trifluoroimidate25,26 using 2,2,2-trifluoro-N-phenyl acetimidoyl chloride in the presence of caesium carbonate in CH2Cl2 afforded donor 2 in a yield of 96%.
23 having an amino masking azido functionality at the C-2 position. The C-4 and C-6 hydroxyls of this compound can be readily protected by Nap ethers by Williamson ether synthesis using 2-methylnaphthyl bromide (NapBr) in the presence of NaH in DMF to give compound 8. We also attempted to install Nap ethers on a similar compound having Troc at the C-2 position under acid conditions using O-(2-naphthylmethyl) trichloroacetamidate; however, these efforts were unsuccessful.24 Next, the azido group of 8 was reduced using Zn in a mixture of THF and acetic acid and the resulting amine was immediately protected as Troc carbamate by reaction with 2,2,2-trichloroethoxycarbonyl chloride (TrocCl) in the presence of N,N-diisopropylethylamine (DIPEA). After silica gel column chromatography, 9 was obtained in a yield of 59% over two steps. The allyl ether at the C-3 position of 9 was removed using PdCl2 in a mixture of CH2Cl2 and MeOH to give 10. In the next step, the C-3 hydroxyl group of 10 was modified by the Steglich acylation using lipid 4 in the presence of N,N-dicyclohexylcarbodiimide (DCC) and a catalytic amount of 4-(dimethylamino)pyridine (DMAP) to give 11. The anomeric thexyldimethylsilyl (TDS) group of 11 was removed by HF in pyridine and the resulting 12 could be isolated by precipitation from water (Scheme 1). Finally, conversion of the anomeric hydroxyl group of 12 into N-phenyl trifluoroimidate25,26 using 2,2,2-trifluoro-N-phenyl acetimidoyl chloride in the presence of caesium carbonate in CH2Cl2 afforded donor 2 in a yield of 96%.
 | ||
| Scheme 1 Preparation of glycosyl donor 2. | ||
Glycosyl acceptor 3 could easily be prepared from 1327 and 5 (Scheme 2). Thus, the carbohydrate building block 13 was acylated with 5 using DCC in the presence of a catalytic amount of DMAP to give 14. Next, Troc carbamate of 14 was replaced by Fmoc by a two-step procedure entailing the reductive removal of Troc by Zn in THF and acetic acid to give the free amine 15 that was reacted with 9-fluorenylmethyloxycarbonyl chloride to give 16 in an overall yield of 61%. Reductive opening of the 4,6-acetal of 16 using dichlorophenylborane and triethylsilane26,28 in CH2Cl2 at −78 °C afforded acceptor 3 in a yield of 51%.
 | ||
| Scheme 2 Preparation of glycosyl acceptor 3. | ||
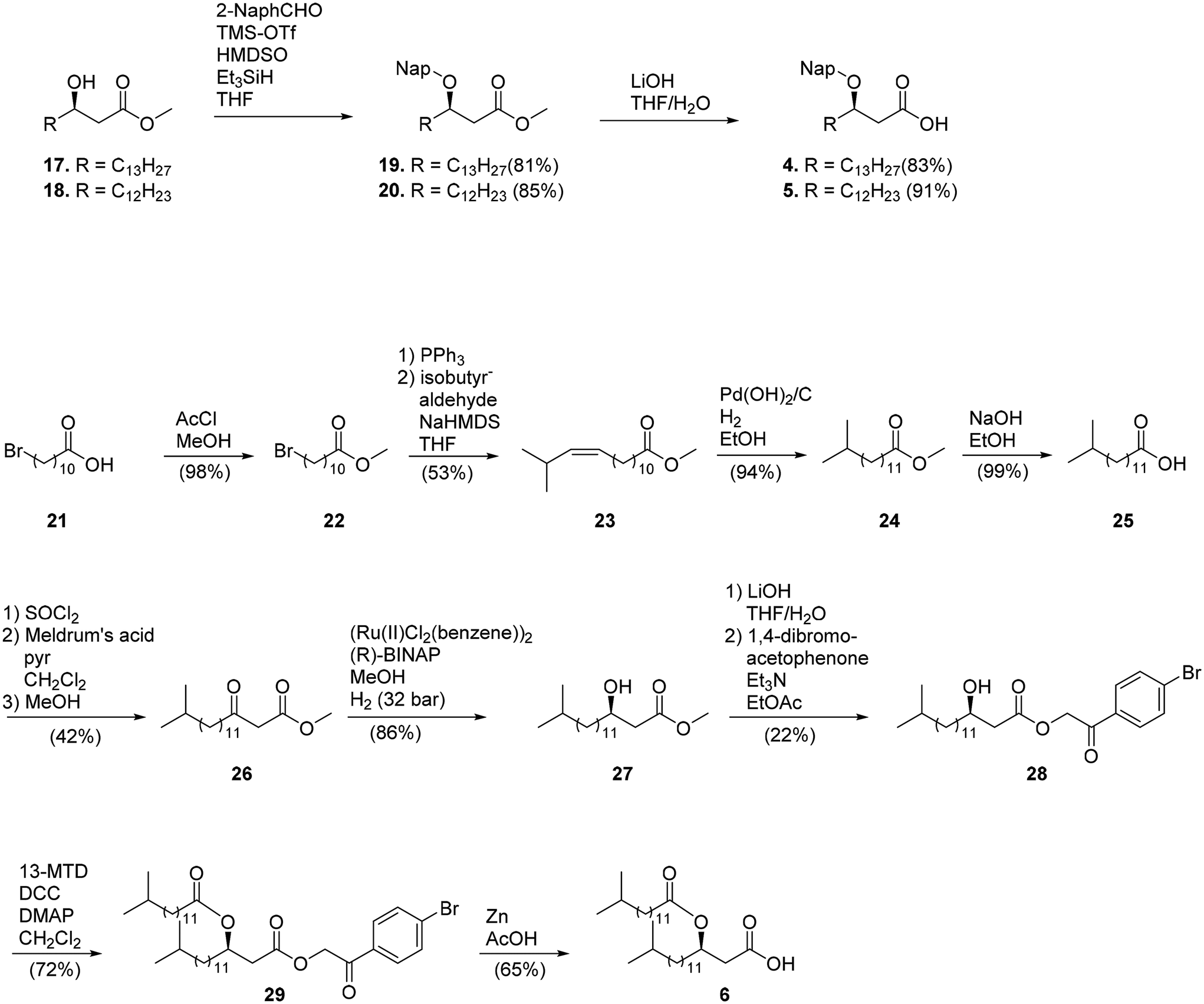
The preparation of 3-(R)-2-naphthylmethoxyhexadecanoic acid (4) and 3-(R)-(2-naphthylmethoxy)pentadecanoic acid (5) started from 1729,30 and 18,31 respectively. Reductive conditions were employed to install the Nap ether32 to prevent epimerization of the chiral center under basic conditions, resulting in 19 and 20. Saponification of the methyl ester afforded the protected acids 4 and 5. 13-Methyltetradecanoic acid (13-MTD; 25) was prepared by a five-step procedure starting from commercially available 11-bromoundecanoic acid (Scheme 3). Esterification of this compound using acetyl chloride in methanol gave 22. The latter compound was treated with triphenylphosphine (neat) at 140 °C to give the corresponding triphenylphosphonium bromide salt that was used without purification in a Wittig reaction with isobutyraldehyde to provide methyl 14-methyltetradecano-11-enate 2 mainly as the cis-isomer. Hydrogenation of the double bond of 23 using Pd(OH)2 gave 24 that was treated with NaOH to saponify the ester to provide 25.
 | ||
| Scheme 3 Preparation of lipids 4 and 5 (top) and bi-antennary lipid 6 (bottom). | ||
Having 25 in hand, attention was focused on the preparation of lipid 6. Thus, 25 was converted into an acyl chloride by reflux in thionyl chloride and then coupled with Meldrum's acid (2,2-dimethyl-1,3-dioxane-4,6-dione) in a mixture of pyridine and dichloromethane to give an intermediate malonate that was subjected to decarboxylation in methanol to provide β-ketoester 26. The ketone of the latter compound was enantioselectively reduced by catalytic hydrogenation in the presence of freshly prepared (R)-BINAP-RuCl2 to give 3-hydroxyl fatty acid 27.33 The enantiomeric excess was determined to be 99% by optical rotation measurement. The methyl ester was saponified and the resulting carboxylic acid was protected as a 2-(4-bromophenyl)-2-oxoethyl ester by reaction with 4-bromoacetophenone to give 28. Esterification of the C-3 hydroxyl of 28 with 25 using DCC in the presence of catalytic DMAP resulted in the formation of 29. The 2-(4-bromophenyl)-2-oxoethyl ester could be selectively cleaved by activated Zn in AcOH to give, after purification by silica gel chromatography, compound 6.
A trifluoromethanesulfonic acid (TfOH) catalyzed glycosylation of glycosyl acceptor 3 and donor 2 resulted in the selective formation of β-glycoside 30 in a yield of 71% (Scheme 4). Next, the Troc group of 30 was removed by activated Zn to give a free amine. Multiple reaction conditions were explored to acylate the amine with 6 to give 31. Low yields were obtained using DCC in combination with K-OxymaPure or hydroxybenzotriazole (HOBt), which in part was due to coupling of the secondary carboxylate to the C-2 amine. Gratifyingly, the desired compound 31 was obtained in a yield of 38% using DCC alone and under these conditions, no side product formation was observed. The Fmoc group of 31 was deprotected using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and the resulting amine was acylated with 4 using DCC and K-OxymaPure to afford 32 in a yield of 72%. The anomeric TDS group was removed using HF in a mixture of pyridine and THF to give pyranose 33. Treatment of the latter compound with di-allyl N,N-diisopropylphosphoramidite in the presence of 1H-tetrazole resulted in the stereoselective installation of an alpha-anomeric phosphite that was in situ oxidized using the mild oxidant tert-butyl hydroperoxide to give α-anomeric phosphotriester 34. The complete deprotection of 34 was carried out in two steps, from which the first was cleavage of the allyl esters of the phosphotriester using PdCl2 in a mixture of CH2Cl2 and MeOH. In the second step, the obtained anomeric phosphate 35 was subjected to catalytic hydrogenation over Pd black in THF for cleavage of the Nap protecting groups. Initial attempts at room temperature and low pressure H2 (g) (hydrogenation balloon) were not successful, leading to only partial deprotection. After varying the reaction temperature and pressure, the optimal conditions were found to be at 50 °C and 50 bar of H2. Target compound 1 was obtained in a yield of 30% after purification by Sephadex LH-20 size exclusion column chromatography and trituration from petroleum ether. The anomeric phosphate was found to be prone to cleavage when a 1:1 mixture of dichloromethane and methanol was used as the eluent for Sephadex LH-20 column chromatography. NMR analysis indicated that at least 60% of the phosphate was cleaved. Fortunately, when employing a mixture of CHCl3/methanol/H2O (2/3/1), no cleavage of the anomeric phosphate was observed, even after 5 days. The stability of an anomeric phosphate in this solvent mixture was found to be comparable with other anomeric phosphates in the literature.34 The structure was confirmed by NMR (1H, 31P and 2D NMR) and MALDI-TOF MS analysis.
 | ||
| Scheme 4 Assembly and deprotection of B. fragilis lipid A (1). | ||
Biological evaluation
Next, we evaluated the immunomodulatory properties of lipid A from B. fragilis and compared them with the similar properties of E. coli 055:B5 LPS, and lipid A and monophosphoryl lipid A from E. coli.35 Release of IL-6 was evaluated in primary human monocytes by applying a wide concentration range (0.01 nM–1000 nM) of the compounds for 16 h and unstimulated cells served as the negative control. As expected, E. coli 055:B5 LPS showed the highest potency with an EC50 (concentration producing 50% activity) of 0.005 nM. Lipid A from E. coli showed a somewhat lower potency (EC50 of 0.14 nM) and efficacy (maximum or plateau level). On the other hand, monophosphoryl lipid A from E. coli and lipid A from B. fragilis (1) only triggered low levels of pro-inflammatory cytokine production at a concentration of 10 nM and 300 nM, respectively (Fig. 3A) and unstimulated human monocytes did not release any IL-6. | ||
| Fig. 3 Biological activity of lipid A from B. fragilis to induce pro- and anti-viral cytokine secretion. (a) E. coli 055:B5 LPS, (b) B. fragilis lipid A, (c) E. coli lipid A, and (d) E. coli monophosphoryl lipid A were administered to (A) human primary monocytes (n = 4, 16 h) and (B) the hTLR4 SEAP HEK-Blue reporter cell line (control: Null2 SEAP HEK-Blue, n = 2, 24 h) using a wide concentration range (0.0001 nM–1000 nM); 100 ng mL−1 LPS corresponds to 10 nM in accordance with an average molecular mass of 10 kDa.36,37 Representative data, expressed as relative NF-κB activation, are shown as the mean of two biological replicates. EC50 values were calculated by sigmoidal curve fitting with a constrained hill slope. (C) The human colon epithelial cancer HT29 cell line (n = 3) and human primary monocytes (n = 2) were exposed to (a) E. coli 055:B5 LPS (10 nM), (b) B. fragilis lipid A (1000 nM), (c) E. coli lipid A (100 nM), and (d) E. coli monophosphoryl lipid A (1000 nM) and poly(I:C) HMW LyoVec (10 μg mL−1) for 24 and 16 h, respectively. IFN-β release is shown as the means of biological duplicates. (D) Human primary monocytes (n = 2) were pre-incubated with 300 nM B. fragilis lipid A for 30 min and subsequently exposed to a wide concentration range (0.0001–100 nM) of E. coli 055:B5 LPS and E. coli lipid A. The data are shown as the mean of biological duplicates from one representative donor. | ||
To confirm that the observed activities were induced by binding to the TLR4/MD2 complex followed by NF-κB activation, the secreted embryonic alkaline phosphatase (SEAP) HEK-Blue reporter cell line expressing the human TLR4 (hTLR4) receptor in combination with MD2 and CD14 was stimulated with LPS and the lipid A derivatives and compared with the Null2 SEAP HEK-Blue reporter cell line lacking the hTLR4/MD2/CD14 complex (Fig. 3B). Indeed, the hTLR4 SEAP HEK-Blue cell line responded strongly to E. coli 055:B5 LPS (EC50 of 0.048 nM) and lipid A (EC50 of 1.7 nM). Monophosphoryl lipid A from E. coli only gave a response at high concentration (100 nM) while B. fragilis did not show any responsiveness. The stimulated Null2 SEAP HEK-Blue reporter cell line and unstimulated cells did not induce SEAP activity.
It has been reported that lipid As derived from commensal bacteria including B. fragilis can induce antiviral responses (i.e. IFN-β secretion).4 Therefore, the human colon epithelial cancer HT29 cell line that is capable of producing and secreting IFN-β was exposed to a wide concentration range of lipid A from B. fragilis and LPS and lipid A derivatives from E. coli for 24 h. Encapsulated high molecular weight (HMW) poly(I:C) (LyoVec), which triggers IFN-β secretion through TLR3 activation, was employed as a positive control. HT29 cells responded in a dose-dependent manner to these stimuli – in the same manner as primary human monocytes – as shown by the quantification of the pro-inflammatory cytokine IL-8 (Fig. S1†). B. fragilis lipid A was not capable of inducing IFN-β secretion even at a concentration of 1000 nM, while HMW poly(I:C) (10 μg mL−1) treatment resulted in a secretion of 137 pg mL−1 IFN-β (Fig. 3C). A similar experiment was performed in primary human monocytes and in this case, encapsulated HMW poly(I:C) also induced the production of IFN-β, whereas no response was measured for lipid A from B. fragilis (Fig. 3C) without any effect on viability (Fig. S2†). To further underpin this observation, the mouse macrophage cell line RAW 264.7 (NO-) was stimulated in the same manner. While LPS and lipid A derived from E. coli were able to induce IFN-β secretion, the synthesised lipid A from B. fragilis was not able to induce IFN-β secretion (Fig. S3†). In all experiments, unstimulated cells did not show any cytokine secretion.
To further elucidate the effect of lipid A from B. fragilis on TLR4 activation, primary human monocytes were pre-incubated for 30 min with a high concentration of lipid A from B. fragilis (300 nM) and subsequently stimulated with a wide concentration range (0.0001–10 nM) of E. coli 055:B5 LPS or E. coli lipid A. It was found that B. fragilis lipid A was able to inhibit the efficacy of IL-6 induced by E. coli 055:B5 LPS and lipid A by 31% and 38%, respectively (Fig. 3D). Application of lipid A from B. fragilis at a lower concentration did not result in inhibition and thus B. fragilis lipid A is a rather weak antagonist.
Conclusions
We have developed a synthetic approach for the preparation of the major lipid A derivative from B. fragilis. It includes an efficient synthetic approach for the unusual (R)-3-(13-methyltetradecanoyloxy)-1.5-methylhexadecanoic acid by the Wittig olefination to install a terminal isopropyl moiety. Furthermore, 2-methylnaphthyl ethers (Nap) were employed as permanent protecting groups because they can be more readily removed by hydrogenation compared to the conventionally employed benzyl ether. By protection of the C-4 hydroxyl of the acceptor and the C-4 and C-6 hydroxyls of the donor by Nap ethers, a disaccharide could be prepared that after protecting group manipulations and installation of fatty acids could be subjected to selective anomeric phosphorylation. Furthermore, Troc and Fmoc were employed as orthogonal amino protecting groups to selectively acetylate the amine of the proximal and distal glucosamine moieties. Cell biological experiments indicate that this synthetic lipid A derivative from B. fragilis does not induce the production of pro-inflammatory (IL-6) and anti-viral (IFN-β) cytokines. It can, however, partially antagonize the production of pro-inflammatory cytokines induced by E. coli LPS or lipid A. The lack of induction of proinflammatory cytokines and antagonizing properties may contribute to a gut homeostasis. In this study, we prepared the major species of lipid A derived from B. fragilis and it cannot be excluded that minor lipid A derivatives of heterogeneous native preparations were responsible for inducing the detected production of IFN-β. Thus, future studies will focus on the preparation of other lipid A derivatives from B. fragilis to evaluate the lipid A derivative responsible for anti-viral cytokine (IFN-β) production. Earlier studies examined the responsiveness of mouse dendritic cell populations (cDC2, CD103+CD11b+ and CD103CD11b+ cells) that reside in the colon to examine the induction of IFN-β.4 It is possible that the ability to induce this cytokine in a TLR4/MD2-dependent manner relies on this cell type.Data availability
The data underlying this study are available in the article and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research was supported by a grant from NWO TA PPS fund (ENPPS.TA.019.008 to G. J. B.).References
- H. Zafar and M. H. Saier Jr., Gut Microbes, 2021, 13, 1–20 CrossRef PubMed.
- M. D. Pither, et al. , Carbohydr. Polym., 2022, 297, 120040 CrossRef CAS PubMed.
- S. Saha, E. Pupo, A. Zariri and P. van der Ley, Microlife, 2022, 3, uqac011 CrossRef PubMed.
- K. L. Stefan, M. V. Kim, A. Iwasaki and D. L. Kasper, Cell, 2020, 183, 1312–1324 CrossRef CAS PubMed.
- A. Steimle, et al. , Mol. Ther., 2019, 27, 1974–1991 CrossRef CAS PubMed.
- F. Di Lorenzo, et al. , ACS Cent. Sci., 2020, 6, 1602–1616 CrossRef CAS PubMed.
- A. Molinaro, et al. , Chem. – Eur. J., 2015, 21, 500–519 CrossRef CAS PubMed.
- A. Ciesielska, M. Matyjek and K. Kwiatkowska, Cell. Mol. Life Sci., 2021, 78, 1233–1261 CrossRef CAS PubMed.
- B. S. Park, D. H. Song, H. M. Kim, B. S. Choi, H. Lee and J. O. Lee, Nature, 2009, 458, 1191–1195 CrossRef CAS PubMed.
- A. Weintraub, U. Zahringer, H. W. Wollenweber, U. Seydel and E. T. Rietschel, Eur. J. Biochem., 1989, 183, 425–431 CrossRef CAS PubMed.
- N. Okahashi, M. Ueda, F. Matsuda and M. Arita, Metabolites, 2021, 11, 197 CrossRef CAS PubMed.
- T. W. Cullen, et al. , Science, 2015, 347, 170–175 CrossRef CAS PubMed.
- S. R. Coats, et al. , Infect. Immun., 2011, 79, 203–210 CrossRef CAS PubMed.
- F. Di Lorenzo, K. A. Duda, R. Lanzetta, A. Silipo, C. De Castro and A. Molinaro, Chem. Rev., 2022, 122, 15767–15821 CrossRef CAS PubMed.
- J. X. Yang, et al. , Pharmaceutics, 2022, 14, 423 CrossRef CAS PubMed.
- M. Imoto, H. Yoshimura, N. Sakaguchi, S. Kusumoto and T. Shiba, Tetrahedron Lett., 1985, 26, 1545–1548 CrossRef CAS.
- A. Shimoyama, et al. , Angew. Chem., Int. Ed., 2021, 60, 10023–10031 CrossRef CAS PubMed.
- C. Qin, G. Tian, J. Hu, X. Zou and J. Yin, Curr. Opin. Chem. Biol., 2024, 78, 102424 CrossRef CAS PubMed.
- Y. Fujimoto, et al. , Mol. Biosyst., 2013, 9, 987–996 RSC.
- M. J. Gaunt, J. Q. Yu and J. B. Spencer, J. Org. Chem., 1998, 63, 4172–4173 CrossRef CAS.
- P. O. Adero, D. R. Jarois and D. Crich, Carbohydr. Res., 2017, 449, 11–16 CrossRef CAS PubMed.
- T. Li, et al. , Org. Lett., 2021, 23, 1638–1642 CrossRef CAS PubMed.
- X. Wu, L. Cui, T. Lipinski and D. R. Bundle, Chem. – Eur. J., 2010, 16, 3476–3488 CrossRef CAS PubMed.
- S. D. Markad and R. R. Schmidt, Eur. J. Org. Chem., 2009, 5002–5011 CrossRef CAS.
- B. Yu and H. C. Tao, J. Org. Chem., 2002, 67, 9099–9102 CrossRef CAS PubMed.
- E. C. J. M. Verpalen, A. J. Brouwer and G. J. Boons, Carbohydr. Res., 2020, 498, 108152 CrossRef CAS PubMed.
- K. F. Mo, et al. , J. Am. Chem. Soc., 2011, 133, 14418–14430 CrossRef CAS PubMed.
- M. Sakagami and H. Hamana, Tetrahedron Lett., 2000, 41, 5547–5551 CrossRef CAS.
- R. Hollaus, et al. , Chem. – Eur. J., 2015, 21, 4102–4114 CrossRef CAS PubMed.
- S. Gotze, et al. , J. Am. Chem. Soc., 2023, 145, 2342–2353 CrossRef PubMed.
- J. Bauer, K. Brandenburg, U. Zahringer and J. Rademann, Chem. – Eur. J., 2006, 12, 7116–7124 CrossRef CAS PubMed.
- S. Hatakeyama, H. Mori, K. Kitano, H. Yamada and M. Nishizawa, Tetrahedron Lett., 1994, 35, 4367–4370 CrossRef CAS.
- O. Labeeuw, P. Phansavath and J. P. Genet, Tetrahedron Lett., 2003, 44, 6383–6386 CrossRef CAS.
- Z. Zhou, A. A. Ribeiro and C. R. Raetz, J. Biol. Chem., 2000, 275, 13542–13551 CrossRef CAS PubMed.
- E. Verpalen, A. J. Brouwer, M. A. Wolfert and G. J. Boons, Chem. – Eur. J., 2024, 30, e202400429 CrossRef CAS PubMed.
- D. Petsch and F. B. Anspach, J. Biotechnol., 2000, 76, 97–119 CrossRef CAS PubMed.
- A. C. Fux, et al. , Int. J. Mol. Sci., 2023, 24, 8395 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01340a |
| This journal is © The Royal Society of Chemistry 2024 |