
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic stereoselective synthesis of cyclopentanones from donor–acceptor cyclopropanes and in situ-generated ketenes†
Shubhanjan Mitraa,
Sophie M. Connollya,
Saud Ayidia,
Mukulesh Mondalb,
Manashi Pandab,
Brian G. Kellyc and
Nessan J. Kerrigan *a
*a
aSchool of Chemical Sciences and Life Sciences Institute, Dublin City University, Glasnevin, Dublin 9, Ireland. E-mail: nessan.kerrigan@dcu.ie
bDepartment of Chemistry, Oakland University, 2200 N. Squirrel Rd, MI 48309, USA
cKelAda Pharmachem, Belfield, Dublin 4, Ireland
First published on 13th August 2024
Abstract
A dual InBr3–EtAlCl2 Lewis acidic system was found to be optimal for promoting the diastereoselective (3 + 2)-cycloaddition of donor–acceptor cyclopropanes with in situ-generated ketenes to form cyclopentanones. The desired products were formed in good to excellent yields (70–93% for 16 examples) and with good to excellent diastereoselectivity and enantiospecificity.
Cyclopentanones and their derivatives, such as cyclopentan-1,3-diols, serve as vital structural components in various naturally occurring prostaglandins and pharmaceutical analogues like PGE2 and bimatoprost.1 Notable pharmaceuticals including loxoprofen, donepezil, lubiprostone, and premarin also feature the cyclopentanone motif.1 However, current approaches to cyclopentanone synthesis often encounter limitations, particularly in achieving the desired 2,3-disubstituted or 2,3,4-trisubstituted cyclopentanone structures with sufficient substituent versatility and high diastereoselectivity. Most available catalytic technologies facilitate the direct preparation of only 2-substituted, 3-substituted, 2,4-disubstituted, and 3,4-disubstituted cyclopentanones, highlighting the need for more efficient and versatile synthetic strategies.2
In seminal work, Johnson and co-workers demonstrated that Lewis acid catalysis could be employed to promote the (3 + 2)-cycloaddition of donor–acceptor (DA) cyclopropanes with aldehydes to access tetrahydrofurans with excellent enantiospecificity, enantioselectivity and diastereoselectivity.3,4
A few years ago, our group and Lu's group independently reported that Pd(0)-catalyzed (3 + 2)-cycloaddition of vinylcyclopropanes with ketenes could afford highly substituted tetrahydrofurans.5 Shortly after, as part of a program on the development of new reactions of ketenes, we reported the dual Lewis acid-catalyzed (3 + 2)-cycloaddition of DA cyclopropanes with disubstituted ketenes to form cyclopentanones 3a (Scheme 1).6 Werz's group later showed that ketenedithioacetals could undergo (3 + 2)-cycloaddition with DA cyclopropanes under Lewis acid catalysis to access dithiaspiro compounds, which were then elaborated to substituted cyclopentanones, albeit with modest diastereoselectivity.7 In 2022, Studer and co-workers demonstrated that related cycloalkanes (bicyclo[1.1.0]-butane ketones) could also undergo Lewis acid-catalysed reaction with disubstituted ketenes to access bicyclo[2.1.1]hexane-2-ones.8 Recently, Punniyamurthy's group proposed the involvement of a vinyl ketene intermediate in the synthesis of bicyclic cyclopentapyrans from 2,4-dienals and DA cyclopropanes.9
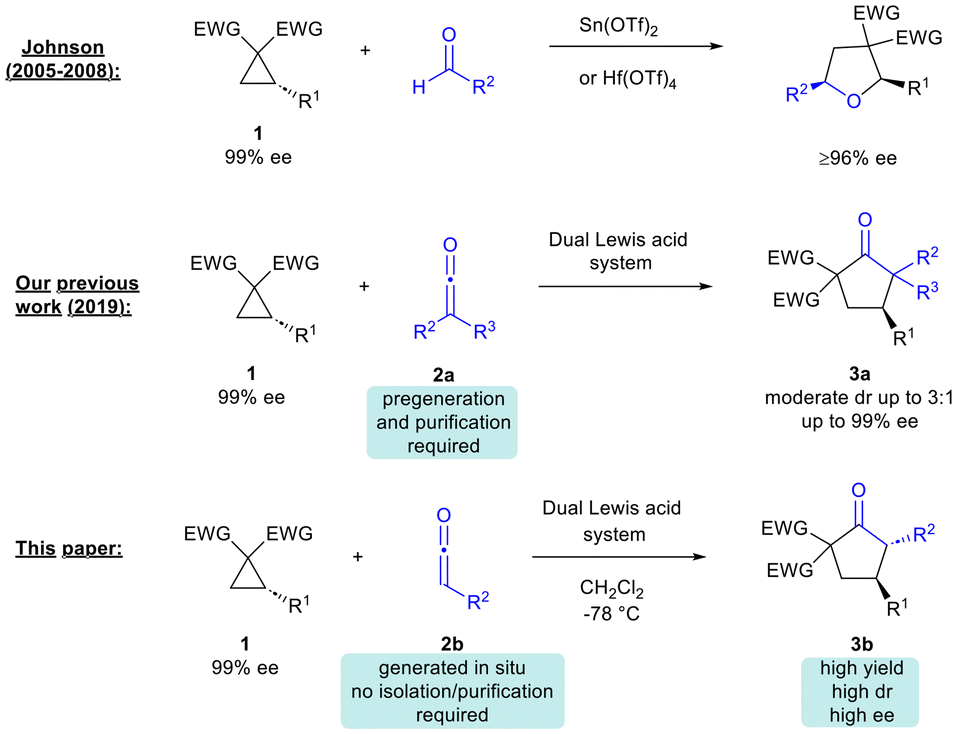 | ||
| Scheme 1 Literature precedent and this work. | ||
A drawback of our 2019 methodology was the need to use pre-generated stable ketenes that are considered technically difficult to work with.6 Furthermore, the reaction diastereoselectivity was at a moderate level (dr up to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). In this paper we describe the development of an efficient methodology for the synthesis of cyclopentanones from in situ-generated ketenes and readily available DA cyclopropanes.10 The current method significantly improves on prior reports of cyclopentanone synthesis from ketenes/ketene surrogates in that it caters for unstable, in situ-generated ketenes, without any need for prior isolation or purification.7,10 Moreover, the desired products are obtained with generally good to excellent diastereoselectivity (dr up to 37:1). Efficient transfer of chirality from starting enantioenriched cyclopropanes to cyclopentanone products was also verified.
:1). In this paper we describe the development of an efficient methodology for the synthesis of cyclopentanones from in situ-generated ketenes and readily available DA cyclopropanes.10 The current method significantly improves on prior reports of cyclopentanone synthesis from ketenes/ketene surrogates in that it caters for unstable, in situ-generated ketenes, without any need for prior isolation or purification.7,10 Moreover, the desired products are obtained with generally good to excellent diastereoselectivity (dr up to 37:1). Efficient transfer of chirality from starting enantioenriched cyclopropanes to cyclopentanone products was also verified.
We began our studies by exploring the reaction of in situ-generated methylketene 2a (generated in situ through reaction of propionyl chloride 4a with i-Pr2NEt) with phenyl-substituted donor–acceptor cyclopropane 1a in CH2Cl2 (Table 1). A range of Lewis acids were investigated including the dual Lewis acidic system (InBr3–EtAlCl2) we had previously determined to be optimal for reactions of disubstituted ketenes (Table 1).6 Reactions were found to proceed most effectively and cleanly at −78 °C. At higher temperatures, such as at −25 °C and at rt, no desired cyclopentanone product was formed and ketene dimerization was competitive. Solvents other than CH2Cl2, such as BTF and THF, were also investigated but led to no desired product being formed. Other reaction parameters that were found to be critical included order of addition of reagents (acyl chloride, amine base and cyclopropane). The dual Lewis acidic system of InBr3–EtAlCl2 was found to give the best results in terms of yield of the desired cyclopentanone (Table 1, entries 4 and 5), although other In(III) salts also functioned well in combination with EtAlCl2 (Table 1, entries 6 and 7).
|
|||
|---|---|---|---|
| Entry | Lewis acid catalyst (mol%) | Lewis acid Co-catalyst (mol equiv) | Yielda [%] |
| a Isolated yield after flash column chromatography through silica gel. dr was ≥3:1 in all cases. |
|||
| 1 | InBr3 (30) | — | 0 |
| 2 | InBr3 (30) | EtAlCl2 (0.5) | 21 |
| 3 | InBr3 (30) | EtAlCl2 (1.0) | 35 |
| 4 | InBr3 (30) | EtAlCl2 (2.5) | 79 |
| 5 | InBr3 (50) | EtAlCl2 (2.5) | 81 |
| 6 | InCl3 (30) | EtAlCl2 (2.5) | 71 |
| 7 | In(OTf)3 (30) | EtAlCl2 (2.5) | 69 |
| 8 | Sn(OTf)3 (30) | EtAlCl2 (2.5) | 65 |
| 9 | Cu(OTf)3 (30) | EtAlCl2 (2.5) | 42 |
| 10 | CuI (30) | EtAlCl2 (2.5) | 53 |
| 11 | FeCl3 (30) | EtAlCl2 (2.5) | 0 |
| 12 | InBr3 (30) | i-Bu2AlCl (2.5) | 61 |
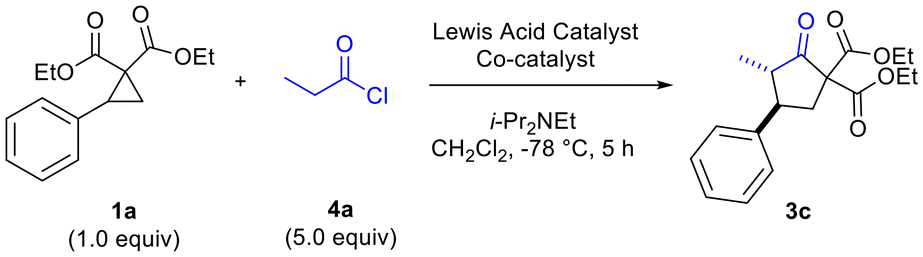
The need to use excess EtAlCl2 (up to 2.5 equiv for optimal yields of 3c) was surmised to be due to quenching/protonation of the co-catalyst by some of the ammonium chloride salt produced by in situ ketene generation. Further optimization, carried out in parallel, demonstrated the need to employ excess mole equivalents of acyl chloride and i-Pr2NEt to achieve optimal yields of 3d (Table 2, entries 1–4). Conventional in situ ketene generation through slow addition of propionyl chloride to a solution containing an amine base and other reagents was found to be completely unsuccessful. Only after addition of the amine to a solution containing the acyl chloride and Lewis acids, followed by slow addition of the cyclopropane solution to the reaction solution, were good results obtained (Table 2, entries 2 and 4 versus entry 3).
|
|||
|---|---|---|---|
| Entry | Propionyl chloridea (mol equiv) | i-Pr2NEta (mol equiv) | Yieldb [%] |
| a Mol equivalents calculated with respect to the starting cyclopropane derivative.b Isolated yield after flash column chromatography through silica gel. dr was ≥6:1 in all cases.c i-Pr2NEt was added to a solution containing the acyl chloride 4a and Lewis acids, followed by slow addition of cyclopropane 1b solution to the reaction solution (see ESI†).d i-Pr2NEt was added to a solution of cyclopropane 1b, acyl chloride 4a, InBr3 and EtAlCl2. |
|||
| 1c | 1.0 | 1.2 | 12 |
| 2c | 3.0 | 3.2 | 54 |
| 3d | 3.0 | 3.2 | 0 |
| 4c | 5.0 | 5.2 | 90 |
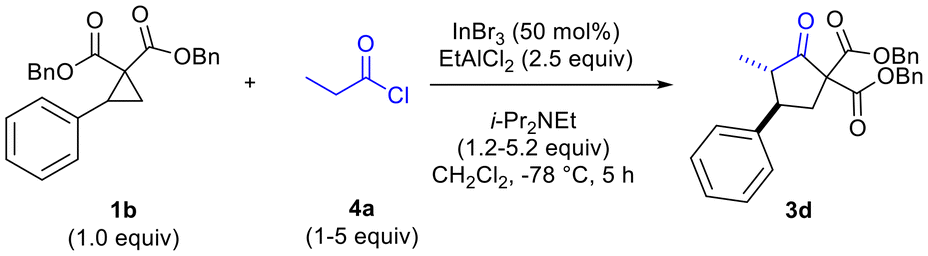
Optimization of diastereoselectivity in the formation of the cyclopentanone was then pursued. We quickly determined that exposure to silica during silica gel purification led to an increase in stereoselectivity, favouring the trans-isomer. To amplify the equilibration process, all crude products were stirred with silica gel in CH2Cl2 for 2 h at 50 °C prior to column chromatographic purification, leading to an increase in trans-diastereroselectivity (e.g. from 3:1 to 31:1 for 3i).
Having determined that the dual Lewis acidic system of InBr3–EtAlCl2 afforded the best results in terms of yield and diastereoselectivity of the desired cyclopentanone, we proceeded to evaluate the scope of the reaction methodology (Table 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yielda [%] | Drb | Compound |
| a Isolated yield after flash column chromatography through silica gel.b dr was determined by 1H NMR analysis of crude after silica gel-mediated isomerization.c Not subjected to isomerization. | ||||||
| 1 | Et | Ph | Me | 79 | 9:1 |
3c |
| 2 | Bn | Ph | Me | 90 | 33:1 |
3d |
| 3 | Bn | Ph | Et | 88 | 4:1 |
3e |
| 4 | Bn | Ph | n-Pr | 93 | 17:1 |
3f |
| 5 | Bn | Ph | Bn | 74 | 7:1 |
3g |
| 6 | Me | Ph | Me | 65 | 3:1 |
3h |
| 7 | Et | 4-FC6H4 | Me | 79 | 31:1 |
3i |
| 8 | Et | 4-MeOC6H4 | Me | 81 | 26:1 |
3j |
| 9 | Et | Styrenyl | Me | 70 | 5:1 |
3k |
| 10 | Et | 2-Furyl | Me | 83 | 13:1 |
3l |
| 11 | Et | 2-Thienyl | Me | 74 | 37:1 |
3m |
| 12 | Et | N-Phthaloyl | Me | 42 | 9:1 |
3n |
| 13 | Bn | Vinyl | Me | 81 | 17:1 |
3o |
| 14 | t-Bu | Vinyl | Me | 59 | 3:1 |
3p |
| 15 | i-Pr | Vinyl | Me | 68 | 6:1 |
3q |
| 16 | Et | Vinyl | Me | 71 | 4:1c |
3r |
| 17 | Me | Vinyl | Me | 56 | 14:1 |
3s |
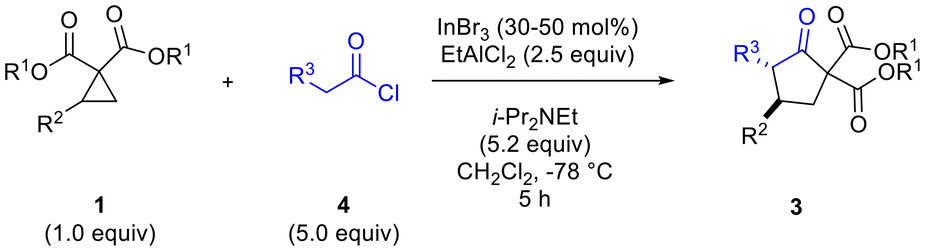
Variation of ketene structure was investigated with methylketene, ethylketene, n-propylketene and benzylketene all giving good results in terms of yield of cyclopentanone product from cyclopropane 1b (Table 3, entries 2–5).10,11 Differences in diastereomeric ratio noted with these ketenes may have been due to isomerization not having gone to completion during 2 h in some cases. Changes in the cyclopropane substitutent (R2) generally did not have a negative impact on diastereoselectivity, with aryl groups bearing electron donating or withdrawing substituents (entries 1 vs. 7 and 8) working equally well.12,13 Heteroaromatic substituents also worked well (entries 10 and 11). A heteroatom substituent (N-phthaloyl, entry 12) directly bonded to the cyclopropane ring at the 2-position also performed quite well. There was an influence of the ester group substituent on diastereoselectivity, with Me and Bn (R1) (entries 13 and 17) found to be superior to t-Bu and i-Pr (entries 14 and 15).
Additionally, it was determined that when enantioenriched 1b (99% ee, (R) or (S)-1b) was used as the substrate that a specific enantiomer of 3d was formed with high enantiomeric excess (96–98% ee), demonstrating the highly enantiospecific nature of the reaction (Scheme 2). Similar results were also observed for formation of enantioenriched 3c (see ESI†).
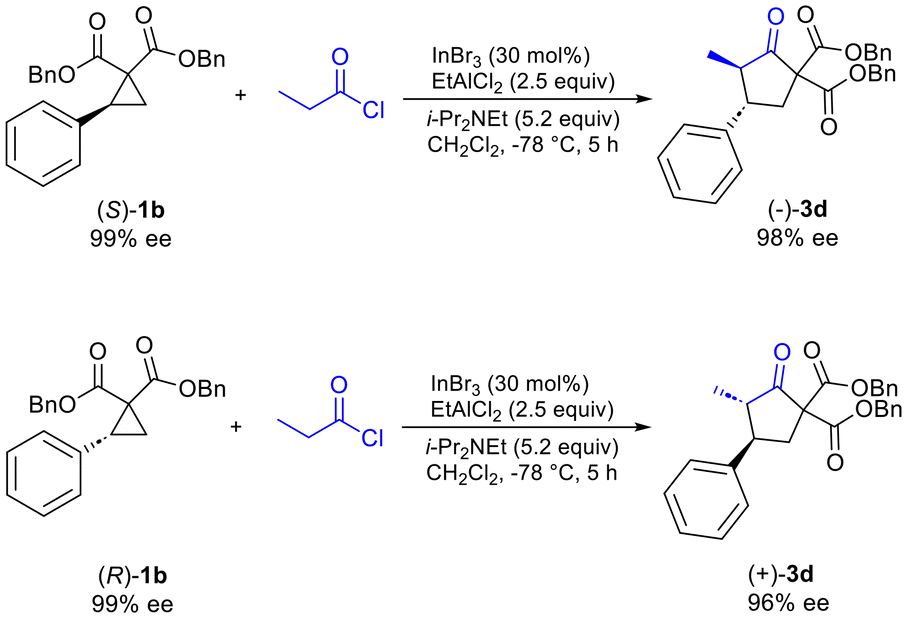 | ||
| Scheme 2 Enantiospecificity of the reaction. | ||
Finally, reduction of 3d using NaBH4 was found to proceed with optimal chemoselectivity (compared to DIBAL-H, L-selectride and CBS reagent) to provide the desired alcohol 5d (68%) with modest diastereoselectivity (dr 2:1) (Scheme 3). Scale-up of the cyclopentanone synthesis was also explored and was found to afford the desired product 3d in 92% yield and with a dr of 28:1 on a 2.6 mmol scale (producing 1.05 g of 3d).
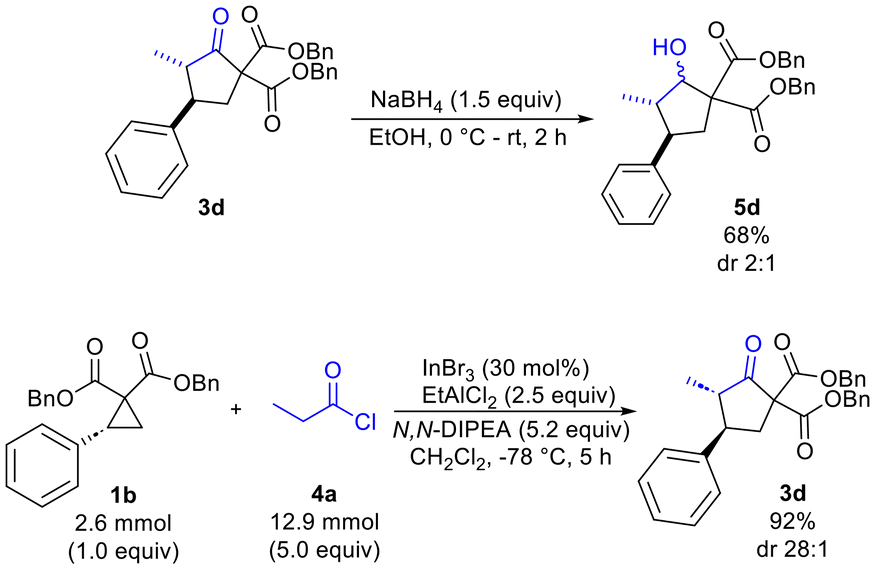 | ||
| Scheme 3 Reduction and scale-up reactions. | ||
We propose that the reaction proceeds through a concerted asynchronous transition state (Scheme 4).3,6 InBr3 is expected to activate the donor–acceptor cyclopropane and weaken the C–C bond between C1 and C2 in the transition state.3 EtAlCl2 is proposed to activate the ketene and enable it to undergo reaction with the DA cyclopropane.14,15 Addition of cyclopropane C1 to the ketene carbonyl in stereoselective fashion (i.e. adjacent to the sterically smaller H substituent) with attack of the ketene α-carbon to C2 of the cyclopropane leads to cyclopentanone formation. Activation by EtAlCl2 is essential to the success of the reaction as in its absence, no desired product is formed (Table 1, entry 1). Alternatively, EtAlCl2 may act by bonding to a bromide ligand in InBr3, thus increasing the Lewis acidity of the In(III) catalyst (Lewis acid-assisted Lewis acidity).16 Regardless, equilibration to provide the trans-isomer as the major diastereomer occurs under the reaction conditions (in the presence of excess base and Lewis acid). Further isomerization to achieve synthetically useful levels of diastereoselectivity (generally ≥8:1) was achieved after exposure to silica in CH2Cl2 (2 h at 50 °C).
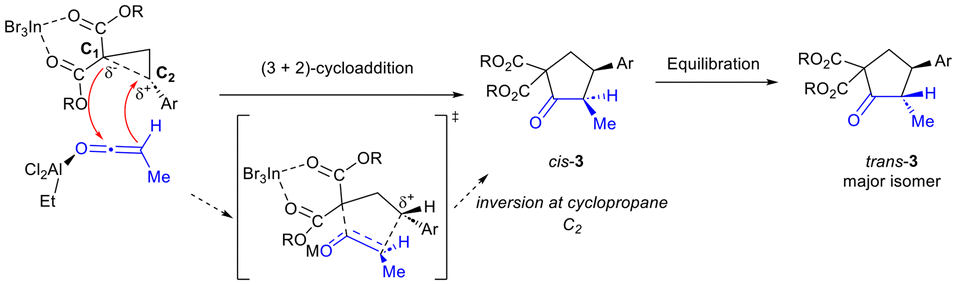 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a synthesis of cyclopentanones from donor–acceptor cyclopropanes and in situ-generated ketenes, in good to excellent yields (up to 93%) and with generally good to excellent diastereoselectivity (dr up to 37:1) and enantiospecificity. Future studies will seek to develop a DyKAT variant of this reaction.
Author contributions
N. J. K. supervised the research; N. J. K., S. M. and M. M. conceived the idea; S. M., S. M. C., S. A., M. M. and M. P. carried out the synthetic experimental work and data acquisition. N. J. K. prepared the manuscript with contributions from S. A. and B. G. K. All authors approved the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support has been provided by Science Foundation Ireland (18/IF/6301 to N. J. K.) and the Irish Research Council (GOIPD/2019/637 to S. M. and GOIPG/2023/4884 to S. A.). Open access funding provided by IReL.References
- (a) E. J. Corey, N. M. Weinshenker, T. K. Schaaf and W. Huber, J. Am. Chem. Soc., 1969, 91, 5675 CrossRef CAS PubMed; (b) T. Hyodo, Y. Kiyotsuka and Y. Kobayashi, Org. Lett., 2009, 11, 1103 CrossRef CAS PubMed; (c) N. Yamakawa, S. Suemasu, Y. Okamoto, K.-i. Tanaka, T. Ishihara, T. Asano, K. Miyata, M. Otsuka and T. Mizushima, J. Med. Chem., 2012, 55, 5143 CrossRef CAS PubMed; (d) H. Sugimoto, Y. Tsuchiya, K. Higurashi, N. Karibe, Y. Limura, A. Sasaki, Y. Yamanashi, H. Ogura, S. Araki, T. Kosasa, A. Kusota, M. Kozasa and K. Yamatsu, U.S. Patent 5100901A, Eisai Co., Ltd., Japan, 1992 Search PubMed.
- (a) S. P. Lathrop and T. Rovis, J. Am. Chem. Soc., 2009, 131, 13628 Search PubMed; (b) J. A. Dabrowski, D. C. Moebius, A. J. Wommack, A. F. Kornahrens and J. S. Kingsbury, Org. Lett., 2010, 12, 3598 CrossRef CAS PubMed; (c) K. E. Ozboya and T. Rovis, Chem. Sci., 2011, 2, 1835 RSC; (d) R. Okamoto and K. Tanaka, Org. Lett., 2013, 15, 2112 CrossRef CAS PubMed; (e) D.-Y. Zhu, M.-H. Xu, Y.-Q. Tu, F.-M. Zhang and S.-H. Wang, Chem. – Eur. J., 2015, 21, 15502 CrossRef CAS PubMed; (f) N. A. White and T. Rovis, J. Am. Chem. Soc., 2015, 137, 10112 Search PubMed; (g) S. Cuadros, L. Dell'Amico and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 11875 Search PubMed; (h) X.-Y. Chen, S. Li, H. Sheng, Q. Liu, E. Jafari, C. von Essen, K. Rissanen and D. Enders, Chem. – Eur. J., 2017, 23, 13042 CrossRef CAS PubMed; (i) W. Liu, S. Rajkumar, W. Wu, Z. Huang and X. Yang, Org. Lett., 2019, 21, 3563 CrossRef CAS PubMed; (j) Z. Chen, Y. Aota, H. M. H. Nguyen and V. M. Dong, Angew. Chem., Int. Ed., 2019, 58, 4705 CrossRef CAS PubMed; (k) S. Kayal, J. Kikuchi, M. Shimizu and M. Terada, ACS Catal., 2019, 9, 6846 Search PubMed.
- (a) P. D. Pohlhaus, S. D. Sanders, A. T. Parsons, W. Li and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 8642 CrossRef CAS PubMed; (b) A. T. Parsons and J. S. Johnson, J. Am. Chem. Soc., 2009, 131, 3122 CrossRef CAS PubMed.
- A. F. G. Goldberg, N. R. O'Connor, R. A. Craig II and B. M. Stoltz, Org. Lett., 2012, 14, 5314 Search PubMed.
- (a) M. Mondal, M. Panda, V. McKee and N. J. Kerrigan, J. Org. Chem., 2019, 84, 11983 Search PubMed; (b) J. Liu, M.-M. Li, B.-L. Qu, L.-Q. Lu and W.-J. Xiao, Chem. Commun., 2019, 55, 2031 RSC.
- (a) M. Mondal, M. Panda, N. W. Davis, V. McKee and N. J. Kerrigan, Chem. Commun., 2019, 55, 13558 RSC; (b) A. A. Ibrahim, S. C. J. O'Reilly, M. Bottarel and N. J. Kerrigan, Chem. Commun., 2024, 60, 3283 RSC; (c) M. Mondal, S. Mitra, D. J. Twardy, M. Panda, K. A. Wheeler and N. J. Kerrigan, Chem. – Eur. J., 2022, 28, e202104391 Search PubMed; (d) A. A. Ibrahim, P.-H. Wei, G. D. Harzmann, D. Nalla, M. Mondal, K. A. Wheeler and N. J. Kerrigan, Tetrahedron, 2021, 78, 131838 Search PubMed; (e) S. Chen, A. A. Ibrahim, N. J. Peraino, D. Nalla, M. Mondal, M. Van Raaphorst and N. J. Kerrigan, J. Org. Chem., 2016, 81, 7824 Search PubMed; (f) M. Mondal, S. Chen, N. Othman, K. A. Wheeler and N. J. Kerrigan, J. Org. Chem., 2015, 80, 5789 CrossRef CAS PubMed.
- (a) A. Lücht, A. Kreft, P. G. Jones and D. B. Werz, Eur. J. Org. Chem., 2020, 2560 Search PubMed; (b) A. U. Augustin, M. Busse, P. G. Jones and D. B. Werz, Org. Lett., 2018, 20, 820 CrossRef CAS PubMed.
- N. Radhoff, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202304771 CrossRef CAS PubMed.
- M. Mishra, K. Verma, S. Banerjee and T. Punniyamurthy, Chem. Commun., 2024, 60, 2788 RSC.
- (a) R. Tennyson and D. Romo, J. Org. Chem., 2000, 65, 7248 CrossRef CAS PubMed; (b) A. E. Taggi, A. M. Hafez, H. Wack, B. Young, W. J. Drury III and T. Lectka, J. Am. Chem. Soc., 2000, 122, 7831 CrossRef CAS; (c) S. G. Nelson, T. J. Peelen and Z. Wan, J. Am. Chem. Soc., 1999, 121, 9742 CrossRef CAS.
- For reviews and leading references on ketene chemistry see: (a) A. D. Allen and T. T. Tidwell, ARKIVOC, 2016, 415 Search PubMed; (b) S. Chen, E. C. Salo and N. J. Kerrigan, in Science of Synthesis Reference Library, Asymmetric Organocatalysis, Vol. 1, Lewis Base and Acid Catalysts, ed. B. List, Thieme, Stuttgart, ch. 1.1.10, 2012, p. 455 Search PubMed; (c) D. H. Paull, A. Weatherwax and T. Lectka, Tetrahedron, 2009, 65, 6771 CrossRef CAS PubMed; (d) T. T. Tidwell, Ketenes, Wiley, New York, 2nd edn., 2006 Search PubMed.
- For reviews and leading references on reactions of donor-acceptor cyclopropanes with electron-rich alkenes/alkynes see: (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504 CrossRef CAS PubMed; (c) H. K. Grover, M. R. Emmett and M. A. Kerr, Org. Biomol. Chem., 2015, 13, 655 RSC; (d) J.-P. Qu, C. Deng, J. Zhou, X.-L. Sun and Y. Tang, J. Org. Chem., 2009, 74, 7684 CrossRef CAS PubMed; (e) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2011, 50, 12075 CrossRef CAS PubMed; (f) J.-P. Qu, Y. Liang, H. Xu, X.-L. Sun, Z.-X. Yu and Y. Tang, Chem. – Eur. J., 2012, 18, 2196 CrossRef CAS PubMed; (g) H. Xu, J.-P. Qu, S. Liao, H. Xiong and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 4004 CrossRef CAS PubMed; (h) Y. A. Volkova, E. M. Budynina, A. E. Kaplun, O. A. Ivanova, A. O. Chagarovskiy, D. A. Skvortsov, V. B. Rybakov, I. V. Trushkov and M. Y. Melnikov, Chem. – Eur. J., 2013, 19, 6586 CrossRef CAS PubMed; (i) F. de Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239 CrossRef CAS PubMed; (j) W. D. Mackay, M. Fistikci, R. M. Carris and J. S. Johnson, Org. Lett., 2014, 16, 1626 CrossRef CAS PubMed; (k) S. Racine, B. Hegedüs, R. Scopelliti and J. Waser, Chem. – Eur. J., 2016, 22, 11997 CrossRef CAS PubMed.
- Synthesis of DA cyclopropanes: (a) R. Ieki, Y. Kani, S. Tsunoi and I. Shibata, Chem. – Eur. J., 2015, 21, 6295 CrossRef CAS PubMed; (b) K. Vermaa and P. Banerjee, Adv. Synth. Catal., 2017, 359, 3848 CrossRef; (c) ref. 3a; ; (d) A. T. Parsons, M. J. Campbell and J. S. Johnson, Org. Lett., 2008, 10, 2541 CrossRef CAS PubMed.
- (a) C. M. Rasik and M. K. Brown, J. Am. Chem. Soc., 2013, 135, 1673 CrossRef CAS PubMed; (b) C. M. Rasik, Y. J. Hong, D. J. Tantillo and M. K. Brown, Org. Lett., 2014, 16, 5168 CrossRef CAS PubMed.
- 13C NMR analysis of CD2Cl2 solutions of DA cyclopropane with InBr3 and, separately, ethylphenylketene with EtAlCl2 displayed a change in the chemical shift values for the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signal(s) indicating strong interactions.
O signal(s) indicating strong interactions. - (a) H. Yamamoto, Proc. Jpn. Acad., Ser. B, 2008, 84, 134 CrossRef CAS PubMed; (b) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01313a |
| This journal is © The Royal Society of Chemistry 2024 |