
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of C-glycoside analogues of isopropyl β-D-1-thiogalactopyranoside (IPTG) and 1-β-D-galactopyranosyl-2-methylpropane. Conformational analysis and evaluation as inhibitors of the lac repressor in E. coli and as galactosidase inhibitors†
Eoin
Hever
a,
Venkatesan
Santhanam
 a,
Sherivan
Alberi
a,
Ashis
Dhara
a,
Mikael
Bols
b,
Heinz-Peter
Nasheuer
a and
Paul V.
Murphy
*ac
a,
Sherivan
Alberi
a,
Ashis
Dhara
a,
Mikael
Bols
b,
Heinz-Peter
Nasheuer
a and
Paul V.
Murphy
*ac
aSchool of Biological and Chemical Sciences, University of Galway, University Road, Galway, Ireland H91TK33. E-mail: paul.v.murphy@universityofgalway.ie
bDepartment of Chemistry, Københavns Universitet, Universitetsparken 5, 2100 København Ø, Denmark
cSSPC – the Science Foundation Ireland Research Centre for Pharmaceuticals, University of Galway, University Road, Galway, Ireland H91TK33
First published on 20th August 2024
Abstract
Isopropyl 1-thio-β-D-galactopyranoside (IPTG, 1) is used widely as an inducer of protein expression in E. coli and 1-β-D-galactopyranosyl-2-methylpropane (2), a C-glycoside analogue of 1, has also been identified as an inducer. Here, synthesis and study of mimetics of 1 and 2, 1-β-D-galactopyranosyl-2-methylpropan-1-ols and two cyclic acetals derivatives, that constrain the presentation of the iPr group in various geometries is described. Conformational analysis of C-glycosides in protic solvent is performed using (i) Desmond metadynamics simulations (OPLS4) and (ii) use of 3JHH values obtained by 1H-NMR spectroscopy. 1-β-D-Galactopyranosyl-2-methylpropane (2) is an effective protein expression inducer when compared to the new mimetics, which were less effective or did not induce expression. 1-β-D-Galactopyranosyl-2-methylpropane (2) led to significantly reduced proteolysis during protein expression, compared to IPTG suggesting that recombinant protein purification will be easier to achieve with 2, yielding proteins with higher quality and activity. IPTG reduced bacterial growth to a greater degree than 2 compared to the control. IPTG's isopropyl group was observed by molecular dynamics (MD) simulations to be flexible in the binding pocket, deviating from its crystal structure binding mode, without impacting other interactions. The MD simulations predicted that 1-β-D-galactopyranosyl-2-methylpropane (2) was more likely than IPTG to bind the repressor with a conformation favoured in protic solvent, while maintaining interactions observed for IPTG. MD simulations predicted that isobutanol derivatives may disrupt interactions associated with IPTG's binding mode. The compounds were also evaluated as inhibitors of galactosidases, with 2 being the more potent inhibitor of the E. coli β-galactosidase. The constrained cyclic acetals showed similar inhibition constants to IPTG indicating E. coli β-galactosidase can recognize galactopyranoses with varying presentation of the iPr group.
1. Introduction
The S- and C-glycosides1 are mimetics of O-glycosides, which are of interest due to their stability to hydrolysis that occurs under acidic conditions or in the presence of enzymes and accordingly have potential as therapeutic agents or as tools where chemically and biochemically stable glycomimetics are needed. Conformational preferences of C-glycosides2 and S-glycosides3 are relevant as they may influence affinity and selectivity for carbohydrate binding receptors.4 Another feature of C-glycosides is that the glycosidic carbon can be substituted more than oxygen, enabling additional interactions with receptors to be facilitated, or which can alter conformation preference around the C–C bond.Many saccharides, such as galactopyranosides/glucopyranosides adopt chair structures for the pyranose ring (4C1). For disaccharides, such as lactose (Galβ1-4Glc), the glycosidic torsion preferences, for ϕ and Ψ, are the main influence on disaccharide conformation. The exo-anomeric effect influences conformation preference about the C–O (glycoside) bond and steric and torsional strain are also factors. For C-glycosides, there is no exo-anomeric effect5 and conformational preference for the C–C bond is primarily governed by minimization of steric and torsional strain. Staggered conformations are generally preferred for the ϕ and Ψ torsions in C-glycosides, as reported by Kishi et al.6 and by Barbero et al.7 In contrast an eclipsed conformation can be preferred for Ψ in O-disaccharides like lactose.
Applications of S- and C-glycosides include lectin inhibitors,8 as decoy substrates for bacterial glycosyltransferases9 or as glycoprocessing inhibitors.10 The topic herein relates to the influence of isopropyl thio-β-D-galactopyranoside (IPTG, 1) and its C-glycoside mimetics on the Escherichia coli (E. coli) lac promoter/repressor.11 Components of the E. coli lac operon, and hybrid promoters containing the lac operator sequence such as TAC and T5-lac promoters, have been modified to enable expression of recombinant proteins, which is in wide use. Allolactose (Galβ1,6Glc), formed in situ from lactose (Galβ1,4Glc), binds to the lac repressor12 and induces a conformational change, that greatly reduces affinity of the repressor for DNA, facilitating protein translation. β-Galactosidase, produced after induction of protein expression, hydrolyses the glycosidic bond present in lactose/allolactose, removing the inducer and thereby allowing the repressor to inhibit protein synthesis. On the other hand, IPTG is a stable mimetic of allolactose, and is not hydrolysed by β-galactosidase, and, thus, is widely used for induction of the lac operon. IPTG induces a conformational change in the repressor, allosterically inhibiting repressor binding to DNA, and enabling protein synthesis. As it is stable to β-galactosidase, the concentration of the IPTG is believed to remain constant and protein synthesis is not inhibited.
Various small molecule inducers of protein expression have been investigated.13 IPTG (1) was identified as a high affinity binder to the lac repressor, with affinity similar to the natural inducer allolactose, and ∼100 fold higher affinity than its O-glycoside counterpart, isopropyl β-D-galactopyranoside (IPG), which is also an inducer. Being an inducer is associated with binding the repressor and a corresponding low affinity of the repressor for DNA. Anti-inducers were also identified, which show affinity for the repressor but these substances interact to increase affinity between the repressor and DNA. Thirdly, nitrophenyl β-D-galactopyranoside (ONPG) is a non-inducer, capable of binding to the repressor, but not influencing its interaction with DNA. Unlike OPNG, nitrophenyl-1-thio-β-D-galactopyranoside (T-ONPG), the S-glycoside mimetic, is a potent inducer and it was speculated by Lewis and co-workers that this could be due to flexibility of the S-glycoside compared to the O-glycoside. The crystal structure of IPTG 1 at 2 Å resolution indicated that the galactopyranoside's 6-OH is involved in stabilizing interactions between the repressor's Ser193 and Asp149, which is believed to induce an inactive conformation of the repressor.14
The C-glycosyl compound, 1-(β-D-galactopyranosyl)-2-methyl-propane (2)15 was earlier shown by Pohl and co-workers to have similar or improved induction properties compared to IPTG.16 Increased ligand preorganization into a bioactive conformation can reduce the entropic penalty when binding, which is in turn reflected in an affinity increase, and so, ligand conformational preorganization may be important.17 Here, we synthesised of analogues of 2 (isobutanols) with a hydroxyl substituent at the anomeric carbon, such as 4 & 5, and also prepared 3 and 6 to probe the role of increasing conformation restriction; we evaluated how these C-glycosides compared to 1 and 2, in inhibiting (i) the lac repressor and (ii) inhibiting galactosidases, to provide biological context for the research.18 We included 7 for the induction experiments. We did find an advantage in using 2 compared to 1 in protein expression in that there was reduced proteolysis of the expressed protein, which may be an indicator of reduced stress. While the newly synthesised C-glycosides did not induce or only weakly induced protein synthesis, they did show comparable activity as inhibitors of β-galactosidase compared to IPTG (Chart 1). We performed conformational analysis of 1–6 and computational methods were used to investigate interaction of ligands to the lac repressor in attempt to rationalize results.
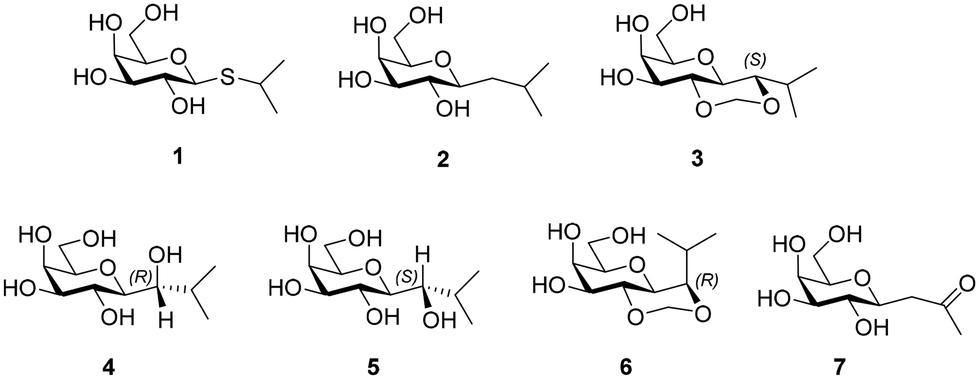 | ||
| Chart 1 Chemdraw structures of 1–7. | ||
2. Results and discussion
2.1 Synthesis of 1–7
Several methods exist for C-glycoside synthesis and this topic has been comprehensively reviewed.19 Samarium iodide (SmI2) promoted C-glycoside synthesis was used in this work.20 Thus, the synthesis started with preparation of a glycosylsulfone 11 from tri-O-benzyl-D-galactal, which underwent epoxidation using dimethyldioxirane generated in situ21 to afford 8. The epoxide 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 is sensitive to hydrolysis and, avoiding chromatographic purification, was subjected to nucleophilic reaction with 2-mercaptopyridine in presence of sodium hydride to give β-substituted thioglycoside 9 in 86% yield. Reaction of 9 with p-methoxybenzyl chloride in presence of sodium hydride (60% in mineral oil) gave the PMB ether 10 (80%), which on subsequent oxidation by m-chloroperoxybenzoic acid gave sulfone 11 in 85% yield (Scheme 1).
22 is sensitive to hydrolysis and, avoiding chromatographic purification, was subjected to nucleophilic reaction with 2-mercaptopyridine in presence of sodium hydride to give β-substituted thioglycoside 9 in 86% yield. Reaction of 9 with p-methoxybenzyl chloride in presence of sodium hydride (60% in mineral oil) gave the PMB ether 10 (80%), which on subsequent oxidation by m-chloroperoxybenzoic acid gave sulfone 11 in 85% yield (Scheme 1).
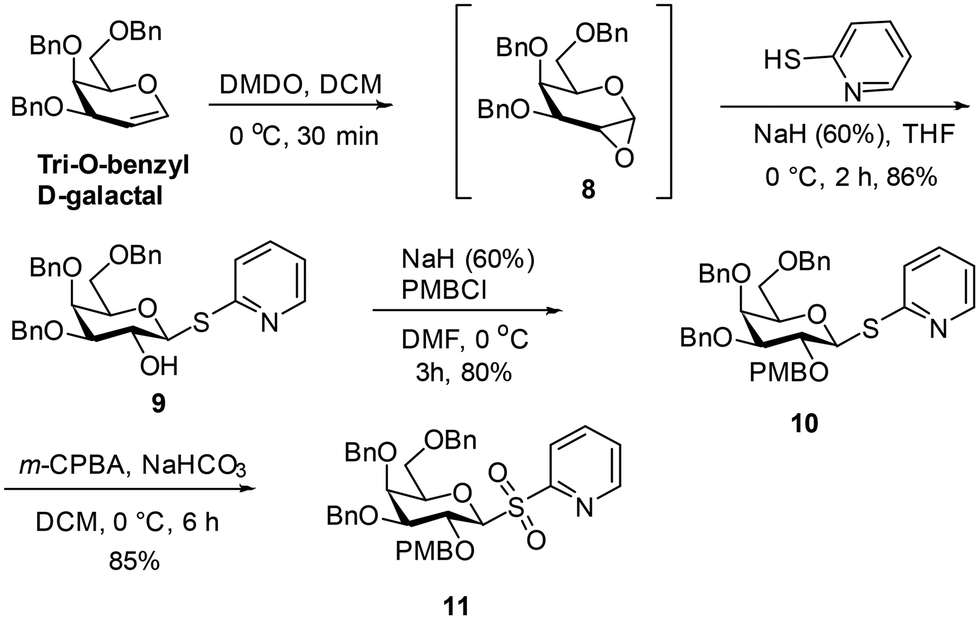 | ||
| Scheme 1 Preparation of the sulfone 11. | ||
The reaction of a freshly prepared SmI223 with sulfone 11 in the presence of isobutyraldehyde gave the secondary alcohol 12 in 42% yield. The NMR spectral analysis indicated that the presence of the 1-deoxygalactopyranose 12a as an impurity from reaction of 11, which was inseparable from the product (ratio 12a : 12 =∼1:3). Removal of the PMB ether from the mixture of 12 and 12a using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)24 followed by chromatography led to isolation of diol 13 in 61% yield. Debenzylation of 13 by catalytic hydrogenation in the presence of 10% Pd–C gave 4 in 53% yield. Treatment of 13 with dimethoxymethane in the presence of camphorsulfonic acid25 provided the acetal 14 in 60% yield. Debenzylation of 14 was carried out using catalytic hydrogenation in the presence of 10% Pd–C to afford 6 in 65% yield (Scheme 2).
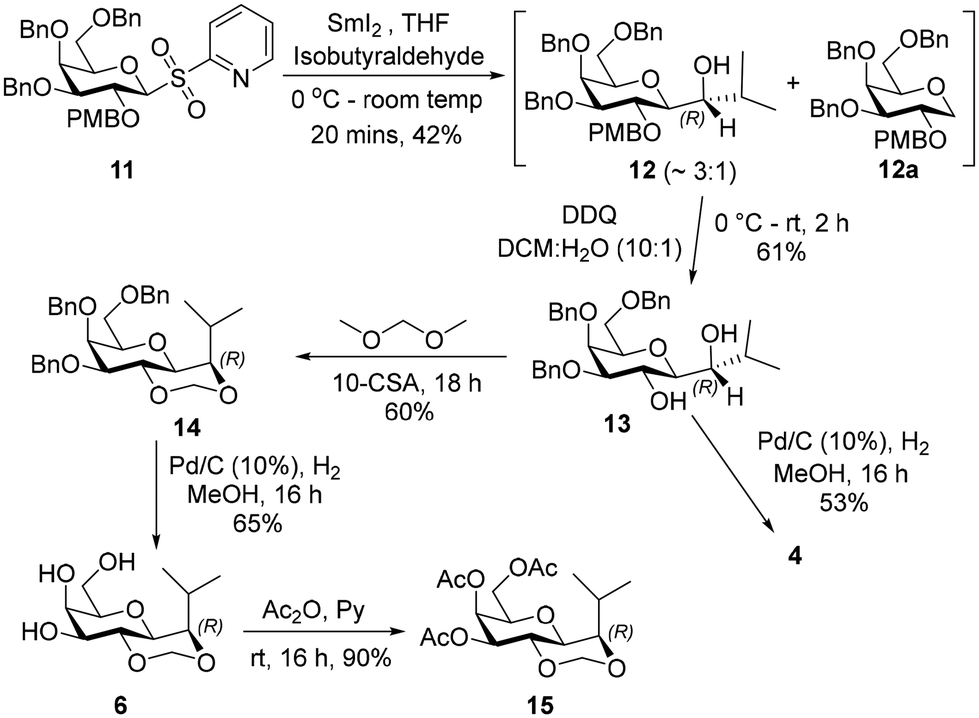 | ||
| Scheme 2 Synthesis of 4 and 6. | ||
Product 6 was peracetylated, to give 15, which enabled stereochemical configuration determination as 1H-NMR spectrum of 15 showed reduced signal overlap compared to 6. The 1H-NMR spectrum of 15 displayed a 3J value of 5.7 Hz for the coupling between the galactopyranosyl H-1 (δ 3.68 ppm) and the isopropyl group H (δ 3.72 ppm), which is consistent with H-1 being axial and the isopropyl H being equatorial in the acetal containing ring. This implied the configuration of each of the intermediates 12–14 and of products 4 and 6 at the carbon indicated in Scheme 2 is (R).
The (S)-diastereiosomer, 17, was obtained (85%) in a two-step oxidation reduction sequence, after treating alcohol 12 with Dess–Martin periodinane, followed by reduction of the resulting ketone with sodium borohydride. Removal of the PMB group from 17 using trifluoroacetic acid afforded the expected diol in 78% yield which was further treated with dimethoxymethane in the presence of CSA to give 18 in 63% yield. Debenzylation of 18 afforded 3 in 76% yield. Debenzylation of 17 by catalytic hydrogenation in the presence of 10% Pd–C gave 5 in 62% yield (Scheme 3).
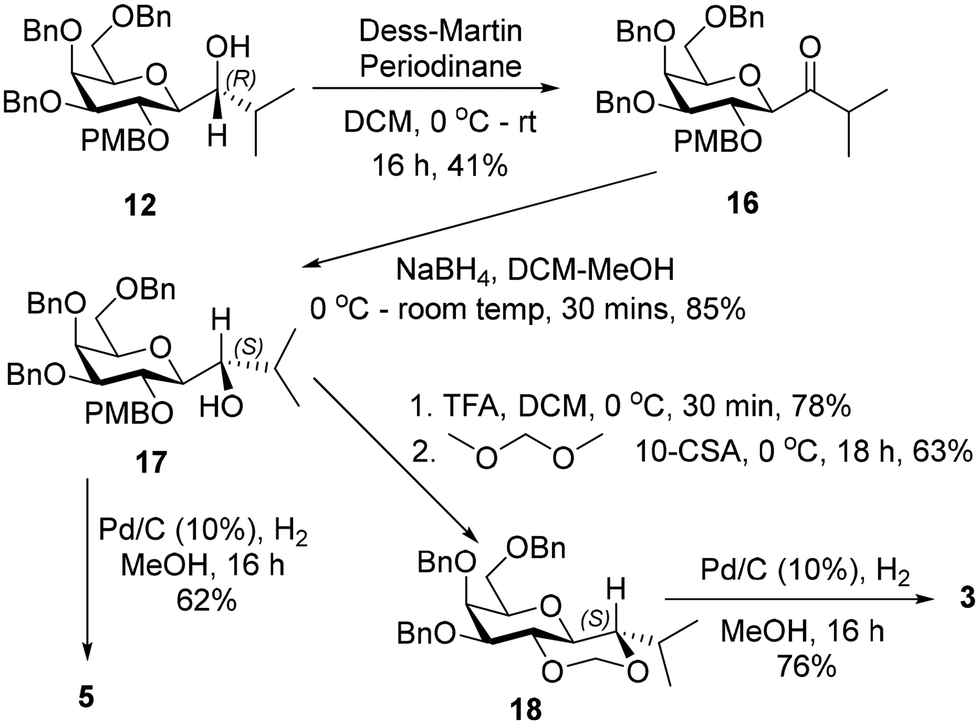 | ||
| Scheme 3 Synthesis of 3 & 5. | ||
Compound 2 was synthesized according to the 3-step procedure previously reported by Pohl et al. (see Scheme S1†). Compound 7 was also prepared as described previously.26
2.2 Conformational analysis of 1–6
|
|
||||
|---|---|---|---|---|
| Compd | 3 J H1, HX | 3 J H1, HY | 3 J HX, HiPr | 3 J HY, HiPr |
| 2 | 10.0 | 2.1 | 4.2 | 9.7 |
| 2 (D2O) | 9.9 | 2.1 | 4.3 | 9.9 |
| 3 | 9.1 | — | 2.8 | — |
| 4 (R) | 1.6 | — | 8.7 | — |
| 5 (S) | 5.2 | — | 5.2 | — |
| 6 | n.d. | — | 10.0 | — |
| 15 | 5.7 | — | 10.3 | — |
 | ||
| Fig. 1 Chemdraw structures and Newman projections for conformers of isopropyl β-D-galactopyranoside (IPG). The plot on bottom left shows the output from coordinate scan plots (OPLS4 force field) obtained from Macromodel while applying the water GBSA model. Contours are those within 5 kcal mol−1 of the lowest energy conformer and there are 0.25 kcal mol−1 differences between contour lines, with energy increasing from red to blue. The plot on the right is the energy surface from 10 ns metadynamics simulation performed in water. Energy (kcal mol−1) increases from blue/navy to yellow. The dihedral angle ϕ is defined by atoms H1–C1–O–CiPr and ψ by C1–O–CiPr–HiPr. The metadynamics and coordinate scans predict similar global minimum conformers (ϕ = exo–syn or + gauche; ψ = +gauche). The term exo refers to the conformers where the exo-anomeric effect occurs. | ||
The energy plots obtained from metadynamics for 1–6, with water as solvent are shown in Fig. 2 and geometries for the lowest energy conformer are displayed in Fig. 3. The metadynamics was performed in CH3OH also for 1–6, as this was the solvent used for NMR, with the same lowest energy conformer and similar energy surfaces found as in water. For IPTG 1, the ϕ +gauche and ψ = +gauche conformer was lowest in energy, like that found in its crystal structure.32 Other low energy conformers identified from the metadynamics simulation had ϕ and ψ angles approximating to those of 1a, 1b, 1d and 1e found by DFT. Metadynamics of 2 showed a preference for the ϕ = +gauche and ψ = +gauche conformation which is consistent with the 3JHH values measured experimentally. For 4 the ϕ = +gauche and ψ = +gauche is preferred based on the metadynamics and this is also supported by the 3JHH values. For 5, in the metadynamics simulations, the anti-conformer, like in 5c, was lowest in energy, not 5a as predicted by DFT in the gas phase, most likely due to intramolecular H-bonding being disrupted by interaction with solvent33 and this conformer geometry agrees with 3JHH values measured. Metadynamics predicted the ϕ = +gauche for 3 and ϕ = anti for 6, due to the cyclic acetal constraint; the 1H-NMR data show that the pyranose rings adopt 4C1 geometry in 3 and 6. Thus 3 is a highly constrained mimetic of the preferred ϕ = exo–syn conformation of the O-glycoside. For 6 the 3JHH value of 10.0 Hz observed in the isopropyl CH signal, assigned to the coupling between this proton and the adjacent acetal ring proton, is consistent with the low energy conformer found for 6 by metadynamics (Fig. 3).
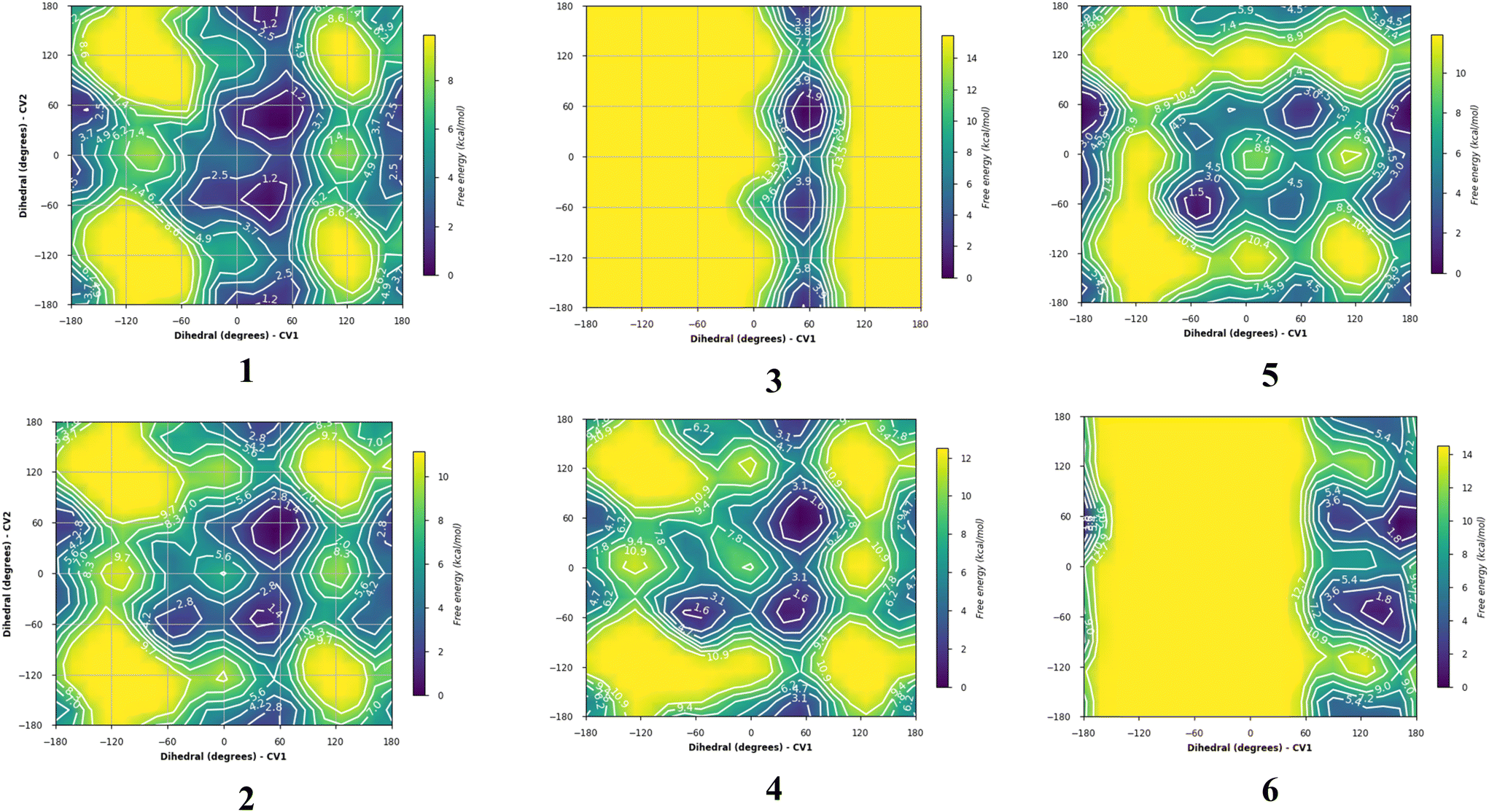 | ||
| Fig. 2 Plots showing relative energy (kcal mol−1) from metadyamics simulations where the collective variables CV1 = ϕ and CV2 = ψ. The simulations were performed in water boxes (with explicit water). | ||
 | ||
| Fig. 3 Low energy conformers for 1–6 obtained from metadynamics simulations in explicit water. | ||
2.3 Induction of protein expression in E. coli and growth by glycomimetic compounds
To determine the activity of glycomimetics in the induction of recombinant proteins in E. coli, the expression of monomeric teal fluorescent protein 1 (mTFP1) was investigated following standard procedures.34 In brief, bacterial cells BL21(DE3) harboring the vector pQLinkHD-mTFP1 were treated with 1 mM of compounds. At the indicated times, bacterial culture samples were collected and OD600 values were determined with a colorimeter 45 (ThermoFisher Scientific). Additionally, mTFP1 expression levels were measured with an Applied Biosystems StepOnePlus™ PCR System (ThermoFisher) by determining fluorescence in the blue channel. To this end, bacterial culture material, V = 30 μl, was centrifuged at 13500 rpm for 5 min at 4 °C. The supernatant was removed, and the cell pellets were suspended in 90 μl of phosphate-buffered saline yielding a 1/3 dilution of the cell concentration and frozen at −20 °C until fluorescence measurement was ready to be taken. After thawing of the cell suspension, 20 μl of the cell suspension were transferred into Applied Biosystems MicroAmp™ 96-Well Reaction plates (ThermoFisher Scientific) for measuring the amount of fluorescence at 25 °C in the blue channel of an Applied Biosystems StepOnePlus™ quantitative PCR System (ThermoFisher Scientific). Then the fluorescence value for the samples were normalized to the OD600 value and the highest value of the 24 h time point was arbitrarily set to 1.
Additionally, the mTFP1 expression was investigated by western blotting. At the indicated times, 500 μl of bacteria cultures were collected and centrifuged. The cell pellets were suspended in 50 μl of PBS (yielding a 10-fold concentration). For SDS PAGE, 5 μl of a culture with 1 OD600 was loaded on a 10% SDS polyacrylamide gel (loading volumes were adjusted to OD600 values). SDS PAGE, electrotransfer of proteins on PVDF membrane, and protein detection were carried out as previously described35 using Fisher BioReagents™ EZ-Run™ Pre-stained Rec Protein Ladder as a marker. The horseradish peroxidase-labelled monoclonal antibody (A7058, Sigma) recognizing the His6-mTFP1 fusion protein and Pierce™ 1-Step Ultra TMB Blotting solution (ThermoFisher Scientific) were used to develop the western blot. A representative western blot of three independent experiments is shown in Fig. 4, middle panel. In parallel, the same bacterial samples were analysed by SDS PAGE and stained with Coomassie Brilliant Blue to verify equal loading samples (data not shown). The lower panel shows the growth of E. coli Rosetta strain harboring the pET28 vector expressing green fluorescent protein in the absence and presence of 1 mM of IPTG 1 and 2. At the indicated time points, culture samples were collected and the OD600 were measured.
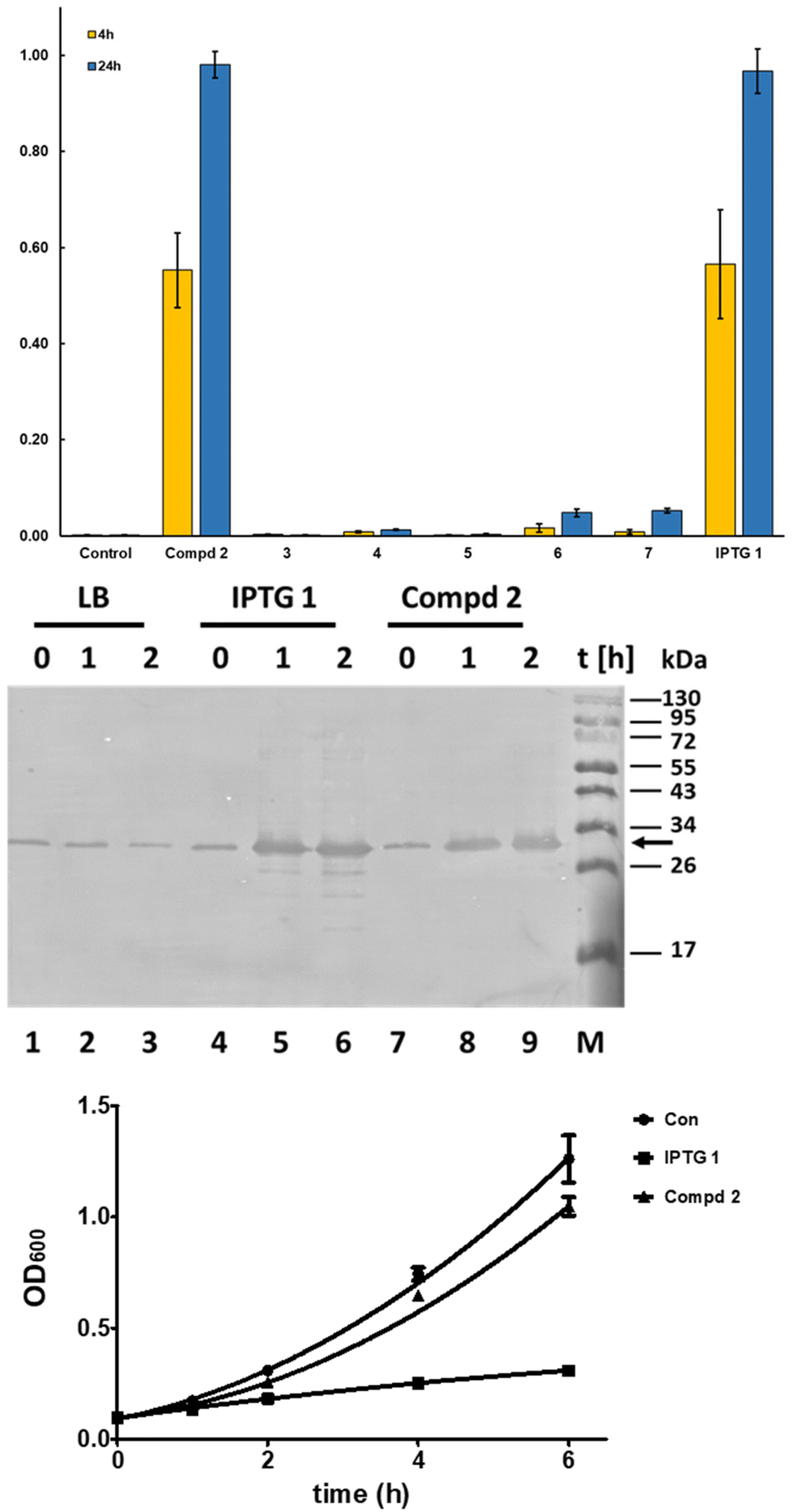 | ||
| Fig. 4 Compd 2 induces expression of recombinant proteins in bacteria with high efficiency. In the top panel, the time-dependent mTFP1 expression as determined after treatment of E. coli with glycomimetics is shown. The mTFP1 expression was verified by western blotting (middle panel). The arrow on the right indicates the full length recombinant His6-mTFP1. The lower panel shows the growth of E. coli BL21(DE3) Rosetta strain harboring the pET28 vector expressing GFP in the absence and presence of 1 mM of IPTG and 2. At the indicated time points, culture samples were collected and their OD600 values were measured. The results, top and lowest panel, are the average and standard deviation of three experiments. The middle panel presents a representative western blot of three independent experiments. | ||
The results (Fig. 4, top and middle panel) show that 2 is an effective inhibitor of the lac repressor protein, leading to induction of protein expression with a similar expression level at 4 h and 24 h of addition as with IPTG (top panel), whereas the other C-glycosides are much less effective. Cyclic acetal 6 and the ketone 7 showed the capability to induce protein production but to a lesser degree than 1 and 2. Similar results were found using bacteria expressing green fluorescent protein (GFP) using the vector pET28 BL21(DE3) Rosetta as a host (data not shown).
The mTFP1 expression was verified using western blotting (Fig. 4, middle panel). The western blot shows that in the absence of IPTG and 2 a minimal amount of mTFP1 is produced, which does not increase during culturing. In contrast, both compounds, 1 and 2, efficiently induce the production of recombinant protein as early as 1 h after their addition to bacterial cultures and the protein expression increases with time (Fig. 4, middle panel). Interestingly, IPTG-treated samples show partial proteolytic degradation of mTFP1 as soon as 1 h, which was not seen in the 2-treated samples even after 2 h (Fig. 4, middle panel compare lanes 5 and 6 with lanes 8 and 9, respectively).
To further analyze the influence of 1 and 2 on E coli, bacteria cultures were treated with 1 mM of 1 and 2 and their growth was compared with bacteria grown in the absence of these compounds. The growth curves of E. coli treated with 1 showed a significant reduction of the cell numbers over time in comparison to untreated and 2-treated cells (Fig. 4, lower panel).
In summary, these findings suggest that 1-β-D-galactopyranosyl-2-methylpropane 2 is an improved inducer of protein expression using the lac repressor compared to IPTG 1. Compound 2 induces similar levels of proteins as determined by two independent methods (Fig. 4 top and middle panel) but with less proteolytic degradation of the recombinant protein as seen with mTFP1 (Fig. 4, middle panel) and an additional recombinant protein LSSmOrange (data not shown). A reduced level of proteolysis allows an easier purification of full-length protein and thus is a marker for improved quality of the recombinant protein important for biochemical, biotechnological and pharmaceutical products and studies. It is known that the addition of IPTG 1 reduces bacterial growth; this has been proposed to be due to stressing the bacteria due to metabolic burden caused by the increased protein synthesis but recently it was shown that metabolic degradation products of IPTG 1 cause additional stress to the bacteria expressing proteins. In contrast, the natural effector lactose36 and 1-β-D-galactopyranosyl-2-methylpropane 2, a subject of this work, caused less stress to the bacteria than 1.
2.4 Investigation of repressor binding to IPTG and C-glycosides by molecular simulations
The interactions of the C-glycoside compounds with the lac repressor were next explored by molecular modelling methods, to try to rationalise the different properties displayed by 1–6.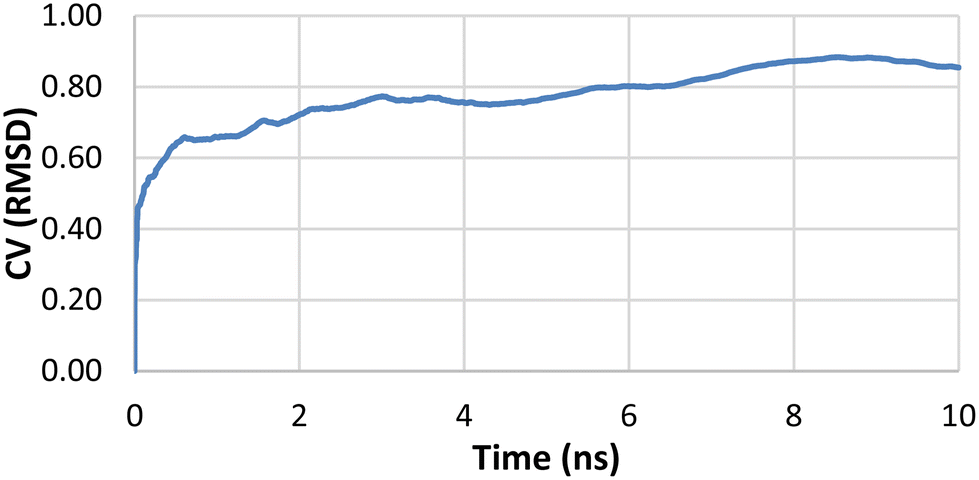 | ||
| Fig. 5 A plot of average RMSD from 10 trials of IPTG heavy atoms from binding pose metadynamics. | ||
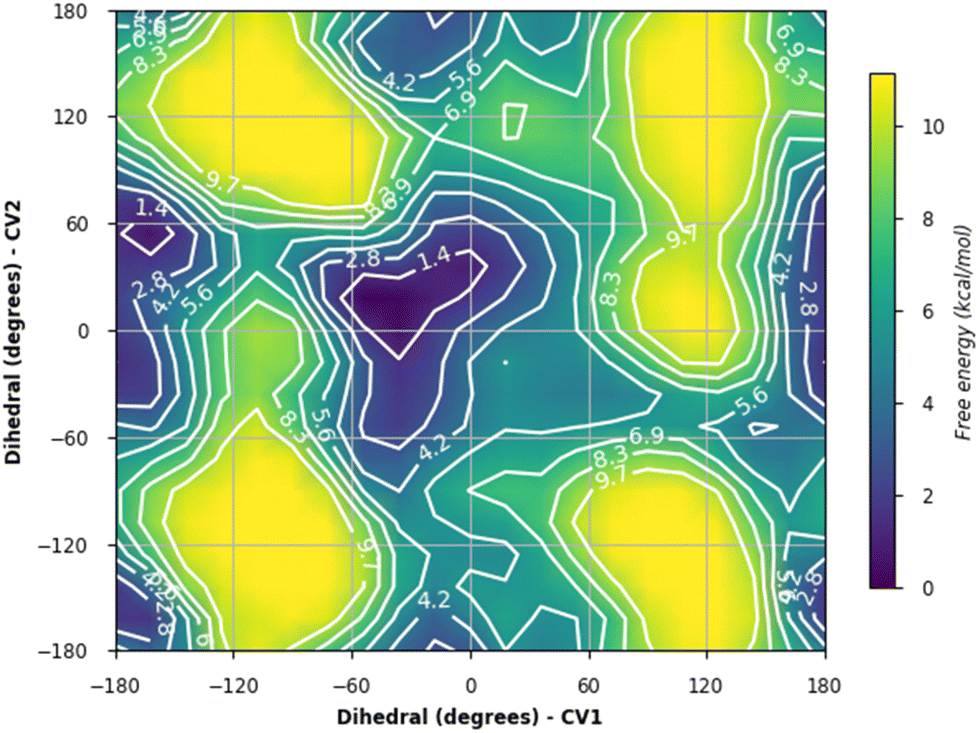 | ||
| Fig. 6 Energy surface plot from metadynamics of IPTG 1 bound to the lac repressor where ϕ (CV1) and ψ (CV2) were the collective variables. | ||
The Desmond analysis reports for each 100 ns simulation is provided in the SI. These reports include monitoring of (i) protein–ligand RMSD values (ii) protein root mean square fluctuation (RMSF), (iii) secondary structure elements (iv) ligand root mean square fluctuation (v) protein–ligand contacts (vi) ligand torsional profiles and (vi) ligand properties such as solvent accessible surface areas and polar surface areas. The root mean square deviation (RMSD) measured the average change in displacement of protein atoms with respect to the starting structure and did not exceed 2 Å in the presence of the ligands studied, indicating there was no major conformational change to protein during the simulation. In the final 10 ns of the simulation for IPTG and the protein, the protein RMSD stabilised at <1.25 Å relative to the starting structure. For protein bound to 2, 4 and 5, the protein RMSD stabilised at <2 Å in the final 20 ns relative to the starting structure, again barely exceeding 2 Å at any stage in the simulations.
The fraction of interactions for each ligand for the first 10 ns (0–10 ns) was compared with the final 10 ns (90–100 ns) during the simulation in Fig. 7. The analysis for IPTG 1 showed that all main direct and indirect H-bonding interactions between protein and ligand mediated by water, as well as the CH–Pi interaction with Trp220 were preserved throughout the 100 ns simulation. For inducer 2, most of the same interactions as IPTG 1 persisted throughout and the direct H-bond interaction between Ser193 and Asp149 occurred frequently for both. For 4 there was the emergence of a direct H-bond of the isobutyl OH with either Asp149 or with Asn125, these being different outcomes from two independent simulations. When the interaction with Asn125 occurred, there was a significant reduction in other interactions, such as removal of direct H-bond between Asn246 and the galactopyranosyl 3-OH and the distance between Ser193 and Asp149 increased, precluding H-bonding between these residues. We, thus, speculate that the additional OH group may disrupt the complex network of interactions that are required for inhibition of the repressor such as the interaction between Ser193 and Asp149. Direct H-bonding from the aglycon OH was seen to occur during the simulation for 5 although it was not as frequent as for 4 and the interaction between Ser193 and Asp149 occurred more frequently for 5. The simulations for 5 did show direct H-bond interaction between Ser193 and Asp149 for much of the simulation comparable to 1 and 2.
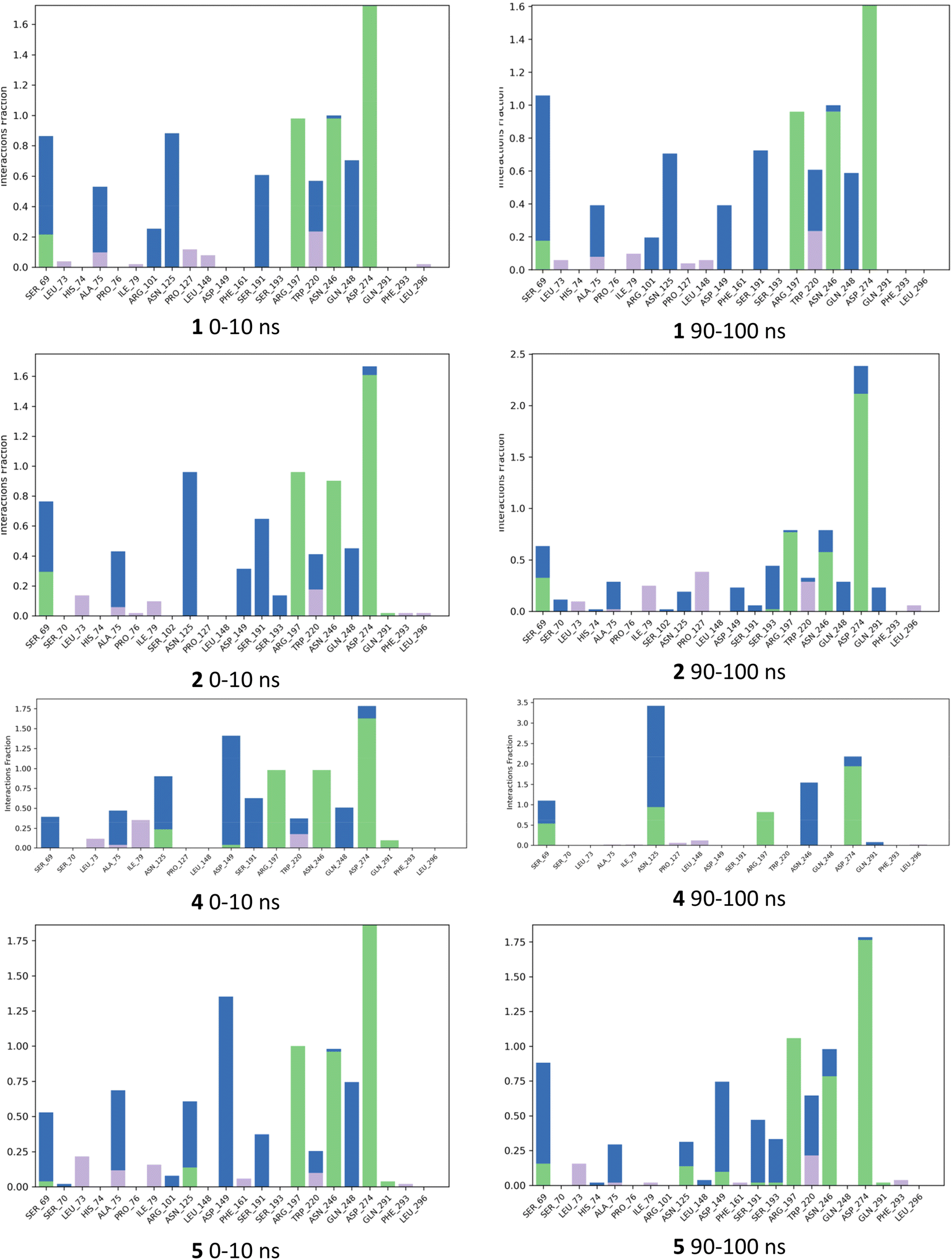 | ||
| Fig. 7 Histograms summarizing fraction of occurrence of ligand protein contacts from selected molecular dynamics simulations. Green = direct H bonds, blue = indirect H bonds, purple = hydrophobic interactions, including CH–Pi interactions. | ||
The values for ϕ and ψ during the simulations were also examined. For 1, the conformation changed from that of the crystal structure; this had occurred within the first 10 ns and persisted for the full length of the simulation. While 1 and 2 showed similar interactions, there was a difference in terms of ϕ and ψ in conformers sampled during the simulations (Fig. 8); 2 was more likely than 1 to adopt ϕ +gauche and ψ +gauche conformer, with 2 being preorganized for binding in this manner; although 2 showed at 90–100 ns sampling of ϕ eclipsed to ϕ +gauche and ψ −gauche conformers like 1, as well as the ϕ +gauche and ψ +gauche. The values for ϕ and ψ for 4, depended on whether there was a direct H-bond to Asp149 or Asn125; a direct H-bonding interaction to Asp149 at 90–100 ns leads to the bound conformation being ϕ +gauche and ψ anti; if the interaction occurred with Asn125 then the ϕ +gauche and ψ −gauche is bound; in the 0–10 ns timeframe before these H-bonding interactions arose there was adoption of the ϕ +gauche and ψ +gauche. For 5, ϕ = −gauche conformers were mostly bound throughout, showing +/− gauche for ψ at 90–100 ns indicating that 5, bound or unbound, does not mimic favoured conformation of 1 or 2.
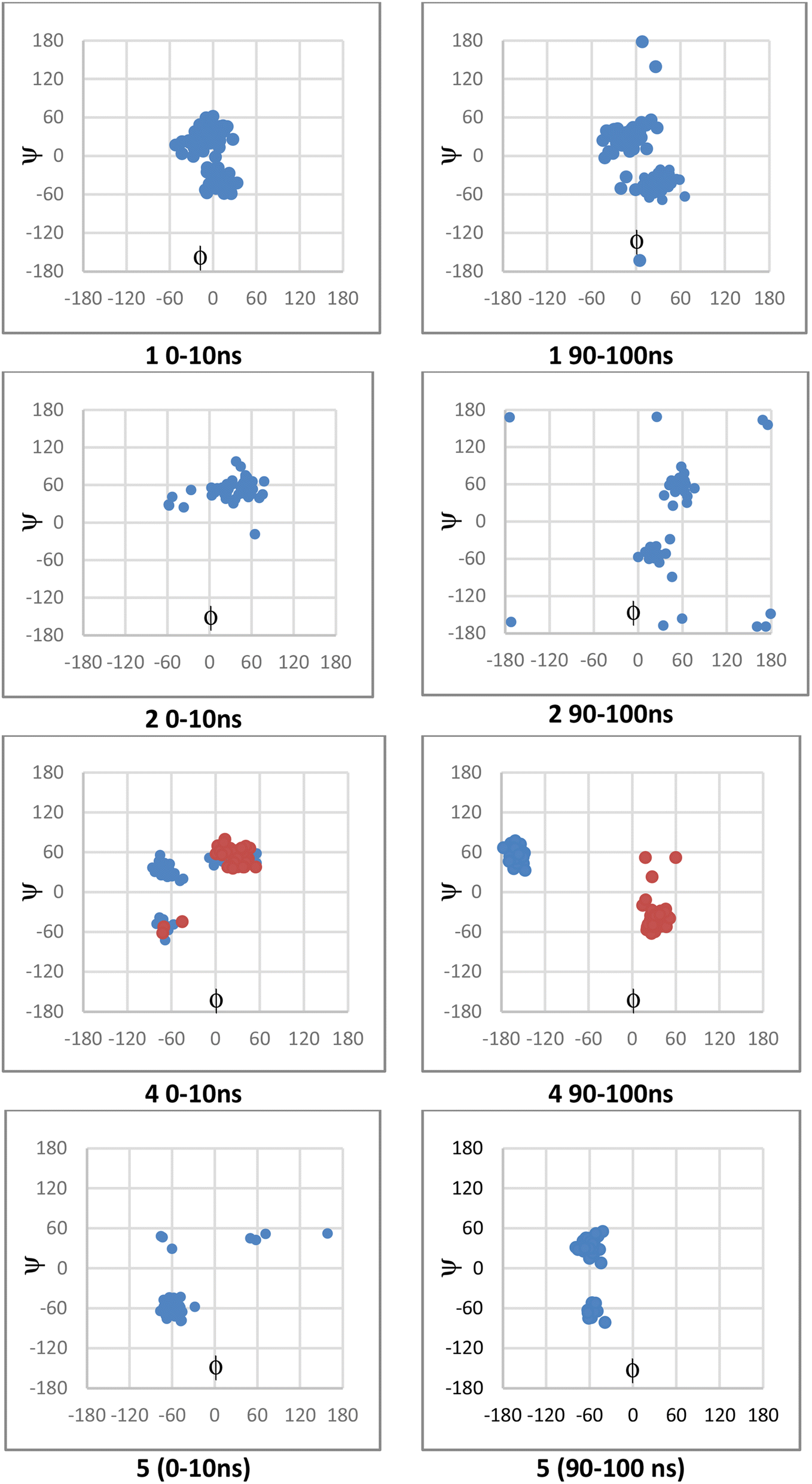 | ||
| Fig. 8 Scatter plots showing ϕ and ψ from structures sampled during molecular dynamics simulations on the left are from 0–10 ns and on right from 90–100 ns. The red and blue data for 4 correspond to data from two simulations; the red occurred when there is direct H-bond interaction of the isobutanol OH with Asn125, whereas the blue there is direct interaction of this OH with Asn149. | ||
2.5 Evaluation of compounds as β-galactosidase inhibitors
The C-glycosides were evaluated as inhibitors of β-galactosidases according to the procedure published previously by Bols et al.42 and the data is shown in Table 2. IPTG 1 is an inhibitor and has a previously reported inhibition constant (Ki) value of 0.08 mM for the native E. coli (lacZ) β-galactosidase;43 the value we obtained was 0.199 mM. This type of inhibitor is weaker than the iminosugar type.44 The best inhibitor in this series was 2, with ∼2-fold improvement seen relative to 1. Compound 2 showed a 6-fold improvement over 1 as an inhibitor of the β-galactosidase from A. niger, while 6 showed improved Ki value compared to that of 2 for the α-galactosidase from A. niger. The crystal structure of IPTG 1 bound to the E. coli β-galactosidase has been determined (PDB ID: 1JYX),45 where the IPTG is bound as the conformer where ϕ = +77° (+gauche) and ψ = −41° (−gauche), the closest conformer to this from the DFT study is 1g. When the coordinates for 1JYX were subjected to protein preparation the ϕ changed to +52° (+gauche) and ψ = −35° (−gauche). The IPTG 1 showed direct H-bond donation from the 2-OH to Glu461 and from 3-OH to Glu537, while the ring oxygen accepted a H-bond from Asn102 and 6-O from His540 and the 6-O also coordinates to a sodium ion; there was also indirect H-bonding (Fig. 9). The isopropyl residue contributes to CH–Pi interactions with Trp999 which is also close to CH protons of the galactose. Similar Ki values especially between 1, 3 and 6 indicate that the enzyme has some flexibility and can accommodate different orientations of the iPr group. It maybe that the iPr of 2 is preorganised similarly to 3, while 2 can also have the H-bond between its 2-OH and Glu461, that explains 2's higher affinity. Although 4 would also be preorganised, there is no benefit to the presence of its extra OH.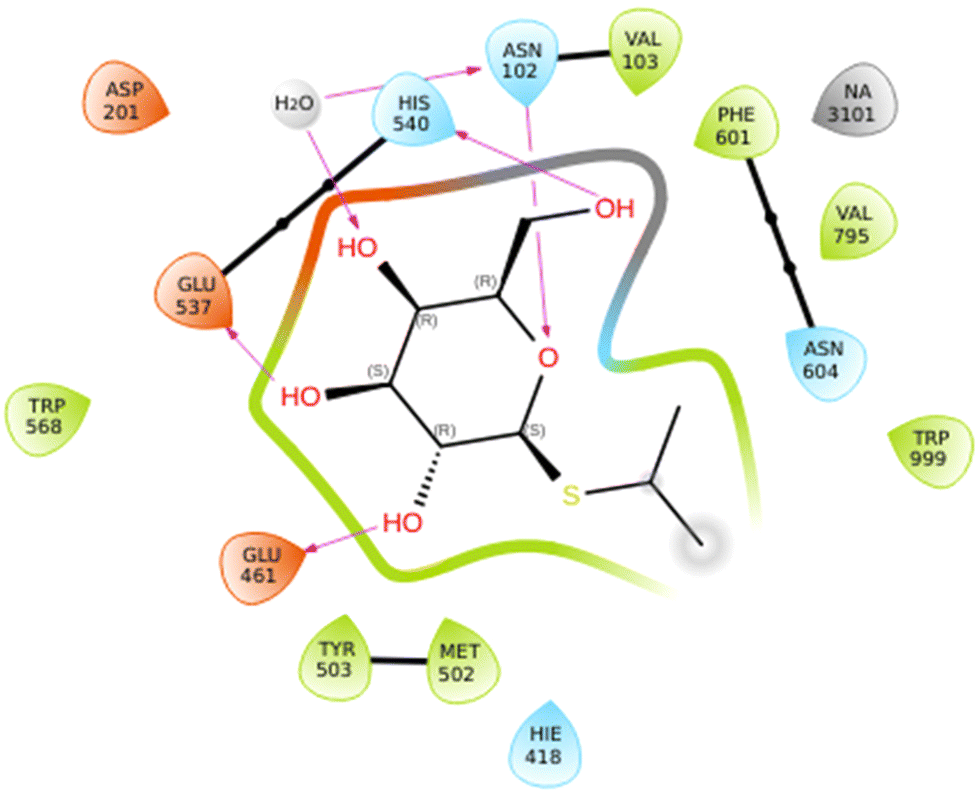 | ||
| Fig. 9 Ligand interaction diagram for IPTG 1 binding to E. coli β-galactosidase. This diagram was generated in Maestro after subjecting coordinates for PDB 1JYX to protein preparation wizard implemented in Maestro. | ||
| Enzyme | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| β-Galactosidase (E. coli) | 0.199 ± 0.066 | 0.0882 ± 0.024 | 0.176 ± 0.094 | 0.230 ± 0.138 | 0.372 ± 0.233 | 0.198 ± 0.086 |
| β-Galactosidase (A. Niger) | 12.1 ± 4.7 | 2.00 ± 1.03 | 3.27 ± 1.52 | 0.832 ± 0.430 | — | — |
| α-Galactosidase (A. Niger) | — | 24.8 ± 12.3 | — | — | — | 2.92 ± 1.30 |
3. Summary and conclusions
Small C-glycoside mimetics of IPTG, 2–7, have been synthesised to investigate their ability to induce protein synthesis via allosteric binding to E. coli's lac repressor and influence the repressor's interaction with DNA. Galactopyranose is an essential component of inducer structure, making strong direct H-bonding interactions with the repressor, with its 6-OH is involved in H-bonding network involving indirect H-bonding via water molecules, that may be involved in stabilizing a repressor conformation that has low affinity for DNA. The nature of galactopyranose's anomeric substituent also determines whether a substance reduces (inducer) or increases (anti-inducer) affinity of the lac repressor for DNA or whether it is a binder but does not increase rate of protein synthesis. Newly synthesized C-glycosides 3–7 did not induce protein synthesis effectively when compared to the standard inducer IPTG 1 and another known inducer 1-β-D-galactopyranosyl-2-methylpropane 2. Interestingly, 1-β-D-galactopyranosyl-2-methylpropane 2 yielded reduced proteolysis of the expressed test proteins compared to 1; the latter observation could be significant in terms of generating proteins with more straightforward purification protocols and improved quality of the purified protein for biochemical, biotechnological, and pharmaceutical purposes.The study of conformation of 1–6 showed parallels between metadynamics simulations with conformation derived by the analysis of the 3JHH values obtained by 1H-NMR in methanol-d4 and for 2 in water. For inducer 2, the major conformation was analogous to that calculated for the O-glycoside, isopropyl β-D-galactopyranoside (ϕ +gauche and ψ +gauche). The S-glycoside 1 appears to be more flexible than C- or O-glycosides based on the metadynamics, evident in energy contour plots. Thus, molecular dynamics simulations (BPMD or longer MD simulation) indicated that 1 shows flexibility in the binding pocket of the lac repressor, that the iPr orientation changes from that observed in the crystal structure; the conformational change for 1 does not appear to significantly disrupt the interaction between repressor's Ser193 and Asp149 or H-bonding network that may be required to adopt a conformation that reduces affinity for DNA. While the orientation of the iPr group in the binding site was different for 2 compared to 1, 2 sustained interactions like those observed for IPTG 1 over 100 ns MD simulations including that between Ser193 and Asp149, linked with low affinity conformation of the repressor for DNA binding. For the cyclic acetal derivatives 3 and 6, the loss of 2-OH mediated H-bonding to the repressor would be predicted to lead to reduced affinity of these substances; however, 6 sustained weak inducer activity and its mode of binding is unclear. For the isobutanols 4 and 5, MD simulation led to prediction that generation of new H-bonding interactions could disrupt the H-bonding network. Clearly the factors which determine whether a substance can be an inducer or negative inducer/neutral effector are complex and will need further investigation. Synthesis of further constrained C-glycosides lacking H-bond disrupting group in the isobutyl residue, and/or determination of further co-crystal structures of C-glycosides with the lac repressor will be needed to give further insight. Finally, the insight from evaluation of various constrained C-glycosides as galactosidase inhibitors shows that there may be flexibility in how this enzyme recognizes the aglycon group in its binding pocket. The synthesis and analysis, described here, provide the basis for synthesis of conformationally constrained galactopyranoside mimetics, which have wide biological relevance, such as for lectins. Our research in galectin inhibitor identification based on related C-glycoside scaffolds as β-galactoside mimetics, will be disclosed in due course.
4. Experimental section
4.1 1-(β-D-Galactopyranosyl)-2-methylpropane (2)
Compound 20 (350 mg, 0.90 mmol) was dissolved in MeOH and Pd–C (10%) was added to the reaction mixture. The flask was fitted with a H2 balloon and stirred overnight, and the mixture was then filtered through Celite and the solvent was removed under reduced pressure. Flash chromatography (cyclohexane–EtOAc 4:1) provided the acetylated intermediate (327 mg, 93%) as a white solid; Rf 0.56 (cyclohexane–EtOAc 4:1); HRMS calcd for C18H32NO9 [M + NH4]+ 406.2071 found m/z 406.2021. The NMR data (see ESI†) for the intermediate obtained was in good agreement with that reported previously.16 The intermediate (550 mg, 1.42 mmol) was dissolved in dry methanol (5.5 mL) and sodium methoxide (8 mg, 0.15 mmol) was added and the mixture was then stirred at room temp for 15 minutes. Amberlite (H+) was added to neutralise the reaction mixture after which the solvent was evaporated under reduced pressure and chromatography of the residue (silica gel, elution with 10:1 acetonitrile–ammonium hydroxide) gave the title compound 2 (240 mg, 77%) as a white solid. 1H-NMR (400 MHz, CD3OD) δ 3.84 (dd, J = 3.3, 1.1 Hz, 1H, H-4), 3.66 (dd, J = 11.3, 6.7 Hz, 1H, H-6a), 3.63 (dd, J = 11.2, 5.8 Hz, 1H, H-6b), 3.41–3.36 (overlapping signals, 2H, H-3 & H-5), 3.31 (d, J = 9.2 Hz, 1H, H-2), 3.11 (td, J = 10.0, 9.1, 2.1 Hz, 1H, H-1), 1.89 (m, 1H, isopropyl CH), 1.59 (ddd, J = 14.0, 9.7, 2.1 Hz, 1H, CHH), 1.39 (ddd, J = 14.2, 10.0, 4.3 Hz, 1H, CHH), 0.91 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H); 13C-NMR (126 MHz, CD3OD) δ 78.61 (C-5), 78.34 (C-1), 75.08 (C-3), 71.80 (C-2), 69.36 (C-4), 61.26 (C-6), 40.67 (C-1′), 24.12 (C-2′), 22.91, 20.69 (each CH3). The NMR data obtained was in good agreement with that reported previously.16 Selected 1H-NMR data for 2 obtained from sample dissolved in D2O (500 MHz): δ 3.30 (ddd (apt td), J = 9.7, 9.9, 2.1 Hz, 1H, H-1), 1.85 (dheptd, J = 9.9, 6.8 (×6), 4.3 Hz, 1H, isopropyl CH), 1.58 (ddd, J = 14.4, 9.9, 2.2 Hz, 1H, CH(H)CHMe2), 1.42 (ddd, J = 14.3, 9.9, 4.3 Hz, 1H, CH(H)CHMe2).
4.2 (4S,4aS,6R,7R,8S,8aR)-6-(Hydroxymethyl)-4-isopropylhexahydropyrano[3,2-d][1,3]dioxine-7,8-diol (3)
Benzylated 18 (115 mg, 0.22 mmol) was dissolved in dry methanol (5 mL), and Pd–C (10%; 115 mg, 100% w/w) was added. The round-bottom flask was fitted with a gas bag filled with hydrogen gas and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was then filtered through Celite, and the solvent was removed under reduced pressure. Chromatography (silica gel, eluting with 10:1 acetonitrile–aqueous ammonium hydroxide) gave 3 (42 mg, 76%) as a white solid; Rf 0.4 (CH2Cl2–MeOH 9:1); 1H-NMR (500 MHz, CD3OD) δ 5.03 (d, J = 6.1 Hz, 1H, methylene CHH), 4.70 (d, J = 6.1 Hz, 1H, methylene CHH), 3.91 (dd, J = 3.5, 1.3 Hz, 1H, H-4), 3.73–3.64 (overlapping signals, 3H, H-3, H-6a & H-6b), 3.56 (t, J = 9.5 Hz, 1H, H-2), 3.53 (ddd, J = 6.6, 5.3, 1.3 Hz, 1H, H-5), 3.37 (dd, J = 9.3, 2.8 Hz, 1H, H-1′), 3.08 (t, J = 9.1 Hz, 1H, H-1), 2.08 (heptet of doublets, J = 7.0, 2.8 Hz, 1H, isopropyl CH), 1.00 (d, J = 7.0 Hz, 3H, CH3), 0.92 (d, J = 7.0 Hz, 3H, CH3); 13C-NMR (126 MHz, CD3OD) δ 93.45 (methylene CH2), 82.61 (C-1′), 79.60 (C-5), 78.19 (C-2), 73.56 (C-1), 71.74 (C-3), 69.61 (C-4), 61.24 (C-6), 27.77 (C-2′), 18.23, 14.70 (each CH3); HRMS calcd for C12H23O6Na [M + Na]+ 271.1152 found m/z 271.1154.
4.3 (2S,3R,4S,5R,6R)-2-((R)-1-Hydroxy-2-methylpropyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (4)
Compound 13 (130 mg, 0.256 mmol) was dissolved in dry methanol (5 mL), and Pd–C (10%; 130 mg, 100% w/w) was added. The round-bottom flask was fitted with a gas bag filled with hydrogen gas and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was then filtered through Celite, and the solvent was removed under reduced pressure. Chromatography (silica gel, eluting with acetonitrile and aqueous ammonium hydroxide solution (10:1)) gave 4 (32 mg, 53%) as a white solid; 1H-NMR (500 MHz, CD3OD) δ 3.85 (dd, J = 3.3, 1.1 Hz, 1H, H-4), 3.79 (t, J = 9.6 Hz, 1H, H-2), 3.70 (dd, J = 11.4, 6.6 Hz, 1H, H-6a), 3.67 (dd, J = 11.4, 5.6 Hz, 1H, H-6b), 3.47–3.41 (overlapping signals, 2H, H-3 & H-5), 3.38 (dd, J = 8.8, 1.6 Hz, 1H, H-1′), 3.22 (dd, J = 9.5, 1.6 Hz, 1H, H-1), 1.94 (doublet of heptets, J = 8.7, 6.7 Hz, 1H, isopropyl CH), 1.02 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H) (each CH3); 13C NMR (126 MHz, CD3OD) δ 79.16 (C-1), 78.80 (C-5), 75.46 (C-3), 74.15 (C-1′), 69.66 (C-4), 67.10 (C-2), 61.25 (C-6), 30.60 (C-2′), 18.80, 18.12 (each CH3); HRMS calcd for C10H20O6Na [M + Na]+ 259.1152 found m/z 259.1153.
4.4 (2S,3R,4S,5R,6R)-2-((S)-1-Hydroxy-2-methylpropyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (5)
Alcohol 17 (150 mg, 0.239 mmol) was dissolved in dry methanol (5 mL), and Pd–C (10%; 150 mg, 100% w/w) was added. The round-bottom flask was fitted with a gas bag filled with hydrogen gas and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was then filtered through Celite, and the solvent was removed under diminished pressure. Column chromatography (silica gel eluting with acetonitrile and aqueous ammonium hydroxide solution (10:1)) gave 5 (35 mg, 62%) as a white solid; 1H-NMR (500 MHz, CD3OD) δ 3.89 (d, J = 3.2 Hz, 1H, H-4), 3.78 (t, J = 9.3 Hz, 1H, H-2), 3.74 (dd, J = 11.5, 6.7 Hz, 1H, H-6a), 3.69 (dd, J = 11.3, 4.9 Hz, 1H, H-6b), 3.64 (t, J = 5.2 Hz, 1H, H-1′), 3.53–3.46 (overlapping signals, 2H, H-3 & H-5), 3.28 (dd, J = 9.4, 5.8 Hz, 1H, H-1), 2.09 (heptet of doublets, J = 6.9, 4.6 Hz, 1H, isopropyl CH), 0.97 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H) (each CH3); 13C-NMR (126 MHz, CD3OD) δ 79.10 (C-1), 78.58 (C-5), 77.94 (C-1′), 75.00 (C-3), 70.04 (C-2), 69.55 (C-4), 61.66 (C-6), 29.11 (C-2′), 18.89, 15.80 (each CH3); HRMS calcd for C12H23O6Na [M + Na]+ 271.1152 found m/z 271.1158.
4.5 (4S,4aS,6R,7R,8S,8aR)-6-(Hydroxymethyl)-4-isopropylhexahydropyrano[3,2-d][1,3]dioxine-7,8-diol (6)
Compound 14 (90 mg, 0.173 mmol) was dissolved in dry methanol (5 mL), and Pd–C (10%; 90 mg, 100% w/w) was added. The round-bottom flask was fitted with a gas bag filled with hydrogen gas and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was then filtered through Celite, and the solvent was removed under reduced pressure. Chromatography (silica gel, eluting with 10:1 acetonitrile and aqueous ammonium hydroxide solution) gave 6 (28 mg, 65%) as a white solid; Rf 0.4 (CH2Cl2–MeOH 9:1); 1H-NMR (500 MHz, CD3OD) δ 4.82 (d, J = 6.5 Hz, 1H, methylene CHH), 4.74 (d, J = 6.4 Hz, 1H, methylene CHH), 3.91 (dd, J = 3.5, 1.3 Hz, 1H, H-4), 3.86 (t, J = 9.3 Hz, 1H, H-2), 3.69 (dd, J = 11.2, 6.5 Hz, 1H, H-6a), 3.66 (dd, J = 11.3, 5.5 Hz, 1H, H-6b), 3.65–3.59 (overlapping signals, 3H, H-1, H-3 & H-1′), 3.48 (td, J = 6.6, 5.4, 1.2 Hz, 1H, H-5), 2.45 (doublet of heptets, J = 10 Hz, 6 × 6.5 Hz, 1H, isopropyl CH), 1.05 (d, J = 6.3 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H) (each CH3); 13C-NMR (126 MHz, CD3OD) δ 87.57 (methylene CH2), 79.72 (C-5), 78.82 (C-1′), 75.88 (C-1), 72.90 (C-2), 72.53 (C-3), 69.50 (C-4), 61.40 (C-6), 24.57 (C-2′), 19.86, 18.64 (each CH3); HRMS calcd for C12H23O6Na [M + Na]+ 271.1152 found m/z 271.1154.
4.6 3,4,6-Tri-O-benzyl-β-thiopyridine-D-galactopyranose (9)
Glycal 7 (1.5 g, 3.60 mmol) was dissolved in dichloromethane (30 mL), acetone (3 mL) and saturated aqueous solution of sodium bicarbonate (50 mL) were added at 0 °C. The mixture was vigorously stirred and a solution of Oxone (6 g, 9.76 mmol) in water (25 mL) was added dropwise over 15 min. The reaction mixture was vigorously stirred at 0 °C for 30 min and was then warmed to room temperature until TLC indicated complete consumption of the starting material. The organic phase was separated, and the aqueous phase was extracted with dichloromethane (30 mL). The combined organic phases were dried with anhydrous sodium sulphate and concentrated under reduced pressure to afford the epoxide 846 (1.6 g), which is sensitive to hydrolysis and was reacted without further purification. 2-Mercaptopyridine (1.03 g, 9.26 mmol) was dissolved in dry THF (10 mL) and the solution was cooled to 0 °C. Sodium hydride (370 mg, 9.26 mmol, 60% w/w in mineral oil) was added and the mixture was stirred at 0 °C for 15 minutes. Then a solution of freshly prepared epoxide 8 (2 g, 4.62 mmol) in dry THF (20 mL) was slowly added to the reaction mixture at 0 °C. The reaction mixture was then warmed to room temperature and stirred for 2 h. Upon completion of reaction using TLC, 10% aqueous solution of HCl (10 mL) was added. The reaction mixture was then extracted with ethyl acetate (30 mL × 2). The combined organic layers were washed with water (20 mL × 2) and dried with anhydrous sodium sulphate. The solvent was evaporated under reduced pressure, and chromatography (silica gel, eluting with 5:1 cyclohexane and ethyl acetate) gave 9 (2.16 g, 86%) as a pale yellow solid; 1H-NMR (500 MHz, CDCl3) δ 8.42 (d, J = 4.5 Hz, 1H), 7.47–7.22 (m, 17H), 7.04 (td, J = 5.7, 1.6 Hz, 1H) (each Ar–H), 5.22 (d, J = 9.8 Hz, 1H, H-1), 4.94 (d, J = 11.4 Hz, 1H), 4.81 (d, J = 12.7 Hz, 1H), 4.78 (d, J = 12.7 Hz, 1H), 4.62 (d, J = 11.4 Hz, 1H), 4.49 (d, J = 11.8 Hz, 1H), 4.43 (d, J = 11.7 Hz, 1H) (each Bn-CH2), 4.20 (t, J = 9.5 Hz, 1H, H-2), 4.02 (d, J = 2.8 Hz, 1H, H-4), 3.76 (t, J = 6.5 Hz, 1H, H-5), 3.63 (d, J = 6.4 Hz, 2H, H-6a & H-6b), 3.58 (dd, J = 9.3, 2.8 Hz, 1H, H-3); 13C-NMR (126 MHz, CDCl3) δ 156.6, 149.4, 138.6, 138.3, 137.8, 136.7, 128.5, 128.4, 128.2, 128.1, 128.0, 127.8, 127.7, 127.7, 127.6, 124.4, 120.8 (each Ar–C/C–H), 85.4 (C-1), 83.6 (C-3), 78.0 (C-5), 74.6 (Bn-CH2), 73.6 (C-4), 73.5, 72.7 (each Bn-CH2), 70.3 (C-2), 68.6 (C-6). HRMS calcd for C32H33NO5NaS [M + Na]+ 566.1977 found m/z 566.1959.
4.7 2-p-Methoxybenzyl-3,4,6-tri-O-benzyl-β-thiopyridine-D-galactopyranose (10)
To 9 (1.5 g, 2.76 mmol) in dry dimethylformamide (15 mL), sodium hydride (221 mg, 5.52 mmol, 60% w/w in mineral oil) was added at 0 °C. After stirring for 10 minutes, 4-methoxybenzyl chloride (0.6 mL, 4.42 mmol) was added at 0 °C, and the reaction mixture was stirred at room temperature for 3 h. Ice-cold water (20 mL) was then added to the mixture and it was extracted with ethyl acetate (30 mL × 2). The organic layer was washed with water (20 mL × 2), dried over anhydrous sodium sulphate, and filtered. Removal of the solvent under reduced pressure and chromatography (silica gel, eluting with 5:1 cyclohexane and ethyl acetate) gave 10 (1.46 g, 80%) as a pale yellow solid; Rf 0.83 (cyclohexane–EtOAc, 1:1); 1H-NMR (500 MHz, CDCl3) δ 8.42 (dd, J = 5.0, 1.9 Hz, 1H), 7.47–7.21 (m, 19H), 6.99 (br t, J = 6.1 Hz, 1H), 6.80 (d, J = 8.6 Hz, 2H) (each Ar–H), 5.31 (d, J = 9.9 Hz, 1H, H-1), 5.00 (d, J = 11.4 Hz, 1H), 4.79 (d, J = 9.6 Hz, 3H), 4.73 (d, J = 9.9 Hz, 1H), 4.64 (d, J = 11.4 Hz, 1H), 4.48 (d, J = 11.7 Hz, 1H), 4.42 (d, J = 11.4 Hz, 1H) (each Bn-CH2), 4.06 (t, J = 9.5 Hz, 1H, H-2), 4.04 (d, J = 2.6 Hz, 1H, H-4), 3.78 (s, 3H, OCH3), 3.74 (t, J = 6.6 Hz, 1H, H-5), 3.69 (dd, J = 9.2, 2.7 Hz, 1H, H-3), 3.65 (d, J = 6.4 Hz, 2H, H-6a & H-6b); 13C-NMR (126 MHz, CDCl3) δ 159.2, 157.8, 149.5, 138.7, 138.4, 137.9, 136.4, 130.3, 130.1, 128.4, 128.4, 128.2, 128.1, 127.9, 127.8, 127.7, 127.6, 127.5, 123.3, 120.2, 113.7 (each Ar–C/C–H) 84.2 (C-1), 84.2 (C-3), 77.5 (C-5), 77.0 (C-2), 75.4 (Bn-CH2), 74.6 (Bn-CH2), 73.8 (C-4), 73.5 (Bn-CH2), 72.8 (Bn-CH2), 68.7 (C-6), 55.3 (OCH3); HRMS calcd for C40H41NO6NaS [M + Na]+ 686.2552 found m/z 686.2539.
4.8 2-p-Methoxybenzyl-3,4,6-tri-O-benzyl-β-sulfonylpyridine-D-galactopyranose (11)
Compound 10 (1.8 g, 2.71 mmol) was dissolved in dichloromethane (30 mL), and the solution was cooled to 0 °C. Sodium bicarbonate (1.37 g, 16.3 mmol) and m-chloroperbenzoic acid (55–70% purity; 2.8 g) were added, and the reaction mixture was warmed to room temperature and stirred for 6 h. On completion of the reaction (TLC), the reaction mixture was diluted with dichloromethane (30 mL), and the organic layer was washed with aqueous sodium sulfite solution (30 mL × 2). The solvent was evaporated under reduced pressure, chromatography (silica gel, eluting with 4:1 cyclohexane and ethyl acetate) gave 11 (1.6 g, 85%) as a white solid; Rf 0.37 (cyclohexane–EtOAc 3:2); 1H-NMR (500 MHz, CDCl3) δ 8.62 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H), 8.02 (bd, J = 7.8 Hz, 1H), 7.73 (td, J = 7.8, 1.7 Hz, 1H), 7.38–7.11 (m, 19H), 6.81 (d, J = 8.7 Hz, 2H) (each Ar-H), 4.98 (d, J = 9.6 Hz, 1H), 4.91 (d, J = 10.3 Hz, 1H) (each Bn-CH2), 4.89 (d, J = 8.4 Hz, 1H, H-1), 4.82 (d, J = 9.7 Hz, 1H, Bn-CH2), 4.72 (s, 2H, each Bn-CH2), 4.55 (d, J = 7.9 Hz, 1H, H-2), 4.53 (d, J = 9.9 Hz, 2H), 4.23 (s, 2H) (each Bn-CH2), 3.87 (d, J = 2.6 Hz, 1H, H-4), 3.79 (s, 3H, OCH3), 3.66 (dd, J = 9.4, 2.7 Hz, 1H, H-3), 3.58 (t, J = 6.3 Hz, 1H, H-5), 3.42 (dd, J = 9.7, 6.7 Hz, 1H, H-6a), 3.37 (dd, J = 9.7, 5.8 Hz, 1H, H-6b); 13C-NMR (126 MHz, CDCl3) δ 159.2, 156.1, 149.9, 138.4, 138.0, 137.7, 137.3, 130.3, 130.0, 129.8, 128.5, 128.4, 128.2, 127.9, 127.5, 127.0, 123.9, 113.6 (each Ar–C/C–H), 89.5 (C-1), 83.9 (C-3), 78.2 (C-5), 74.9 (Bn-CH2), 74.4 (Bn-CH2), 73.9 (C-2), 73.4 (Bn-CH2), 73.1 (C-4), 72.9 (Bn-CH2), 68.4 (C-6), 55.3 (OCH3); HRMS calcd for C40H41NO8NaS [M + Na]+ 718.2451 found m/z 718.2433.
4.9 1-((2S,3R,4S,5S,6R)-4,5-Bis(benzyloxy)-6-((benzyloxy)methyl)-3-((p-methoxybenzyl)oxy)tetrahydro-2H-pyran-2-yl)-2-methylpropan-1-ol (12)
Sulfone 11 (500 mg, 0.718 mmol) and isobutyraldehyde (0.26 mL, 2.86 mmol) were dissolved in dry tetrahydrofuran (10 mL) and cooled to 0 °C. A solution of samarium iodide in dry tetrahydrofuran (55 mL, freshly prepared sing samarium metal (1.65 g) and iodine (1.4 g)) was added at 0 °C. The resulting green coloured solution was warmed to room temperature and stirred for 20 minutes. Saturated ammonium chloride solution (30 mL) was then added and the mixture was extracted with ethyl acetate (40 mL × 2). The combined organic layer was dried over anhydrous sodium sulphate and filtered. Evaporation of the solvent under reduced pressure and subsequent chromatography (silica gel, eluting with 6:1 cyclohexane and ethyl acetate) gave 12 (190 mg, 42%) as a pale-yellow viscous oil, along with an impurity believe to be the 1-deoxygalactopyranose derivative. Selected data for 121H-NMR signals δ 3.98 (d, J = 2.7 Hz, 1H, H-4 major) HRMS calcd for C41H52O7Na [M + Na]+ 541.2595 found m/z 541.2566.
4.10 (2S,3R,4R,5S,6R)-4,5-Bis(benzyloxy)-6-((benzyloxy)methyl)-2-((R)-1-hydroxy-2-methylpropyl)tetrahydro-2H-pyran-3-ol (13)
To alcohol 12 (190 mg, 0.303 mmol) in dichloromethane and water (10:1, 2 mL) at 0 °C was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 90 mg, 0.40 mmol). The reaction mixture was then stirred at room temperature for 2 h. The reaction mixture was then diluted with dichloromethane (10 mL) and washed with saturated aqueous solution of sodium bicarbonate (10 mL). The organic layer was dried with anhydrous sodium sulphate and filtered and the solvent was removed under reduced pressure. Chromatography of the residue (silica gel, eluting with 5:1 cyclohexane and ethyl acetate) gave 13 (93 mg, 61%) as a pale yellow oil; Rf 0.55 (cyclohexane–EtOAc 3:2); 1H-NMR (500 MHz, CDCl3) δ 7.43–7.21 (overlapping signals, 15H, each Ar–H), 4.88 (d, J = 11.5 Hz, 1H), 4.76 (d, J = 11.8 Hz, 1H), 4.57 (d, J = 12.2 Hz, 1H), 4.55 (d, J = 12.6 Hz, 1H), 4.50 (d, J = 11.7 Hz, 1H), 4.46 (d, J = 11.7 Hz, 1H) (each Bn-CH2), 4.22 (t, J = 9.4 Hz, 1H, H-2), 4.01 (d, J = 2.8 Hz, 1H, H-4), 3.67–3.54 (overlapping signals, 3H, H-5, H-6a & H-6b), 3.47–3.41 (overlapping signals, 2H, H-3 & H-1′), 3.31 (bd, J = 9.4 Hz, 1H, H-1), 1.86 (h, J = 7.1 Hz, 1H, isopropyl CH), 1.02 (d, J = 6.7 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H) (each CH3); 13C-NMR (126 MHz, CDCl3) δ 138.6, 137.9, 137.9, 128.6, 128.4, 128.2, 127.9, 127.9, 127.9, 127.8, 127.7, 127.6 (each Ar–C/C–H), 84.4 (C-1′), 79.0 (C-1), 77.2 (C-5), 74.5 (C-3), 74.4 (Bn-CH2), 73.6 (Bn-CH2), 72.8 (C-4), 71.8 (Bn-CH2), 68.9 (C-6), 66.7 (C-2), 31.2 (C-2′), 19.3, 19.2 (each CH3); HRMS calcd for C31H38O6Na [M + Na]+ 529.2566 found m/z 529.2571.
4.11 (4S,4aS,6R,7S,8S,8aR)-7,8-Bis(benzyloxy)-6-((benzyloxy)methyl)-4-isopropylhexahydropyrano[3,2-d][1,3]dioxine (14)
Diol 13 (180 mg, 0.35 mmol) was dissolved in dimethoxymethane (4.00 mL, 45.6 mmol), camphorsulfonic acid (165 mg, 0.71 mmol) was added and the reaction was allowed to heat at reflux overnight while stirring vigorously. The solvent was then removed under reduced pressure and chromatography (silica gel, eluting with cyclohexane–EtOAc 6:1) provided 14 (110 mg, 60%) as a colourless oil; Rf 0.88 (cyclohexane–EtOAc 3:2); 1H-NMR (500 MHz, CDCl3) δ 7.46–7.22 (overlapping signals, 15H, each Ar–H), 4.96 (d, J = 11.8 Hz, 1H), 4.89–4.79 (overlapping signals, 3H), 4.71 (d, J = 12.2 Hz, 1H), 4.59 (d, J = 11.7 Hz, 1H) (each Bn-CH2), 4.45 (s, 2H, methylene CHH & CHH), 4.21 (t, J = 9.2 Hz, 1H, H-2), 3.92 (d, J = 3.0 Hz, 1H, H-4), 3.69 (overlapping signals, 2H, H-1 & H-1′), 3.63–3.48 (overlapping signals, 4H, H-3, H-5, H-6a & H-6b), 2.42 (doublet of heptets, J = 6.6, 2.5 Hz, 1H, isopropyl CH), 1.03 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.5 Hz, 3H) (each CH3); 13C-NMR (126 MHz, CDCl3) δ 139.0, 138.5, 138.0, 128.5, 128.4, 128.4, 128.2, 128.1, 127.8, 127.8, 127.7, 127.7, 127.6, 127.5, 127.4 (each Ar–C/C–H), 87.9 (Bn-CH2), 81.2 (C-3), 78.8 (C-1′), 78.0 (C-5), 76.3 (C-1), 75.1 (C-4), 74.6 (Bn-CH2), 73.5 (methylene CH2), 73.4 (C-2), 72.8 (Bn-CH2), 69.1 (C-6), 25.0 (C-2′), 20.6, 19.7 (each CH3); HRMS calcd for C32H38O6Na [M + Na]+ 541.2566 found m/z 541.2595.
4.12 (4S,4aS,6R,7S,8R,8aS)-6-(Acetoxymethyl)-4-isopropylhexahydropyrano[3,2-d][1,3]dioxine-7,8-diyl diacetate (15)
Compound 6 (30 mg, 0.12 mmol) was dissolved in Ac2O (4 mL) and pyridine (4 mL) and stirred overnight. The reaction mixture was diluted with EtOAc, washed with 1 M HCl, water and brine, dried over Na2SO4, filtered and the solvent was removed. Chromatography (silica gel, eluting with cyclohexane–EtOAc 3:2) gave 15 (41 mg, 90%) as a white solid; Rf 0.41 (cyclohexane–EtOAc 3:2); 1H-NMR (500 MHz, CDCl3) δ 5.40 (dd, J = 3.4, 1.3 Hz, 1H, H-4), 5.01 (dd, J = 9.9, 3.4 Hz, 1H, H-3), 4.77 (d, J = 6.5 Hz, 1H, methylene CHH), 4.75 (d, J = 6.4 Hz, 1H, methylene CHH), 4.06 (dd, J = 11.2, 6.5 Hz, 1H, H-6a), 4.00 (dd, J = 11.3, 6.7 Hz, 1H, H-6b), 3.97 (d, J = 9.7 Hz, 1H, H-2), 3.85 (td, J = 6.6, 1.4 Hz, 1H, H-5), 3.78 (dd, J = 10.0, 5.7 Hz, 1H, H-1), 3.68 (dd, J = 10.3, 5.7 Hz, 1H, H-1′), 2.37 (heptet of doublets, J = 10.1, 6.6 Hz, 1H, isopropyl CH), 2.10 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H), 0.99 (d, J = 6.4 Hz, 3H), 0.96 (d, J = 6.6 Hz, 3H); 13C-NMR (126 MHz, CDCl3) δ 170.40, 170.13, 170.04 (each C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 87.78 (methylene CH2), 78.29 (C-1′), 75.97 (C-1), 74.73 (C-5), 72.00 (C-3), 69.88 (C-2), 67.87 (C-4), 61.46 (C-6), 24.90 (C-2′), 20.70, 20.62, 20.57 (each OAc), 20.31, 19.47 (each CH3).
O), 87.78 (methylene CH2), 78.29 (C-1′), 75.97 (C-1), 74.73 (C-5), 72.00 (C-3), 69.88 (C-2), 67.87 (C-4), 61.46 (C-6), 24.90 (C-2′), 20.70, 20.62, 20.57 (each OAc), 20.31, 19.47 (each CH3).
4.13 1-((2R,3R,4S,5S,6R)-4,5-Bis(benzyloxy)-6-((benzyloxy)methyl)-3-((4-methoxybenzyl)oxy)tetrahydro-2H-pyran-2-yl)-2-methylpropan-1-one (16)
Alcohol 12 (750 mg, 1.2 mmol) was dissolved in CH2Cl2 and cooled to 0 °C. Dess–Martin Periodinane (1.14 g, 2.69 mmol) was added and the mixture stirred overnight. The reaction mixture was filtered through Celite, washed with satd. Na2S2O3 solution, satd. NaHCO3 solution, water, and brine. The organic layer was dried over MgSO4, filtered and the solvent removed under reduced pressure. The resulting ketone product (305 mg, 41%) was obtained after chromatography (silica gel, eluting with cyclohexane–EtOAc 6:1); Rf 0.66 (cyclohexane–EtOAc 6:1); 1H-NMR (500 MHz, CDCl3) δ 7.45–7.24 (overlapping signals, 17H), 7.18 (d, J = 8.6 Hz, 1H, Ar–H), 6.83 (d, J = 8.6 Hz, 1H, Ar–H), 4.98 (d, J = 11.8 Hz, 1H), 4.81–4.76 (overlapping signals, 2H), 4.73 (d, J = 11.7 Hz, 1H), 4.64 (d, J = 11.7 Hz, 1H), 4.59 (d, J = 10.1 Hz, 1H), 4.48 (d, J = 11.8 Hz, 1H), 4.44 (d, J = 11.6 Hz, 1H) (each Bn-CH2), 4.18 (t, J = 9.6 Hz, 1H, H-2), 3.98 (d, J = 2.8 Hz, 1H, H-4), 3.91 (d, J = 9.6 Hz, 1H, H-1), 3.79 (s, 3H, OCH3), 3.65 (dd, J = 9.5, 2.8 Hz, 1H, H-3), 3.63–3.56 (overlapping signals, 3H, H-5, H-6a & H-6b), 3.00 (hept, J = 7.0 Hz, 1H, isopropyl CH), 1.14 (d, J = 7.0 Hz, 3H), 1.12 (d, J = 7.0 Hz, 3H) (each CH3); 13C-NMR (126 MHz, CDCl3) δ 209.36 (CO), 159.24, 138.68, 138.32, 137.87, 130.55, 129.90, 129.50, 128.48, 128.47, 128.44, 128.43, 128.41, 128.34, 128.27, 128.24, 128.14, 128.02, 128.01, 127.97, 127.95, 127.89, 127.83, 127.80, 127.77, 127.69, 127.62, 127.60, 127.57, 127.55, 127.52, 127.48, 127.36, 113.82, 113.74 (each Ar–C/C–H), 84.52 (C-3), 81.01 (C-1), 77.88 (C-5), 75.39 (C-2), 74.90, 74.43, 73.57 (each Bn-CH2), 73.51 (C-4), 72.50 (Bn-CH2), 69.04 (C-6), 55.27 (OCH3), 38.55 (C-2′), 18.23, 17.82 (each CH3).
4.14 (S)-1-((2S,3R,4S,5S,6R)-4,5-Bis(benzyloxy)-6-((benzyloxy)methyl)-3-((4-methoxybenzyl)oxy)tetrahydro-2H-pyran-2-yl)-2-methylpropan-1-ol (17)
Ketone 16 (269 mg, 0.43 mmol) was dissolved in CH2Cl2–MeOH (1:1) and NaBH4 (22 mg, 0.59 mmol) was added to the reaction mixture and allowed to stir for 30 min, after which TLC analysis indicated complete consumption of the starting material. The solvent was then removed under reduced pressure. Flash chromatography (cyclohexane–EtOAc 6:1) provided the product (230 mg, 85%) as a white solid; Rf 0.29 (cyclohexane–EtOAc 4:1); 1H-NMR (400 MHz, CDCl3) δ 7.44–7.15 (overlapping signals, 17H, aromatic H), 6.90–6.75 (overlapping signals, 2H, Ar–H), 5.01 (d, J = 10.3 Hz, 1H), 4.94 (d, J = 11.6 Hz, 1H), 4.78 (d, J = 11.7 Hz, 1H), 4.67 (d, J = 10.4 Hz, 1H), 4.67 (d, J = 11.6 Hz, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.48 (d, J = 11.7 Hz, 1H), 4.42 (d, J = 11.6 Hz, 1H) (each Bn-CH2), 4.04 (t, J = 9.4 Hz, 1H, H-2), 3.99 (d, J = 2.7 Hz, 1H, H-4), 3.78 (s, 3H, OCH3), 3.68 (dd, J = 9.4, 2.7 Hz, 1H, H-3), 3.64 (dd, J = 6.7, 3.7 Hz, 1H, H-1′), 3.55 (s, 3H, H-5, H-6a & H-6b), 3.23 (dd, J = 9.2, 6.8 Hz, 1H, H-1), 2.02 (tdd, J = 10.5, 5.2, 2.6 Hz, 1H, isopropyl CH), 0.95 (d, J = 7.0 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H) (each CH3); 13C-NMR (101 MHz, CDCl3) δ 130.02, 129.93, 128.65, 128.55, 128.37, 128.19, 127.97, 127.92, 127.89, 127.74, 127.69, 114.03 (each aromatic C/C–H), 85.49 (C-3), 78.72 (C-1), 78.48 (C-2), 77.54 (C-1′), 77.51 (C-5), 75.08, 74.65, 73.63 (each Bn-CH2), 73.61 (C-4), 72.19 (Bn-CH2), 69.08 (C-6), 55.38 (OCH3), 28.83 (C-2′), 19.66, 15.64 (each CH3); HRMS calcd for C41H52O7Na [M + Na]+ 649.3136 found m/z 649.3134.
4.15 (4S,4aS,6R,7S,8S,8aR)-7,8-Bis(benzyloxy)-6-((benzyloxy)methyl)-4-isopropylhexahydropyrano[3,2-d][1,3]dioxine (18)
Alcohol 17 (200 mg, 0.32 mmol) was dissolved in CH2Cl2 and cooled to 0 °C. Trifluoroacetic acid (0.32 mL, 4.23 mmol) was added to the reaction mixture and allowed to stir for 30 min, after which time TLC analysis indicated complete consumption of the starting material. The solvent was then removed under reduced pressure and chromatography (cyclohexane–EtOAc 4:1) provided the intermediate diol (126 mg, 78%) as a white solid; Rf 0.3 (cyclohexane–EtOAc 3:2); 1H-NMR (500 MHz, CDCl3) δ 7.41–7.24 (overlapping signals, 15H, each Ar–H) 4.86 (d, J = 11.6 Hz, 1H), 4.74 (d, J = 11.7 Hz, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.56 (d, J = 11.8 Hz, 1H), 4.50 (d, J = 11.7 Hz, 1H), 4.45 (d, J = 11.8 Hz, 1H) (each Bn-CH2), 4.13 (t, J = 9.3 Hz, 1H, H-2), 3.99 (d, J = 2.8 Hz, 1H, H-4), 3.72 (dd, J = 7.7, 3.1 Hz, 1H, H-1′), 3.61 (dd, J = 12.4, 5.4 Hz, 1H, H-5), 3.57–3.53 (overlapping signals, 2H, H-6a & H-6b) 3.46 (dd, J = 9.4, 2.9 Hz, 1H, H-3), 3.19 (dd, J = 9.1, 7.6 Hz, 1H, H-1), 2.09 (pd, J = 6.9, 3.1 Hz, 1H, isopropyl CH), 0.98 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H) (each CH3); 13C-NMR (126 MHz, CDCl3) δ 138.47, 137.91, 137.87, 128.64, 128.60, 128.44, 128.27, 128.18, 127.96, 127.87, 127.82, 127.76, 127.67, 113.96 (each Ar–C/C–H), 83.86 (C-3), 78.29 (C-1), 78.27 (C-1′), 77.42 (C-5), 74.46, 73.55 (each Bn-CH2), 72.69 (C-4), 72.02 (Bn-CH2), 71.61 (C-2), 68.96 (C-6), 28.74 (C-2′), 19.30, 15.02 (each CH3); HRMS calcd for C32H41O6Na [M + Na]+ 529.2560 found m/z 529.2563. The intermediate (126 mg, 0.25 mmol) was dissolved in dimethoxymethane (4.00 mL, 45.6 mmol), camphorsulfonic acid (115 mg, 0.49 mmol) was added and the mixture was heated at reflux for 18 h while stirring vigorously. The solvent was removed under reduced pressure and flash chromatography (cyclohexane–EtOAc 6:1) provided 18 (81 mg, 63%) as a colourless oil; Rf 0.88 (cyclohexane–EtOAc 3:2); 1H-NMR (500 MHz, CDCl3) δ 7.44–7.23 (overlapping signals, 15H, Ar–H), 5.13 (d, J = 6.2 Hz, 1H, methylene CHH), 4.96 (d, J = 11.4 Hz, 1H), 4.84 (d, J = 12.2 Hz, 1H) (each Bn-CH2), 4.77 (d, J = 6.3 Hz, 1H, methylene CHH), 4.71 (d, J = 12.3 Hz, 1H), 4.63 (d, J = 11.4 Hz, 1H), 4.47 (d, J = 11.8 Hz, 1H), 4.42 (d, J = 11.8 Hz, 1H) (each Bn-CH2), 4.00–3.93 (overlapping signals, 2H, H-2 & H-4), 3.65 (dd, J = 9.8, 3.0 Hz, 1H, H-3), 3.61 (dd, J = 12.4, 5.4 Hz, 1H, H-5), 3.57–3.53 (overlapping signals, 2H, H-6a & H-6b), 3.44 (dd, J = 9.3, 2.9 Hz, 1H, H-1′), 3.12 (t, J = 9.2, 9.1 Hz, 1H, H-1), 2.05 (pd, J = 7.0, 2.8 Hz, 1H, isopropyl CH), 1.00 (d, J = 7.0 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H) (each CH3); 13C-NMR (126 MHz, CDCl3) δ 138.47, 137.93, 128.50, 128.42, 128.23, 127.85, 127.78, 127.67, 127.59, 127.53 (each Ar–C or Ar C–H), 93.82 (methylene CH2), 82.50 (C-1′), 80.51 (C-3), 78.59 (C-2), 78.30 (C-5), 74.99 (Bn-CH2), 74.88 (C-4), 74.26 (C-1), 73.52, 72.81 (each Bn-CH2), 68.87 (C-6), 27.99 (C-2′), 19.11, 15.71 (each CH3).
4.16 Measurements of galactosidase inhibition
Each galactosidase assay was performed according to a procedure previously described by Bols et al. preparing eight 250 μL samples in cuvettes containing 100 μL of either sodium phosphate buffer (0.1 M) containing 10 mM MgCl2 and 1 mg mL−1 BSA of pH 6.5, or sodium acetate buffer (0.1 M) containing 1 mg mL−1 BSA of pH 4.5, along with 10 to 80 μL of a 5 or l0 mM solution of either 4-nitrophenyl α-D-galactopyranoside or 4-nitrophenyl β-D-galactopyranoside in water, and 20 μL of a solution of either the potential inhibitor (1–6) or water, and topped up to a total volume of 250 μL with distilled water. All the test samples were made up to 230 μL, containing the potential inhibitor at a fixed concentration but with varying concentrations of nitrophenyl glycoside. The control samples were made up to 250 μL, they contained no inhibitor, but also varying concentrations of the nitrophenyl glycoside. Finally, the reaction was started by adding 20 μL of a diluted solution of either β-galactosidase from E. coli (EC 3.2.1.23, Megazyme E-ECBGAL), β-galactosidase from A. niger (EC 3.2.1.23, Megazyme E-BGLAN) or α-galactosidase from A. niger (EC 3.2.1.22, Megazyme E-AGLAN). The formation of 4-nitrophenol was monitored for 2–10 min at 26 °C by measurement of the absorbance at 400 nm. Initial velocities were calculated from the slopes for each of the eight reactions and used to construct two Hanes plots, one with and one without inhibitor, From the two Michaelis–Menten constants (Km) thus obtained, the inhibition constant (Ki) was calculated.4.17 Computational methods
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of to declare.Acknowledgements
Research presented herein was financially supported by Science Foundation Ireland (Grant No. 16/IA/4419). SA obtained an Erasmus+ mobility fellowship. PVM acknowledges the Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support. The authors thank Andrew Flaus for providing the pET28-eGFP vector Daniel Pritko and Marc R Birtwistle for providing the vector pQLinkHD-mTFP1.References
- H. Yuasa and H. Hashimoto, Trends Glycosci. Glycotechnol., 2001, 13, 31–55 CrossRef.
- (a) P. G. Goekjian, T. C. Wu and Y. Kishi, J. Org. Chem., 1991, 56, 6412–6422 CrossRef CAS; (b) V. García-Aparicio, M. Sollogoub, Y. Blériot, V. Colliou, S. André, J. L. Asensio, F. J. Cañada, H. J. Gabius, P. Sinay and J. Jiménez-Barbero, Carbohydr. Res., 2007, 342, 1918–1928 CrossRef.
- (a) F. Strino, J. H. Lii, H. J. Gabius and P. G. Nyholm, J. Comput.-Aided Mol. Des., 2009, 23, 845–852 CrossRef PubMed; (b) E. Montero, A. García-Herrero, J. L. Asensio, K. Hirai, S. Ogawa, F. Santoyo-González, F. J. Cañada and J. Jiménez-Barbero, Eur. J. Org. Chem., 2000, 1945–1952 CrossRef.
- L. M. Mikkelsen, M. J. Hernáiz, M. Martín-Pastor, T. Skrydstrup and J. Jiménez-Barbero, J. Am. Chem. Soc., 2002, 124, 14940–14951 CrossRef.
- (a) R. U. Lemieux, A. A. Pavia, J. C. Martin and K. A. Watanab, Can. J. Chem., 1969, 47, 4427 CrossRef CAS; (b) I. Tvaroŝka and T. Bleha, Anomeric and Exo-Anomeric Effects in Carbohydrate Chemistry, ed. R. S. Tipson and D. Horton, Advances in Carbohydrate Chemistry and Biochemistry, Academic Press, 1989, vol. 47, pp. 45–123 Search PubMed.
- T. C. Wu, P. G. Goekjian and Y. Kishi, J. Org. Chem., 1987, 52, 4819–4823 CrossRef CAS.
- (a) J. F. Espinosa, F. J. Cañada, J. L. Asensio, M. Martín-Pastor, H. Dietrich, R. R. Schmidt and J. Jiménez-Barbero, J. Am. Chem. Soc., 1996, 118, 10862–10871 CrossRef CAS; (b) M. Martín-Pastor, A. Canales, F. Corzana, J. L. Asensio and J. Jiménez-Barbero, J. Am. Chem. Soc., 2005, 127, 3589–3595 CrossRef.
- R. Sommer, K. Rox, S. Wagner, D. Hauck, S. S. Henrikus, S. Newsad, T. Arnold, T. Ryckmans, M. Brönstrup, A. Imberty, A. Varrot, R. W. Hartmann and A. Titz, J. Med. Chem., 2019, 62, 9201–9216 CrossRef CAS PubMed.
- I. L. Quintana, A. Paul, A. Chowdhury, K. D. Moulton, S. S. Kulkarni and D. H. Dube, ACS Infect. Dis., 2023, 9, 2025–2035 CrossRef PubMed.
- F. Stauffert, A. Bodlenner, T. M. N. Trinh, M. I. García-Moreno, C. O. Mellet, J. F. Nierengarten and P. Compain, New J. Chem., 2016, 40, 7421–7430 RSC.
- A. Marbach and K. Bettenbrock, J. Biotechnol., 2012, 157, 82–88 CrossRef PubMed.
- M. Lewis, G. Chang, N. C. Horton, M. A. Kercher, H. C. Pace, M. A. Schumacher, R. G. Brennan and P. Lu, Science, 1996, 271, 1247–1254 CrossRef PubMed.
- (a) B. Müller-Hill, H. V. Rickenberg and K. Wallenfels, J. Mol. Biol., 1964, 10, 303–318 CrossRef PubMed; (b) M. D. Barkley, A. D. Riggs, A. Jobe and S. Bourgeois, Biochemistry, 1975, 14, 1700–1712 CrossRef PubMed.
- R. Daber, S. Stayrook, A. Rosenberg and M. Lewis, J. Mol. Biol., 2007, 370, 609–619 CrossRef PubMed.
- In ref. 16 2 is referred to as IBCG, abbreviated from ‘isobutyl C-galactoside’ although the latter is not an IUPAC name.
- K. S. Ko, J. Kruse and N. Pohl, Org. Lett., 2003, 5, 1781–1783 CrossRef PubMed.
- C. P. Sager, B. Fiege, P. Zihlmann, R. Vannam, S. Rabbani, R. P. Jakob, R. C. Preston, A. Zalewski, T. Maier, M. W. Peczuh and B. Ernst, Chem. Sci., 2018, 9, 646–654 RSC.
- R. Caraballo, M. Sakulsombat and O. Ramström, ChemBioChem, 2010, 11, 1600–1606 CrossRef CAS PubMed.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS PubMed.
- N. Miquel, G. Doisneau and J. M. Beau, Angew. Chem., Int. Ed., 2000, 39, 4111–4114 CrossRef CAS PubMed.
- W. Adam, J. Bialas and L. Hadjiarapoglou, Chem. Ber., 1991, 124, 2377 CrossRef CAS.
- C. H. Marzabadi and C. D. Spilling, J. Org. Chem., 1993, 58, 3761–3766 CrossRef CAS.
- (a) M. Szostak, M. Spain and D. J. Procter, J. Org. Chem., 2012, 77, 3049–3059 CrossRef CAS PubMed; (b) C. Beemelmanns and H. U. Reissig, Angew. Chem., Int. Ed., 2010, 49, 8021–8025 CrossRef CAS.
- H. Ando, S. Manabe, Y. Nakahara and Y. Ito, Angew. Chem., Int. Ed., 2001, 40, 4725–4728 CrossRef CAS PubMed.
- G. Veeresa and A. Datta, Tetrahedron Lett., 1998, 39, 3069–3070 CrossRef.
- L. Liu, B. A. Motaal, M. Schmidt-Supprian and N. L. B. Pohl, J. Org. Chem., 2012, 77, 1539–1546 CrossRef CAS PubMed.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS.
- C. Lu, C. Wu, D. Ghoreishi, W. Chen, L. Wang, W. Damm, G. A. Ross, M. K. Dahlgren, E. Russell, C. D. V. Bargen, R. Abel, R. A. Friesner and E. D. Harder, J. Chem. Theory Comput., 2021, 17, 4291–4300 CrossRef CAS.
- K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida, 2006, November 11–17 ( DOI:10.1109/SC.2006.54).
- Schrödinger Release 2023-4 Maestro version 13.8.135, Schrödinger, LLC, New York, NY, 2024 Search PubMed.
- P. M. Matias and G. A. Jeffrey, Carbohydr. Res., 1986, 153, 217–226 CrossRef CAS.
- K. N. Kirschner and R. J. Woods, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10541–10545 CrossRef CAS.
- (a) H. Y. Holzapfel, A. D. Stern, M. Bouhaddou, C. M. Anglin, D. Putur, S. Comer and M. R. Birtwistle, ACS Comb. Sci., 2018, 20, 653–659 CrossRef CAS; (b) K. Weisshart, P. Pestryakov, R. W. P. Smith, H. Hartmann, E. Kremmer, O. Lavrik and H. P. Nasheuer, J. Biol. Chem., 2004, 279, 35368–35376 CrossRef CAS PubMed.
- H. P. Nasheuer and F. Grosse, J. Biol. Chem., 1988, 263, 8981–8988 CrossRef CAS PubMed.
- P. Dvorak, L. Chrast, P. I. Nikel, R. Fedr, K. Soucek, M. Sedlackova, R. Chaloupkova, V. de Lorenzo, Z. Prokop and J. Damborsky, Microb. Cell Fact., 2015, 14, 201–215 CrossRef.
- R. Daber, S. Stayrook, A. Rosenberg and M. Lewis, J. Mol. Biol., 2007, 370, 609–619 CrossRef.
- (a) M. Tamres, J. Am. Chem. Soc., 1952, 74, 3375–3378 CrossRef; (b) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS.
- G. Rastelli, A. D. Rio, G. Degliesposti and M. Sgobba, J. Comput. Chem., 2010, 31, 797–810 CrossRef CAS PubMed.
- A. J. Clark, P. Tiwary, K. Borrelli, S. Feng, E. B. Miller, R. Abel, R. A. Friesner and B. J. Berne, J. Chem. Theory Comput., 2016, 12, 2990–2998 CrossRef CAS PubMed.
- M. Bols, R. G. Hazell and I. B. Thomsen, Chem. – Eur. J., 1997, 3, 940–947 CrossRef CAS.
- M. L. Dugdale, D. L. Dymianiw, B. K. Minhas, I. D. Angelo and R. E. Huber, Biochem. Cell Biol., 2010, 88, 861–869 CrossRef CAS PubMed.
- A. Biela-Banaś, F. Oulaïdi, S. Front, E. Gallienne, K. Ikeda-Obatake, N. Asano, D. A. Wenger and O. R. Martin, ChemMedChem, 2014, 9, 2647–2652 CrossRef.
- D. H. Juers, T. D. Heightman, A. Vasella, J. D. McCarter, L. Mackenzie, S. G. Withers and B. W. Matthews, Biochemistry, 2001, 40, 14781–14794 CrossRef.
- P. Cheshev, A. Marra and A. Dondoni, Carbohydr. Res., 2006, 341, 2714–2716 CrossRef.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef.
- Schrödinger Release 2024-2: LigPrep, Schrödinger, LLC, New York, NY, 2024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures and analytical data; 1H and 13C NMR spectra of 1–6 and intermediates; geometry after protein preparation of IPTG bound to lac repressor; MD simulations analysis for compounds 1, 2, 4 and 5 with the lac repressor. See DOI: https://doi.org/10.1039/d4ob01286k |
| This journal is © The Royal Society of Chemistry 2024 |