
gem-Difluoroallene (DFA): an emerging versatile synthetic precursor to enable diverse fluorinated molecules
Kota
Sathish
 ,
Swati
Jain
,
Naveen
Sihag
and
M. Ramu
Yadav
*
,
Swati
Jain
,
Naveen
Sihag
and
M. Ramu
Yadav
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi, 110016, India. E-mail: ramuyadav@chemistry.iitd.ac.in; Tel: (+91) 11-2659-1506
First published on 18th September 2024
Abstract
Organofluorine compounds are increasingly found in diverse fields, such as pharmaceuticals, agrochemicals, and materials science. gem-Difluoroallenes, which have gem-difluoro alkenes and allenes-like properties, offer a distinct and adaptable platform for novel synthetic transformations. Their distinctiveness is highlighted by various strategies, where the gem-difluoro group's presence plays a pivotal role in successful reactions. This review article presents a comprehensive overview of the latest progress in utilizing gem-difluoroallenes for selective additions, defluorination, as well as cycloaddition and cyclization reactions. We also discuss the limitations and future directions in this area, inspiring further exploration and innovation.
 Kota Sathish | Kota Sathish was born in Warangal, India. He completed MSc in Chemistry from Kakatiya University Warangal in 2014 and received a Ph.D. Degree in 2021 from National Institute of Technology, Warangal under the supervision of Prof. D. Kashinath. Subsequently, he worked as a Senior Research Associate at Curia Global and Aragen Life Sciences, Hyderabad. Currently, he is an ANRF (DST-SERB)-National Postdoctoral fellow in Dr M. Ramu Yadav's research group in the Department of Chemistry, Indian Institute of Technology, Delhi. His research mainly focuses on gem-difluoroallenes (DFA) and their synthetic transformations in organic synthesis. |
 Swati Jain | Swati Jain obtained her bachelor's and master's degrees in chemistry with honors from Miranda House, University of Delhi, in 2018 and 2020, respectively. In 2020, she joined Dr M. Ramu Yadav's research group at Indian Institute of Technology, Delhi to pursue her doctoral studies. Her research interests center on designing and developing novel catalytic systems for C–C bond formation, with a particular focus on artificial metalloenzymes. |
 Naveen Sihag | Naveen Sihag received his bachelor's degree from Govt. College Hisar, Haryana, India. Subsequently, he pursued his master's education in chemistry from Chaudhary Devi Lal University, Sirsa. Since, December 2020, he is pursuing his doctoral studies with Dr M. Ramu Yadav at Indian Institute of Technology, Delhi. His research work involves the development of enantioselective trifluoromethylated oxindoles from CF3–acryl amides. |
 M. Ramu Yadav | M. Ramu Yadav received his master's degree in chemistry from Kakatiya University in 2008. He obtained a Ph.D. degree in 2014, working with Prof. Akhila K. Sahoo at the School of Chemistry, University of Hyderabad. Subsequently, he pursued post-doctoral studies with Prof. Yoshiaki Nakao, Kyoto University, Japan, and later with Prof. Ryan Altman, The University of Kansas, USA. Since 2019, he has been working as an assistant professor at the Department of Chemistry, IIT-Delhi. The current research interest of his group is the development of novel fluorination strategies and asymmetric catalysis. |
1. Introduction
Allenes, characterized by two cumulated π-bonds, have sp2-hybridized terminal carbons and an sp-hybridized central carbon, leading to a unique reactivity distinct from alkenes and alkynes.1 These allenes are valuable for various organic transformations: carbometallation, electrophilic addition, nucleophilic addition, and radical addition reactions. Consequently, they have been recognized as valuable intermediates for synthesizing complex molecular targets, revealing novel applications in natural product synthesis, pharmaceutical chemistry, and materials science.2 Therefore, the development of novel functionalized allenes and their potential synthetic transformations plays a vital role in synthetic organic and medicinal chemistry.In recent years, there has been a substantial increase in interest in fluorinated organic compounds due to their widespread applications in pharmaceuticals and agrochemicals.3 This increasing fascination comes from the distinct physicochemical and biological properties arising from the presence of fluorine (Fig. 1a). In particular, gem-dilfuoroalkene is considered as a bio-isostere of carbonyl and amide group4 and undergoes preferential nucleophilic attack at α-position due to the two σ-withdrawing fluorine atoms which enabled wide synthetic transformations such as addition/elimination, coupling reaction, hydro-defluorination, etc. Consequently, the fusion of conventional allene and gem-difluoroalkene generates gem-difluoroallene (DFA), a unique molecular template with distinct characteristics, that demonstrates potential as a special fluorinated framework for new synthetic reactions.
 | ||
| Fig. 1 (a) CF2-containing drug molecules and (b) background of DFA. | ||
gem-Difluoroallenes (DFA) feature orthogonal cumulative double bonds, similar to normal allenes, but are distinguished by two vinylic fluorine atoms at the α-position, which confers a unique reactivity profile5 (Fig. 1b). The regioselectivity of DFA is determined by the heightened positive electrostatic charge at the α-position, indicative of its electrophilic nature, as well as the augmented LUMO coefficient at the γ-position, due to the presence of two electron-withdrawing fluorine substituents.6 Consequently, Dolbier Jr., Lentz, and Ichikawa groups have independently made substantial advancements in both synthesis and exploring the reactivity of DFA.
Dolbier Jr., in particular, was an early pioneer in recognizing the potential of DFA, showcasing various cycloaddition reactions, including regioselective [2 + 4] and [2 + 3] cycloadditions involving the non-fluorinated double bonds, and [2 + 2] cycloadditions featuring fluorinated double bonds.7 Subsequently, Lentz and colleagues have built upon the hydrometalation of DFA, resulting in the generation of (2,2-difluorovinyl)metals and [1-(difluoromethyl)vinyl]metals through reactions with nonfluorinated and fluorinated double bonds, respectively.8 The recent research by the Ichikawa group has been centred on investigating the mechanisms and applications of electrophilic cyclization and nucleophilic reactions of DFA,9 leading to the development of a variety of intriguing organic transformation reactions, including cycloadditions, nucleophilic substitutions, and addition reactions.
1.1. Scope and urgency of the review
Despite recent advancements, the full potential of DFA in novel synthetic organic transformations remains underexplored. To gain insight into the current significance of this area, it is crucial to highlight the evolution and scope of DFA reactivity. This article aims to provide a comprehensive overview of the significant recent progress in DFA, a topic that has not been thoroughly reviewed until now. The present review is primarily focused on the reactivity and selectivity at the α- and γ-sites, fluorination and defluorination, cyclization, and ring construction strategies utilizing DFA, while excluding early cycloaddition reactions. We believe that this article will serve as a valuable reference for further advancements in this field.2. Synthesis and reactions of gem-difluoroallenes (DFA)
2.1. Synthetic approaches to DFA
Despite the significance of DFA as versatile synthons in numerous organic transformations, very few methods have been reported for their synthesis. Although they were first identified in 1957, challenges in scaling up DFA preparation have hindered extensive research in this domain. In 1982, more than two decades later, Dolbier Jr. devised the first synthetic route for the preparation of DFA in high yields utilizing 3,3,3-trifluoropropene. The reaction of 2-bromo-3,3,3-trifluoropropene with alkyl lithium reagent enables unsaturated organofluorine lithium compound, that, upon heating produces 1,1-difluoroallenes by β-F elimination i.e. lithium fluoride (Scheme 1a).7a Later in 1988, Xu's group utilized trifluoromethylketone and 2,4,6 triisopropylbenzenesulfonylhydrazones to synthesize several 1,1-difluoroallenes via Shapiro reaction (Scheme 1b).10 Hammond et al. described the synthesis of DFA using Cu mediated difluoropropargyl bromides with Grignard reagent through magnesium organocuprate SN2′ substitution (Scheme 1c).11 Afterwards, the Ichikawa group presented two highly effective methods for synthesizing DFA (Scheme 1d). In the first approach, 1-bromo-2,2-difluorovinyllithium, obtained from 1,1-dibromo-2,2-difluoroethylene and n-butyllithium through lithium-bromide exchange, reacted with carbonyl compounds, followed by acetylation to yield 2-bromo-3,3-difluoroallylic acetates. The resulting bromo acetates, in the presence of BuLi, undergo elimination to produce DFA.12a In the second-generation synthesis, 1,1-difluoroallenes were synthesized using readily available 1,1,1-trifluoro-2-iodoethane (CF3CH2I) as a difluorovinylation agent. In this scenario, 3,3-difluoro-2-iodoallylic acetates were generated from carbonyl compounds by treating them with 1,1,1-trifluoro-2-iodoethane and lithium diisopropylamide, followed by acetylation. These acetates then readily undergo elimination with zinc metal to yield 1,1-difluoroallenes.12b,c Currently, the second method is more widely utilized for the preparation of various DFA and has spurred further research in this area. | ||
| Scheme 1 Synthetic methods for the preparation of DFA. | ||
In the following sections, the DFA reactivity and regioselectivity which is governed by higher positive electrostatic charge at α-position (electrophilic character) and higher LUMO coefficient at γ-position due to the presence of two electron-withdrawing fluorine substituents, will be discussed.
2.2. γ-Selective addition reaction of DFA
Inspired by these precedents, in 1995, Huang's group demonstrated the feasibility of γ-selective thiol addition of DFA under basic conditions (Scheme 2).14 They successfully obtained γ,γ-difluoroallyl sulphides (3) via nucleophilic addition reaction of aryl- or alkyl-thiols (2) to DFA (1) in the presence of a base such as KOH or Et3N. The γ-selective addition product formation is attributed to the lowering of energy of LUMO in the non-substituted double bond of DFA. When aryl-thiols (2) reacted with DFA (1) in the presence of a catalytic amount of KOH, they yielded γ,γ-difluoroallyl sulfides (3) in good yields (81–94%). However, o-aminophenylthiol required a stoichiometric amount of Et3N to produce the corresponding product. On the other hand, alkyl-thiols like benzylthiol, in the presence of a stoichiometric amount of KOH, exhibited a lower yield (31%) of γ,γ-difluoroallyl sulfide and a significant amount of defluorinated compound (61%). It is worth noting that despite this significant finding, the scope of the reaction is limited to aryl thiols.
 | ||
| Scheme 2 γ-Selective nucleophilic addition of thiols to DFA. | ||
After a prolonged period of inactivity, in 2022, Shi and coworkers advanced on Huang's methodology and accomplished a highly regioselective addition of thiols (2) to monosubstituted DFA (1) at the β or γ-position by tuning the reaction condition by utilizing bidentate N and P ligands and rhodium catalysts (Scheme 3).15 A range of nitrogen and phosphorus ligands were tested to optimize both the regioselectivity and enantioselectivity of the reaction. Application of the powerful combination of rhodium catalyst and ligand (L1) enabled the asymmetric γ-addition of thiols with DFA in good to excellent yields. Both aromatic and aliphatic thiols generated the corresponding allylic sulfanes (4) with moderate to good yields (56–96%) and enantiomeric excess of up to 99%. In addition to this, preliminary investigations with other nucleophiles, such as anilines, resulted in γ-addition with DFA in 69–88% yields using ligand (L3), showcasing exceptional regioselectivity and enantioselectivity. Concurrently, the combination of rhodium catalyst with ligand (L2) selectively facilitated the β-addition of thiols (2) to DFA (1) with 58–77% yields. The developed system showed impressive chemoselectivity. It allowed thiols, even with potentially competitive nucleophilic sites such as 4-mercaptophenol, 4-aminobenzenethiol, 4-mercaptobenzoic acid, and L-cysteine, to exclusively produce β- and γ-thiol addition products. The addition of OH, NH2, or COOH groups was not observed under the reaction conditions. The resulting products underwent significant synthetic transformations, including hydrogenation, oxidation, and stereoselective hydrodefluorination. The regio- and stereoselectivity of the reaction has been determined by conducting isotopic labeling experiments and DFT calculations. Initially, the Rh(I) species undergoes oxidative addition with thiols (2) to form the Rh(III) species A. The Rh(III) intermediate A, with a P-ligand (L1), tends to bind more to the electron-rich, non-fluorinated double bond of DFA (1) to give B, which is 1.1 kcal mol−1 lower in energy than binding to fluorinated double bond. The insertion process through a four-membered rhodacyclic transition state creates two potential η3-allylrhodium complexes, and the Rh-Complex C (R-) is more stable by 4.0 kcal mol−1 than the (S-) transition state. Reductive elimination from C produces the γ,γ-difluoroallyl sulfane (4) with excellent enantioselectivity. However, for N-ligated Rh(III) intermediate A, interaction with the electron-poor α,β-difluoroallene double bond B′ is 8.5 kcal mol−1 more stable than the interaction with non-fluorinated double bond. Subsequently, insertion occurs through a more stable Z-rhodacyclic transition state C′, followed by reductive elimination, leading to the desired β-selective addition product (5).
 | ||
| Scheme 3 Regio- and enantioselective nucleophilic addition to DFA. | ||
In 2021, Xu and colleagues disclosed a copper catalyzed methodology for the efficient protoborylation and protosilylation of DFA (1), leading to the formation of gem-difluoroallyl-boronates/silanes (7) (Scheme 4).17 The unusual selectivity is achieved by reacting Nu–Cu (borylcopper or silylcopper) with mono- or di-substituted DFA. The methodology has a wide range of applications and accommodates various functional groups. The resulting gem-difluoroallyl boronates, though unstable, can be oxidized to gem-difluoro-allyl alcohols using H2O2. Moreover, a gram-scale reaction also proceeded with good to excellent yields, indicating the potential utility of this protocol.
 | ||
| Scheme 4 Copper catalyzed γ-addition of DFA. | ||
Mechanistically, the reaction starts with the formation of intermediate A (Cu-R3), which is driven by the interaction between Lewis acidic Bpin moiety and alkoxide of Cu-OR in a σ-bond metathesis step. The resulting [Cu]-R3 species A then reacts with the DFA (1) to form intermediate B, which subsequently abstracts a proton from ROH to produce the desired product (7) and species C. Finally, the [Cu]-R3 species A is regenerated from the [Cu]-OR through σ-bond metathesis. The selective addition of [Cu]-R3 to the more distant double bond from the fluorine atom is due to the LUMO of the DFA being positioned on the remote double bond.
At the same time, Feng et al. introduced a similar type of Cu/NHC-catalyzed approach for protoborylation and protosilylation of DFA (1), leading to multi-functionalized tertiary and secondary gem-difluoroallylboronates (7) and gem-difluoroallylsilanes (10) (Scheme 5).18 Consequently, these gem-difluoroallylboronates (7) can be converted into carbinol derivatives (8) via oxidation of H2O2/NaOH.
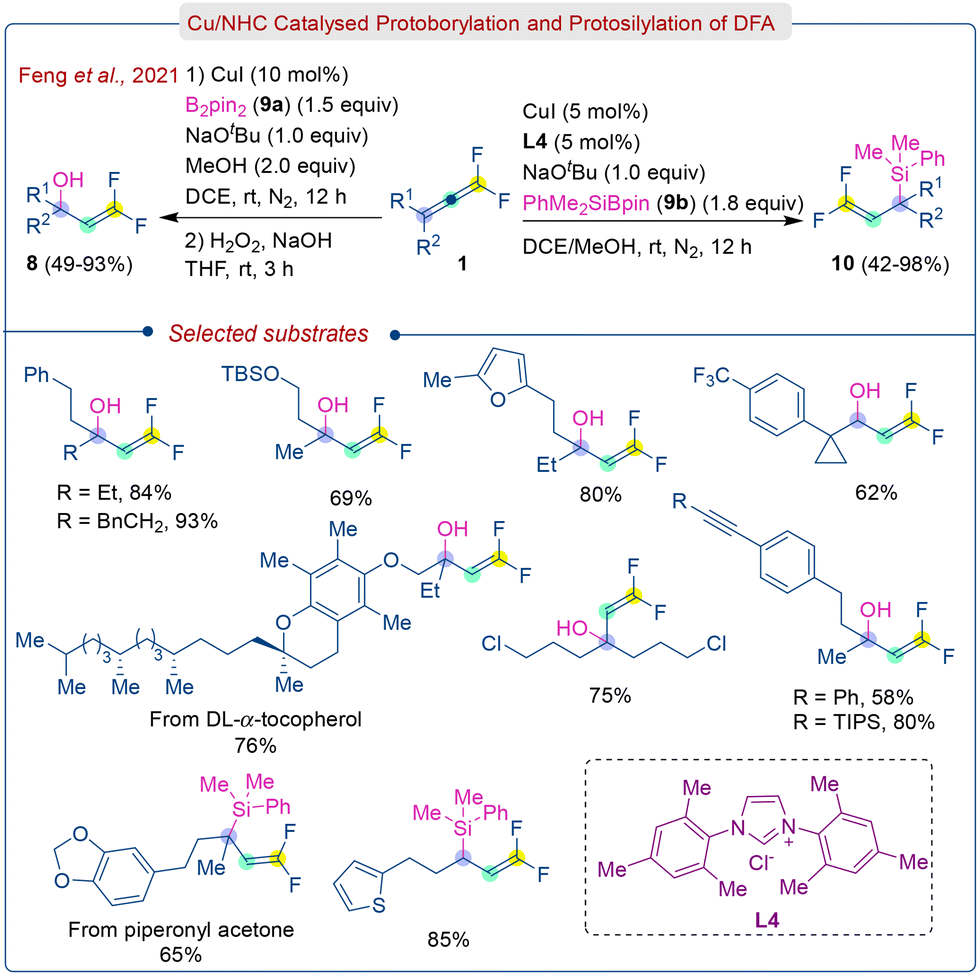 | ||
| Scheme 5 γ-Addition protoboration/protosilylation of DFA. | ||
 | ||
| Scheme 6 γ-Selective asymmetric hydroamination of DFA with anilines. | ||
The authors proposed a mechanism based on isotopic labeling experiments and computational studies. The control experiments using aniline-d7, PPTS (pyridin-1-ium-4-methylbenzenesulfonate)-d1, and EtOD, resulted in deuterium incorporation at the β-position. This supports the exclusive formation of the π-allyl Rh complex via the hydrometallation pathway and H/D exchange between aniline, PPTS, and EtOH. The reaction commences with the in situ generation of cationic Rhodium complex A, which then undergoes oxidative addition with N–H bond to form Rh–H species B. Subsequently, complex B inserts into fluorinated double bond of DFA (1) to give intermediate C-Z which is energetically favored due to the α-cation stabilization by fluorine. Finally, the product (12) is formed via reductive elimination following an inner-sphere nucleophilic attack pathway through a gem-difluoro π-allyl rhodium intermediate (viaD-S and D-TS-S).
In 2023, Gao and colleagues utilized sulfoximines (13) to facilitate the hydroamination of allenes and a limited number of DFA (1) (Scheme 7).21 The reaction occurred in the presence of [Rh(cod)Cl]2 (2 mol%), BF3·Et2O as additive and rac-BINAP as the ligand, yielding the desired products (14). However, this method presents notable drawbacks: a restricted substrate scope with DFA and moderate product yields. Furthermore, the investigation of an asymmetric variant using the ligand (L1) resulted in low yield (27%) and poor enantioselectivity (30% ee). Mechanistic studies suggest that the initial activation step involves the complexation between BF3·Et2O and the sulfonimidoyl imine group. This coordination complex A, facilitates the formation of the active Rh(III)–H species, which then efficiently couples with allene or DFA to yield the respective products.
 | ||
| Scheme 7 Rodium catalyzed γ-selective N-allylation of sulfoximines to DFA. | ||
 | ||
| Scheme 8 Palladium catalyzed γ-selective hydrophosphination of DFA. | ||
The deuterium labeling experiment using D-PPh2 (90% D) furnished the allylic phosphine product with 40% deuteriation incorporation at the β-position. Based on the investigation, the proposed reaction mechanism is depicted in Scheme 8. Initially, ligand exchange between L6PdX2 and HPPh2 forms the Pd(II)–PPh2 species A. This species then reacts with DFA (1), generating intermediate B, with the R group of allene pointing away from the chiral ligand and the metal centre for minimized steric repulsions. Next, the enantiodetermining and regioselective migratory insertion of PPh2 into the γ-carbon of DFA occurs via transition state C to yield intermediate D. Protonolysis of the Pd–vinyl bond or σ-bond metathesis then produces the desired product and regenerates the Pd(II) catalyst.
Based on orbital-control and DFT calculations, in 2012, Ichikawa originally reported γ-selective addition to DFA (1) using alkylcopper reagents (Scheme 9).6a They explored various organometallic species to study the reactivity of DFA at γ-position. Use of MeLi and EtMgBr resulted in complex mixtures, while Et2Zn led to the formation of monofluoroallene (MFA) through an undesired α-attack. On the other hand, the combination of EtMgBr (17) and CuBr·SMe2 successfully facilitated γ-addition via in situ generated ethylcopper as a soft nucleophile. The resulting difluorovinylcopper intermediates served as coupling partners and reacted with various electrophiles to yield functionalized gem-difluoroalkenes (18). Treatment of the copper intermediate with NIS, NBS, or NCS yielded the corresponding 2-halogenated 1,1-difluoroalkenes in 75–84% yield, which are effective partners for Suzuki coupling. Additionally, difluorovinylcopper intermediates can integrate with palladium-catalyzed cross-coupling, leading to a three-component coupling sequence that produces 2,2-disubstituted 1,1-difluoroalkenes in moderate to good yields (53–90%). However, its broad applicability is limited by the need for stoichiometric quantities of copper reagents and highly reactive Grignard reagents.
 | ||
| Scheme 9 Three-component synthesis of disubstituted gem-difluoroalkenes. | ||
A decade later, in 2022, Feng et al. disclosed Rh(III) catalyzed hydroarylation of DFA (1) with N-methoxy benzamides (19) under mild conditions for the synthesis of quaternary benzylic center-tethered gem-difluoro alkenes (20) (Scheme 10).25 The protocol offers several advantages, including atom economy, room-temperature operation, C–H functionalization, excellent regioselectivity, and the production of diverse gem-difluoroalkene products. Control experiments revealed that the Rh(III) catalyst and additive NaOAc (or CsOPiv) is essential for the success of the reaction. Interestingly, the replacement of NaOAc with HOAc or K2CO3 and Rh(III) with Ir(III) or Ru(III) catalyst did not produce the hydroarylation product. The methodology showed good scope with various N-methoxybenzamides and DFA.
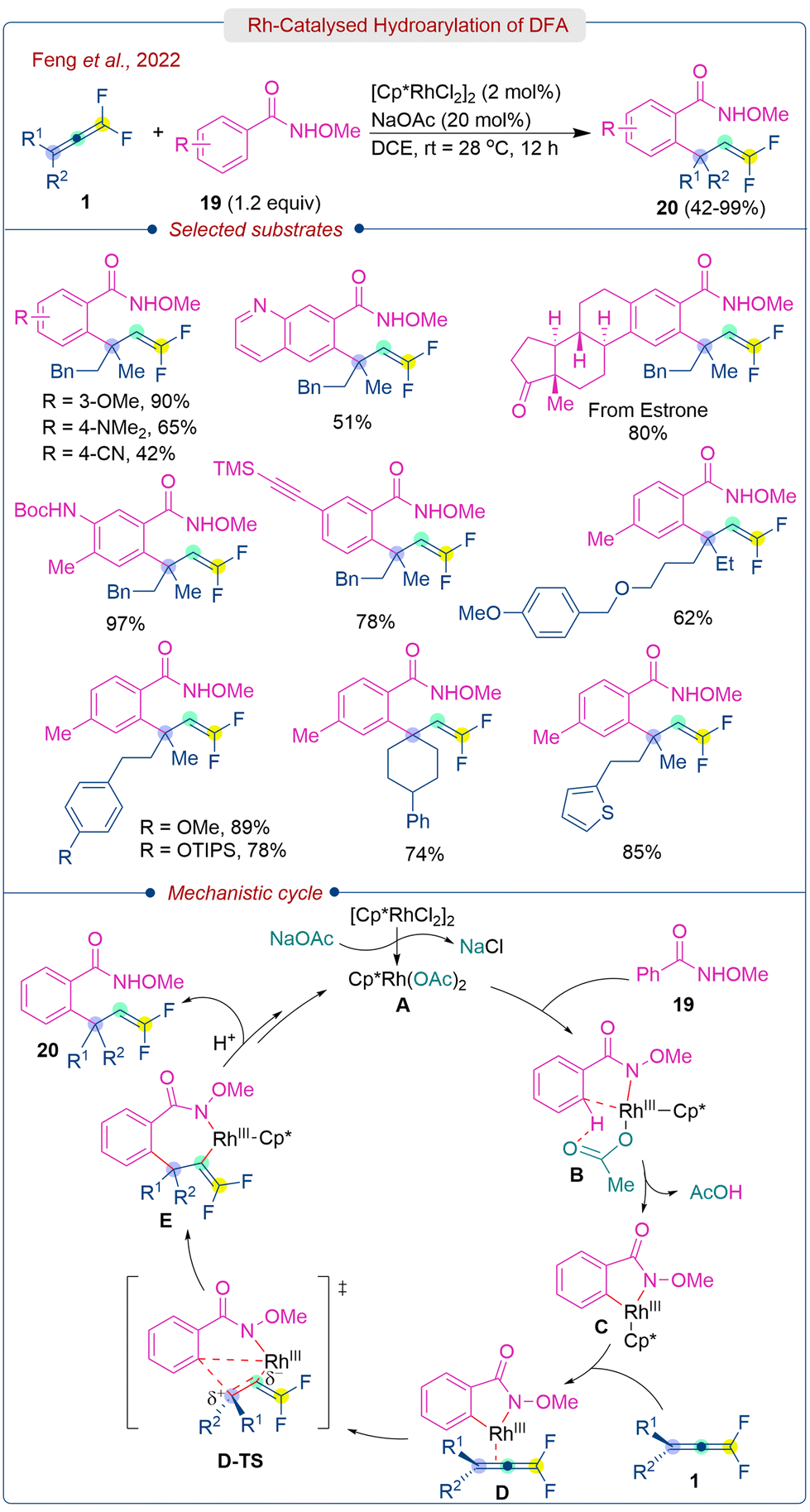 | ||
| Scheme 10 Regioselective hydroarylation of DFA. | ||
A combination of mechanistic experiments and DFT calculations has been employed to explain the regioselectivity of the reaction. Intermolecular kinetic isotope effect experiments suggested that C–H bond cleavage is involved in the turnover-limiting step. Additionally, conducting the reaction with 5 equiv. of D2O led to 24% of deuterium incorporation at the olefinic position of the product, indicating the involvement of proto-demetallation in releasing the product from the rhodium center. The mechanistic pathway involves Rh(III) catalyzed C–H activation to form an electron-deficient rhodacyclic intermediate C, which coordinates preferentially to the electron-rich dialkyl substituted π bond of DFA (1) to form D. Subsequent insertion viaD-TS generates the quaternary centre E, which is followed by two successive proto-demetallations to results in the formation of product (20) and liberation of free catalyst A.
More recently, Cheng et al. reported copper-catalysed regioselective proto-arylation of DFA (1) with aryl and alkenyl boronic esters (21), leading to a variety of gem-difluoroalkenes (22) in high yields (Scheme 11).26 In this case, the CuCl accompanied by bidentate phosphine ligand (L7) and KOtBu as the base exhibited good catalytic efficiency in iPrOH (proton source) and 1,4-dioxane at 45 °C. The substrate scope of this process was quite broad, including benzyl ether, terminal alkene, thioether, acetal, alcohol, heterocycles, as well as various electron-donating and electron-withdrawing groups on arylboronic esters. Noticeably, however, no reaction occurred for tertiary alkyl difluoroallene substrate, due to steric hindrance. The use of chiral bidentate phosphine ligand (L8) enabled chiral gem-difluoroalkenes bearing γ-carbon stereogenic centres in good yield with up to 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 er. The reaction commences with the formation of LCu-R2A through the transmetalation between LCuCl and aryl boronic ester (21) in the presence of a base (KOtBu). Subsequently, L-CuAr A undergoes γ-selective addition of the aryl group to DFA (1), followed by the protonation of intermediate B with isopropanol (iPrOH), yielding the final product (22) and regenerating the catalyst.
:11 er. The reaction commences with the formation of LCu-R2A through the transmetalation between LCuCl and aryl boronic ester (21) in the presence of a base (KOtBu). Subsequently, L-CuAr A undergoes γ-selective addition of the aryl group to DFA (1), followed by the protonation of intermediate B with isopropanol (iPrOH), yielding the final product (22) and regenerating the catalyst.
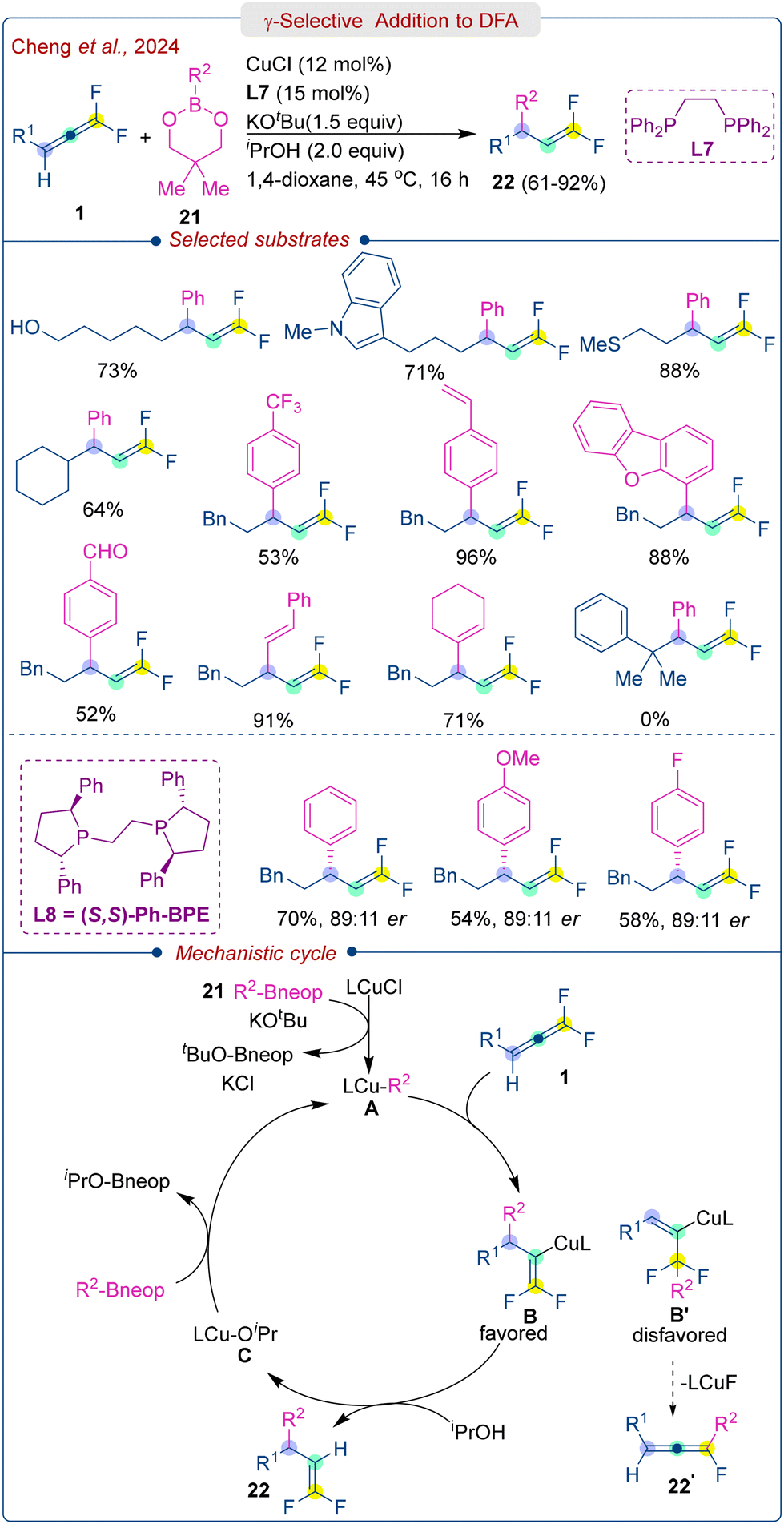 | ||
| Scheme 11 γ-Selective copper-catalyzed protoarylation of DFA. | ||
2.3. α-Selective addition on DFA
 | ||
| Scheme 12 α- and γ-Selective addition of heteroatom to DFA. | ||
In 2020, Shi et al. revealed a Pd/DPEPhos-catalyzed, stereoselective method for synthesizing trifluoromethylated alkenes (27), utilizing DFA (1) with aryl halides (26) and AgF as the fluorine source (Scheme 13).29 Alternative phosphine ligands such as Xantphos, DPPF, or RuPhos, and fluoride sources like KF or CsF, were ineffective for this transformation or resulted in low product yields. The transformation exhibited good tolerance to various aryl iodides, including electron-donating and electron-withdrawing groups, as well as aryl/alkyl substituents on DFA, delivering products in moderate to good yields and excellent E-selectivity. However, disubstituted DFA afforded a mixture of desired products in a 68% yield with poor selectivity (E/Z = 1/1). The radical process in the mechanism was ruled out using radical scavengers (TEMPO, BHT and 1,1-diphenyl ethylene). Based on 19F-NMR studies, the proposed mechanism begins with the oxidative addition of aryl iodide to form an aryl-palladium species A. Simultaneously, the DFA (1) reacts with AgF, yielding predominantly E-configured trifluoromethylated vinyl silver intermediate B. The coordination of silver or palladium atom with MeCN solvent increases the E/Z ratio of the compound due to steric repulsion between coordinated MeCN and Vinyl-H. Subsequent transmetalation of B with intermediate A generates palladium complex C, which undergoes reductive elimination, yielding the trifluoromethylated alkene (27) and regenerating the palladium catalyst.
 | ||
| Scheme 13 Pd-Catalysed stereoselective fluoroarylation of DFA. | ||
Afterward, Lou and colleagues demonstrated the synthesis of Z-selective α-trifluoromethyl arylenes (28) using DFA (1) and aryl iodides (26) as starting materials. The use of bulky monophosphine ligand (XPhos) is the key to the success of this transformation, which produces a Z-selective product,30 complementing Shi’s report on E-selectivity (Scheme 14).29 A wide variety of DFA and aryl iodides participated in this transformation to give the corresponding Z-configured α-trifluoromethyl arylenes in good yields (64–93%) and excellent stereoselectivity (Z/E > 97/3). Nevertheless, the presence of a quaternary alkyl group is necessary for DFA to enable high stereoselectivity.
 | ||
| Scheme 14 Pd-Catalyzed fluoroarylation of DFA. | ||
In the oxidative addition step, the Pd species reacts with an aryl iodide to form an aryl-palladium(II) iodide species A. Then, selective nucleophilic insertion of AgF into DFA (1) produces an E-vinyl silver species B. This species undergoes transmetallation to yield an E-vinylpalladium(II) species C through halide abstraction. The formation of E-selective vinyl metal species B and C, rather than the Z-isomeric configurations, is caused by steric repulsion between the ligand and the substrate, which defines the underlying source of the reaction stereoselectivity. Finally, reductive elimination results in the formation of the desired product with Z-selectivity (28) and the regeneration of the Pd catalyst.
Very recently, Feng's research group demonstrated an interesting strategy of a visible-light-promoted gold-catalyzed system for the fluorarylation of DFA (1) with xanthone as a photocatalyst.31 They used Et3N·3HF as the cost-effective fluoride source and aryl diazonium salt (29) as the aryl precursor to provide E-selective trifluoromethylated alkene products (30) under the optimized conditions. The use of other nucleophilic fluoride sources like CsF, nBu4NF, and pyridine·xHF proved to be ineffective for this transformation. Control experiments showed that the gold catalyst was crucial for the success of the reaction.
Several mechanistic studies have been conducted to elucidate the mechanism of the reaction. Both Au(I) and Au(III) catalysts can activate the DFA, however, Au(III) proved superior due to its strong Lewis acidic character. The addition of AgBF4 and PPh3 was found to be beneficial, indicating the involvement of π-acidic cationic Au(III) species is more potent.
The proposed Au-catalyzed mechanism is illustrated in Scheme 15. Initially, aryldiazonium salt (29) undergoes oxidative addition with the Au(I) catalyst A, facilitated by photoredox catalysis, resulting in the formation of the cationic Ar–Au(III) species B. Subsequently, DFA (1) coordinates with the Au(III) center, forming intermediate C. The α-carbon of intermediate C is more electron-deficient due to stabilization by two fluorine atoms, making it susceptible to nucleophilic attack by fluoride, leading to the formation of trifluoromethyl vinyl Au(III) complex D. Finally, reductive elimination of D yields the final product (30) and regenerates the catalyst A. The 1,3-allylic strain in intermediate D accounts for the E-stereoselectivity in the transformation.
 | ||
| Scheme 15 Au-Catalyzed fluoroarylation of DFA. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCF2) structure under certain reaction conditions.
CCF2) structure under certain reaction conditions.
The first defluoroalkylation process to produce monofluoroallenes (33) was reported by the Hammond group (Scheme 16). This was done during the preparation of DFA using gem-difluoropropargyl bromides (31) with alkyl Grignard reagents (32) in the presence of copper salts.11a However, the use of a substantial excess of copper salts and only two substrates restrict the scope of the reaction, highlight the need for further investigation.
 | ||
| Scheme 16 Defluoroarylation by magnesium organocuprate reagents. | ||
In 2023, Wu et al. developed a Cu/bipyridine catalytic system for the defluoroarylation of DFA (1) in the presence of KOtBu base (Scheme 17).33 This system employs cost-effective and readily available aryl boronic esters (34) to prepare arylated monofluoroallenes (35) via a selective β-fluorine elimination process. The reaction condition demonstrated a broad scope with respect to the DFA and boronic esters. However, pyridine-based boronic esters were found to be unsuccessful in this reaction. It was noted that trisubstituted or aryl-tethered tetrasubstituted DFA was inactive in the transformation due to the instability of the expected products. The preliminary experiment of asymmetric reaction showed that the use of a chiral phosphine-oxazoline ligand (L10) afforded 87% yield with low enantioselectivity (32% ee). The possible reaction pathway is depicted in Scheme 16. The reaction begins with the formation of the LCu-Ar species B through the transmetalation of a copper catalyst A with an aryl boronic ester (34) in the presence of a base. This step is followed by a regioselective addition to the difluorinated double bond of DFA (1), leading to the creation of alkenyl copper intermediates C. These intermediates subsequently undergo facile β-F elimination, producing the final product (35) and regenerating the catalyst A.
 | ||
| Scheme 17 Cu-Catalyzed defluoroarylation of DFA. | ||
Later, same group further explored defluoroalkylation or defluoroarylation of DFA (1) using commercially available Grignard reagents (32) under Cu-catalysis (Scheme 18).34 The study found that CuCl worked best, while CuBr, CuTC, and CuCl2 were less efficient and PdCl2 or NiCl2 completely stopped the reaction. The amount of Grignard reagent also plays a critical role in this transformation, an excess can lead to further conversion of the monofluoroallene to non-fluorinated allene. The protocol was applied to various DFAs bearing electron donation and withdrawing groups, heteroaryls, cycloalkyls and long alkyl chain molecules. Gratifyingly, even tetrasubstituted DFA successfully yielded the persubstituted monofluoroallene product in 81% yield. Alkyl Grignard reagents were well reacted, but aryl Grignard reagents afforded low to moderate efficiency (36–61%) at low temperatures (−60 °C) due to the instability of the resulting aryl allenyl fluorides. Initial efforts to implement an asymmetric reaction using chiral carbene ligand (L11) resulted in moderate enantioselectivity (36% ee) with a lower yield (24%). The amount of Grignard reagent and the substituents on DFA were found to play crucial roles. The reaction mechanism was shown to proceed via an organocopper intermediate followed by rapid β-fluorine elimination to produce monofluoroallenes (36), similar to the mechanism in Scheme 17.
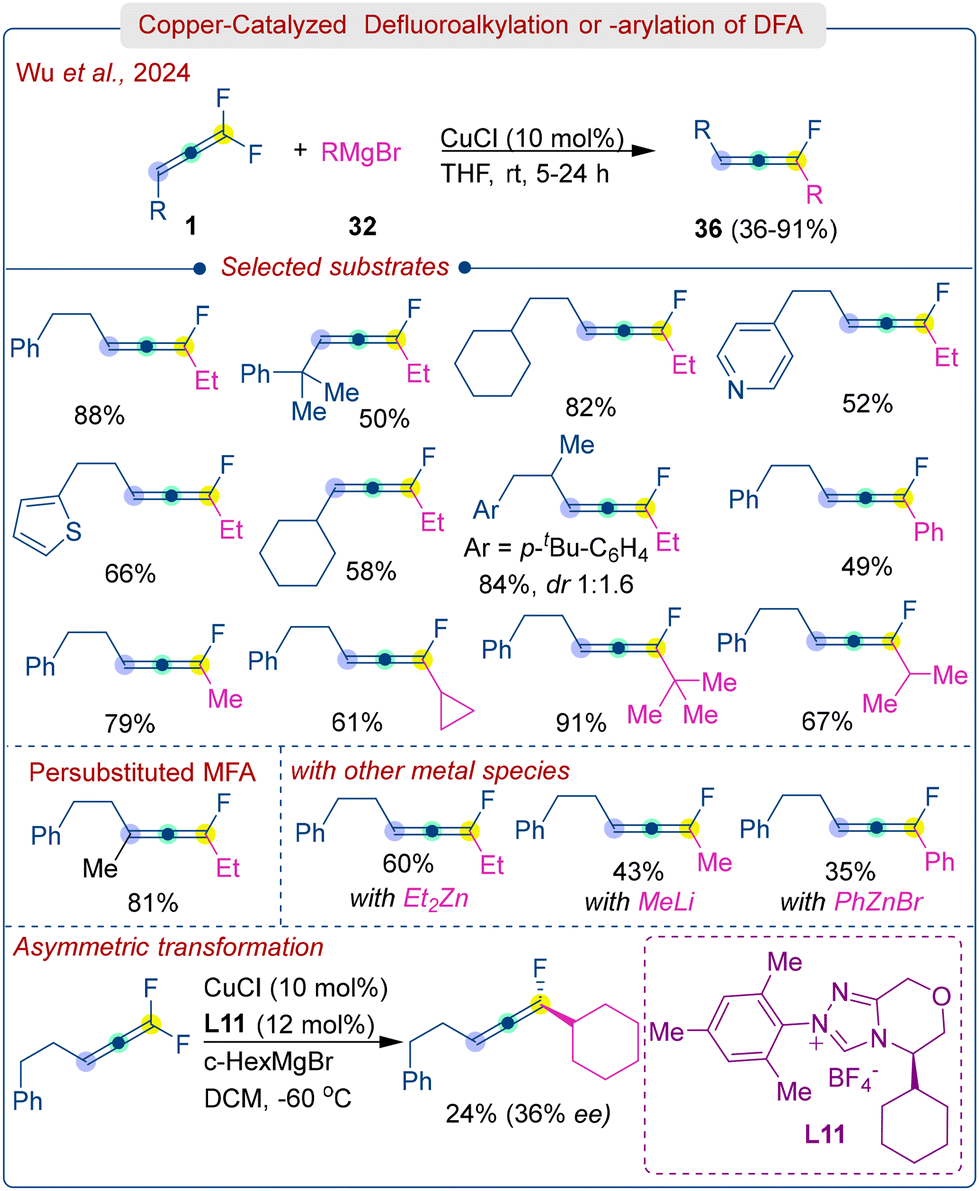 | ||
| Scheme 18 Copper-catalyzed stereoselective defluoroalkylation or arylation of DFA. | ||
2.4. Cycloaddition reactions of DFA
gem-Difluoroallenes are significantly more reactive in cycloaddition reactions similar to allenes and difluoroalkenes. In the early stages, Dolbier Jr. extensively studied various cycloaddition reactions of DFA with nitrile oxides and proceeded in a β,γ-selective fashion under metal-free conditions.7e,g,35 In contrast, Ichikawa et al. developed an Au-catalyzed α,β-selective [2 + 3] cycloaddition of DFA (1) with nitrile oxides and imine oxides (37) to produce ring fluorinated isoxazole derivatives (Scheme 19).36 The use of an AuCl catalyst as a Lewis acid results in preferred regioselectivity by generating localised difluoroallylic cations from DFA, which facilitates directly α-selective bond-forming reactions. The β,γ-selective cycloadduct can be obtained without any catalyst or with InBr3, AuCl(PPh3), PdCl2, and PtCl2 catalysts. However, AuCl results in the production of an α,β-selective cycloadduct with an E-configuration. A wide variety of aromatic-substituted DFA participated in the reaction, producing the corresponding (E)-4-alkylidene-5,5-difluoroisoxazolines (38) in good to excellent yields. Subsequently, the 5,5-difluoroisoxazolines could be efficiently aromatized to 5-fluoroisoxazole through 1,4-elimination of HF using Me3SiOEt/n-Bu4NF. The proposed mechanism is illustrated in Scheme 19. The reaction initiates with the generation of the aurated difluoroallylic cations Avia activation of DFA (1) with AuCl. Subsequently, the oxygen atom of the nitrile oxides (37) attacks the electrophilic α-position of A, producing B followed by ring closure to furnish the α,β-[2 + 3] cycloaddition products (38). | ||
| Scheme 19 Au-catalyzed cycloaddition reaction of nitrile oxides and imine oxides with DFA. | ||
2.5. Ring construction strategy of DFA
Pin-point fluorinated polycyclic aromatic hydrocarbons (F-PAHs) hold great potential as fundamental building blocks for the development of advanced materials, including semiconductors and pharmaceuticals, because of the unique options of fluorine substituents.37 The high electronegativity of fluorine lowers the energy level of their HOMO, thereby enhancing the resistance of PAHs to aerial oxidation. Furthermore, these compounds exhibit high solubility in polar organic solvents, making them suitable for printable organic electronics. Therefore, the development of efficient methods for F-PAHs is highly desirable.Ichikawa et al. was the first research group to achieve the In(III)-catalyzed domino synthesis of F-PAHs (39) with DFA (1) containing an aryl group and a cyclopentene moiety (Scheme 20).9a The methodology involves generating allylic CF2 cation through the π-coordination interaction between the metal catalyst and the non-fluorinated, electron-rich double bond of DFA, followed by cyclization. The reaction conditions were compatible with DFA bearing various substituents and produced the F-PAHs (39) in good to excellent yields (66–98%). Treatment of NBS or NIS with (1) aided in the halogenation of the C–In bond, resulting in ortho-fluoro(halo)phenanthrenes through sequential addition of DDQ, which were efficiently participated in the Suzuki-Miyuara coupling reactions. The proposed mechanism is illustrated in Scheme 19. Initially, InBr3 reacts with difluoroallene (1) to form an allylic CF2 cation A, which undergoes Friedel–Crafts-type cyclization at the positively charged CF2 carbon, resulting in the organoindium intermediate B. This intermediate further undergoes protonolysis of the C–In bond and elimination of fluoride, producing the second cationic intermediate C, followed by 1,2-migration and ring expansion to yield fluorodihydrophenanthrenes D. Afterwards, one-pot dehydrogenation with DDQ affords the desired F-PAHs (39) in good yields.
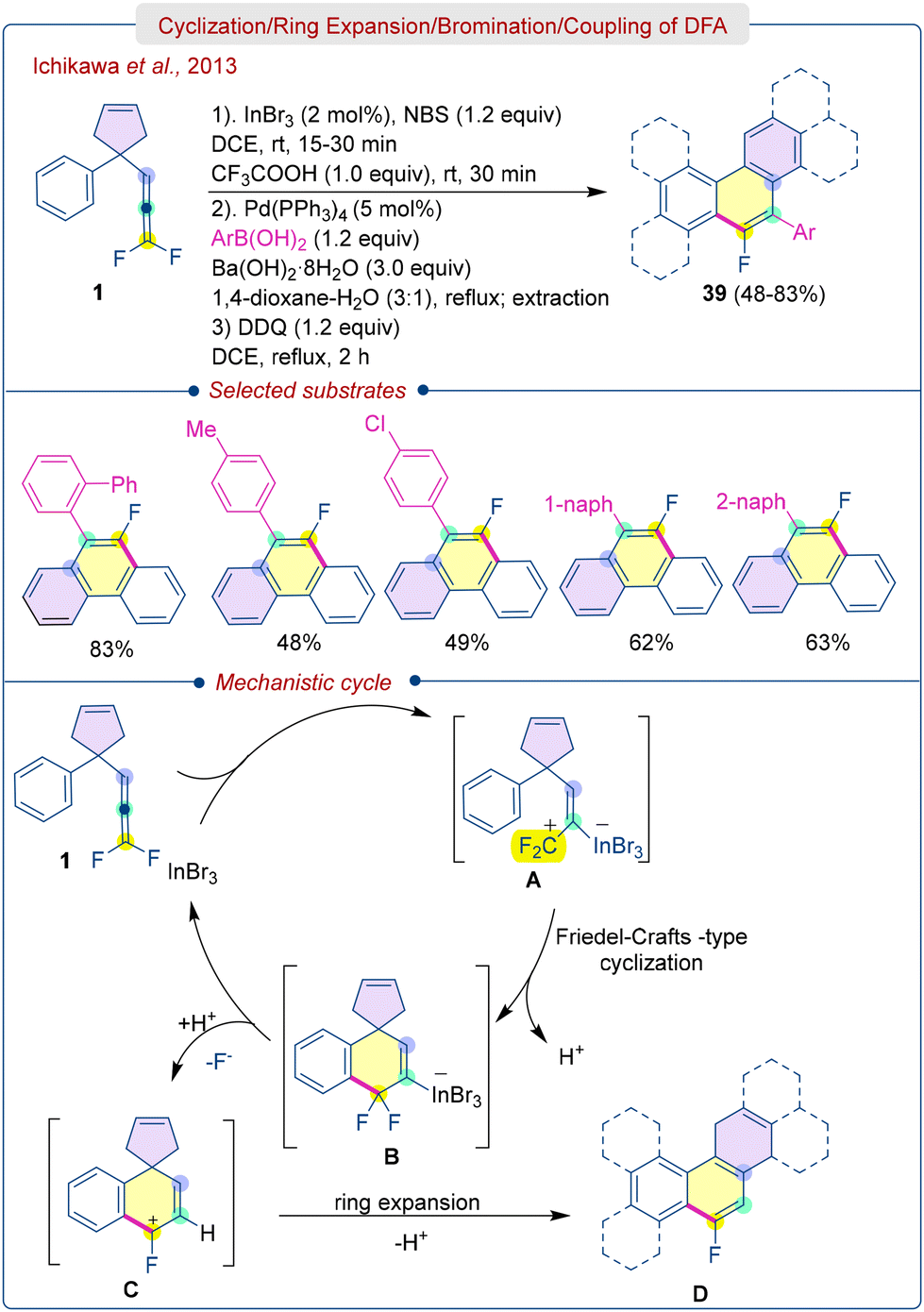 | ||
| Scheme 20 In(III)-catalyzed synthesis of o-arylated F-phenanthrenes from DFA. | ||
Later, in 2014, the same group developed a method for the regioselective synthesis of 1-fluoronaphthalenes (40) through the InBr3-catalyzed cyclization of DFA (1) (Scheme 21).38 DFT calculations indicate that a positive charge is localized on the CF2 carbon, which is stabilized by hyperconjugation of the C–In σ bond due to the presence of fluorine substituents. Dialkylated gem-difluoroallenes afforded fluorodimethylnaphthalenes via a cyclization/1,2-migration/deprotonation sequence with 56–85% yields, while monoalkylated gem-difluoroallenes underwent deprotonation followed by cyclization to furnish the corresponding fluoro naphthalene products with 65–83% yields. A plausible mechanism for the transformation is shown in Scheme 21. The mechanistic studies indicate that In(III) bromide forms π-coordination A with non-fluorinated double bond of DFA (1) to generate allylic CF2 cation B, which undergoes a Friedel–Crafts-type cyclization to give intermediate C. Protonolysis of the C–In bond, followed by In(III)-promoted elimination of a fluoride ion generates the second cationic intermediate D. Successive 1,2-migration and deprotonation finally produce 1-fluoronaphthalenes (40).
 | ||
| Scheme 21 Synthesis of 1-fluoronaphthalenes via InBr3-catalyzed ring construction of DFA. | ||
Next, in 2017, same group also synthesized non-vic-F2-PAHs (42 and 43) (Fluorinated Polycyclic Aromatic Hydrocarbons) using the In(III)-catalyzed domino ring construction in a tandem fashion (Scheme 22).39 The bis(gem-difluoroallenyl) compound derived from p-xylene (1), was transformed into 6,7-difluoropicene (42), producing a single isomer with a 75% yield. The in situ generated dihydrophenanthrene intermediate (41) undergoes a second cyclization reaction at the α-position of the newly formed naphthalene substructure, followed by one-pot dehydrogenation using DDQ. Similarly, bis(gem-difluoroallene), prepared from m-xylene (1′), underwent tandem cyclization, resulting in F2-dibenzanthracene (43) as a single isomer with a 76% yield. In this case, severe steric repulsion likely encourages cyclization at the electronically less reactive β-position of the naphthalene substructure.
 | ||
| Scheme 22 In(III)-catalyzed synthesis of F2-PAHs from bis(1,1-difluoroallenyl) compounds. | ||
Additionally, the Ichikawa group expanded their In(III)-catalyzed domino reaction for synthesizing higher-order F-PAHs, sequentially starting from fluoroarenes (44) (Scheme 23).40 To accomplish this, a range of fluorinated arenes were reacted with aliphatic nitriles via nucleophilic aromatic substitution (SNAr) under microwave irradiation, resulting in the formation of alkyl-substituted naphthalene derivatives (45). The nitrile group was subsequently converted into a formyl group through partial reduction with DIBAL-H, yielding the corresponding aldehydes (46). These aldehydes were readily transformed into DFA (1) via difluorovinylidenation. Finally, the In(III)-catalyzed cyclization of DFA (1) produced benzene ring-extended F-PAHs (47). Further repeating the cycles on the obtained fluorophenanthrenes (47) produced pinpoint fluorinated chrysenes (48) and subsequently, pinpoint fluorinated picenes (49). Both terminal and internal fluoroarenes underwent benzene ring extension to furnish higher-order pinpoint fluorinated PAHs.
 | ||
| Scheme 23 Benzene ring extension of fluoroarenes. | ||
Continuing their exploration of the ring construction reactivity of DFA, Ichikawa, in 2019, developed palladium-catalyzed regioselective insertion of o-bromophenyl-bearing gem-difluoroallenes (1), resulting in the formation of (difluoromethyl)naphthalenes (50) (Scheme 24).9b The difluromethyl group is regarded as a bioisostere of the hydroxyl group, acting as a hydrogen bond donor while also exhibiting hydrophobic properties. This methodology facilitates the C–C bond formation at the β-position, distinguishing it from previously explored α- and γ-additions. Moderate yields (43–83%) were achieved with electron-withdrawing and electron-donating groups attached to the benzene ring of DFA. Disubstituted difluoroallenes, on the other hand, yielded lower amounts of naphthalene product (36%). The reaction sequence begins with the formation of aryl palladium(II) bromides Avia oxidative addition of the Pd catalyst with bromoallenes (1). These intermediates then undergo regioselective insertion, resulting in more stable π-allylpalladium(II) intermediates B and forming a C–C bond at the β-position relative to the fluorine substituents. The removal of β-hydrogen from σ-allylpalladium(II) intermediates C leads to the formation of cyclic 1,1-difluoro-1,3-dienes D, which isomerizes to generate the final product (50).
 | ||
| Scheme 24 Synthesis of (difluoromethyl)naphthalenes via β-selective Pd-catalysed intramolecular insertion of o-bromophenyl-bearing 1,1-difluoroallenes. | ||
Furthermore, in 2020, Ichikawa and group effectively utilized 1,1-difluoroallenes that contain cyclopentene moiety (1) and an aryl group for the synthesis of three-ringed ortho-fluoro(halo)phenanthrenes, four-ringed ortho-fluoro(halo)tetraphenes, ortho-fluoro(halo)chrysenes and fluoro[4]helicenes (52), which were obtained via NBS or NIS trapped intermediate of InBr3 ring construction methodology as aryne precursors (Scheme 25).9c The metalation process involving either n-BuLi or Me2(TMP)ZnLi with these precursors (52) results in the subsequent elimination of a fluoride ion, leading to the formation of the corresponding arynes (53). Diels–Alder reactions of these arynes with substituted isobenzofurans (54) furnished the corresponding fully aromatized benzotriphenylenes (56) in good yields via reductive aromatization in SnCl2/HBr. In addition, the Scholl reaction, an oxidative aryl/aryl coupling, has also been demonstrated to synthesize “half HBCs” (hexabenzocoronenes) (57) from these benzotriphenylenes (56).
 | ||
| Scheme 25 Synthesis of half-HBCs via π-extended arynes generated from o-brominated F-PAH. | ||
2.6. Miscellaneous
CF2-enriched acyclic, cyclic, bicyclic and heterocyclic systems facilitate the development of enzyme-activated irreversible inhibitors and other bioactive compounds. Replacing the methylene group with CF2 can prevent metabolic oxidation and increase the reactivity of nearby groups because of the strong electron-withdrawing effect of fluorine.24b,41 Compounds containing vinyl CF2, such as allenes, dienes, or dienynes, are well-suited for building carbocycles using annulation and cycloaddition strategies. However, there are few reported syntheses of these molecules.In 2001, G. B. Hammond's group developed an efficient method for the synthesis of 1,1-difluoro-2-substituted-1,3-dienes (59) and 1,1-difluoro-2-ethylidene-1,3-enynes (59) from homoallenyl bromides (1) using a palladium catalyst (Scheme 26).42 The homoallenyl bromides (1) were prepared through the protection of the hydroxyl group of homoallenyl alcohol, followed by nucleophilic substitution with LiBr. Treatment of homoallenyl bromides with zinc in DMF led to the formation of organozinc allenes A, which quickly isomerized into organozinc dienes B. Under Pd catalysis, the intermediates cross-coupled with aryl halides to form 1,1-difluoro-2-aryl-1,3-dienes (59), with good to excellent yields. Homoallyl bromides produced similar products in good yields through a palladium-catalyzed Suzuki reaction with aryl boronic acids. Additionally, conjugated enynes were obtained in nearly quantitative yields by cross-coupling of homoallenyl bromides with terminal alkynes under Sonogashira reaction conditions.
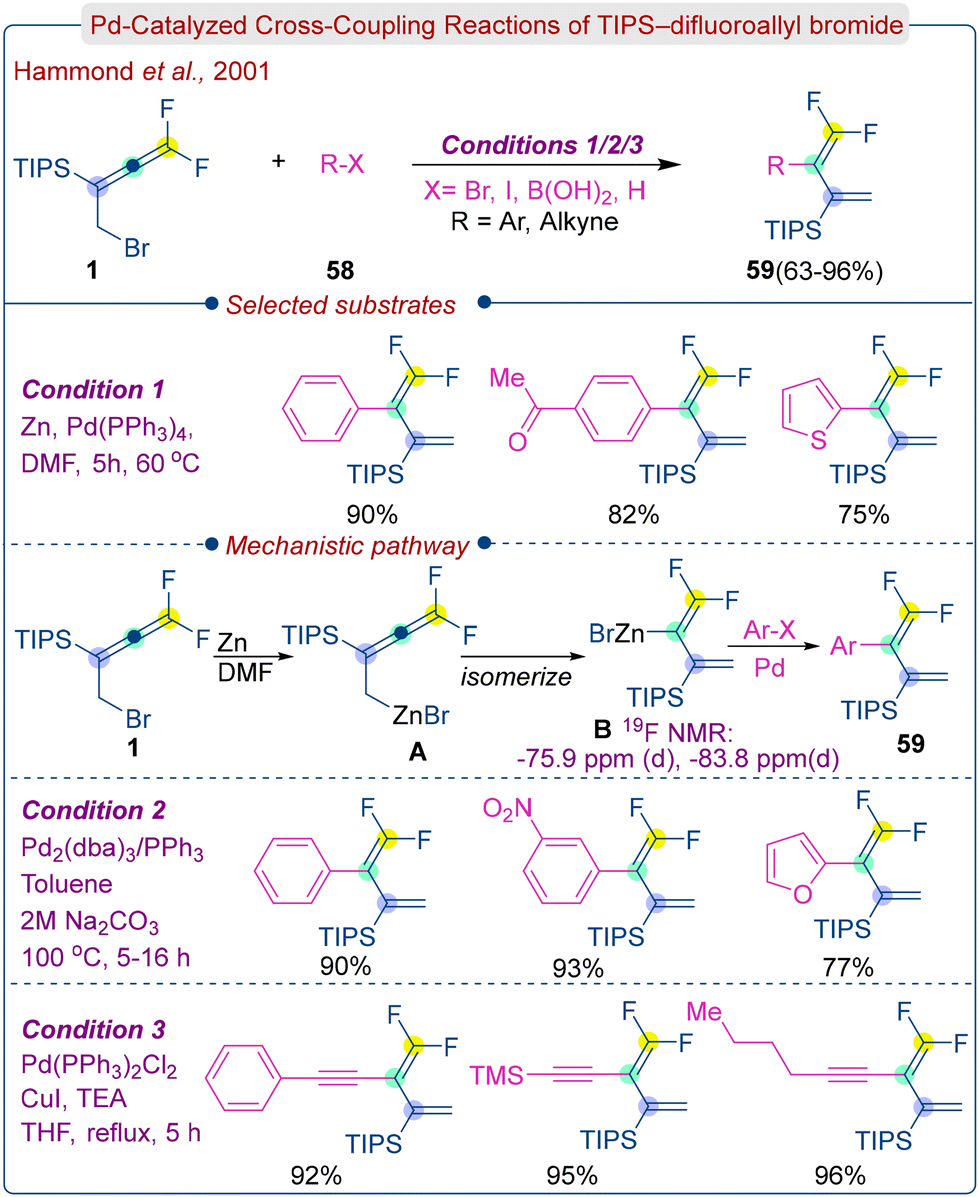 | ||
| Scheme 26 Pd-Catalyzed cross-coupling reactions of TIPS–difluoroallyl bromides. | ||
Later, the same group reported the synthesis of substituted gem-difluoroallenyl amines (60) from difluoroallenyl alcohols (1) under Mitsunobu condition (Scheme 27).43 The initial gem-difluoroallenyl alcohol was synthesized by reacting TIPS-difluoropropargyl bromide with indium in a H2O/THF mixture, followed by overnight sonication at room temperature with an excess of aqueous formaldehyde. The standard Mitsunobu redox system using DEAD (diethyl azodicarboxylate)/PPh3 resulted in low yields but using DIAD (diisopropyl azodicarboxylate)/PPh3 at low temperature produced gem-difluoroallenyl amines (60) in good yields. Unsubstituted N-tosyl propargyl amines yielded the product in high yields, but alkyl substituents on the triple bond or simple imide substrates resulted in reduced yields (32–44%). The protected sulfonamide required higher amounts of reagent quantities [PPh3 (2 equiv.) and DEAD (4 equiv.)], resulting in the desired gem-difluoroallenyl amine in up to 61% yield.
 | ||
| Scheme 27 Mitsunobu reaction of TIPS–difluoroallyl alcohols. | ||
Further, the Hammond group explored cycloaddition reactions using DFA (1). Initially, they planned to carry out Pauson–Khand type [2 + 2 + 1] cycloadditions of alkyne-tethered DFA derivative with Mo(CO)6. However, they observed that [2 + 2] cycloaddition occurred instead, leading to the formation of CF2-containing fused cyclobutenes (61), a previously unattained structure (Scheme 28).44 When various functionalized DFAs were subjected to the reaction conditions, bicyclo- and heterobicyclo-gem-difluorocyclobutenes were obtained with yields ranging from 72% to 92%. The cleavage of TIPS from (1) using TBAF resulted in the formation of CF2H-diyne with an 86% yield. Control experiments indicated that the reaction proceeded through a [2 + 2] cycloaddition, with the molybdenum metallocycle undergoing reductive elimination rather than CO insertion. The rate of reductive elimination was found to be significantly faster than the rate of carbonylation.
 | ||
| Scheme 28 Mo-Catalyzed intramolecular [2 + 2] cycloaddition reaction of functionalized gem-difluoroallenes. | ||
3. Conclusions
This review delves into the synthetic methods and reactivity profile of DFA. DFAs, with their significant potential, serve as powerful synthons, opening doors to functionalized intermediates and a diverse range of fluorine-containing carbocycles and heterocycles. The electronegative vinylic fluorine substituents give these DFAs unique physicochemical properties and diverse reactivity, thanks to their multiple reactive centres (α-, β-, and γ-positions). The regioselectivity of DFA is determined by the higher positive electrostatic charge at α-position and the higher LUMO coefficient at γ-position. This makes the mode of activation and the nature of the nucleophile crucial factors in reaction design. Generally, hard nucleophiles primarily attack the α-position, while soft nucleophiles prefer γ-addition. Consequently, several research groups contributed to developing interesting reactions such as regio-selective nucleophilic additions, defluorination, cycloaddition, and cyclization reactions. Among all these reactions, one notable observation is the presence of the gem-difluoro group of DFA, which is crucial for developed reactions. Despite these promising results, the full potential of DFA chemistry remains underexplored and requires further development. For instance, compared to α and γ selective addition reactions, the β-addition reactions are scarce; hence, future studies should be focused in this direction. Further, the enantioselective defluoroarylation of DFA for mono-fluoroallenes has been very limited in success, and the defluoroalkylation strategy should go beyond the Grignard reagent. Developing more general catalytic systems with diverse reacting coupling partners should be investigated, as current strategies mainly use boron reagents. Further, these DFAs should be integrated with other substrates through C–H activation strategies and cyclization approaches to generate functional fluorinated molecules of interest.In summary, this review comprehensively discusses the typical transformations, reaction mechanisms, substrate range, and subsequent applications of DFA chemistry. We believe that researchers will find this review article to be an invaluable resource, helping in addressing challenges and navigating potential future advancements. These advancements could lead to the creation of value-added substances, such as new drugs or materials with unique properties, and the introduction of innovative applications in materials, such as advanced electronics or high-performance polymers.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Indian Institute of Technology (IIT)–Delhi, India, and DST–SERB (CRG/2023/006833) for providing research facilities and financial support. Dr K. Sathish thanks SERB for NPDF (PDF/2023/001841), S. J. thanks UGC, India and N. S. thanks CSIR, India, for the research fellowship.References
- (a) S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef PubMed; (b) S. Kitagaki, F. Inagaki and C. Mukai, Chem. Soc. Rev., 2014, 43, 2956–2978 RSC; (c) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067–3126 CrossRef CAS PubMed; (d) N. Krause and A. S. K. Hashmi, Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004 CrossRef.
- (a) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed; (b) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (b) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (d) H. Kawai and N. Shibata, Chem. Rec., 2014, 14, 1024–1040 CrossRef CAS PubMed; (e) R. R. Nair, E. W. Seo, S. Hong, K. O. Jung and D. Kim, ACS Appl. Bio Mater., 2023, 6, 4081–4099 CrossRef CAS PubMed.
- C. Leriche, X. He, C. W. T. Chang and H. W. Liu, J. Am. Chem. Soc., 2003, 125, 6348–6349 CrossRef CAS PubMed.
- K. Fuchibe and J. Ichikawa, Science of Synthesis: Knowledge Updates 2014/2, Georg Thieme Verlag KG, Stuttgart, 2014, pp. 217–231 Search PubMed.
- (a) K. Fuchibe, M. Ueda, M. Yokota and J. Ichikawa, Chem. Lett., 2012, 41, 1619–1621 CrossRef CAS; (b) K. Fuchibe, M. Abe, M. Sasaki and J. Ichikawa, J. Fluorine Chem., 2020, 232, 109452 CrossRef CAS.
- (a) W. R. Dolbier Jr., C. R. Burkholder and C. A. Piedrahita, J. Fluorine Chem., 1982, 20, 637–647 CrossRef; (b) W. R. Dolbier Jr., C. R. Burkholder and W. R. Winchester, J. Org. Chem., 1984, 49, 1518–1522 CrossRef; (c) W. R. Dolbier Jr. and G. E. Wicks, J. Am. Chem. Soc., 1985, 107, 3626–3631 CrossRef; (d) W. R. Dolbier Jr., C. R. Burkholder, G. E. Wicks, G. J. Palenik and M. Gawron, J. Am. Chem. Soc., 1985, 107, 7183–7184 CrossRef; (e) W. R. Dolbier Jr. and C. R. Burkholder, Isr. J. Chem., 1985, 26, 115–119 CrossRef; (f) W. R. Dolbier Jr., G. E. Wicks and C. R. Burkholder, J. Org. Chem., 1987, 52, 2196–2201 CrossRef; (g) W. R. Dolbier Jr., Acc. Chem. Res., 1991, 24, 63–69 CrossRef.
- (a) M. F. Kühnel and D. Lentz, Dalton Trans., 2009, 24, 4747–4755 RSC; (b) M. F. Kuehnel, T. Schlöder, S. Riedel, B. Nieto-Ortega, F. J. Ramirez, J. T. L. Navarrete, J. Casado and D. Lentz, Angew. Chem., Int. Ed., 2012, 51, 2218–2220 CrossRef CAS PubMed; (c) D. Lentz and S. Willemsen, Organometallics, 1999, 18, 3962–3964 CrossRef CAS; (d) D. Lentz, N. Nickelt and S. Willemsen, Chem. – Eur. J., 2002, 8, 1205–1217 CrossRef CAS PubMed; (e) D. Lentz, J. Fluorine Chem., 2004, 125, 853–861 CrossRef CAS.
- (a) K. Fuchibe, Y. Mayumi, N. Zhao, S. Watanabe, M. Yokota and J. Ichikawa, Angew. Chem., Int. Ed., 2013, 52, 7825–7828 CrossRef CAS PubMed; (b) K. Fuchibe, S. Watanabe, G. Takao and J. Ichikawa, Org. Biomol. Chem., 2019, 17, 5047–5054 RSC; (c) K. Fuchibe, M. Abe, H. Idate and J. Ichikawa, Chem. – Asian J., 2020, 15, 1384–1392 CrossRef CAS PubMed; (d) K. Fuchibe, M. Abe, K. Oh and J. Ichikawa, Org. Synth., 2016, 93, 352–366 CrossRef CAS.
- G. Shi and Y. Xu, J. Fluorine Chem., 1989, 44, 161–166 CrossRef CAS.
- (a) M. Mae, J. A. Hong, B. Xu and G. B. Hammond, Org. Lett., 2006, 8, 479–482 CrossRef CAS PubMed; (b) B. Xu and G. B. Hammond, Chem. – Eur. J., 2008, 14, 10029–10035 CrossRef CAS PubMed.
- (a) M. Yokota, K. Fuchibe, M. Ueda, Y. Mayumi and J. Ichikawa, Org. Lett., 2009, 11, 3994–3997 CrossRef CAS PubMed; (b) K. Oh, K. Fuchibe and J. Ichikawa, Synthesis, 2011, 881–886 CAS; (c) K. Fuchibe, M. Abe, K. Oh and J. Ichikawa, Org. Synth., 2016, 93, 352–366 CrossRef CAS.
- (a) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed; (b) K. A. Scott and J. Njardarson, Top. Curr. Chem., 2018, 376, 1–34 CrossRef CAS PubMed.
- Y. Y. Xu, F. Q. Jin and W. Y. Huang, J. Fluorine Chem., 1995, 70, 5–6 CrossRef CAS.
- X. Han, M. Wang, Y. Liang, Y. Zhao and Z. Shi, Nat. Synth., 2022, 1, 227–234 CrossRef.
- (a) T. Hiyama and M. Oestreich, Organosilicon Chemistry: Novel Approaches and Reactions, Wiley-VCH, Weinheim, Germany, 2019 CrossRef; (b) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC.
- C. C. Shan, K. Y. Dai, M. Zhao and Y. H. Xu, Eur. J. Org. Chem., 2021, 4054–4058 CrossRef CAS.
- C. Q. Wang, Y. Li and C. Feng, Cell Rep. Phys. Sci., 2021, 2, 100461–100474 CrossRef CAS.
- (a) M. Kolb, J. Barth, J. G. Heydt and M. J. Jung, J. Med. Chem., 1987, 30, 267–272 CrossRef CAS PubMed; (b) Y. Pan, J. Qiu and R. B. Silverman, J. Med. Chem., 2003, 46, 5292–5293 CrossRef CAS PubMed.
- X. Han, Y. Zhao, Z. Shi and M. Wang, Tetrahedron Chem, 2002, 3, 100023 CrossRef.
- Y. Y. Li and B. Gao, Org. Lett., 2023, 25, 2756–2760 CrossRef CAS PubMed.
- (a) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed; (b) G. Mallesham, C. Swetha, S. Niveditha, M. E. Mohanty, N. J. Babu, A. Kumar, K. Bhanuprakash and V. J. Rao, J. Mater. Chem. C, 2015, 3, 1208–1224 RSC; (c) G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704–5783 CrossRef CAS PubMed.
- D. Ji, Z. Qi and X. Li, Org. Lett., 2023, 25, 5957–5962 CrossRef CAS PubMed.
- (a) G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 Search PubMed; (b) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS.
- C. Q. Wang, Z. Q. Li, L. Tian, P. J. Walsh and C. Feng, Cell Rep. Phys. Sci., 2022, 3, 101117–101131 Search PubMed.
- W. Li, C. Wang, M. Xiao and L. J. Cheng, Org. Lett., 2024, 26, 525–529 CrossRef CAS PubMed.
- (a) R. Filler, Y. Kobayashi and L. M. Yagupolskii, Organofluorine Compounds in Medicinal Chemistry and Biological Applications, Elsevier, Amsterdam, 1993 Search PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 Search PubMed; (c) D. Barnes-Seeman, M. Jain, L. Bell, S. Ferreira, S. Cohen, X. H. Chen, J. Amin, B. Snodgrass and P. Hatsis, ACS Med. Chem. Lett., 2013, 4, 514–516 CrossRef CAS PubMed.
- (a) T. Kobayashi, T. Eda, O. Tamura and H. Ishibashi, J. Org. Chem., 2002, 67, 3156–3159 Search PubMed; (b) T. Hanamoto, N. Morita and K. Shindo, Eur. J. Org. Chem., 2003, 4279–4285 CrossRef CAS; (c) S. M. Landge, D. A. Borkin and B. Torok, Lett. Org. Chem., 2009, 6, 439–443 CrossRef CAS; (d) A. T. Parsons, T. D. Senecal and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 2947–2950 Search PubMed; (e) E. J. Cho and S. L. Buchwald, Org. Lett., 2011, 13, 6552–6555 CrossRef CAS PubMed; (f) A. Hafner and S. Bräse, Adv. Synth. Catal., 2011, 353, 3044–3048 CrossRef CAS.
- H. Luo, Y. Zhao, D. Wang, M. Wang and Z. Shi, Green Synth. Catal., 2020, 1, 134–142 Search PubMed.
- L. Chen, Z. F. Luo, P. Ye, Y. J. Mao, Z. Y. Xu, D. Q. Xu and S. J. Lou, Org. Biomol. Chem., 2023, 21, 8979–8983 RSC.
- Z. Q. Li, H. J. Tang, Z. Wang, C. Q. Wang and C. Feng, Chem. Sci., 2024, 15, 3524–3529 RSC.
- (a) J. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O'Hagan, J. Fluorine Chem., 2020, 239, 109639 CrossRef CAS; (b) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (c) J. Han, L. Kiss, H. Mei, A. M. Remete, M. Ponikvar-Svet, D. M. Sedgwick, R. Roman, S. Fustero, H. Moriwaki and V. A. Soloshonok, Chem. Rev., 2021, 121, 4678–4742 CrossRef CAS PubMed.
- Y. You, J. Hu and T. Wu, Org. Lett., 2023, 25, 4546–4550 CrossRef CAS PubMed.
- Y. Tan, K. Chen, J. Hu, S. Lin and T. Wu, Eur. J. Org. Chem., 2024, e202300869 CrossRef CAS.
- (a) W. R. Dolbier Jr., G. D. Purvis III, M. J. Seabury, G. E. Wicks and C. R. Burkholder, Tetrahedron, 1990, 46, 7991–8004 CrossRef.
- K. Fuchibe, K. Sakon, K. Suto, R. Eto, S. Nakazono and J. Ichikawa, Org. Lett., 2023, 25, 7258–7262 CrossRef CAS PubMed.
- (a) Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Fukai, Y. Inoue, F. Sato and S. Tokito, J. Am. Chem. Soc., 2004, 126, 8138–8140 CrossRef CAS PubMed; (b) K. Fuchibe, T. Fujita and J. Ichikawa, Bull. Chem. Soc. Jpn., 2024, 97, uoad024 CrossRef.
- K. Fuchibe, Y. Mayumi, M. Yokota, H. Aihara and J. Ichikawa, Bull. Chem. Soc. Jpn., 2014, 87, 942–949 CrossRef CAS.
- K. Fuchibe, K. Shigeno, N. Zhao, H. Aihara, R. Akisaka, T. Morikawa, T. Fujita, K. Yamakawa, T. Shimada and J. Ichikawa, J. Fluorine Chem., 2017, 203, 173–184 CrossRef CAS.
- K. Fuchibe, H. Imaoka and J. Ichikawa, Chem. – Asian J., 2017, 12, 2359–2363 CrossRef CAS PubMed.
- M. J. Tozer and T. F. Herpin, Tetrahedron, 1996, 52, 8619–8683 CrossRef CAS.
- Q. Shen and G. B. Hammond, Org. Lett., 2001, 3, 2213–2215 CrossRef CAS PubMed.
- Q. Shen, C. H. Chen and G. B. Hammond, J. Fluorine Chem., 2002, 117, 131–135 CrossRef CAS.
- Q. Shen and G. B. Hammond, J. Am. Chem. Soc., 2002, 124, 6534–6535 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |