
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating chemical diversity: o-propargylphenols as key compounds in the divergent synthesis of 2-substituted benzofurans and chromenes†
Alessandra
Gritti
 ab,
Elisa
Brambilla
*a,
Ilaria
Nania
a,
Federico
Turba
a,
Valentina
Pirovano
a and
Giorgio
Abbiati
*a
ab,
Elisa
Brambilla
*a,
Ilaria
Nania
a,
Federico
Turba
a,
Valentina
Pirovano
a and
Giorgio
Abbiati
*a
aDipartimento di Scienze Farmaceutiche, Sezione di Chimica Generale e Organica “A. Marchesini”, Università degli Studi di Milano, Via Golgi, 19, 20133 Milano, Italy. E-mail: giorgio.abbiati@unimi.it
bDipartimento di Chimica, Università degli Studi di Milano, Via Golgi, 19, 20133 Milano, Italy
First published on 5th September 2024
Abstract
In this study, we explored and optimized a MW-enhanced divergent approach for the synthesis of 2-substituted benzofurans and chromenes, starting from seventeen substituted o-propargylphenols characterized by a monoaryl substitution on the propargylic sp3 carbon. Firstly, we developed a robust platform for the preparation of a library of o-propargylphenols. Under basic conditions, o-propargylphenols reacted regioselectively to yield benzofurans in yields ranging from 43% to 100%. Conversely, under cationic gold catalysis, we were able to obtain the corresponding 4H-chromenes, albeit in more variable yields (from 25% to 93%) and slightly lower regioselectively. We also proposed plausible mechanisms to explain the divergent outcomes observed. Our findings underscore the potential of diversity-oriented synthesis in the investigation of molecular complexity. Our neglected o-propargylphenols have proven to be versatile and strategic starting materials for accessing oxygen-containing heterocyclic scaffolds through intramolecular cyclization reactions.
Introduction
Small organic molecules can exert powerful effects on the functions of macromolecules that compose and regulate the living systems. Synthetic organic chemists can access these compounds by exploiting different approaches. Target-Oriented Synthesis1 (TOS) is the approach of choice when the aim is to prepare small molecules with predetermined macromolecule–perturbing properties. For example, a natural compound can be extracted from natural matrices, purified, and then structurally characterized. Once its activity has been well-defined, it can become a target for a chemical synthesis. Thus, the objective of TOS is to prepare a molecule able to access a precise region of the chemical space of the macromolecular target. For example, TOS is the key strategy in total synthesis studies.A similar approach, aware of medicinal and combinatorial chemistry, aims to explore a dense region of the chemical space in proximity to a region known to have useful properties. The template for the identification of lead compounds may be a natural product, a known drug, or an in-silico-designed structure developed from a mechanistic hypothesis and/or a crystal structure determination of the macromolecular target of interest. This strategy can also be considered a different sort of TOS.
However, as pointed out by Schreiber and Buke in their pivotal review,2 despite these approaches have led to great advances in the chemical and life sciences fields, a question remains unanswered: “Are the regions of chemistry space defined by natural products and known drugs, which have been so intensely scrutinized to date, the best or most fertile regions for discovering small-molecules that modulate macromolecular function in useful ways?”
The first step to answer this question is to recognize that the problem of exploring broader regions of the chemical space is different than the problem of accessing precise or dense regions, so different chemical tools and distinct solutions are necessary. Diversity-Oriented Synthesis3 (DOS) is a strategy that allows obtaining a broad distribution of different compounds, resulting in a wider exploration of the chemical space, generating, for example, skeletal diversity. Two main approaches allow the generation of skeletal diversity; the first is the Reagent-Based Approach, which involves the use of different reagents to transform a common substrate with the potential for diverse reactivity into structurally diverse molecules (Fig. 1A). The second, called Substrate-Based Approach, foresees the transformation of different substrates characterized by the presence of different substituents that pre-encode skeletal information (called σ elements) into a collection of products having distinct molecular skeletons by using common reaction conditions (Fig. 1B).
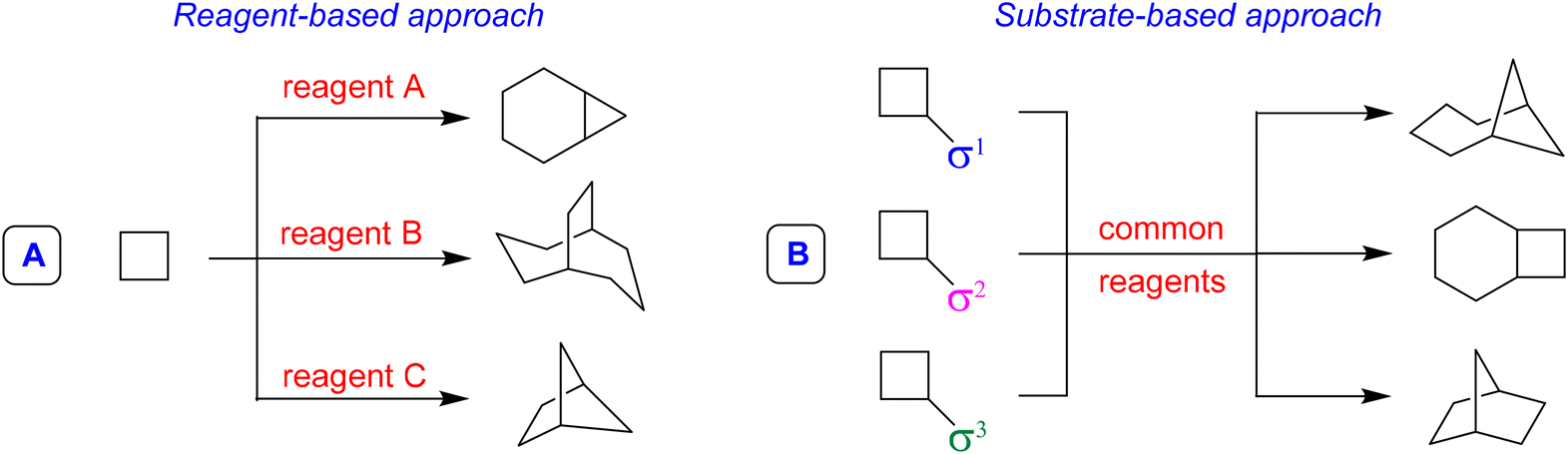 | ||
| Fig. 1 Two general approaches for generating skeletal diversity. | ||
The Reagent-Based Approach (A)4 is probably the most used tool in DOS to achieve skeletal diversity.
An example of the application of the reagent-based approach (A) from our research group is the divergent synthesis of isobenzofurans and isochromenes starting from 2-alkynylbenzaldehydes and alcohols. By changing the reaction conditions, it was possible to selectively obtain the two different regioisomeric heterocycles. Isobenzofurans have been obtained in good yields by a base-promoted domino nucleophilic addition/5-exo-dig cyclization sequence, under microwave heating at 70–110 °C (Scheme 1, left).5 Starting from the same substrates, the reaction catalyzed by an original [Ag(I)(Pc-L)] complex at 30 °C resulted in the selective 6-endo-dig cyclization with formation of the regioisomeric isochromene derivatives (Scheme 1, right).6
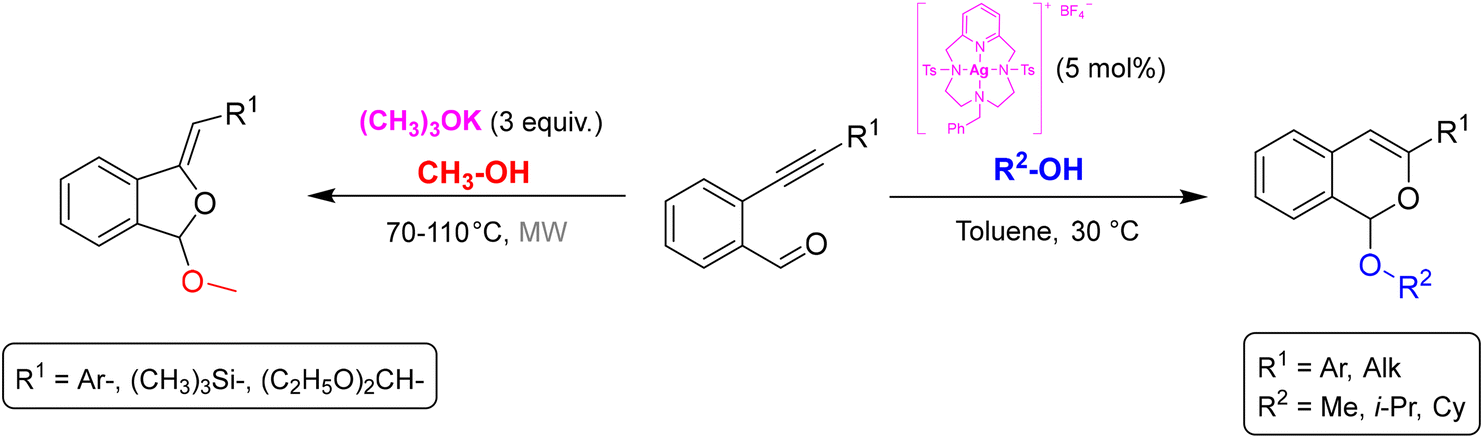 | ||
| Scheme 1 Divergent synthesis of isobenzofurans and isochromenes from 2-alkynylbenzaldehydes. | ||
Based on these premises, and in connection with the research interests of our group in diversity-oriented synthesis and discovery of novel strategies for the preparation of heterocycles starting from arylalkynes bearing a proximate nucleophile,7 in this work we developed a divergent approach for the regioselective synthesis of 3-unsubstituted-benzofuran and 4-unsubstituted-chromene nuclei starting from different 2-propargylphenols (Scheme 2).
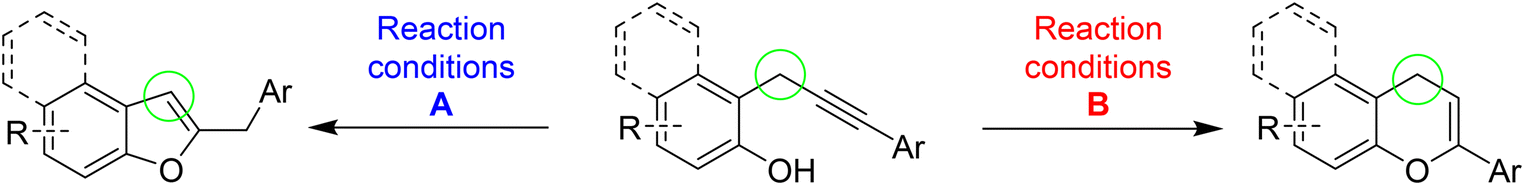 | ||
| Scheme 2 This work: divergent synthesis of benzofurans and chromenes from 2-propargylphenols. | ||
Simple 2-propargylphenols, i.e. characterized by the presence of a monoarylsubstitution on the propargylic sp3 carbon, have been selected as strategic starting material for the following reasons: (1) their chemistry has not received enough focus,8 (2) their structure displays great potential as versatile precursors for different reactions and cyclization paths, (3) the nucleophilic attitude of phenol oxygen is modulable by changing the reaction conditions, (4) cyclization products could represent relevant nuclei in the field of biologically active compounds. In the literature, there are some examples of base-promoted cyclization of propargylphenols to give the corresponding benzofurans through a 5-exo-dig cyclization.9 On the other hand, only one example of gold-catalyzed10 6-endo-dig cyclization of propargylphenol to give the corresponding six-member oxygenated heterocycles has been reported, and regarding other metal catalysts, another isolated example describes this cyclization promoted by a ruthenium-based catalyst.11 Interestingly, should be underlined that in all the above-mentioned examples the starting propargylphenols are always characterized by a double substitution on the propargylic sp3 carbon. These compounds are strongly different from our propargylphenols regarding both the synthesis and the reactivity. Conversely, to the best of our knowledge, starting from simple monoarylsubstuituted 2-propargylphenols such as those used in this study, there are only a few examples of synthesis of simple oxygen-containing heterocycles such as dihydrobenzofurans,12 whereas no example regarding the preparation of benzofurans and chromenes. Thus, in this work, three main essays have been developed: (a) the optimization of a general and robust protocol to generate a library of differently substituted 2-propargylphenols, (b) the development of a selective approach to 3-unsubstituted-benzofurans and (c) the development of a divergent approach to isomeric 4-unsubstituted-chromenes. In this paper, we describe the results of our efforts.
Results and discussion
Firstly we prepared a library of sixteen 2-propargylphenols 6a–p, characterized by the presence of different ED and EW groups in different positions on both the arene moieties. The general synthetic pathway is described in Scheme 3.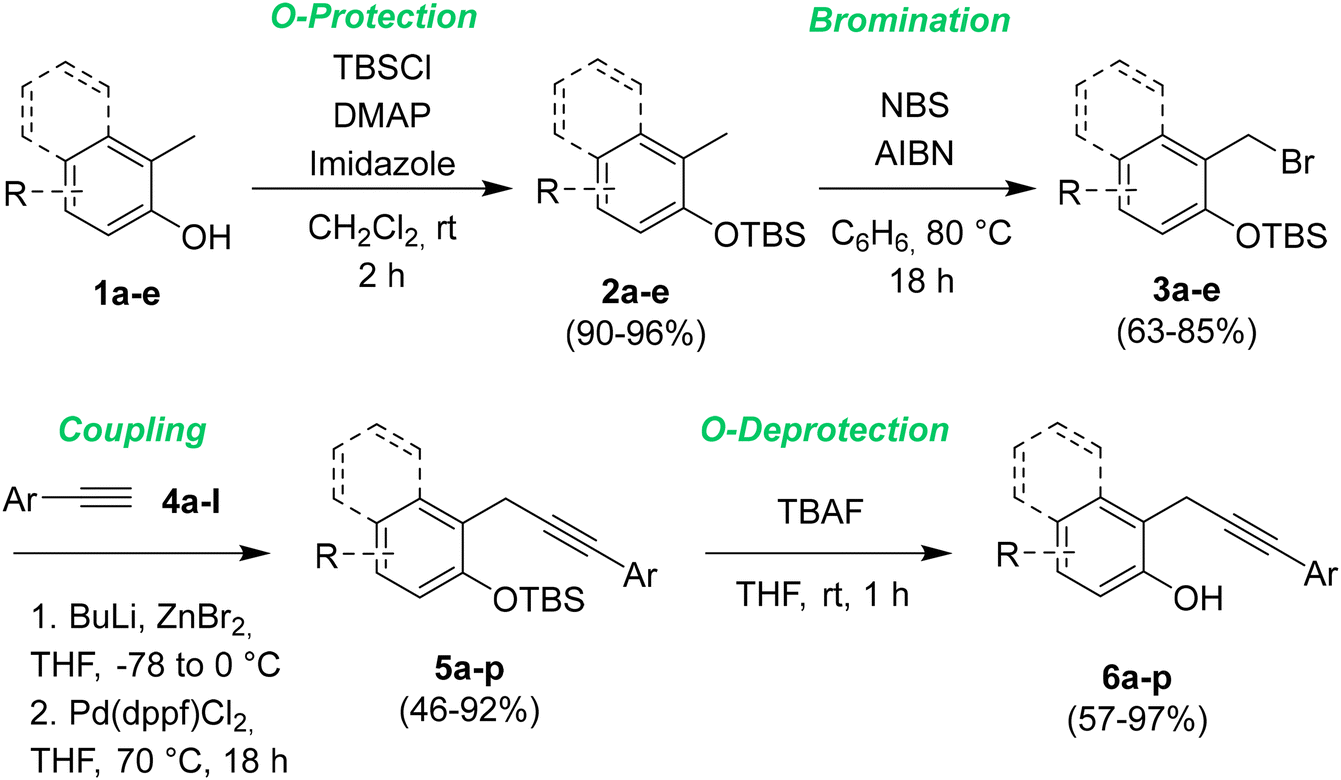 | ||
| Scheme 3 General synthetic path to 2-propargylphenols 6. | ||
The protection of phenol derivatives 1a–e was obtained by the reaction of o-cresols 1a–e with tert-butyldimethylsilylchloride (TBSCl) in the presence of 4-dimethylaminopyridine (DMAP) and imidazole in DCM at rt. By this approach, we were able to synthesize five different substituted tert-butyldimethyl(2-tolyloxy)silanes 2a–e in excellent yields, which were brominated by NBS and AIBN in refluxing benzene for 18 h to give intermediates 3a–e in very good yields.13 The cross-coupling of compounds 3a–e with different terminal alkynes 4a–l has been optimized based on a literature procedure reported by Negishi.14 The alkynylzinc bromide intermediates have been obtained by treatment of 4a–l with butyllithium and ZnCl2. Next, a Pd(dppf)Cl2 catalyzed cross-coupling with (2-(bromomethyl)phenoxy)(tert-butyl)dimethylsilanes 3a–e in refluxing THF overnight gave the sixteen protected o-propargyl phenols 5a–p in yields ranging from 40 to 91%. The following deprotection15 by treatment with TBAF in THF at rt gave the desired 2-propargylphenols 6a–p in yields ranging from good to excellent. 4-Amino-2-(3-phenylprop-2-yn-1-yl)phenol 6q was obtained by the chemical reduction of 6p employing iron powder in water (see ESI† for detailed procedures and full characterization data).
The screening of the most favourable reaction conditions for the base-promoted cyclization of 2-propargylphenols was performed using 2-(3-(p-tolyl)prop-2-yn-1-yl)phenol (6a) as a model substrate. The reactions were performed on a 0.2 mmol scale testing different organic and inorganic bases, in different solvents and modifying the reaction temperature and the energy source. Table 1 displays a selection of the most representative results of this screening.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Base (10 mol%) | T (°C) | Energy source | t | 7a yielda (%) | 6a rec. (%) |
| a Referred to pure isolated product. b TLC analysis displays the presence of a single spot referred to unreacted starting material 6a; this result is also confirmed by 1H-NMR spectra of the reaction crude. | |||||||
| 1 | DMF | CsCO3 | RT | Oil bath | 16 h | — | Quant.b |
| 2 | CH3CN | CsCO3 | 70 | Oil bath | 16 h | 46 | — |
| 3 | CH3CN | K2CO3 | 70 | Oil bath | 16 h | 75 | — |
| 4 | CH3CN | KOH | 70 | Oil bath | 16 h | 35 | — |
| 5 | CH3CN | TEA | 70 | Oil bath | 16 h | — | Quant.b |
| 6 | DCE | K2CO3 | 70 | Oil bath | 16 h | — | Quant.b |
| 7 | THF | K2CO3 | 70 | Oil bath | 20 h | — | Quant.b |
| 8 | MeOH | K2CO3 | 70 | Oil bath | 24 h | 12 | 76 |
| 9 | DMSO | K2CO3 | 70 | Oil bath | 2 h | 79 | — |
| 10 | DMF | K2CO3 | 70 | Oil bath | 4.5 h | 82 | — |
| 11 | DMF | K2CO3 | 90 | Oil bath | 1 h | 93 | — |
| 12 | DMF | K2CO3 | 90 | MW | 30 min | 64 | — |
| 13 | DMF | K2CO3 | 90 | MW | 10 min | 83 | — |
| 14 | DMF | K2CO3 | 90 | MW | 5 min | 83 | — |
| 15 | DMF | K 2 CO 3 | 70 | MW | 5 min | 89 | — |
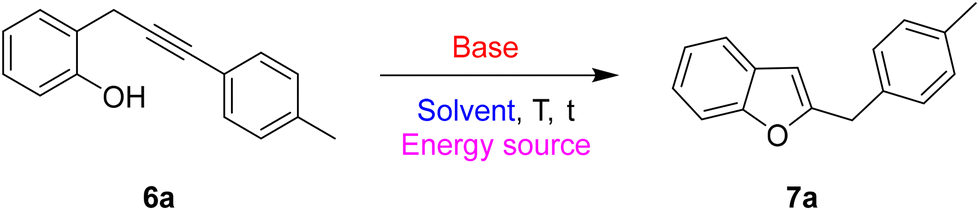
We started the study by using already established conditions for related base-promoted cyclizations, i.e. DMF as the solvent and cesium carbonate as the base.16 After overnight stirring at rt the starting material 6a was quantitatively recovered unreacted (Table 1, entry 1). By changing the solvent (acetonitrile) and raising the reaction temperature to 70 °C, the desired product 7a was obtained in an encouraging 46% yield (Table 1, entry 2). Then, we tested the activity of some other organic and inorganic bases (Table 1, entries 3–5); potassium carbonate was demonstrated to be the base of choice, yielding the desired product in 75% yield (Table 1, entry 3). Next, we evaluated the effect of the solvent (Table 1, entries 6–9), discovering that the presence of a polar aprotic media is mandatory for the success of the reaction. So, while the reaction in dichloromethane and THF failed (Table 1, entries 6 and 7), and a polar protic solvent such as methanol gave very poor results (Table 1, entry 8), dimethyl sulfoxide gave an interesting 76% yield of the desired product in 2 hours (Table 1, entry 9). The best results in terms of yield, time, and cleanness of the reaction have been obtained with DMF (82% yield) in 4.5 hours (Table 1, entry 10). Yields could be further increased by raising the reaction temperature to 90 °C; under these conditions, the reaction was complete in only 1 hour with a satisfactory 93% yield (Table 1, entry 11). The promising best reaction conditions (polar aprotic solvent and heating) seemed to be ideal for testing the approach under microwave heating17 (entries 12–15). It is well recognized that MAOS (Microwave Assisted Organic Synthesis) can lead to some important advantages, such as a reduction of reaction times and by-product formation. In the transformation under study, the efficiency of dielectric heating allowed a dramatic drop in reaction times and a slight reduction in reaction temperature. Under dielectric heating, the best results were obtained in a very short time (5 min) at 70 °C (Table 1, entry 15), hence with an overall gain in terms of sustainability of the process.
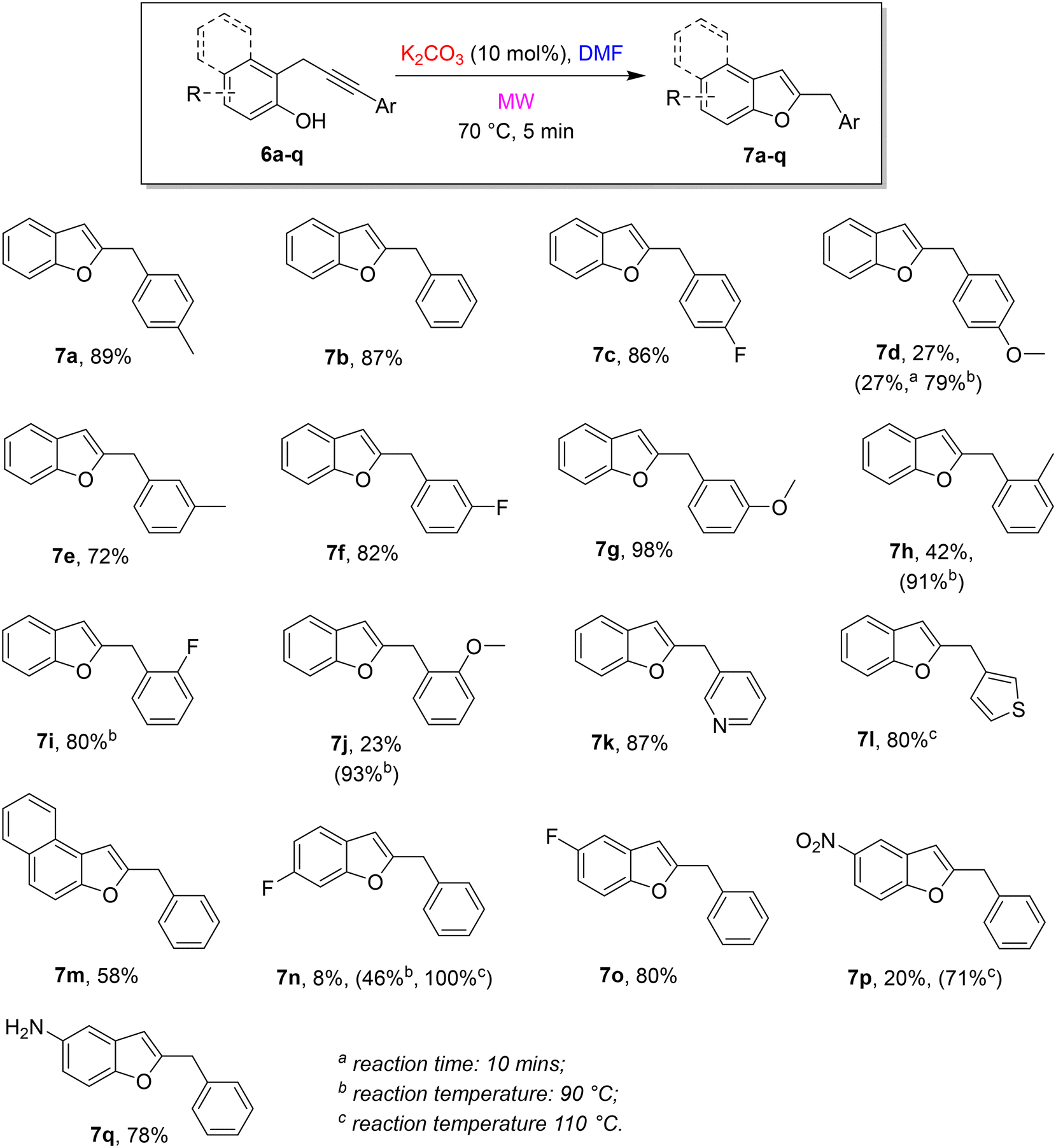
With optimal reaction conditions in hands, the scope and limitations of the approach were explored by changing the substitution on the terminal alkyne and the phenol moiety. The results are summarized in the following Scheme 4.
 | ||
| Scheme 4 Scope and limitation in the synthesis of benzofurans. | ||
We made a systematic study with different substitutions on the aryl group at the alkyne terminus. Steric and electronic features have been investigated by changing the electronic nature of the substituent (neutral, electron-donating, and electron-withdrawing) and its position (para, meta, and ortho) on the phenyl ring. The approach was demonstrated to be robust and high-yielding, despite the different substitutions on the aryl group at the alkyne terminus. In the presence of a neutral or an EW group, the cycloisomerization gave the corresponding benzofurans in very good yields under the standard conditions (7a, 7b, 7c, 7e, 7f, 7g). Conversely, an ED group on the aryl group in a conjugate position resulted in poor yields (see 7d and 7j). This behaviour is probably related to the effect of the electronic nature of the substituent on the electrophilicity of the distal sp carbon involved in the cyclization.18 In these cases, a rise of the reaction time to 10 minutes at 70 °C did not have any beneficial effect, whereas an increase of the temperature to 90 °C resulted in a dramatic increase in yields (7d). A related behaviour was observed also in the presence of a heteroaryl substituent on the alkynyl terminus: an electron-poor heterocycle such as the 3-pyridyl group did not affect the reaction under standard conditions (7k, 87%), on the contrary, to obtain good yields in the presence of an electron-rich heterocycle such as 3-thiophenyl, the reaction temperature should be raised to 110 °C (7l, 80%). Finally, we tested some modifications on the phenol unit. The shift from ortho-propargyl phenol to 1-propargyl-2-naphtol was well-tolerated (7m). The results in the presence of a fluorine atom on the phenol moiety depend on the position on the ring (7n and 7o) and an increase of the reaction temperature was required when the fluorine atom is in meta to the hydroxy group. A nitro group in para to the hydroxy group required a higher temperature to reach a satisfying yield (7p) whereas the amino group in the same position was tolerated under standard reaction conditions (7q).
Next, to develop a divergent synthesis starting from the same starting materials, we explored the possibility of preparing the regioisomeric chromenes through a metal-catalyzed 6-endo dig cyclization. Also in this case, the 6a was chosen as the model compound and we tested different catalysts, solvents, and conditions to obtain the best reaction results (Table 2). All the reactions were conducted with 0.2 mmol of 6a, 5 mol% loading of the metal catalyst in 2 mL of anhydrous solvent (c = 0.1 M), under a nitrogen atmosphere.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (5 mol%) | T (°C) | Energy source | t | 8a yielda (%) | 9a yielda (%) | 10a yielda (%) |
| a Referred to pure isolated product. b The reaction was performed without 4 Å MS. c Starting material was almost quantitatively recovered. d 15% of 7a was obtained. e Beside unidentified by-products. f 10% of 7a was obtained. | ||||||||
| 1 | DCE | JohnPhosAuNTf2 | RT | Oil bath | 24 h | 21b | — | 13 |
| 2 | DCE | JohnPhosAuNTf2 | 70 | Oil bath | 2 h | 75 | 5 | — |
| 3 | DCE | AgSbF6 | 70 | Oil bath | 24 h | NRc | — | — |
| 4 | DCE | CuBr | 70 | Oil bath | 24 h | NRc | — | — |
| 5 | DCE | Fe(OTf)3 | 70 | Oil bath | 24 h | NRc | — | — |
| 6 | DCE | NaAuCl4 | 70 | Oil bath | 24 h | NRc | — | — |
| 7 | DCE | Ph3PAuNTf2 | 70 | Oil bath | 24 h | 26 | — | — |
| 8 | DCE | IPrAuNTf2 | 70 | Oil bath | 3 h | 31d | — | — |
| 9 | DCE | P(OAr)3AuNTf2 | 70 | Oil bath | 2 h | 47e | 7 | — |
| 10 | Toulene | JohnPhosAuNTf2 | 70 | Oil bath | 2 h | 68f | — | — |
| 11 | THF | JohnPhosAuNTf2 | 70 | Oil bath | 2 h | 60 | 27 | — |
| 12 | DMF | JohnPhosAuNTf2 | 70 | Oil bath | 1 h | 52e | — | — |
| 13 | DCE | JohnPhosAuNTf2 | 70 | MW | 15 min | 61 | 6 | — |
| 14 | DCE | JohnPhosAuNTf2 | 85 | MW | 15 min | 68 | 5 | — |
| 15 | DCE | JohnPhosAuNTf 2 | 100 | MW | 15 min | 79 | 9 | — |
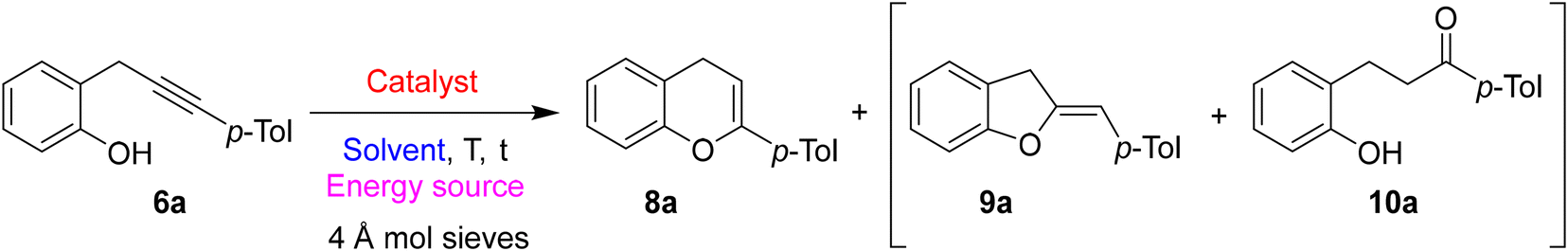
The first reaction was performed in the presence of JohnPhosAuNTf2 (5 mol%) in DCE at rt (Table 2, entry 1). However, after 24 hours, we obtained only a 21% yield of the desired product 8a beside a series of by-products and a 13% yield of the 2-hydroxydihydrochalcones 10a probably arising from the hydrolysis of 8a triggered by adventitious water in the reaction mixture. To overcome the formation by-product 10a, 4 Å molecular sieves were added to the reaction mixture, and to increase the yield the temperature was raised to 70 °C (Table 2, entry 2). Under these conditions, the desired product 8a was obtained in 75% yield beside a small amount of the corresponding 2,3-dihydrobenzofuran with exocyclic double bond 9a.
Other metal catalysts were tested; however, neither Cu(I), Ag(I), Fe(III), or Au(III) salts gave any positive results, and the starting material was recovered unreacted after 24 hours of reaction at 70 °C (Table 2, entries 3–6).
We next screened other cationic gold(I) catalysts (i.e. Ph3PAuNTf2, IPrAuNTf2, and P(OAr)3AuNTf2; Ar = 2,4-di-tert-butylphenyl) were tested (Table 2, entries 7–9). In all cases, the results were inferior to JohnPhosAuNTf2, moreover using IPrAuNTf2 a small amount of regioisomeric benzofuran 7a was observed (Table 2, entry 8).
Three different solvents were used in the presence of JohnPhosAuNTf2 as the catalyst at 70 °C. The use of toluene resulted in a slight decrease in the reaction yield with an increase in the formation of isomeric benzofuran compound 7a (Table 2, entry 10). Conversely, the use of THF increased the conversion rate but strongly reduced the regioselectivity, giving rise to a huge amount of the regioisomeric 2,3-dihydrobenzofuran 9a (Table 2, entry 11). The more polar DMF allowed to recover the regioselectivity of the reaction but with modest yields (Table 2, entry 12).
We also tried to shift from traditional to dielectric heating to reduce the reaction time and increase yields. Firstly, the reaction was performed at 70 °C for 15 minutes giving rise to a good 61% yield of 8a and 6% yield of 9a (Table 2, entry 13). Increasing the temperature to 85 °C a slight increase of the yield of 8a to 68% was observed (Table 2, entry 14) beside a reduced amount of by-product 9a. Finally, a reaction temperature of 100 °C allowed to obtain the best result in terms of conversion (88%) and yield of desired 8a (79%), with only a little increase of by-product 9a (Table 2, entry 15).
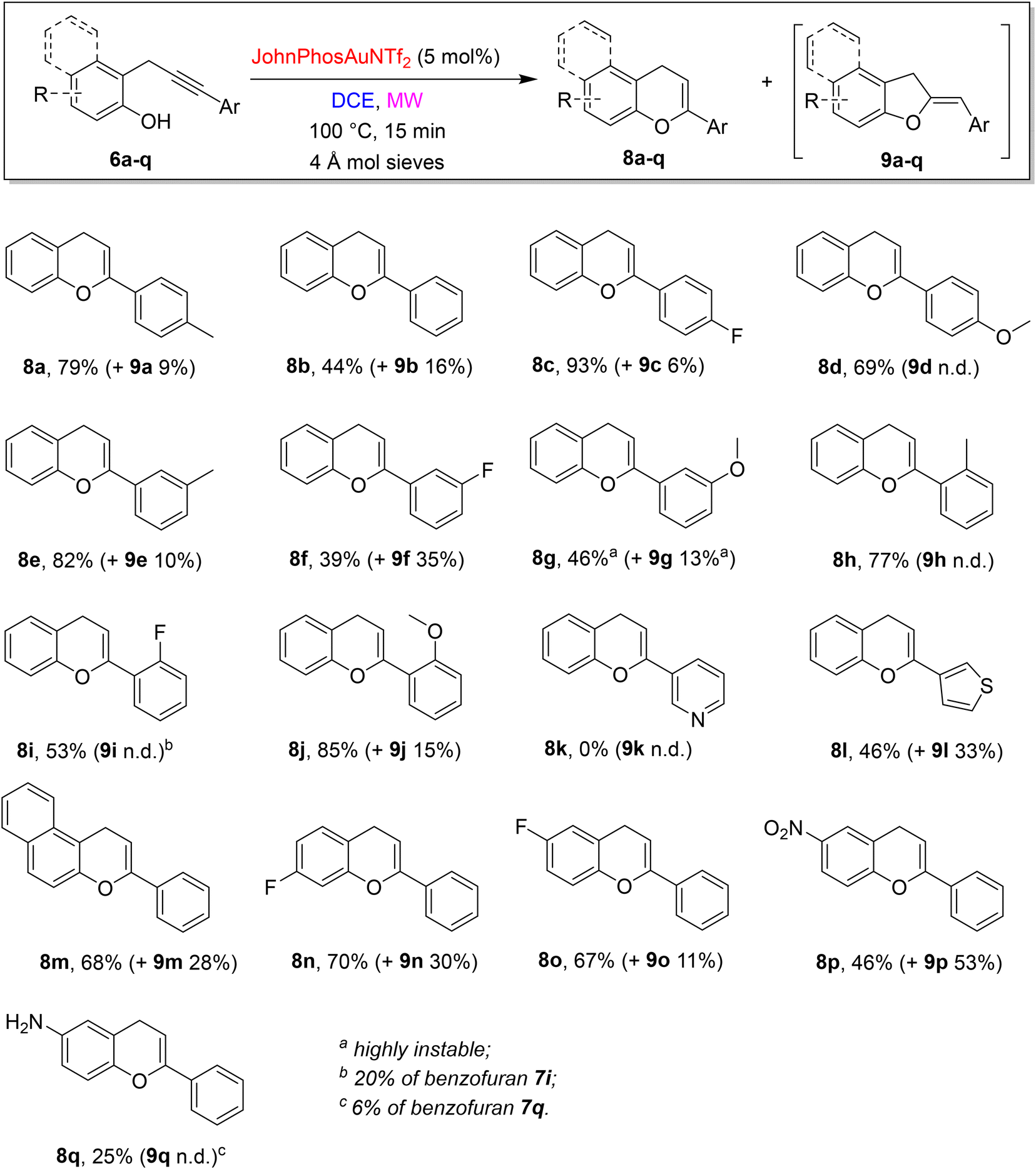
Thus, with optimal reaction conditions in hands, the scope and limitations of the divergent approach were explored (Scheme 5).
 | ||
| Scheme 5 Scope and limitation in the synthesis of chromenes. | ||
Different substituents showed different effects based on their positions and steric/electronic properties. However, it is difficult to find a rationale to describe accurately the differences in yields and selectivity. The substitution in the para position of the phenyl at the alkyne terminus gave rise to the desired chromenes 8a,c,d in very good yields and selectivity (isomeric dihydrobenzofurans 9 absent or <10% yield). Shifting to the substitution in meta position, the methyl group allowed the formation of 8e in 82% yield beside a 10% of 9e, while both ED and EW groups caused a decrease in the yields (8f 39%, and 8g 46%, respectively). Moreover, the regioselectivity of the reaction of 6f is one of the poorest, giving rise to the formation of 9f in 35% yield. When the phenyl ring at the alkyne terminus is substituted at the ortho position, the outcomes strongly depend on the electronic feature of the substituent: EWG showed worse results than EDG, but in general, the selectivity is high (8h–j). The substitution of the aryl ring with a heteroaryl moiety gave on the whole modest results: propargylphenol 6k, bearing a pyridyl 3-moiety on the alkyne terminus was unreactive and the starting material was fully recovered, while the presence of a 3-thiophenyl substituent induces the formation of the corresponding chromene derivative 8l in a moderate 46% yield, besides a consistent 33% yield of 9l. The failure of the reaction of 6k could be ascribed to a plausible inactivation/complexation of the gold catalyst from the pyridine nitrogen.
The investigation of the scope and limitations proceeded with modifications on phenol moiety. The reaction of 1-propargyl-2-naphtol 6m was high-yielding but poorly selective and led to the formation of a mixture of 8m and 9m in 68% and 28% yield, respectively. A similar behaviour was observed starting from fluorine-containing propargylphenols 6n, whereas a simple change in the position of the fluorine on the phenol ring (6o) resulted in a slight reduction of the amount of dihydrobenzofuran by-product formed (9o).
A reversed regioselectivity was obtained with 6p, bearing a nitro group, with the formation of 9p as the main product (53% yield) and 8p in only 46% yield. Finally, the presence of the amino group showed a quite high selectivity, being 8q the main product obtained, but a low reaction yield (25%), due to the presence of some unidentified by-products in the reaction mixture. The Z stereochemistry of dihydrobenzofurans 9 was determined through a NOESY experiment on product 9p and extended to the entire series by analogy (see ESI† for details).
Taking into account the previous literature findings and based on the experimental results, plausible reaction mechanisms for the divergent formation of isomeric benzofurans 7 chromenes 8 and dihydrobenzofurans 9 are proposed in Scheme 6.
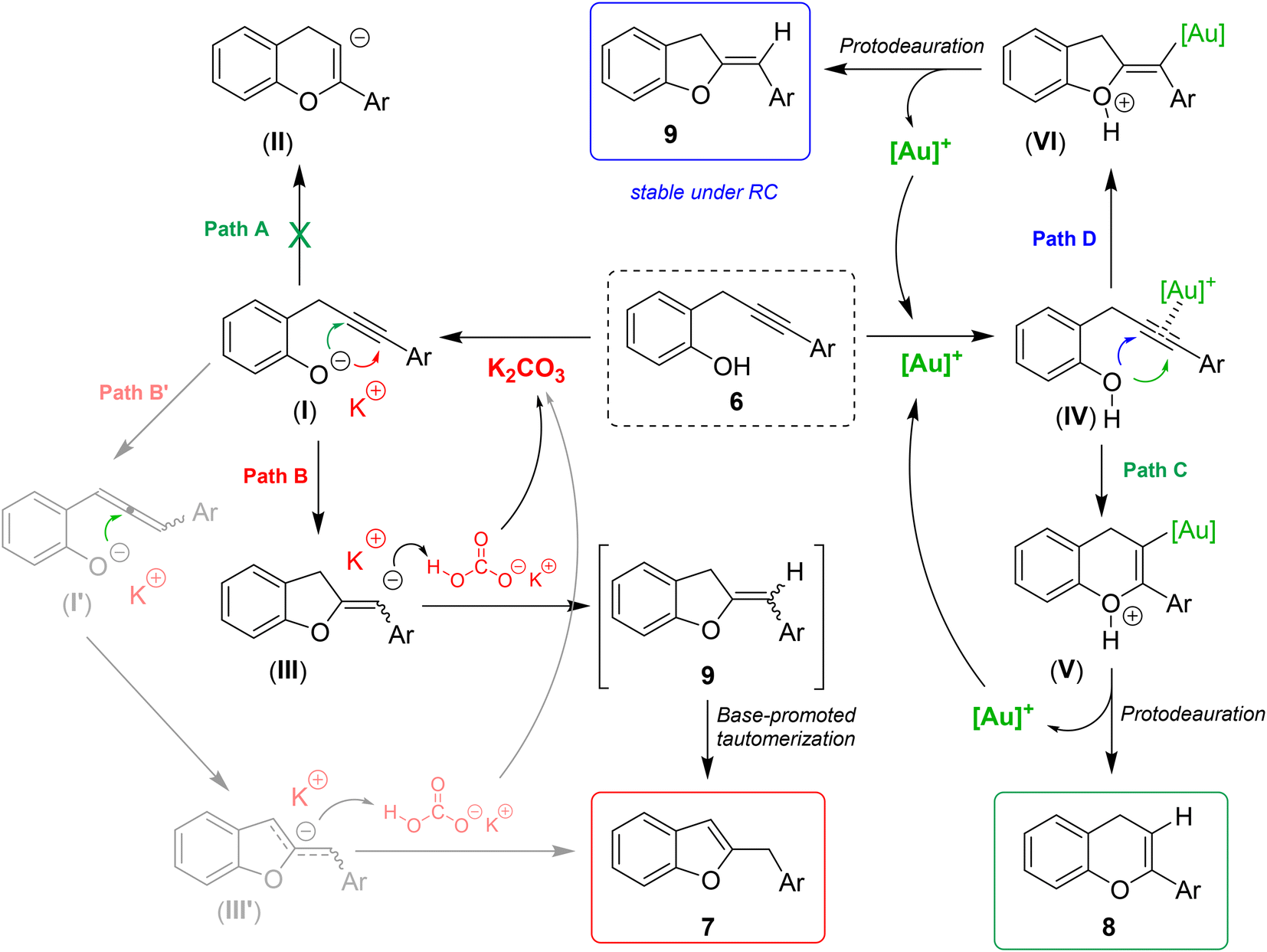 | ||
| Scheme 6 Proposed divergent reaction mechanisms. | ||
The formation of benzofurans 7 can be justified as follows: potassium carbonate (pKb = 3.75) is a base strong enough to partially deprotonate the 2-propargylphenol 6 (pKa ≅ 10) to give the phenoxide intermediate I. The latter can theoretically follow two cyclization paths. Path A involves a 6-endo-dig cyclization to give a 4H-chromene anion intermediate (II), whereas path B, through a 5-exo-dig cyclization mode, provides a 2-methylene-2,3-dihydrobenzofuran anion (III). As already observed in similar base-promoted cyclizations,19 the 5-exo-dig mode is preferred, probably because the alkenyl α-anion (III) is stabilized by resonance from the conjugated aryl group. Next, potassium bicarbonate, arising from the phenol deprotonation, can provide the proton to give the neutral 2-methylene-2,3-dihydrobenzofuran derivative 9, which under the basic reaction conditions undergoes a quick base-mediated tautomerization with formation of the aromatic benzofuran nucleus 7 stabilized by resonance. We cannot exclude a priori an alternative mechanism that involves the base-promoted formation of an allene intermediate (I′),20 which could evolve by intramolecular nucleophilic attack of the oxy anion to the central carbon of the allene to a delocalized benzylic/allylic anionic intermediate (III′) and then directly to benzofuran 7 by protonation (Path B′). However, this alternative path seems to be more unlikely because, as already reported by Arai and Shioiri in 2000, moderate bases such as K2CO3 are quite ineffective in efficiently promoting such transformation on these systems.21
The formation of chromenes involves the activation of the triple bond by the cationic gold to give the π-complex (IV).22 The complexation with the metal enhances the electrophilic properties of Csp carbons that undergo a nucleophilic attack from phenolic oxygen through a 6-endo-dig or a 5-exo-dig mechanism to give oxo-cationic intermediates (V) (Path C)23 and/or (VI) (Path D), respectively. The following protodemetallation gives the neutral product chromene 8 and/or the 2,3-dihydrobenzofuran 9 and regenerates the cationic gold catalyst. Normally, the formation of the 6-member heterocycle is preferred, nevertheless, when the electron density of the triple bond is strongly perturbed by the presence of electron-withdrawing substituents on the distal alkyne terminus, the 5-exo-dig cyclization mode becomes a competitive path and the isomeric 2,3-dihydrobenzofurans 9, are obtained as by-products in not negligible amounts. It is worth noting that under these neutral reaction conditions, the 2,3-dihydrobenzofurans 9 with exocyclic double bond are stable and the base-promoted tautomerization to give the corresponding aromatic benzofurans 7 was not observed. On the other hand, the treatment of isolated 2,3-dihydrobenzofurans 9 under basic conditions resulted in a quick and quantitative isomerization to the aromatized benzofurans 7 (see ESI† for details).
Conclusions
In this work, a MW-enhanced divergent approach for the synthesis of 2-substituted benzofurans and chromenes starting from variously substituted o-propargylphenols has been studied and optimized. Overall, we developed a robust protocol to synthesize o-propargylphenols (seventeen examples) as resourceful precursors for the preparation of seventeen benzofurans and sixteen chromenes. Most of them are new entities with no precedents in the literature. The reactions of o-propargylphenols under base conditions, give regioselectively the benzofurans (7) in yields ranging from 43 to 98%. Conversely, under cationic gold catalytic conditions, chromenes (8) were achieved in more variable yields (ranging from 25 to 93%), often beside small amounts of the corresponding 2,3-dihydrobenzofuran isomers (9), thus with lower regioselectivity.The results of this work demonstrated once again the potential of diversity-oriented synthesis for the exploration of molecular complexity. Neglected o-propargylphenols were demonstrated to be versatile and strategic starting materials to access simple nuclei and complex heterocyclic scaffolds by intramolecular cyclization reactions. The potential of these intriguing substrates will be further investigated in our lab.
Author contributions
A. G.: investigation, validation, writing – original draft, data curation. E. B.: conceptualization, investigation, validation, writing – original draft, data curation. I. N. and F. T.: investigation, data curation. V. P.: conceptualization, writing – review & editing. G. A.: conceptualization, funding acquisition, methodology, supervision, writing – original draft.Data availability
All data supporting the findings of this study, including experimental details, spectroscopic characterization data, and spectra for all compounds, are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Mrs Donatella Nava for the NMR analyses, and Dr Lucia Feni and Mr Stefano Pandini for the MS analyses.This research was supported by the MUSA – Multilayered Urban Sustainability Action – project, funded by the European Union – NextGenerationEU, under the National Recovery and Resilience Plan (NRRP) Mission 4 Component 2 Investment Line 1.5: Strengthening of research structures and creation of R&D “innovation ecosystems”, set up of “territorial leaders in R&D”.
References
- S. L. Schreiber, Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery, Science, 2000, 287, 1964–1969 CrossRef CAS.
- M. D. Burke and S. L. Schreiber, A planning strategy for diversity-oriented synthesis, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobert and D. R. Spring, Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules, Nat. Commun., 2010, 1, 80 CrossRef.
- S. Chen, L. Vaccaro and Y. Gu, Recent advances of versatile reagents as controllable building blocks in organic synthesis, Chin. Chem. Lett., 2024, 35, 109152 CrossRef CAS.
- M. Dell'Acqua, D. Facoetti, G. Abbiati and E. Rossi, Selective Base Promoted Synthesis of Dihydroisobenzofurans by Domino Addition/Annulation Reactions of ortho-Alkynylbenzaldehydes, Synthesis, 2010, 2367–2378 Search PubMed.
- M. Dell'Acqua, B. Castano, C. Cecchini, T. Pedrazzini, V. Pirovano, E. Rossi, A. Caselli and G. Abbiati, Mild Regiospecific Synthesis of 1-Alkoxy-isochromenes Catalyzed by Well-Defined [Silver(I)(Pyridine-Containing Ligand)] Complexes, J. Org. Chem., 2014, 79, 3494–3505 CrossRef.
- (a) E. Brambilla, A. Gritti, V. Pirovano, A. Arcadi, R. Germani, M. Tiecco and G. Abbiati, Acidic Deep Eutectic Solvents as Active Media for Sustainable Synthesis of Biindoles Starting from 2,2′-Diaminotolanes and Aldehydes, Eur. J. Org. Chem., 2023, e202300204 CrossRef CAS; (b) V. Pirovano, E. Brambilla, G. Fanciullacci and G. Abbiati, Cooperative Photoredox/gold Catalysed Cyclization of 2-Alkynylbenzoates with Arenediazonium Salts: Synthesis of 3,4-Disubstituted Isocoumarins, Org. Biomol. Chem., 2022, 20, 8065–8070 RSC; (c) E. Brambilla, M. Giannangeli, V. Pirovano, E. Rossi, A. Caselli and G. Abbiati, Synthesis and Photophysical Evaluation of Polarity Sensitive Push-pull Isoquinolines and their alkynyl precursors, Org. Biomol. Chem., 2021, 19, 4958–4968 RSC; (d) D. Garanzini, V. Pirovano, I. Menghi, G. Celentano, S. Rizzato, E. Rossi, A. Caselli and G. Abbiati, [Ag(PcL)]-Catalysed Domino Approach to 6-Substituted Benzoxazino Isoquinolines, Eur. J. Org. Chem., 2020, 3660–3670 CrossRef CAS; (e) V. Pirovano, G. Hamdan, D. Garanzini, E. Brambilla, E. Rossi, A. Caselli and G. Abbiati, [Ag(PcL)]-Catalyzed Domino Reactions of 2-Alkynylbenzaldehydes with Electron-Poor Anilines: Synthesis of 1-Aminoisochromenes, Eur. J. Org. Chem., 2020, 2592–2599 CrossRef CAS.
- T. C. Berg, V. Bakken, L.-L. Gundersen and D. Petersen, Cyclization and Rearrangement Products from Coupling Reactions Between Terminal o-Alkynylphenols or o-Ethynyl(hydroxymethyl)benzene and 6-Halopurines, Tetrahedron, 2006, 62, 6121–6131 CrossRef CAS.
- (a) Z. Liu, L. Liu, Z. Shafiqa, Y.-C. Wua, D. Wanga and Y.-J. Chen, InCl3-Catalyzed Propargylation of Indoles and Phenols with Propargylic Acetates: Application to the Syntheses of Benzofurans and Naphthofurans, Synthesis, 2007, 1961–1969 CrossRef CAS; (b) F.-Q. Yuan and F.-S. Han, Iron-Catalyzed Direct Synthesis of Densely Substituted Benzofurans and Naphthopyrans from Phenolic Compounds and Propargylic Alcohols, Adv. Synth. Catal., 2013, 355, 537–547 CrossRef CAS; (c) W.-T. Li, W.-H. Nana and Q.-L. Luo, Metal-free Sequential Reaction via a Propargylation, Annulation and Isomerization Sequence for the one-pot Synthesis of 2,3-Disubstituted Benzofurans, RSC Adv., 2014, 4, 34774–34779 RSC; (d) M. Sun, J. Song, L. Wang, W. Yin, M. Miao and H. Ren, Direct Propargylation of ortho-Quinone Methides with Alkynyl Zinc Reagents: An Application to the One-Pot Synthesis of 2,3-Disubstituted Benzofurans, Synlett, 2020, 818–822 CAS.
- M. Montesinos-Magraner, C. Lluna-Galán, F. Cernicharo-Toledo, C. Vila, G. Blay and J. R. Pedro, Enantioselective Friedel–Crafts Reaction of Hydroxyarenes with Nitroenynes to Access Chiral Heterocycles via Sequential Catalysis, Org. Biomol. Chem., 2021, 19, 6990–6994 RSC.
- K. Kanao, Y. Miyake and Y. Nishibayashi, Ruthenium-Catalyzed Enantioselective [3 + 3] Cycloaddition of Propargylic Alcohols with 2-Naphthols, Organometallics, 2010, 29, 2126–2131 CrossRef CAS.
- (a) F.-T. Luo, I. Schreuder and R.-T. Wang, Intramolecular Oxypalladation and Cross-Coupling of Acetylenic Alkoxides, J. Org. Chem., 1992, 57, 2213–2215 CrossRef CAS; (b) V. Klaus and J. S. Clark, Thioether-catalysed Tandem Synthesis of Furans and Cyclic Ethers or Lactones, Synlett, 2017, 1358–1362 CAS; (c) R. Quach, D. P. Furkert and M. A. Brimble, Synthesis of Benzannulated Spiroacetals Using Chiral Gold–phosphine Complexes and Chiral Anions, Tetrahedron Lett., 2013, 54, 5865–5868 CrossRef CAS.
- Q. Zhang and J. M. Takacs, Click-Connected Ligand Scaffolds: Macrocyclic Chelates for Asymmetric Hydrogenation, Org. Lett., 2008, 10, 545–548 CrossRef CAS PubMed.
- M. Qian and E.-I. Negishi, Palladium-catalyzed cross-coupling reaction of alkynylzincs with benzylic electrophiles, Tetrahedron Lett., 2005, 46, 2927–2930 CrossRef CAS.
- S. Kobayashi, K. Morino and E. Yashima, Macromolecular helicity inversion of an optically active helical poly(phenylacetylene) by chemical modification of the side groups, Chem. Commun., 2007, 2351–2353 RSC.
- M. T. Herrero, J. Díaz de Serralde, R. SanMartin, L. Bravo and E. Domínguez, Cesium Carbonate-Promoted Hydroamidation of Alkynes: Enamides, Indoles and the Effect of Iron(III) Chloride, Adv. Synth. Catal., 2012, 354, 3054–3064 CrossRef CAS.
- Microwaves in Organic Synthesis, ed. A. Loupy and A. de la Hoz, Wiley-VCH, Weinheim, 3rd edn, 2013 Search PubMed.
- M. Alfonsi, M. Dell'Acqua, D. Facoetti, A. Arcadi, G. Abbiati and E. Rossi, Microwave-Promoted Synthesis of N-Heterocycles by Tandem Imination/Annulation of γ- and δ-Ketoalkynes in the Presence of Ammonia, Eur. J. Org. Chem., 2009, 2852–2862 CrossRef CAS.
- (a) M.-J. Wu, L.-J. Chang, L.-M. Wei and C.-F. Lin, A Direct Anionic Cyclization of 2-Alkynylbenzonitrile to 3-Substituted-1(2H)-Isoquinolones and 3-Benzylideneisoindol-2-Ones Initiated by Methoxide Addition, Tetrahedron, 1999, 55, 13193–13200 CrossRef CAS; (b) W.-D. Lu, C.-F. Lin, C.-J. Wang, S.-J. Wang and M.-J. Wu, Substituent Effect on Anionic Cycloaromatization of 2-(2-Substituted ethynyl)benzonitriles and Related Molecules, Tetrahedron, 2002, 58, 7315–7319 CrossRef CAS.
- (a) D. J. Pasto, Recent Developments in Allene Chemistry, Tetrahedron, 1984, 40, 2805–2827 CrossRef CAS; (b) D. R. Taylor, The Chemistry of Allenes, Chem. Rev., 1967, 67, 317–359 CrossRef CAS; (c) V. K. Brel, Synthesis and Intramolecular Cyclization of Diethylphosphono-Substituted Allenic Glycols, Synthesis, 2001, 1539–1545 CAS.
- M. Oku, S. Arai, K. Katayama and T. Shioiri, Catalytic Synthesis of Allenes via Isomerization of Alkynes under Phase-Transfer Catalyzed Conditions, Synlett, 2000, 493–494 CAS.
- (a) R. Dorel and A. M. Echavarren, Gold(I)-catalyzed Activation of Alkynes for the Construction of Molecular Complexity, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS; (b) C. J. V. Halliday and J. M. Lynam, Gold–alkynyls in Catalysis: Alkyne Activation, Gold Cumulenes and Nuclearity, Dalton Trans., 2016, 45, 12611–12626 RSC.
- For a related mechanism with propargyl anilines see: Z.-Y. Han, H. Xiao, X.-H. Chen and L.-Z. Gong, Consecutive Intramolecular Hydroamination/Asymmetric Transfer Hydrogenation Under Relay Catalysis of an Achiral Gold Complex/Chiral Brønsted Acid Binary System, J. Am. Chem. Soc., 2009, 131, 9182–9183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01272k |
| This journal is © The Royal Society of Chemistry 2024 |