
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the reactivity of 2H-(thio)pyran-2-(thi)ones in cycloaddition reactions with strained alkynes through density functional theory studies†
Wei
Huang
a,
Kangqiao
Wen
a,
Scott T.
Laughlin
 *a and
Jorge
Escorihuela
*b
*a and
Jorge
Escorihuela
*b
aDepartment of Chemistry, Stony Brook University, 100 Nicolls Road, Stony Brook, NY 11790, USA. E-mail: scott.laughlin@stonybrook.edu
bDepartamento de Química Orgánica, Universitat de València, Avda. Vicente Andrés Estellés s/n, Burjassot, 46100 Valencia, Spain. E-mail: jorge.escorihuela@uv.es
First published on 18th September 2024
Abstract
Over the past two decades, click chemistry transformations have revolutionized chemical and biological sciences. Among the different strain-promoted cycloadditions, the inverse electron demand Diels–Alder reaction (IEDDA) has been established as a benchmark reaction. We have theoretically investigated the IEDDA reaction of endo-bicyclo[6.1.0]nonyne (endo-BCN) with 2H-pyran-2-one, 2H-thiopyran-2-one, 2H-pyran-2-thione and 2H-thiopyran-2-thione. These 2H-(thio)pyran-2-(thi)ones have displayed different reactivity towards endo-BCN. Density functional theory (DFT) calculations show, in agreement with experiments, that endo-BCN reacts significantly faster with 2H-thiopyran-2-one compared to other 2H-(thio)pyran-2-(thi)one derivatives because of the lower distortion energy. Experimentally determined second-order rate constants for the reaction of a 2H-pyran-2-thione with different strained derivatives, including a 1-methylcyclopropene derivative and several cycloalkynes (exo-BCN, (1R,8S)-bicyclo[6.1.0]non-4-yne-9,9-diyl)dimethanol, dibenzocycylooctyne and a light activatable silacycloheptyne, were used to validate the computational investigations and shed light on this reaction.
Introduction
Since the seminal 2001 work of Sharpless and coworkers,1 click chemistry has blossomed to include new reactions optimized for diverse applications. For example, after reports showing the toxicity of Cu(I),2 strain-promoted cycloadditions were developed as an alternative to the copper(I)-catalysed azide alkyne cycloaddition (CuAAC).3 Among the strain-promoted cycloadditions, the strain-promoted azide–alkyne cycloaddition (SPAAC),4 the inverse electron demand Diels–Alder reaction (IEDDA),5 and more recently, the strain-promoted oxidation-controlled quinone cycloaddition (SPOCQ),6 have emerged as powerful organic transformations. Due their high rates and efficiency, strain promoted cycloadditions have been successfully applied in bioconjugate functionalization,7 surface chemistry,8 and polymer science.9 Theoretical studies on the mechanism of SPAAC,10 IEDDA11 and SPOCQ,12 have led to a deeper understanding of these widely used organic reactions.Among the different dienes used in IEDDA reactions, 1,2,4,5-tetrazines have been widely investigated as counterparts for reacting with alkynes/alkenes.13 Conversely, reactivity studies of 2H-pyran-2-ones and analogues, the focus of this report, are scarce. Kranjc and Kočevar reported in 2005 the Diels–Alder reaction of 2H-pyran-2-ones with alkynes under thermal conditions and at high pressures to afford aniline derivatives.14 In 2022, Xian and coworkers reported a click-and-release strategy based on the reaction of 2H-pyran-2-thiones and other analogues with a strained alkyne, (1R,8S,9S)-bicyclo[6.1.0]non-4-yn-9-ylmethanol (endo-BCN, 1).15 In that report, computational studies based on the nucleus-independent chemical shift (NICS) were used to predict the cycloaddition efficiency of endo-BCN with the 2H-(thio)pyran-2-(thi)one analogues. In a recent work, we synthesized substituted pyrones and pyranthiones and studied their biorthogonality, demonstrating their utility in protein labelling and subcellular delivery of hydrogen sulfide in living cells.16 A deeper understanding of these reactions can help in the design of 2H-pyran-2-thiones and heterocyclic analogues with enhanced reactivity for potential applications in chemical biology.
Previous studies have shown that dienes 2–5 can react with strained alkynes through an IEDDA reaction to form an unstable non-aromatic intermediate 6–9viaTS1 (Scheme 1).15 This intermediate then undergoes a retro Diels–Alder reaction through TS2 to provide the aromatic derivative 10 and release a molecule of CO2, COS or CS2 as a byproduct (Scheme 1). Understanding the trends in their reactivity regarding the 2H-(thio)pyran-2-(thi)one derivative will be key for the further development of this click reaction.
 | ||
| Scheme 1 Schematic representation of the IEDDA reaction of exo-BCN 1 with 2H-pyran-2-one (2), 2H-thiopyran-2-one (3), 2H-pyran-2-thione (4) and 2H-thiopyran-2-thione (5). | ||
Herein, a systematic theoretical study of cycloadditions of endo-BCN with 2H-pyran-2-one (2), 2H-thiopyran-2-one (3), 2H-pyran-2-thione (4) and 2H-thiopyran-2-thione (5) was performed using the activation strain model. A combined experimental-computational study was conducted to evaluate reactivity of a 2H-pyran-2-thione analogue towards different strained dienophiles including 1-methylcyclopropene (MeCp) and cycloalkynes such as endo-BCN, ((1R,8S,9R)-bicyclo[6.1.0]non-4-yn-9-yl)methanol (exo-BCN), ((1R,8S)-bicyclo[6.1.0]non-4-yne-9,9-diyl)dimethanol (DMBCN), dibenzocycylooctyne (DBCO) and a light activated silacycloheptyne (Si7HT).
Results and discussion
As mentioned above, oxygen and sulphur-containing heterocycles such as 2H-pyran-2-one (2), 2H-thiopyran-2-one (3), 2H-pyran-2-thione (4) and 2H-thiopyran-2-thione (5) can react with a strained cycloalkyne such as endo-BCN to form a non-aromatic intermediate (6–9) viaTS1. Thus, we performed a computational study in order to rationalize the observed reactivity of the different oxygen and sulphur-containing heterocycles. To this end, the effect on the reactivity of the oxygen and sulphur atoms in these heterocycles was investigated by means of DFT calculations at the M06-2X/6-311+G(d,p) level of theory. Initially, a conformational search was performed to analyse the multiple conformations of endo-BCN (1) and the conformation with the lowest energy and an energy gap of 4.0 kcal mol−1 towards the other conformations was selected for the mechanistic study.The formation of the aromatic derivative 10 can be rationalized in terms of the following general reaction mechanism, which can be divided into two main steps: [4 + 2] cycloaddition and retro Diels–Alder reaction with the elimination COS, CO2 or CS2 depending on the oxygen or sulphur-containing heterocycle. The Gibbs energy profile for the reaction of endo-BCN with the different dienes 2, 3, 4 and 5 is shown in Fig. 1. The first step of the mechanism involves the [4 + 2] cycloaddition and is exergonic. In line with similar Diels–Alder reactions, a pre-reaction complex with higher energy than both separated reactants was located.17 The interaction between the diene and endo-BCN (1) led to the formation of the molecular pre-reaction complex (RC). A decrease in the enthalpy of the reaction system of around 6 kcal mol−1 was computed of this transformation accompanied with substantial reduction in the entropy (Fig. S2, ESI†). Therefore, Gibbs energy (ΔG) of the formation of pre-reaction complex (RC) has a positive value, which excluded the possibility of pre-reaction complexes as stable intermediates. As inferred from Fig. 1, 2H-pyran-2-thione (4) has the lower activation Gibbs energy (21.2 kcal mol−1) of the four heterocyclic derivatives (2–5). For 2H-pyran-2-one (2), the computed activation Gibbs energy was higher (22.7 kcal mol−1), whereas for 2H-thiopyran-2-thione (5) the barrier was up to 25.3 kcal mol−1.
 | ||
| Fig. 1 Gibbs energy profile (in kcal mol−1) for the cycloaddition of endo-BCN (1) with dienes 2, 3, 4 and 5. | ||
Finally, 2H-thiopyran-2-one (3) exhibited the highest barrier (26.5 kcal mol−1) for the [4 + 2] cycloaddition among the four derivatives. Fig. 2 shows the transition state structures and activation Gibbs free energies (in kcal mol−1) for the cycloaddition of BCN (1) with heterocyclic structures 2, 3, 4 and 5. Given the lack of symmetry in the heterocyclic dienes, all cycloadditions occurred via non-synchronous transition state structures with C⋯C bond forming distances within the range of 2.13–2.47 Å. The pattern of reactivity was 3 < 5 < 2 < 4. Carbonyl derivatives, i.e., 2H-pyran-2-one (2) and 2H-thiopyran-2-one (3), had activation Gibbs free energies of 22.7 and 26.5 kcal mol−1, respectively, whereas for thiocarbonyl compounds, 2H-pyran-2-thione (4) and 2H-thiopyran-2-thione (5), activation Gibbs free energies of 21.2 and 25.3 kcal mol−1, respectively, were computed. The analysis of transition state structures reveals that 2H-pyran-2-thione (4) has the longest forming bond distance (2.47 Å), and therefore the earliest early transition state leading to the lowest activation energy. The forming bond distances are almost identical for the oxygen and sulphur-containing heterocyclic structures 2, 3, 4 and 5. According to the experimental data, 2H-pyran-2-thione (4) reacts 3.7 times faster than 2H-pyran-2-one (2) in a PBS buffer (pH 7.4 with 10% DMSO).15 Computational data predict a higher reactivity for 2H-pyran-2-thione (4) compared to the other oxygen and sulphur-containing heterocycles.
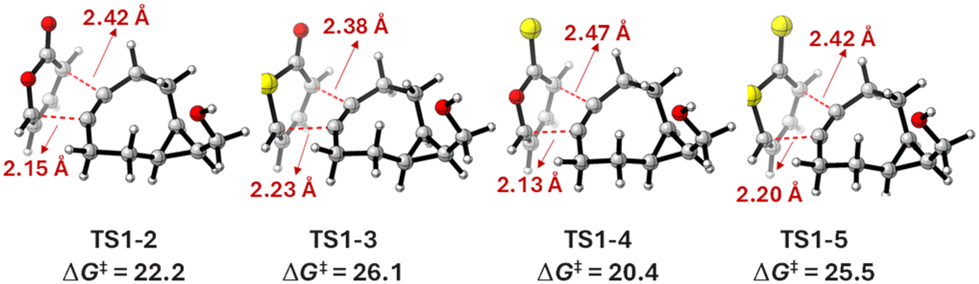 | ||
| Fig. 2 Optimized transition state structures for the [4 + 2] cycloaddition. Activation Gibbs energies (ΔG‡) are given in kcal mol−1, and distances are shown in Angstroms. | ||
From non-aromatic intermediates 6–9, a retro Diels–Alder reaction affords the aromatic product 10 and releases a molecule of COS for 7 and 8, CO2 for 6 or CS2 for 9. Fig. 3 shows the transition state structures and activation Gibbs free energies (in kcal mol−1) for the retro Diels–Alder reaction to yield the final product 10. The lowest activation Gibbs energy for the retro Diels–Alder reaction was found for intermediate 8 (TS2-4) (coming from 2H-thiopyran-2-thione (4)), with a barrier of 13.4 kcal mol−1, which delivered the final product 10 with an exergonicity of −49.3 kcal mol−1. Thiopyran-derivatives 7 and 9 (coming from 2H-thiopyran-2-one (3) and 2H-thiopyran-2-thione (5)) had barriers of 18.9 and 18.8 kcal mol−1, respectively; whereas intermediate 6 (TS2-2) showed a Gibbs activation barrier of 17.5 kcal mol−1. After the second transition state (TS2), the formation of the aromatic product 10 is highly exergonic, acting as the thermodynamic driving force of this process. The high exergonicity observed from the retro Diels–Alder reaction is due to the recovery of the aromaticity from non-aromatic intermediates 6–9 to deliver the final aromatic product 10 and the release of a molecule of COS, CO2, or CS2. In general, the O⋯C and S⋯C distances for the breaking bonds were around 2.1 and 2.6 Å, respectively, whereas the C⋯C distance for the breaking bond in the case of pyran-derivatives 6 and 8 (TS2-2 and TS2-4) was found to be shorter than that of thiopyran-derivatives 7 and 9 (TS2-3 and TS2-5). Overall, the type of atom at the heterocycle becomes a key factor with a significant effect the thermodynamic reaction feasibility (ΔG°). In this regard, those reactions involving the breaking of a C⋯O bond (TS2-2 and TS2-4) are more exergonic than those in which a C⋯S is being broken (TS2-3 and TS2-5).
 | ||
| Fig. 3 Optimized transition state structures for retro Diels–Alder reaction. Activation Gibbs energies (ΔG‡) and free energies of reaction (ΔG°), and distances are shown in Angstroms. Energies are given in kcal mol−1. | ||
Comparing the two transient steps (TS1 and TS2), the [4 + 2] cycloaddition is the rate determining step (RDS) for the IEDDA reaction between endo-BCN and 2H-(thio)pyran-2-(thio)ones, as also observed for IEDDA reactions of 1,2,4,5-tetrazines.18 According to the computed activation free energies for both transition states, and the high exergonicity of the overall process, the cycloadduct (10) would be formed after the reaction with the release of CO2, COS or CS2. Finally, a comparison between M06-2X/6-311+G(d,p), M06-2X/Def2TZVPP and ωB97xD/Def2TZVPP methods was performed to validate the results and each method yielded very similar results in terms of transition state barriers (ΔG‡) (see Table S3, ESI†).
Next, the activation strain model was used to rationalize the trend in reactivity.19Fig. 4a shows the distortion/interaction–activation strain (D/I-AS) analysis of the transition state structures. According to this model, the optimized transition state structure is separated into two fragments (distorted BCN and distorted heterocyclic structure), and the energy difference between the distorted fragment and the corresponding optimized structure is defined as the distortion energy (ΔEdist). The difference between the activation energy and the total distortion energy is the interaction energy (ΔEint). This model has been successfully applied to rationalize the reactivity of cycloadditions, substitution and elimination reactions, and transition metal catalysed reactions.20 As inferred from the analysis, BCN distortion varies slightly (green arrows, 2.6 to 2.9 kcal mol−1), but the variation in distortion energy for each diene is more significant (blue arrows). The variation of geometrical parameters of distorted endo-BCN at the TS was very similar for the four structures (Table S4, ESI†). Small angle deviations (lower than 3°) from the endo-BCN reactant structure agree with the computed low BCN distortion energies. The high distortion energy for the dienes can be associated to the distorted and bended structure at the TS in comparison with the almost planar structure of the reactant. In this regard, 2H-pyran-2-thione (4) displayed the lowest distortion in line with the lowest activation energy. The substitution of oxygen by sulphur at the pyranthione ring (from 4 to 5) was reflected in a significant increase in the distortion energy of the heterocycle, leading to a higher activation energy. A similar effect was observed in the atomic substitution from 2 to 3.
 | ||
| Fig. 4 (a) Distortion/interaction analysis on the transition structures (blue arrow for distortion energy of heterocyclic compound; green arrow for distortion energy of BCN; red arrow for interaction energy, and black arrow for activation potential energy). Energies are given in kcal mol−1. (b) NCI surfaces for the transition state structures. | ||
Finally, a non-covalent interaction (NCI) analysis was performed on the transition states (Fig. 4b). Upon inspection of the NCI surfaces, it is clear for all compounds a similarly sized contour was computed, with an attractive interaction in the region where C–C bonds are being formed, but also with a strong repulsive interaction.
Fig. 5 shows the activation strain diagram (ASD) of the cycloaddition of endo-BCN with 2H-pyran-2-one (black line), 2H-thiopyran-2-one (red line), 2H-pyran-2-thione (blue line) and 2H-thiopyran-2-thione (green line). The distortion energies for reactions of 2H-pyran-2-one (black line) and 2H-pyran-2-thione (blue line) are quite similar, and significantly lower than that for 2H-thiopyran-2-one (red line), and 2H-thiopyran-2-thione (green line) along the complete reaction coordinate. This trend is also observed for the activation energy and the interaction energy, and the study suggests the reactivity is controlled by the distortion energy. An earlier transition state indicates that the transition state geometry is closer to that of the reactants, which is reflected in a lower distortion energy for 2H-pyran-2-thione (4). It is evident that the reactivity differences between 2H-pyran-2-one (2) and 2H-pyran-2-thione (4) with endo-BCN comes from the significant differences in the distortion energy between reactants along the reaction coordinate as both reactions show similar interaction energies. Furthermore, these type of reactions are inverse electron-demand Diels–Alder reactions, in which the HOMO of the dienophile interacts with the LUMO of the diene. A more effective orbital overlap interaction was computed between the HOMO of endo-BCN and the LUMO of 2H-pyran-2-thione (4) compared to the other set of compounds.
 | ||
| Fig. 5 Activation strain diagrams of the cycloaddition of BCN with the different heterocyclic compounds along the reaction coordinate projected onto the C⋯C bond stretch, computed at M06-2X/6-311+G(d,p). | ||
Given the faster kinetics observed for 2H-pyran-2-thione among the different oxygen and sulphur-containing heterocycles,15,16 we experimentally studied the reactivity of a 2H-pyran-2-thione with several popular dienophiles. For this study, we choose a diester substituted 2H-pyran-2-thione, PyrS2, the current best in class compound based on our previous studies with these reagents in protein labelling experiments.16 The dienophile reaction partners we explored were 1-methylcyclopropene (MeCp) and the cycloalkynes exo-BCN, DMBCN,21 DBCO22 and light activatable Si7HT.23 We conducted kinetics studies for reaction of each of these dienophiles with PyrS2 (see ESI† for further details). Reactivity tests towards exo-BCN, DMBCN, DBCO and MeCp and were carried out at 25 °C in 50% CH3CN/H2O; whereas for Si7HT the kinetic data was determined at 25 °C in CH3CN due to the limited stability of silacycloheptyne after UV illumination (Scheme 2). As expected, PyrS2 exhibited high reactivity with exo-BCN, resulting in a second-order rate constant of 0.36 M−1 s−1. Contrary to other related cycloadditions, endo-BCN and exo-BCN displayed a similar reactivity. Slightly slower kinetics was observed for DMBCN, having a second-order rate constant of 0.15 M−1 s−1. In the case of DBCO, no significant reaction was observed at room temperature after 24 h, whereas the methylcyclopropene derivative (MeCp) had a second-order rate constant of 8 × 10−4 M−1 s−1. Finally, a second-order rate constant of 1.03 M−1 s−1 was determined for Si7HT after light activation. In all cases, the cycloadducts were isolated after the release of COS.
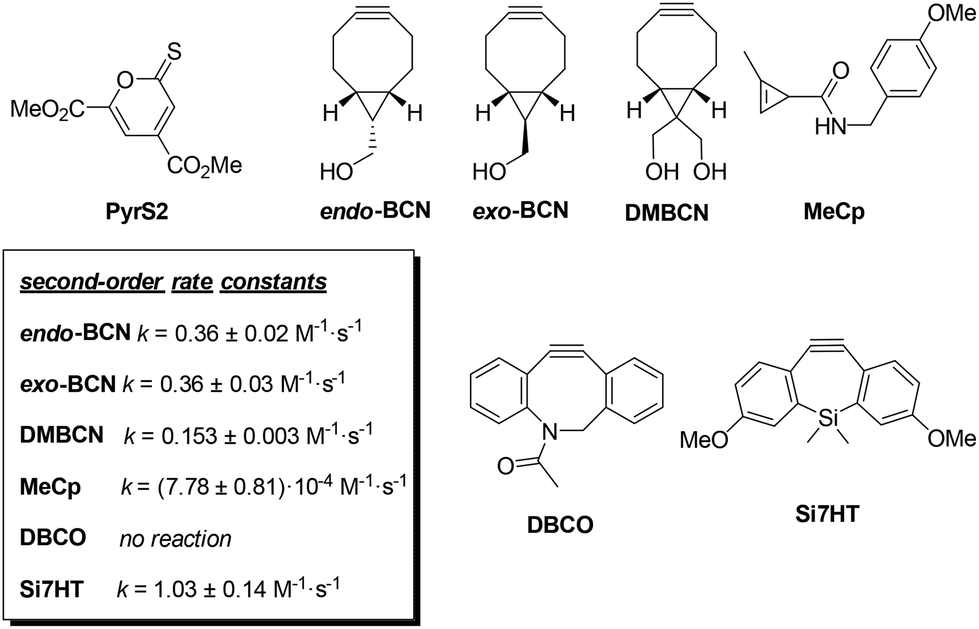 | ||
| Scheme 2 Chemical structures of 2H-pyran-2-thione PyrS2, the PyrS2 reaction partners evaluated here, and their second-order rate constants for the reaction with 2H-pyran-2-thione PyrS2. | ||
To further support our experimental findings, computational investigations using DFT calculations were performed at the M06-2X/6-311+G(d,p) level of theory and including the implicit solvent model SMD. To this purpose, we determined the Gibbs energy barriers for the cycloaddition of 2H-pyran-2-thione PyrS2 with different strained counterparts. The transition state structures and the calculated activation energy (ΔE‡), activation Gibbs energy (ΔG‡), and Gibbs energy of the reaction (ΔGrxn) are displayed in Fig. 6a. It is worth mentioning that activation energy barriers (ΔE‡) are lower than Gibbs energy barriers (ΔG‡). Gibbs energy barriers take into account both enthalpy and entropy changes that occur during a chemical reaction. These differences reveal that entropic contributions on these reactions are significant. The TS structures are all asynchronous with C⋯C bond forming distances ranging from 2.24 to 2.34 Å. The lowest activation Gibbs energy was computed for Si7HT, in agreement with the faster experimentally determined kinetics, having an earlier transition state and a longer forming C⋯C bond distance than other strained counterparts. The computed barriers for the BCN derivatives exo-BCN and DMBCN were found to be 19.0 and 18.8 kcal mol−1, respectively. In contrast, to other related cycloadditions, endo-BCN and exo-BCN displayed a similar reactivity. In the case of DBCO, the high activation Gibbs energy barrier of 25.1 kcal mol−1 is in agreement with the lack of reactivity of this dibenzocycloocytne derivative, as observed for other cycloaddition reactions.12a In agreement with experimental results, the computed rate constants that predict: (1) DMBCN will react faster than endo-BCN and exo-BCN with 2H-pyran-2-thione PyrS2, (2) MeCp has a low reactivity, and that (3) Si7HT will react with PyrS2 around 3 times faster than endo-BCN and exo-BCN. To our surprise, the kinetics observed for DMBCN and PyrS2 were slightly slower than expected from our computational findings, which might imply other factors like solvent effects played some role in reaction kinetics.
 | ||
| Fig. 6 (a) Optimized transition state structures at the M06-2X/6-311+G(d,p) level of theory with bond forming distances in Å. Values of activation energy (ΔE‡), activation Gibbs energy (ΔG‡), and Gibbs energy of the reaction (ΔGrxn) are shown below each structure in black, green, and purple, respectively, in kcal mol−1. (b) Values of distortion energy of pyrones (Edist(11), in blue), distortion energy of strained cycloderivative (Edist(cyclo), in green), interaction energy (Eint, in red) and activation energy (ΔE‡, in black). Energies are given in kcal mol−1. | ||
The D/I-AS analysis of the transition state structures of 2H-pyran-2-thione PyrS2 with the different studied strained counterparts is shown in Fig. 6b. In this case, the D/I-AS analysis was performed at the transition state as a defined point for each reaction under study. It should be noted that performing the D/I-AS analysis at the state geometry can lead to incorrect interpretations if transition states occur at highly different bond forming distances.24 However, here the forming bond lengths are very similar in all cases, warranting the comparison at the transition state. As shown in Fig. 6b, the distortion energy for PyrS2 (blue arrow) decreased with the smaller ring sizes with relatively smaller energy for MeCp and Si-7HT (14.2 and 13.3 kcal mol−1, respectively). In contrast, biphenyl flanked cyclooctyne, DBCO (19.6 kcal mol−1), showed significantly increased distortion energy for PyrS2 compared with non-fused ring cyclooctynes. The relatively larger distortion energy of PyrS2 was possibly resulting from the large steric effect from DBCO. This was further confirmed by the distortion energy for dienophiles. Another biphenyl flanked dienophile, Si7HT showed much smaller distortion energy compared with DBCO owing to the lower steric interactions after ring size contraction. The non-phenyl flanked cyclooctyne dienophiles had even lower distortion energy, providing more support for the vital role of steric effects in dienophiles. Interestingly, MeCp showed the highest distortion energy (10.5 kcal mol−1) with PyrS2, implying that the interaction between alkynes and PyrS2 were much more favorable than that with alkenes. Additionally, the interaction energy was relevant to steric effects with more favorable interactions with cycloocytnes (exo-BCN, endo-BCN, and DMBCN) with non-fused rings compared to fused ring counterparts (DBCO and Si7HT). The interactions between alkynes were also favorable compared that with alkenes.
Conclusions
In this computational study, we have studied the inverse electron-demand Diels–Alder reactions of BCN towards 2H-pyran-2-one, 2H-thiopyran-2-one, 2H-pyran-2-thione and 2H-thiopyran-2-thione. DFT calculations have been carried out to rationalize why 2H-pyran-2-thiones have higher reactivity compared to the other counterparts. The enhanced reactivity of 2H-pyran-2-thiones (4) with respect to the other set of derivatives results from a lower distortion energy. The reaction of 2H-pyran-2-thiones was further extended different strained derivatives and DFT calculations showed a good agreement with experimentally determined second-order rate constants. We envision that this mechanistic study will help in the understanding and development of more reactive 2H-pyran-2-thiones and related analogues in the future.Experimental
Reactivity tests and second order rate constant determination
Reactivity tests between PyrS2 and DBCO were carried out at 25 °C in a 50% mixture of CD3CN/D2O, which was monitored by 1H nuclear magnetic resonance (NMR) spectroscopy using 1,3,5-trimethoxybenzene as the internal standard. Reactions between PyrS2 and MeCp were monitored using HPLC and the cycloadducts were analysed by LC-MS. Second order rate constants were determined by mixing PyrS2 with dienophiles (exo-BCN, DMBCN, MeCp) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 1:15, or 1:20 ratio for pseudo 1st order kinetics and monitoring the characteristic absorption decay of PyrS2 at 412 nm using a plate reader. Uncaging tests with the Si7HT precursor Cp-7HT and PyrS2 were carried out at room temperature in CH3CN with light exposure using a mounted LED (Thorlabs, 365 nm, input power 3 W) and reaction monitoring using HPLC assay. For kinetics analysis, reaction mixtures containing the Si7HT precursor Cp-7HT (2 to 3 mM) in CH3CN were uncaged for 1 min in a quartz test tube and then mixed with PyrS2 (0.2 mM) to make final concentrations of 1 to 1.5 mM for Cp-7HT and 0.1 mM for PyrS2, respectively. After that, the reaction mixtures were aliquoted into the plate reader for absorption monitoring at 412 nm over time. All the cycloadducts were isolated from reaction mixtures after kinetics determination and characterized by 1H NMR, cosy, and HRMS for reaction validation.
:10, 1:15, or 1:20 ratio for pseudo 1st order kinetics and monitoring the characteristic absorption decay of PyrS2 at 412 nm using a plate reader. Uncaging tests with the Si7HT precursor Cp-7HT and PyrS2 were carried out at room temperature in CH3CN with light exposure using a mounted LED (Thorlabs, 365 nm, input power 3 W) and reaction monitoring using HPLC assay. For kinetics analysis, reaction mixtures containing the Si7HT precursor Cp-7HT (2 to 3 mM) in CH3CN were uncaged for 1 min in a quartz test tube and then mixed with PyrS2 (0.2 mM) to make final concentrations of 1 to 1.5 mM for Cp-7HT and 0.1 mM for PyrS2, respectively. After that, the reaction mixtures were aliquoted into the plate reader for absorption monitoring at 412 nm over time. All the cycloadducts were isolated from reaction mixtures after kinetics determination and characterized by 1H NMR, cosy, and HRMS for reaction validation.
Computational details
All calculations were performed using Gaussian 16.25 Geometry optimizations of the minima and transition states were carried out using the M06-2X functional26 with the 6-311+G(d,p) basis set and SMD model27 for mimicking the solvent (50% CH3CN/H2O mixture, χ = 55.69 at 25 °C).28 This functional has proved to give accurate energetics for cycloaddition reactions according to previously reported studies.29 Vibrational frequencies were computed at the same level to verify that optimized structures are energy minima or transition structures and to evaluate zero-point vibrational energies (ZPVE) and thermal corrections at 298 K. Intrinsic reaction coordinate (IRC) calculations were performed to verify the expected connections of the first-order saddle points with the local minima found on the potential energy surface.30 Finally, a comparison between M06-2X/6-311+G(d,p), M06-2X/Def2TZVPP and ωB97xD/Def2TZVPP methods was performed to validate the results and all the methods yield very similar results (see ESI† for details). Distortion/interaction analysis were done at the M06-2X/6-311+G(d,p) level. To this end, the optimized transition state structure is separated into two fragments (distorted BCN and distorted heterocyclic structure), followed by single-point energy calculations on each fragment obtained from the IRC analysis. The energy difference between the distorted fragment and corresponding optimized structure is defined as the distortion energy. The difference between the activation energy and the total distortion energy is the interaction energy. ASD was performed using Gaussian 16 by single point calculations of separate reactants from the IRC analysis. The optimized structures indicated and shown in this study are illustrated using the CYLview program.31 The presence of noncovalent interactions was evaluated using a reduced density gradient (RDG) analysis32 performed with Multiwfn33 and visualized by visual molecular dynamics (VMD).34 Orbital overlap was calculated with Multiwfn.35Author contributions
JE and STL conceived and planned the research. WH and KW performed the kinetic study and analysed the data, and then discussed the results with STL. JE performed DFT calculation and analysed the data. JE and STL wrote the manuscript. All authors contributed to the manuscript and have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computational resources from the Servei d'Informàtica de la Universidad de Valencia (SIUV) are gratefully acknowledged for providing access to supercomputing resources.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- (a) L. M. Gaetke and C. K. Chow, Toxicology, 2003, 189, 147–163 CrossRef CAS PubMed; (b) M. C. Linder, Mutat. Res., 2012, 733, 83–91 CrossRef CAS PubMed.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- (a) J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 100, 9116–9121 Search PubMed; (b) S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed; J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delf, Top. Curr. Chem., 2016, 374, 16 Search PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- (a) A. Borrmann, O. Fatunsin, J. Dommerholt, A. M. Jonker, D. W. P. M. Löwik, J. C. M. van Hest and F. L. van Delft, Bioconjugate Chem., 2015, 26, 257–261 CrossRef CAS PubMed; (b) R. Sen, J. Escorihuela, F. L. van Delft and H. Zuilhof, Angew. Chem., Int. Ed., 2017, 56, 3299–3303 CrossRef CAS PubMed; (c) D. Gahtory, R. Sen, A. R. Kuzmyn, J. Escorihuela and H. Zuilhof, Angew. Chem., 2018, 130, 10118–10122 CrossRef PubMed; (d) B. Albada, J. F. Keijzer, H. Zuilhof and F. L. van Delft, Chem. Rev., 2021, 121, 7032–7058 CrossRef CAS PubMed.
- (a) M. F. Debets, S. S. van Berkel, J. Dommerholt, A. J. Dirks, F. P. J. T. Rutjes and F. L. van Delf, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed; (b) J. J. Bruins, D. Blanco-Ania, V. van der Doef, F. L. van Delft and B. Albada, Chem. Commun., 2018, 54, 7338–7341 RSC.
- J. Escorihuela, A. T. M. Marcelis and H. Zuilhof, Adv. Mater. Interfaces, 2015, 2, 1500135 CrossRef.
- K. Li, D. Fong, E. Meichsner and A. Adronov, Chem. – Eur. J., 2021, 27, 5057–5073 CrossRef CAS PubMed.
- (a) F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121–8133 CrossRef CAS PubMed; (b) Y. Liang, J. L. Mackey, S. A. Lopez, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 17904–17907 CrossRef CAS PubMed; (c) J. Dommerholt, O. van Rooijen, A. Borrmann, C. Fonseca Guerra, F. M. Bickelhaupt and F. L. van Delft, Nat. Commun., 2014, 5, 5378 CrossRef CAS PubMed.
- (a) Y.-F. Yang, Y. Liang, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 1660–1667 CrossRef CAS PubMed; (b) B. J. Levandowski, D. Svatunek, B. Sohr, H. Mikula and K. N. Houk, J. Am. Chem. Soc., 2019, 141, 2224–2227 CrossRef CAS PubMed; (c) P. García-Aznar and J. Escorihuela, Org. Biomol. Chem., 2022, 20, 6400–6412 RSC.
- (a) J. Escorihuela, A. Das, W. J. E. Looijen, F. L. van Delft, A. J. A. Aquino, H. Lischka and H. Zuilhof, J. Org. Chem., 2018, 83, 244–252 CrossRef CAS PubMed; (b) J. Escorihuela, W. J. E. Looijen, X. Wang, A. J. A. Aquino, H. Lischka and H. Zuilhof, J. Org. Chem., 2020, 85, 13557–13566 CrossRef CAS PubMed; (c) J. A. M. Damen, J. Escorihuela, H. Zuilhof, F. L. van Delft and B. Albada, Chem. – Eur. J., 2023, 29, e202300231 CrossRef CAS PubMed.
- (a) H. Wu and N. K. Devaraj, Acc. Chem. Res., 2018, 51, 1249–1259 CrossRef CAS PubMed; (b) S. Mayer and K. Lang, Synthesis, 2017, 830–848 CAS; (c) P. Kumar, O. Zainul, F. M. Camarda, T. Jiang, J. A. Mannone, W. Huang and S. T. Laughlin, Org. Lett., 2019, 21, 3721–3725 CrossRef CAS PubMed; (d) A. Jemas, Y. Xie, J. E. Pigga, J. L. Caplan, C. W. am Ende and J. M. Fox, J. Am. Chem. Soc., 2022, 144, 1647–1662 CrossRef CAS PubMed; (e) L. Liu, D. Zhang, M. Johnson and N. K. Devaraj, Nat. Chem., 2022, 14, 1078–1085 CrossRef CAS PubMed.
- K. Kranjc and M. Kočevar, New J. Chem., 2005, 29, 1027–1034 RSC.
- Q. Cui, T. W. Pan, M. Shieh, S. S. Kelly, S. Xu, W.-J. Qian and M. Xian, Org. Lett., 2022, 24, 7334–7338 CrossRef CAS PubMed.
- W. Huang, N. Gunawardhana, Y. Zhang, J. Escorihuela and S. Laughlin, Chem. – Eur. J., 2024, 30, e202303465 CrossRef CAS PubMed.
- (a) R. Jasiński, M. Kwiatkowska, V. Sharnin and A. Barański, Monatsh. Chem., 2013, 144, 327–335 CrossRef PubMed; (b) E. Dresler, A. Wróblewska and R. Jasiński, Molecules, 2023, 28, 5289 CrossRef CAS PubMed; (c) D. Loco, I. Chataigner, J. Piquemal and R. Spezia, ChemPhysChem, 2022, 23, e202200349 CrossRef CAS PubMed.
- (a) J. Cioslowski, J. Sauer, J. Hetzenegger, T. Karcher and T. Hierstetter, J. Am. Chem. Soc., 1993, 115, 1353–1359 CrossRef CAS; (b) D. Svatunek, M. Wilkovitsch, L. Hartmann, K. N. Houk and H. Mikula, J. Am. Chem. Soc., 2022, 144, 8171–8177 CrossRef CAS PubMed.
- (a) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (b) F. Liu, Y. Liang and K. N. Houk, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed.
- (a) A. M. Sarotti, Org. Biomol. Chem., 2014, 12, 187–199 RSC; (b) W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC; (c) P. Vermeeren, T. Hansen, P. Jannsen, M. Swart, T. A. Hamlin and F. M. Bickelhaupt, Chem. – Eur. J., 2020, 26, 15538–15548 CrossRef CAS PubMed; (d) N. Grimblat and A. M. Sarotti, Org. Biomol. Chem., 2020, 18, 1104 RSC; (e) S. Portela and I. Fernández, Eur. J. Org. Chem., 2021, 6102–6110 CrossRef CAS; (f) T. Hansen, P. Vermeeren, L. de Jong, F. M. Bickelhaupt and T. A. Hamlin, J. Org. Chem., 2022, 87, 8892–8901 CrossRef CAS PubMed; (g) T. Hansen, J. C. Roozee, F. M. Bickelhaupt and T. A. Hamlin, J. Org. Chem., 2022, 87, 1805–1813 CrossRef CAS PubMed; (h) J. Escorihuela and L. M. Wolf, Organometallics, 2022, 41, 2525–2534 CrossRef CAS PubMed; (i) J. Escorihuela, Synthesis, 2023, 1139–1149 CrossRef CAS.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- (a) M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC; (b) A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076–2085 CrossRef CAS PubMed.
- M. Martínek, L. Filipová, J. Galeta, L. Ludvíková and P. Klán, Org. Lett., 2016, 18, 4892–4895 CrossRef PubMed.
- T. A. Hamlin, D. Svatunek, S. Yu, L. Ridder, I. Infante, L. Visscher and F. M. Bickelhaupt, Eur. J. Org. Chem., 2019, 378–386 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- L. G. Gagliardi, C. B. Castells, C. Ràfols, M. Rosés and E. Bosch, J. Chem. Eng. Data, 2007, 52, 1103–1107 CrossRef CAS.
- (a) R. S. Paton, J. L. Mackey, W. H. Kim, J. H. Lee, S. J. Danishefsky and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 9335–9340 CrossRef CAS PubMed; (b) F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef CAS PubMed.
- (a) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS; (b) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- Legault, C. Y. CYLview, 1.0b, Université de Sherbrooke, Canada, 2009, https://www.cylview.org Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Lu and Q. Chen, J. Phys. Chem. A, 2023, 127, 7023–7035 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01263a |
| This journal is © The Royal Society of Chemistry 2024 |