
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Unsubstituted 2- and 3-thiophenimines†
Amavi
Kpoezoun
ab,
Gnon
Baba
b and
Jean-Claude
Guillemin
 *a
*a
aUniv Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR – UMR6226, F-35000 Rennes, France. E-mail: jean-claude.guillemin@ensc-rennes.fr
bUniversité de Lomé, Département de Chimie, Laboratoire de Chimie Organique et des Substances Naturelles, 01 BP 1515 Lomé, Togo
First published on 23rd September 2024
Abstract
N-Unsubstituted 2-thiophenemethanimine and 3-thiophenemethanimine are the simplest derivatives of a family of imines, the thienyl imines. These two thienyl aldimines and the C-methyl derivatives were prepared in a gas solid reaction by dehydrocyanation of the corresponding α-aminonitriles or in the gas phase by a retro-ene reaction from N-allyl derivatives, then characterized by IR and NMR spectroscopy at low temperature and used in transimination reactions. From several angles, these compounds in the free or complexed state have been compared to the corresponding recently synthesized furanimines and to other N-unsubstituted imines, with the aim of studying the specificity of each of them.
Introduction
The synthesis and spectroscopic characterization of N-unsubstituted 2- and 3-furanaldimines have recently been reported.1 These species are the parent compounds of numerous derivatives, including a significant number of drugs containing the 2-furanimine moiety.2–5 For thienylimines also called thiophenimines, the corresponding compounds where the furan moiety has been substituted by a thiophene moiety, biological activity has been found for 2- and 3-derivatives but mainly, for the latter, with a particular type of bicyclic compound (Scheme 1). Thus Etizolam,6 brotizolam7 and clotiazepam8 are thienodiazepines, compounds comparable to benzodiazepines. Apafant acts as a potent, selective inhibitor of the phospholipid mediator platelet-activating factor (PAF).9 Examples of 2-thienylimines include lotilaner,10 an ectoparasiticide (anti-parasitic) medication for the treatment of eyelid inflammation, or motapizone11 for the inhibition of thrombocyte aggregation. The comparison of biological activity between furanimines and thienylimines is limited, since the synthesis of sulphur analogues of cefuroxime axetil or nifuratel, for example, have never been reported, while dantrolene analogues have proved far less effective.12 | ||
| Scheme 1 Some drugs containing the thiophenimine motif. | ||
The aim of this article is to prepare hitherto unknown 2- and 3-thiophenemethanimines, which are the parent compounds of many synthesized 2- and 3-thienylimines, to characterize them spectroscopically, to compare these aldimines with the corresponding C-methylated ketimines and complexed thienyl aldimine derivatives, as well as with the corresponding furanimines and N-unsubstituted imines, in order to determine whether any of their physicochemical properties are peculiar.13
Although often kinetically unstable, the simplest derivative of a family of compounds, such as formaldehyde for aldehydes or ethenamine for enamines, is essential in understanding this group.
Results and discussion
The difficulty of synthesizing N–H imines has already been reported including in the case of ketimines and is essentially associated with the kinetic instability.14–18 For the same kinetic instability, species isolation is easier for volatile compounds than for high-boiling compounds. At similar decomposition temperatures, the former can often be re-vaporized, whereas the latter cannot, as their decomposition temperature is lower than their vaporization temperature. As sulphur derivatives of furanimines, for which revaporisation was limited to ketimines that were more stable than aldimines, thienylaldimines – assuming similar kinetic stability – should again be more difficult to revaporize since the boiling point of thiophene (84 °C) is higher than that of furan (31.3 °C).To synthesize the simpler 2-thiophenemethanimine 1a and 3-thiophenemethanimine 1b and the corresponding C-methyl derivatives, the ketimines 1c and 1d (Chart 1), dehydrocyanation over KOH powder of the corresponding α-aminonitriles was considered by analogy with the synthesis of many kinetically unstable imines (route A).1,14–17 However, due to the difficulty observed in vaporizing this type of precursor as the substituent size increases, retro-ene reactions starting from more volatile and kinetically stable N-allyl derivatives were also investigated (route B) (Scheme 2). Such retro-ene reactions have enabled the synthesis of a wide range of compounds.19
 | ||
| Chart 1 | ||
 | ||
| Scheme 2 Two possible routes for the synthesis of the N-unsubstituted thienylimines 1a–1d. | ||
The thiophenyl α-aminonitriles 2a–2d,20,21 potential precursors of imines 1a–1d, were synthesized in a Strecker reaction in good yields ranging from 86 to 92%, the C-methyl derivatives requiring a longer reaction time as generally observed for ketones (Scheme 3).1 They should be stored in the freezer (−20 °C).
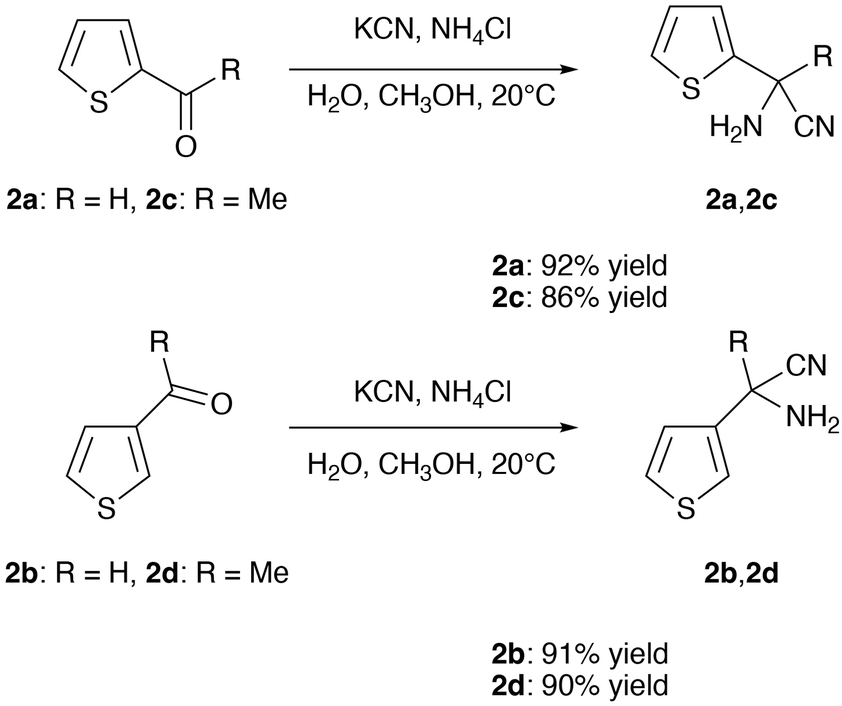 | ||
| Scheme 3 Synthesis of α-aminonitriles 2a–2d. | ||
On the other hand, N-allylamines 4a–4d were easy to synthesize, as is generally the case for many secondary N-allylamines.22–26 The condensation reaction between thienaldehydes or thienylketones and allylamine, followed by reduction of the imine formed with sodium borohydride as reducing agent, gave the N-allylamines 4a–4d in yields ranging from 77 to 90% (Scheme 4).25 Such compounds are stable at room temperature.
 | ||
| Scheme 4 Synthesis of N-allylthiophenamines 4a–4d. | ||
Using a heat gun, α-aminonitriles 2a–2d were slowly vaporized in a vacuum line (0.1 mbar) through a reactor half-filled with powdered KOH heated to 90 °C, where they underwent dehydrocyanation.
Ketimines 1c, 1d were obtained in 53 (1c) and 56% (1d) yields, and N-unsubstituted 2-thienylimine 1a and 3-thienylimine 1b in only 33 (1a) and 38% (1b) yields, probably because the precursors partially decompose during vaporization or the aldimines are less stable on hot KOH (Scheme 5). In addition to imines, we observed, by 1H NMR spectroscopy, the presence of by-products such as ammonia and a few other minor products that were not identified.
 | ||
| Scheme 5 Synthesis of thienylimines 1a–1d. | ||
N-Allylamines 4a–4d were vaporized under vacuum (0.1 mbar) with gentle heating in a quartz tube placed in an oven heated to 800 °C, where the chemical reaction took place. Imine and propene were formed. Ketimines 1c, 1d were obtained in yields of 70 and 73% respectively, but aldimines in yields of only 18 (1a) and 24% (1b), in the presence of propene and numerous minor impurities. Although simpler to implement, this approach is clearly limited to thienylketimines such as 1c and 1d, for which it can easily be scaled up to gram scale.
Imines synthesized by both routes were isolated using the same technique. Ketimines 1c, 1d were selectively trapped in a U-tube cooled to −35 °C to separate them from the more volatile by-products. Revaporization by gentle heating to around 30–40 °C led to pure compounds that could be condensed on a cold finger with a solvent for NMR analysis or on a KBr window cooled to 77 K for IR spectroscopy. Using the same approach, aldimines 1a, 1b led to a complex mixture of products containing small amounts of the expected imine. Consequently, in what follows, only route A was used for aldimines 1a, 1b; they were condensed directly on the cold finger cooled to 77 K, but the presence of ammonia could not be avoided.
NMR analysis of imines 1a–1d was performed at low temperature (−50 °C).
Imines 1a–1d diluted to 5% in deuterated chloroform or dideutero methylene chloride are stable at −50 °C. At rt, a half-life time of 60 min for 1a, 45 min for 1b and 30 h for 1c and 1d was measured by 1H NMR spectroscopy with an internal reference.
As already reported the reaction of triethylborane with imines does not lead to the complexed systems.1,27,28 Complexes 5a, 5b were obtained by reaction of superhydride with the corresponding nitriles but are therefore limited to complexes of aldimines (Scheme 6).‡
 | ||
| Scheme 6 Formation of the thienylaldimine complexes 5a, 5b. | ||
The (E) and (Z) isomers were observed for the 4 imines 1a–1d by 1H NMR spectroscopy for a sample diluted in CDCl3 or CD2Cl2 and cooled to −50 °C and never previously warmed to a higher temperature: E/Z = 9/1 (1a), E/Z = 8/2 (1b), E/Z = 9/1 (1c), E/Z = 7/3 (1d). These ratios were the same at room temperature. Only (E) isomers were observed for complexes 5a, 5b as already observed for other of N–H aldimines complexes.1,27,28
The chemical shift of the proton on the nitrogen atom was observed as a doublet at δH 9.65 (E) and 9.52 (Z) ppm for 1a, δH 9.69 (E) and 9.61 (Z) ppm for 1b but a rather significant difference was observed in the chemical shifts of the proton on the carbon of the imine function: 8.83 and 8.48 ppm for 1a and 8.56 and 8.22 ppm for 1b (Fig. 1). Coupling constant for imines 1a, 1b around 25 Hz (3JHHtrans) and 16 Hz (3JHHcis) are typical.
 | ||
| Fig. 1 1H NMR spectra from 7.0–10.0 ppm of 2-thiophenemethanime 1a (left) and 3-thiophenemethanime 1b (right). E/Z ratio and chemical shifts show differences. | ||
For 1c and 1d, the chemical shifts of the (E) and (Z) isomers were attributed following 2D NOESY NMR experiments (see ESI†). Two singlets at δH 8.99 (E) and 9.26 (Z) ppm were observed for 1c and δH 9.17 (E) and 9.39 (Z) ppm for 1d. The 13C NMR chemical shifts of the corresponding carbon are at δC 163.6 (E) and 162.0 ppm (Z) for 1a and δC 164.7 (E) and 163.3 ppm (Z) for 1b.
The comparison of thienylimines with furanimines shows a difference in the Z/E ratio and chemical shifts of the imine function for 2-thiophene and 2-furan derivatives. For alkylated N–H aldimines, this ratio is around 1/3.15 In the case of 2-furanmethanimine, we observed a Z/E = 53/47 ratio we attributed to the formation of a weak hydrogen bond between the hydrogen on nitrogen and the oxygen of the cycle.1 For thienylimine 1a, this Z/E ratio of 1/9 is much smaller, even smaller than that of alkylaldimines.15 That could be explained by a repulsive effect between the N–H and the sulphur atom for thiophenaldimine 1a in which the proton on the nitrogen of the (E) isomer is at downfield than that of the (Z) isomer unlike the corresponding chemical shifts of the furanimine which are an exception (Fig. 2). The same effect was observed on imines 1c with a similar 1/9 ratio for Z/E. The main difference between thienylimines and furanimines is the higher aromaticity of thiophenes and the nature of the heteroatom, which has a much lower electronegativity than oxygen. However, a role attributed to the cycle based or not on its greater aromaticity can be excluded since this effect was not observed for 3-thiophenimines 1b, 1d.
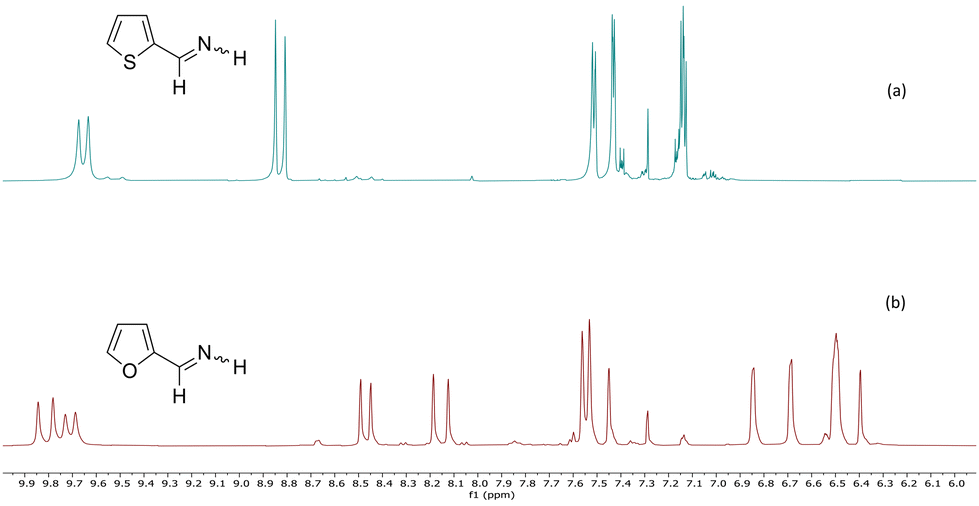 | ||
| Fig. 2 Comparison between 6–10 ppm 1H NMR spectra of 2-thiophenemethanimine 1a (a) and 2-furanemethanimine (b). | ||
The chemical shift of the carbon corresponding to the imine function carbon lies between δC 161.9 and 164.7 ppm, a little at downfield than those of furanimines1 (δC: 156.9–162.3 ppm) and a lot at upfield than those of arylimines.28 These differences are consistent with those of the aldehydes: δC(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of 2-thiophenecarboxaldehyde: 183.1 ppm for δC(CHO) of 2-furaldehyde: 177.0 ppm, δC(CHO) of 3-thiophenecarboxaldehyde: 185.0 ppm for δC(CHO) of 3-furaldehyde: 184.4 ppm while benzaldehyde was observed at δC(CHO) 192.3 ppm.
O) of 2-thiophenecarboxaldehyde: 183.1 ppm for δC(CHO) of 2-furaldehyde: 177.0 ppm, δC(CHO) of 3-thiophenecarboxaldehyde: 185.0 ppm for δC(CHO) of 3-furaldehyde: 184.4 ppm while benzaldehyde was observed at δC(CHO) 192.3 ppm.
Expected shielding effects were observed in the 1H and 13C NMR spectra of both complexes 5a, 5b with a chemical shift difference of ΔδC(CN) of −7 ppm.1,27,28
The infrared spectra of 1a–1d showed characteristic νCN absorptions at 1616 (1a), 1632 (1b), 1607 (1c) and 1615 cm−1 (1d), respectively, close to those observed for furanimines1 or alkylaldimines.15
What about the chemistry of N-unsubstituted thienylimines? In the aim to confirm that transimination reaction can be involved with kinetically unstable imines in good yields, thienylimines 1a–1d were readily involved in transimination reactions with propylamine or aniline to give imines 6a, 6b, 6d–6g, 6i, 6j. We have extended this study to two transimination reactions using cyanamide with 2-thiophenemethanimine 1a and hydroxylamine with 3-thiophenemethanimine 1b to form compounds 6c and 6h, respectively (Scheme 7). On the basis of the yields obtained for thienylimines 1a–1d, the yields of the transimination reaction ranged between 75 and 92%. Compounds 6a–6j were characterized by 1H and 13C NMR spectroscopy and compared with the spectra of authentic samples.23,25 The NMR data of the new compounds are fully consistent with the previous ones.
 | ||
| Scheme 7 Transimination reactions. | ||
Conclusions
The parent compounds 2-thiophenemethanimine and 3-thiophenemethanimine and the corresponding C-methyl ketimines have been synthesized and spectroscopically characterized. The C-methyl derivatives were obtained on a preparative scale by thermolysis of allylamine precursors in a retroene reaction. For the parent compounds, the best purity was achieved by dehydrocyanation of the corresponding α-aminonitriles in a vacuum gas–solid reaction, as thermolysis of the N-allyl(thiophenyl)methanamines did not lead to a more efficient synthesis. Due to the boiling point of the N-unsubstituted thienylaldimines and their kinetic instability, we are at the limit of species generation in the gas phase. Nevertheless, obtaining these compounds in a gas stream is the key to accessing spectroscopic studies such as millimetre or photoelectron spectroscopy, the determination of molecular structures by gas phase electron diffraction (GED) or mass spectrometry.Although some differences were observed, the N-unsubstituted thiophenimines are differed little from the corresponding furanimines. This can probably be attributed to the presence of an heteroaromatic ring in both series, which reduces the role played by the heteroatom despite the greater aromaticity of thiophenes, whereas a thiol or thiocarbonyl group has properties strongly different from those of the corresponding alcohol or carbonyl group, respectively.
We are continuing these studies by seeking to isolate N-unsubstituted pyrrolaldimines and pyridinaldimines, compounds that are even less volatile than the thienylimines studied here, with the aim of providing tools for understanding the role played by the nature of the aromatic substituent on the unsubstituted imino group and its physicochemical consequences.
Experimental
Route A: general procedure for the synthesis of imines ( 1a–1d ) by dehydrocyanation (see ESI† for the picture of the apparatus).A reactor (ϕ = 2.0 cm, L = 40 cm) half-filled with KOH powder (39 g, 0.7 mol) was placed in a vacuum line (0.1 mbar) between a reagent inlet on one side and a solvent inlet, a nitrogen gas inlet and a cold finger on the other. KOH was heated to 90 °C by a circulating bath, the cold finger was filled with liquid nitrogen and α-aminonitrile 2a–2d (2.0 mmol) was slowly vaporised in the reactor. The products formed were condensed on the cold finger and a solvent can be added at this stage. At the end of the addition, the pump was disconnected, the assembly was filled with dry nitrogen and the liquid nitrogen in the cold finger was expelled with compressed air. The imine formed and the solvent drained rapidly as soon as they melted in the NMR tube or in the flask immersed in a liquid nitrogen bath.
This approach, extended to 5 mmol of precursor, leads to yields around half as high.
Route B: general procedure for the synthesis of imines by retro-ene reaction (see ESI† for the picture of the apparatus).
The reactor used in route A was replaced by an oven. Compound 4a–4d (2.0 mmol) was vaporized under vacuum (0.1 mbar) in a quartz tube (L = 35 cm, ϕ = 25 mm) heated in an oven to 800 °C. The thermolyzed products were condensed on a cold finger cooled with liquid nitrogen. CDCl3 (700 μl) was added. At the end of the reaction, the cold finger was isolated from the vacuum line by stopcocks and compounds were isolated by the way reported above for route A.
A very slight decrease in yield was observed from 10 mmol for the synthesis of ketimines 1c, 1d, but this approach is only analytical for imines 1a, 1b.
In the case of ketimines 1c, 1d, the cold finger can be replaced by a U-tube fitted with stopcocks and immersed in a cold bath cooled to −35 °C, enabling the product to be isolated in its pure state.
2-Thiophenemethanimine (1a)
Yield: 33% (route A) (73 mg, 0.66 mmol), 18% (route B), (40 mg, 0.36 mmol) E/Z = 9/1. τ1/2 (5% in CDCl3 or CD2Cl2, 20 °C): 60 min. (E) 1H NMR (400 MHz, CDCl3, 223 K) δ 9.65 (d, 3J = 16.0 Hz, 1H), 8.83 (d, 3J = 16.0 Hz, 1H), 7.51 (d, 3J = 5.0 Hz, 1H), 7.43 (d, 3J = 3.7 Hz, 1H), 7.14 (dd, 3J = 5.0, 3.7 Hz, 1H) ppm. 13C NMR (100 Hz, CDCl3, 223 K) δ 163.6, 143.2, 132.9, 130.0, 127.9 ppm. (Z) 1H NMR (400 MHz, CDCl3, 223 K) δ 9.52 (d, 3J = 24.8 Hz, 1H), 8.48 (d, 3J = 24.8 HZ, 1H); 7.51 (d, 3J = 5.0 Hz, 1H), 7.43 (d, 3J = 3.7 Hz, 1H), 7.14 (dd, 3J = 5.0, 3.7 Hz, 1H) ppm. 13C NMR (100 Hz, CDCl3, 223 K) δ 162.0, 146.3, 132.9, 130.7, 128.5 ppm. IR (film, 77 K, ν cm−1): 3292 (m), 1616 (s, νCN), 1431 (m), 1261 (m).
3-Thiophenemethanimine (1b)
Yield: 38% (route A) (84 mg, 0.76 mmol), 24% (route B), (53 mg, 0.48 mmol). E/Z = 4/1. τ1/2 (5% in CDCl3 or CD2Cl2, 20 °C): 45 min. (E) 1H NMR (400 MHz, CDCl3, 223 K) δ 9.69 (d, 3J = 16.4 Hz, 1H), 8.56 (d, 3J = 16.4 Hz, 1H), 7.57 (d, J = 2.9 Hz, 1H), 7.48 (d, 3J = 5.1 Hz, 1H), 7.21 (dd, J = 5.1, 2.9 Hz, 1H) ppm. 13C NMR (100 Hz, CDCl3, 223 K) δ 164.7, 141.2, 131.1, 127.0, 124.4 ppm. (Z) 1H NMR (400 MHz, CDCl3, 223 K) δ 9.61 (d, 3J = 25.4 Hz, 1H), 8.22 (d, 3J = 25.4 Hz, 1H), 7.57 (d, J = 2.9 Hz, 1H), 7.48 (d, 3J = 5.1 Hz, 1H), 7.21 (dd, J = 5.1, 2.9 Hz, 1H) ppm. 13C NMR (100 Hz, CDCl3, 223 K) δ 163.3, 139.5, 129.9, 127.6, 122.9 ppm. IR (film, 77 K, ν cm−1): 3368 (m), 1632 (m, νCN), 1465 (s), 1262 (s).
α-Methyl-2-thiophenemethanimine (1c)
Yield: 53% (route A) (132 mg, 1.06 mmol), 70% (route B), (175 mg, 1.40 mmol). E/Z = 9/1. τ1/2 (5% in CDCl3 or CD2Cl2, 20 °C): 30 h. (E) 1H NMR (400 MHz, CDCl3, 223 K) δ 8.99 (s, 1H), 7.37 (m, 1H), 7.31 (m, 1H), 7.01 (m, 1H), 2.37 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3, 223 K) δ 169.1, 143.7, 129.4, 129.2, 127.3, 26.0 ppm. (Z) 1H NMR (400 MHz, CDCl3, 223 K) δ 9.26 (s, 1H), 7.35 (m, 1H), 7.28 (m, 1H), 7.00 (m, 1H), 2.53 (s, 3H) ppm. 13C NMR (100 Hz, CDCl3, 223 K) δ 168.1, 142.5, 128.0, 126.5, 124.9, 26.6 ppm. IR (film, 77 K, ν cm−1): 3372 (s), 1607 (vs., νCN), 1430 (s), 1241 (s). HRMS (ASAP) m/z: calculated for C6H8NS+ [M + H]+: 126.0372; found: 126.0371.
α-Methyl-3-thiophenemethanimine (1d)
Yield: 56% (route A) (140 mg, 1.12 mmol), 73% (route B), (182 mg, 1.46 mmol). E/Z = 7/3. τ1/2 (5% in CDCl3 or CD2Cl2): 30 h. (E) 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H), 7.73 (m, 1H), 7.63 (m, 1H), 7.36 (m, 1H), 2.42 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 170.6, 141.9, 128.0, 126.5, 126.3, 27.9 ppm. (Z) 1H NMR (400 MHz, CDCl3) δ 9.39 (s, 1H), 7.73 (m, 1H), 7.63 (m, 1H), 7.36 (m, 1H), 2.56 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 167.6, 140.9, 126.9, 126.3, 124.5, 24.9 ppm. IR (film, 77 K, ν cm−1): 3353 (m), 1671 (m, νCN), 1425 (s), 1259 (m). HRMS (ASAP) m/z: calculated for C6H8NS+ [M + H]+: 126.0372; found: 126.0371.
Data availability
The data underlying this study are available in the published article and its ESI.† Synthesis of all compounds, NMR and IR characterization data, NMR spectra of all new compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A.K. thanks the Togolese government, the French government and the French embassy in Togo for research grants. The authors thank Jean-Paul Guégan for technical assistance.References
- A. Kpoezoun, H. Gazzeh, G. Baba and J.-C. Guillemin, J. Org. Chem., 2024, 89, 11026–11030 CrossRef CAS PubMed
.
- A. E. Glatt, Hosp. Pract., 2016, 21(3), 158A–158L CrossRef PubMed
- A. M. Timperio, H. A. Kuiper and L. Zolla, Xenobiotica, 2003, 3(2), 153–167 CrossRef PubMed
- R. N. Grüneberg and A. Leakey, Br. Med. J., 1976, 2(6041), 908–910 CrossRef PubMed
- T. Krause, M. U. Gerbershagen, M. Fiege, R. Weisshorn and F. Wappler, Anaesthesia, 2004, 59(4), 364–373 CrossRef CAS PubMed
- K. Araki, N. Yasui-Furukori, T. Fukasawa, T. Aoshima, A. Suzuki, Y. Inoue, T. Tateishi and K. Otani, Eur. J. Clin. Pharmacol., 2004, 60(6), 427–430 CrossRef CAS PubMed
- M. S. Langley and S. P. Clissold, Drugs, 1988, 35(2), 104–122 CrossRef CAS PubMed
- T. Cheng, D. M. Wallace, B. Ponteri and M. Tuli, Neuropsychiatr. Dis. Treat., 2018, 14, 1351–1361 CrossRef CAS PubMed
- J. Casals-Stenzel, Lipids, 1991, 26(12), 1157–1161 CrossRef CAS PubMed
- E. A. Kuntz and S. Kammanadiminti, Parasites Vectors, 2017, 10(1), 538 CrossRef PubMed
- V. Schulz, W. Fisher, U. Hanselle, W. Huhmann and V. Zietsch, Eur. J. Clin. Pharm., 1986, 31, 411–414 CrossRef CAS PubMed
- H. R. Snyder Jr, C. S. Davis, R. K. Bickerton and R. P. Halliday, J. Med. Chem., 1967, 10(5), 807–810 CrossRef CAS PubMed
- A spectroscopic detection of 2-thiophenimine par GC-MS has been reported: S. Chakraborty and H. Berke, ACS Catal., 2014, 4, 2191–2194 CrossRef CAS
- J.-C. Guillemin and J.-M. Denis, J. Chem. Soc., Chem. Commun., 1985, 951–952 RSC
- J.-C. Guillemin and J.-M. Denis, Tetrahedron, 1988, 44(14), 4431–4446 CrossRef CAS
- J.-C. Guillemin, W. Nasraoui and H. Gazzeh, Chem. Commun., 2019, 55, 5647–5650 RSC
- H. Ayachi, H. Gazzeh, T. Boubaker and J.-C. Guillemin, J. Org. Chem., 2023, 88(4), 2570–2574 CrossRef CAS PubMed
- K. Morisaki, H. Morimoto and T. Ohshima, ACS Catal., 2020, 10(12), 6924–6951 CrossRef CAS
- J.-L. Ripoll and Y. Vallée, Synthesis, 1993, 659–677 CrossRef CAS
- M. J. Thompson, H. Adams and B. Chen, J. Org. Chem., 2009, 74, 3856–3865 CrossRef CAS PubMed
-
Mitsui Toatsu Chemicals, Inc., Federal Republic of Germany, DE3713774A1, 1987
- J. S. Yadav, C. Madhuri, B. V. S. Reddy, G. S. K. K. Reddy and G. Sabitha, Synth. Commun., 2002, 32(18), 2771–2777 CrossRef CAS
- S.-I. Murahashi, Y. Imada, Y. Taniguchi and Y. Kodera, Tetrahedron Lett., 1988, 29(24), 2973–2976 CrossRef CAS
- F. Palacios, D. Aparicio and J. García, Tetrahedron, 1996, 52(28), 9609–9628 CrossRef CAS
- F. Ghelfi, A. F. Parsons, D. Tommasini and A. Mucci, Eur. J. Org. Chem., 2001, 1845–1852 CrossRef CAS
- N. Choony, A. Dadabhoy and P. G. Sammes, J. Chem.
Soc., Perkin Trans. 1, 1998, 2017–2022 RSC
- P. V. Ramachandran and D. Biswas, Org. Lett., 2007, 9, 3025–3027 CrossRef CAS PubMed
- J. H. Lee, S. Gupta, W. Jeong, Y. H. Rhee and J. Park, Angew. Chem., Int. Ed., 2012, 51(43), 10851–10855 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: General procedure for the syntheses and 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d4ob01315h |
| ‡ The synthesis of complex 5a by a similar approach had already been reported.27 |
| This journal is © The Royal Society of Chemistry 2024 |