
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking nature's antioxidants: a novel method for synthesising plasmalogens†
Jay
Tromans
a,
Bian
Zhang
b and
Bernard T.
Golding
 *a
*a
aSchool of Natural and Environmental Science – Chemistry, Newcastle University, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: bernard.golding@ncl.ac.uk
bBiBerChem Research Ltd, The Biosphere, Draymans Way, Newcastle Helix, Newcastle upon Tyne, NE4 5BX, UK
First published on 5th September 2024
Abstract
Plasmalogens are glycerophospholipids distinguished by their O-(Z)-vinyl ether at the sn-1 position. These lipids are implicated in several disease states requiring analytical, diagnostic and therapeutic interventions, which demand synthetic availability for a variety of structural types. By deploying the new O-protecting group 1,4-dimethoxynaphthyl-2-methyl (‘DIMON’) and a new stereospecific method for accessing Z-vinyl ethers, a reproducible, versatile synthetic route to plasmalogens [plasmenyl phosphocholines] has been developed. A key intermediate is (S,Z)-1-((1,4-dimethoxynaphthalen-2-yl)methoxy)-3-(hexadec-1-en-1-yloxy)propan-2-ol, which in principle, permits plasmalogen synthesis ‘à la carte’ at scale. The methodology compares favourably with all previous synthetic routes by virtue of the very high configurational (>99% Z) and optical purity (>99% ee), including the ability to incorporate polyunsaturated fatty acyl chains (e.g. all Z docosahexaenoic acid) reliably at the sn-2 position.
Introduction
Cells are protected by a phospholipid membrane without which there would be no life on Earth as presently known. As well as providing a portal for the ingress and export of molecules and ions, there are membrane components that protect the cell against assault by reactive species. One such component is a family of ‘ether lipids’ called plasmalogens (Fig. 1), which are glycerophospholipids containing an alkenyl ether group at the sn-1 position, an unsaturated acyl moiety at sn-2 and usually either an ethanolamine or choline head group linked to a phosphate entity at sn-3.1–4 | ||
| Fig. 1 General plasmalogen structure and their constituents. | ||
Plasmalogens have been known for 100 years,5 with the structure elucidation6 of these relatively unstable molecules requiring decades of research after several missteps. The crucial Z-configured vinyl ether, a functionality quite rare in natural products,7 was established by infrared spectroscopy8,9 and NMR for the related ‘neutral plasmalogens’.10 The biosynthesis of plasmalogens differs in aerobes and anaerobes: in the former the vinyl ether is created by dehydrogenation of an alkyl (‘plasmanyl’) precursor, whereas in the latter a sequence of carbonyl reduction and water elimination occurs from an acyl precursor.11 The unique vinyl ether entity of plasmalogens acts sacrificially to trap reactive oxygen species (ROS).12–15
Paradoxically, although human life depends on dioxygen, this molecule can translate into ROS, which are highly damaging to DNA and proteins,16 seeding diseases and limiting human lifespan. Cancers are primarily spawned by DNA damage17 whilst protein oxidation may kindle Alzheimer's disease (AD) and other dementias.18,19 As the vinyl ether of plasmalogens provides an exquisite trap for ROS12,13 (Scheme 1), individuals with a deficiency of cellular plasmalogens may be especially vulnerable to diseases associated with oxidative damage to biomolecules. The complete range of plasmalogen activities is still subject to intensive research, which has accelerated in the last decade because of their connection to AD, certain cancers and rare hereditary diseases.2–4,20
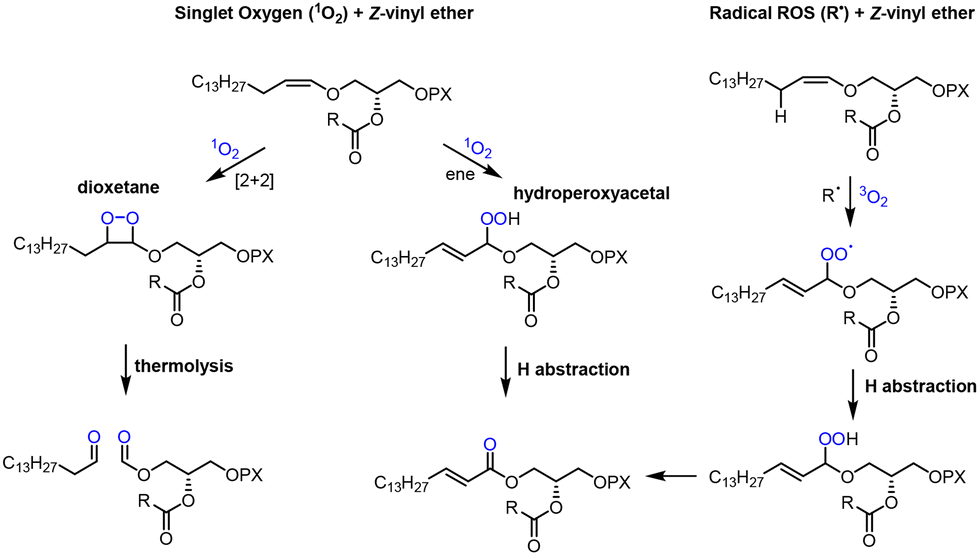 | ||
| Scheme 1 Reactions of plasmalogen Z-vinyl ether with singlet oxygen (1O2) and radical ROS (R˙); R = oleoyl chain, P = phosphate, X = choline. | ||
Plasmalogens can be isolated from natural sources (e.g. scallops or chicken21,22) but the extracts are mixtures, which are not ideal for investigating molecular mechanisms of action. In particular, plasmalogens with a polyunsaturated acyl (PUFA) moiety at the sn-2 position of the glycerol core (e.g. arachidonoyl) are of greatest interest because of their concentration in brain and heart tissues,4,23,24 and the possibility that the polyene component acts synergistically with the vinyl ether to trap ROS.13 To determine whether plasmalogens with polyunsaturated acyl groups are the most important defensive molecules, efficient scalable syntheses are required for all types of plasmalogen to enable their antioxidant characteristics and other properties to be compared. Further, ‘plasmalogen replacement therapy’4,25–27 seeks to remedy deficiency by supplementation and requires a dependable supply of individual plasmalogens with optimal properties. In this paper we describe a reliable synthesis of choline plasmalogens containing 4 and 6 Z double bonds in the PUFA component.
Biomimetic syntheses of plasmalogens are beyond the reach of current synthetic methodology. Nevertheless, ‘standard’ chemical synthesis of polyunsaturated plasmalogens is a challenging obstacle course, requiring judicious ordering of steps and the unavoidable use of protecting groups (PGs). Among the potential pitfalls are acyl migration from sn-2 to sn-1/sn-328 in any glycerol-derived intermediate with a free primary hydroxyl group, which also applies to the structurally related plasmanyl ether lipids,29 and a myriad of unwanted reactions with the vinyl ether including isomerisation, oxidation, reduction, hydrolysis and cyclisation, the latter with a free hydroxyl group giving a cyclic acetal.30 These processes may be acid- and/or base-catalysed, or for isomerisation, induced by a thiyl radical generated from a thiol by irradiation with ultraviolet light.31 Above all, an effective Z-vinyl ether synthesis is essential to access plasmalogens, but the stage of generation of this group needs careful consideration of its compatibility with subsequent steps.
Following many heroic attempts to access plasmalogens,32 the first successful stereoselective synthesis gave one choline plasmalogen in relatively low yield.33 The Z-vinyl ether was installed by reduction of a vinyl phosphonate (58% yield, 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Z:E). Several other strategies towards plasmalogen Z-vinyl ethers have been published (Scheme 2), including Lindlar-catalysed alkynyl-ether reduction (63% yield, 66:1 Z:E),34 reductive cleavage under Barbier conditions (47% yield, 49:1 Z:E),35 and alkylation of lithioalkoxy allyl intermediates (77% yield, 9:1 Z:E).36,37 The latter method for installing the vinyl ether chain was also described in a patent,38 which claimed the synthesis of a phosphoethanolamine plasmalogen containing a docosahexaenoyl group at C-2 of glycerol, although no supporting spectroscopic data was provided. Despite these achievements, at the outset of our research there was no published methodology affording a fully characterised polyunsaturated plasmalogen in high yield and with perfect stereocontrol at the Z-vinyl ether.
:1 Z:E). Several other strategies towards plasmalogen Z-vinyl ethers have been published (Scheme 2), including Lindlar-catalysed alkynyl-ether reduction (63% yield, 66:1 Z:E),34 reductive cleavage under Barbier conditions (47% yield, 49:1 Z:E),35 and alkylation of lithioalkoxy allyl intermediates (77% yield, 9:1 Z:E).36,37 The latter method for installing the vinyl ether chain was also described in a patent,38 which claimed the synthesis of a phosphoethanolamine plasmalogen containing a docosahexaenoyl group at C-2 of glycerol, although no supporting spectroscopic data was provided. Despite these achievements, at the outset of our research there was no published methodology affording a fully characterised polyunsaturated plasmalogen in high yield and with perfect stereocontrol at the Z-vinyl ether.
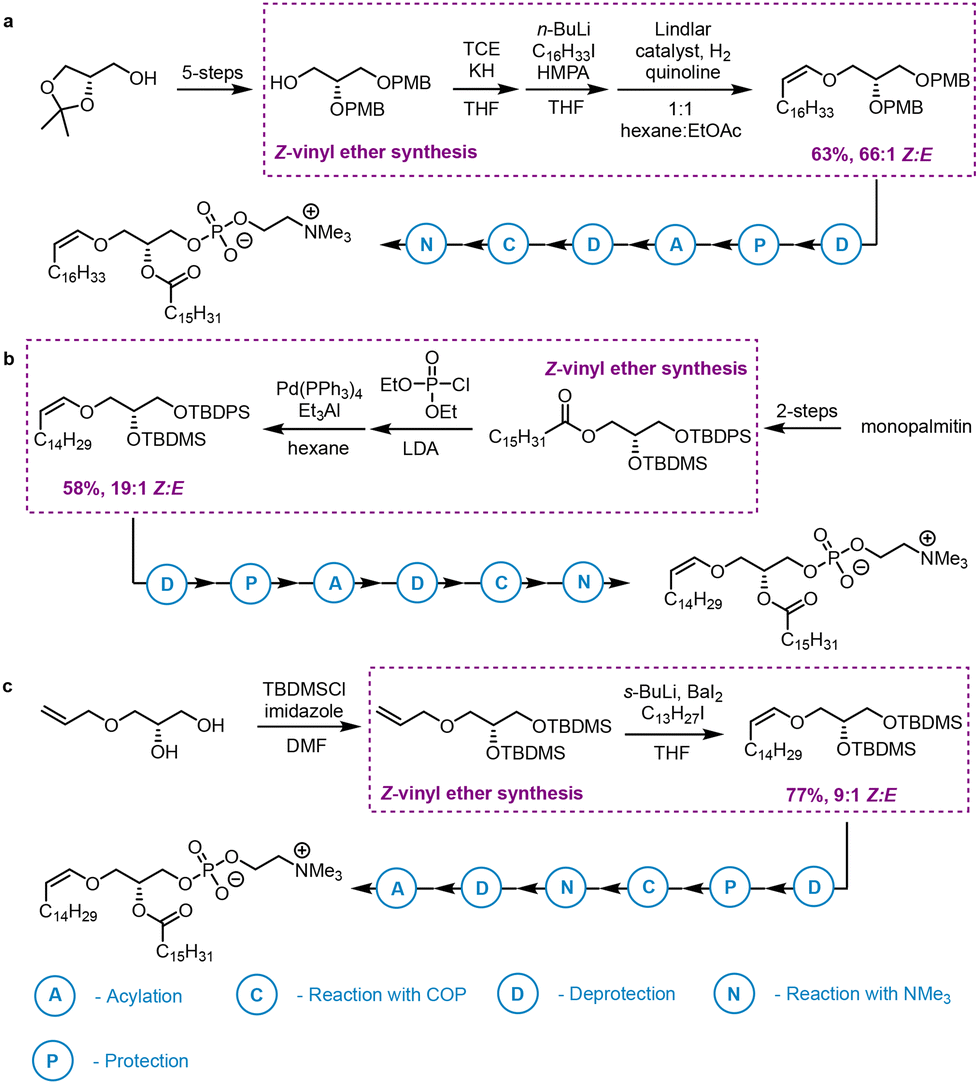 | ||
| Scheme 2 Published strategies for the synthesis of plasmalogen-type Z-vinyl ethers: (a) vinyl phosphonate reduction;33 (b) partial hydrogenation of an alkynyl ether;34 (c) alkylation of a lithioalkoxy allyl intermediate.36 | ||
Results and discussion
Synthesis of plasmalogens
We recently reported a modification of the Peterson olefination that afforded Z-vinyl ethers in high yield and with exceptional stereocontrol (99% Z).39 The reaction of a cis-2-trimethylsilyl-3-alkyl epoxide with an excess of an alcohol under boron trifluoride catalysis gave a 1-alkoxy-2-hydroxylalkylsilane in up to 97% yield. Treatment of the silane with potassium hydride in α,α,α-trifluorotoluene (TFT), triggered a syn-elimination producing Z-vinyl ethers with 99:1 Z:E selectivity and up to 96% yield (Scheme 3a).
 | ||
| Scheme 3 (a) Synthesis of plasmalogen-type Z-vinyl ethers via a modified Peterson reaction;39 (b) selective oxidation of DIMON in the presence of a methylene-skipped polyene.40 | ||
To functionalise glycerol selectively, PGs are required to distinguish the three hydroxyl groups. As silyl ethers require an acidic or basic source of fluoride for their cleavage, such conditions can catalyse acyl migration.28 Electron-rich benzyl ethers may be suitable as this PG class can be cleaved under neutral conditions. However, Bittman et al.34 showed that 4-methoxybenzyl (PMB) was not oxidatively cleavable using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) or ceric(IV) ammonium nitrate (CAN) in the presence of a Z-vinyl ether. Instead, Birch reduction was used to remove the PMB group but partially isomerised the vinyl ether. Benzyl ethers are not oxidatively cleavable in the presence of 1,3- or 1,4-dienes,40 which excludes polyunsaturated plasmalogens. Therefore, prior to our research, no known41hydroxyl PG was compatible with both Z-vinyl ethers and PUFA acyl groups, thus restricting plasmalogen accessibility.
We recently reported40 a novel benzyl-type protecting group, 1,4-dimethoxynapthalene-2-methyl (‘DIMON’, Scheme 3b), that was oxidatively cleavable in the presence of a methylene-skipped polyene (e.g.1Z,4Z,7Z,10Z-tetraene). We have found that DIMON can also be oxidatively removed without perturbing a Z-vinyl ether and is therefore compatible with all plasmalogen functionalities.
With the invention of DIMON40 and the adaptation of the Peterson reaction for synthesising Z vinyl ethers39 we were ready to synthesise natural plasmalogens 1–3 (Fig. 2), for which a retrosynthetic analysis guided our approach (Scheme 4). The plan required that the phosphate group is introduced last, via alcohol 4, to avoid adverse solubility and chromatographic issues posed by this charged entity. Hence, the precursor of 4 is 5, protected at the sn-3 hydroxyl, preceded by alcohol 6, which is acylated. Compound 6 is derived by selective deprotection of 7, which bears the vinyl ether arising from combining 1,2-diprotected glycerol 8 with silyl epoxide 9. DIMON was one of the protecting groups (PG1), whilst the other must be removed under conditions that enable the survival of DIMON and the vinyl ether. DIMON must be the only protecting group after the fatty acyl chain is installed.
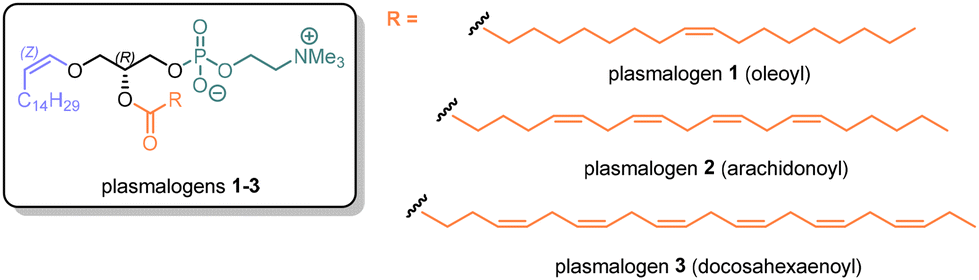 | ||
| Fig. 2 Structure of plasmalogens 1–3. | ||
 | ||
| Scheme 4 Retrosynthetic analysis for plasmalogens. | ||
A variety of protected glycerols or C3 glycerol precursors have been used for the synthesis of glycerophospholipids.42–44 To apply our Z-vinyl ether synthesis required ring-opening of an intermediate cis-silyl epoxide, e.g. trimethyl(-3-tetradecyloxiran-2-yl)silane 9, by a suitable glycerol derivative or surrogate (Scheme 5). Six glycerol moieties were investigated: allyl alcohol 10, glycidol 11, 4-(hydroxymethyl)-1,3-dioxolan-2-one 12, solketal 13, mono-protected glycerol 14, and di-protected glycerol 15.
 | ||
| Scheme 5 Results of epoxide 9 ring-opening reactions with alcohols 10–15. | ||
Allyl alcohol 11 requires an asymmetric epoxidation to introduce a second oxygen into the molecule followed by epoxide ring-opening to introduce a third. 4-(Hydroxymethyl)-1,3-dioxolan-2-one 12 is not commercially available as a single enantiomer corresponding to a natural plasmalogen. Single enantiomers of glycidol 11 and solketal 13 are commercially available, with compound 11 being the more attractive starting material, as 13 would require acid-catalysed hydrolysis of the ketal at a later stage. Reactions of alcohols 10–15 with epoxide 9 to form 1-alkoxy-2-hydroxyalkylsilanes 16–21 were attempted with boron trifluoride (BF3), a commonly used catalysis for reactions of epoxides with nucleophiles. Glycidol 11, 4-(hydroxymethyl)-1,3-dioxolan-2-one 12, and solketal 13 all failed to afford the desired product. Mono-protected glycerol 14 afforded the desired product in 36% yield, whilst 1,2-diprotected glycerol 15 gave product in 63% (0.32 mmol scale) or 77% yield (0.96 mmol scale). Allyl alcohol 11 gave a near quantitative conversion of epoxide to 1-alkoxy-2-hydroxyalkylsilane (6.40 mmol scale). All the reactions described exhibited, as required, very high regioselectivity for attack at the silyl-substituted centre in line with previous observations with amines.45
Despite the efficiency of the ring-opening with allyl alcohol, further elaboration required enantioselective epoxidation of intermediate 16. Many excellent methods have been described for such epoxidations [e.g. ref. 46], and now for terminal alkenes high enantioselectivity can be achieved with synthetically accessible catalysts [see e.g. ref. 47]. However, although we explored this possibility it led to incompatibility of functional groups when attempting to generate the Z vinyl ether. We therefore chose to proceed with the R-enantiomer (R-15) of the DIMON-protected glycerol derivative 15, which was obtained from the readily available precursor (R)-glycidol R-11 in 4 steps with 41% overall yield (Scheme 6).
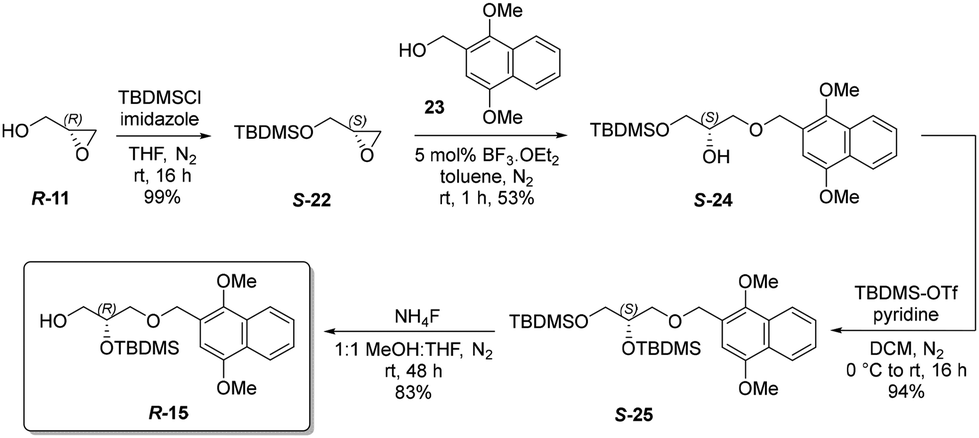 | ||
| Scheme 6 Synthesis of (R)-2-((tert-butyldimethylsilyl)oxy)-3-((1,4-dimethoxynaphthalen-2-yl)methoxy)propan-1-ol R-15. | ||
Thus, (R)-glycidol R-11 was silylated with tert-butyldimethylsilyl chloride (TBDMSCl) to afford protected glycidol S-2248 in quantitative yield. This compound was subjected to acid-catalysed epoxide ring-opening with DIMON alcohol 23 to give the 1,3-diprotected glycerol S-24 in 53% yield. The 1,2-di-protected glycerol isomer was obtained in 17% yield, but readily separated chromatographically. Regioisomer S-24 was silylated with TBDMSCl to give tri-protected glycerol S-25 in 94% yield.
Finally, the primary silyl group was selectively cleaved by ammonium fluoride to afford 1,2-di-protected glycerol R-15 in 83% yield (see ESI for full characterisation of this intermediate†). The epoxide ring-opening was also performed in dichloromethane or with ytterbium triflate as catalyst but did not improve on BF3 in toluene. The optical purity and configuration of R-15 was confirmed by high-performance liquid chiral chromatography analysis (>99% ee, see ESI†). The diastereomeric mixture denoted as compound 21 was subjected to Peterson elimination with potassium hydride in TFT (Scheme 7), which consumed all diastereoisomers and afforded the Z-vinyl ether S-26 in 85% yield and with a Z:E isomeric ratio determined by 1H NMR (see ESI†) to be 99:1. This compound is a convenient, storable intermediate for synthesising a variety of plasmalogens. Compound S-26 was desilylated with tetrabutylammonium fluoride (TBAF) to give intermediate S-27, which was acylated with three fatty acids to afford the corresponding esters in high yields: 95% (S-28), 93% (S-29) and 92% (S-30) (see ESI for representative NMR spectra, which showed that the ratio of Z:E isomers remained at 99:1†).
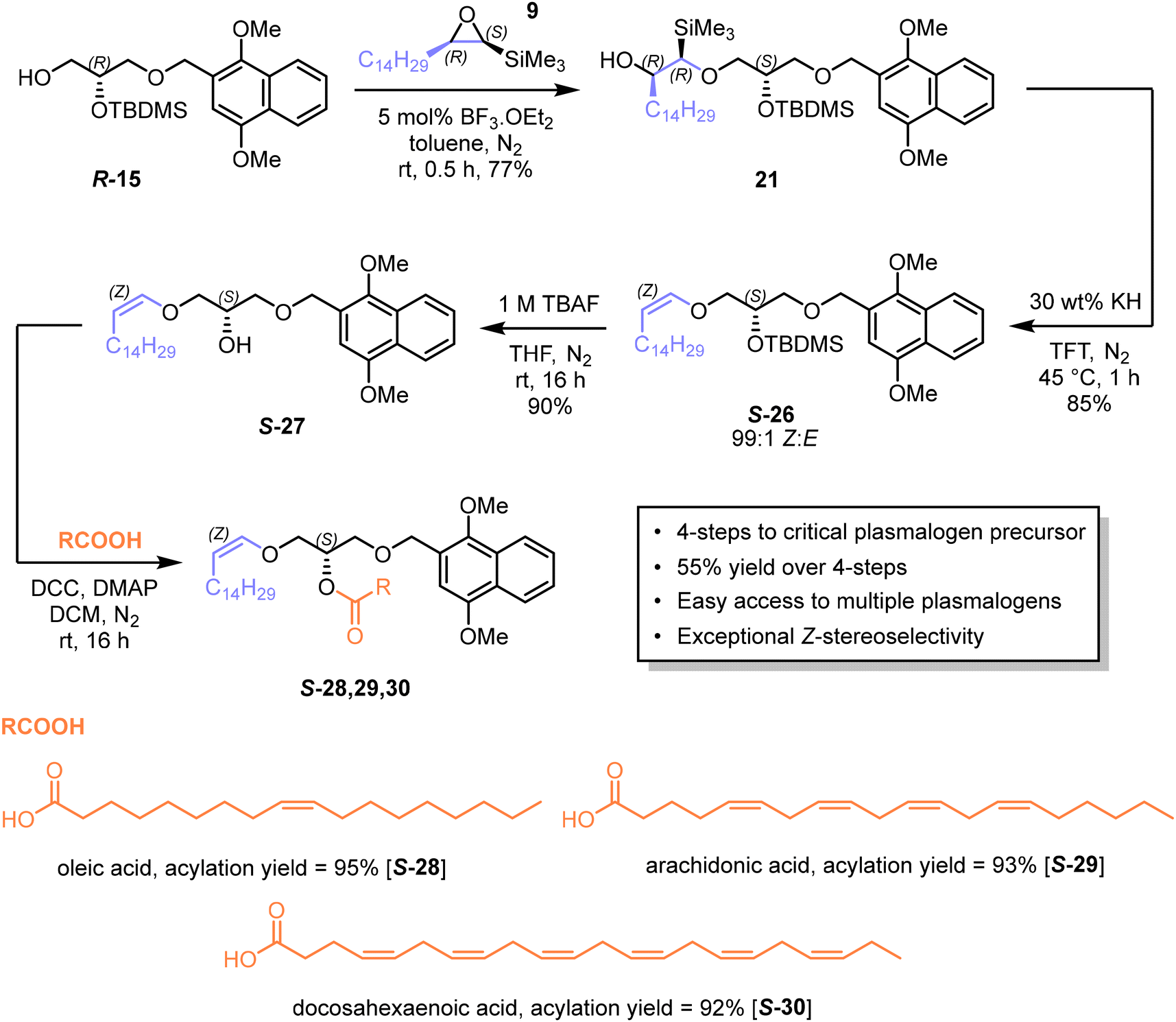 | ||
| Scheme 7 Synthesis of plasmalogen precursors S-28,29,30. | ||
With the sn-1 and sn-2 functionalities installed, it remained to attach a phosphocholine headgroup at the sn-3 position. To install this group, the DIMON was cleaved oxidatively with DDQ (Scheme 8). This was a critical step as success depended on whether the selection of DIMON based on consideration of oxidation potentials40 was valid. Gratifyingly, all three reactions proceeded well to afford the desired primary alcohols in 92% (oleoyl, S-31), 65% (arachidonoyl, S-32), and 61% (docosahexaenoyl, S-33) yield. That the vinyl ether was undisturbed and the acyl group had not migrated, were confirmed by 1H NMR (see ESI†). As there was no evidence of a new aldehyde proton, the Z-vinyl ether had not been affected. For compound S-33 the hydroxyl proton was assigned to the triplet at 1.78 ppm (1 H, J = 6.2 Hz), which was coupled to the doublet of doublets at 3.75 ppm (2 H, J = 6.1, 4.7 Hz) assigned to the neighbouring methylene protons. Therefore, the compound contained a primary alcohol and no acyl migration had occurred. Further, the resonance at 5.04 ppm for the CH at the chiral centre was consistent with a CH group bearing an OCOR moiety. The very specific, near netral conditions for DIMON removal are therefore perfectly compatible with the susceptible functionalities in plasmalogens and related glycerophospholipids.
 | ||
| Scheme 8 Synthesis of plasmalogens R-1,2,3. | ||
The final challenge was to install the phosphate head group for which several methods were explored including the use of phosphorus oxychloride49 and tris(2,2,2-trifluoroethyl)phosphate.50 In each case sequential substitutions were attempted (with e.g. tert-butyl (2-hydroxyethyl)carbamate, di-protected glycerol) but did not proceed satisfactorily beyond monosubstitution (see ESI†). We therefore employed the well-established reagent 2-chloro-1,3,2-dioxaphospholane 2-oxide 34 (COP51). The compatibility of this reagent with a model compound (35) containing a Z vinyl ether was explored (Fig. 3), showing that COP could only be used in the presence of an excess of pyridine provided this base was added first.
 | ||
| Fig. 3 Vinyl ether stability to COP, triethylamine, and pyridine. | ||
Applying this methodology to S-31 afforded cyclic phosphate R-36 quantitatively, which on reaction with an excess of trimethylamine afforded plasmalogen R-1 in 55% yield, with no evidence of vinyl ether hydrolysis according to 1H NMR. The plasmalogen was characterised by 1H, 31P, 1H–1H COSY, 1H–13C HMBC and 1H–13C HSQC NMR, as well as LCMS and HRMS (ESI†).
Plasmalogen precursors S-32 and S-33 were reacted with COP and pyridine affording intermediates R-37 and R-38, respectively, in quantitative yield. The proton-decoupled 31P NMR analysis showed both products had a single, singlet peak at 17.5 ppm, which agreed with the observed resonance for compound R-36. The 1H NMR spectra exhibited the phosphate ethylene peak at 4.41 ppm (multiplet, 4 H) and showed no evidence of vinyl ether hydrolysis. The intermediates were subjected to the NMe3 ring-opening reaction, which afforded plasmalogens R-2 and R-3 in 45% and 41% yield, respectively.
To the best of our knowledge, this is the first time that plasmalogens R-1, R-2 or R-3 have been synthesised and fully characterised with respect to both the integrity of the vinyl ether configuration and enantiomeric purity. The overall yields were 28% (oleoyl R-1), 16% (arachidonyl R-2), and 14% (docosahexaenoyl R-3) over 7 steps from compound R-15. While the two-step phosphorylation reaction was lower yielding than desired, it was the best method found for installing the phosphocholine functionality.
Conclusions
We have shown that the combination of the novel O-protecting group DIMON and a modified Peterson reaction provides a new general synthetic route to plasmalogens, illustrated by the synthesis of three such molecules differing with respect to the sn-2 acyl group. Previous studies have only reported plasmalogens containing either a saturated (e.g. palmitoyl) or mono-unsaturated (oleoyl) acyl moiety or have lacked full characterisation of the polyunsaturated molecules described. The present studies provide a new adaptable method for the synthesis of the most important natural plasmalogens, i.e. those containing polyunsaturated acyl groups. This should assist the development of novel diagnostic, prognostic, and therapeutic tools for diseases implicating plasmalogens, including AD.Author contributions
Jay Tromans: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing - original draft, Writing - review & editing. Bian Zhang: Funding acquisition, Project administration, Supervision, Writing - review & editing. Bernard T. Golding: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing - original draft, Writing - review & editing.Data availability
All data reported in this paper is available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.References
- E. L. Gottfried and M. M. Rapport, J. Biol. Chem., 1962, 237, 329 CrossRef CAS PubMed [first in a series of papers].
- M. Vítova, A. Palyzov and T. Rezanka, Prog. Lipid Res., 2021, 83, 101111 CrossRef PubMed.
- F. Dorninger, J. Berger and M. Honsho, Front. Cell Dev. Biol., 2023, 11, 1137868 CrossRef PubMed : introducing a series of papers in an e-book collection.
- F. Dorninger, E. R. Werner, J. Berger and K. Watschinger, Front. Cell Dev. Biol., 2022, 10, 946393 CrossRef PubMed.
- R. Feulgen and K. Voit, Pfluegers Arch. Gesamte Physiol., 1924, 206, 389 CrossRef CAS.
- J. Koch, K. Watschinger, E. R. Werner and M. A. Keller, Front. Cell Dev. Biol., 2022, 10, 864716 CrossRef PubMed.
- T. B. Poulsen, Acc. Chem. Res., 2021, 54, 1830 CrossRef CAS PubMed.
- W. T. Norton, E. L. Gottfried and M. M. Rapport, J. Lipid Res., 1962, 3, 456 CrossRef CAS.
- H. R. Warren and W. E. M. Lands, J. Am. Chem. Soc., 1963, 85, 60 CrossRef.
- H. H. O. Schmid, W. J. Baumann and K. Mangold, J. Am. Chem. Soc., 1967, 89, 4797 CrossRef CAS PubMed.
- H. Goldfine, Prog. Lipid Res., 2010, 49, 493 CrossRef CAS PubMed.
- O. Skaff, D. I. Pattison and M. J. Davies, Biochemistry, 2008, 47, 8237 CrossRef CAS PubMed.
- R. C. Murphy, Chem. Res. Toxicol., 2001, 14, 463 Search PubMed.
- A. M. Luoma, F. Kuo, O. Cakici, M. N. Crowther, A. R. Denninger, R. L. Avila, P. Brites and D. A. Kirschner, Free Radicals Biol. Med., 2015, 84, 296 CrossRef CAS PubMed.
- N. Jimenez-Rojo and H. Riezman, FEBS Lett., 2019, 593, 2378 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Acc. Chem. Res., 2018, 51, 1850 CrossRef CAS PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461(7267), 1071 CrossRef CAS PubMed.
- W. R. Markesbery and M. A. Lovell, Arch. Neurol., 2007, 64, 954 CrossRef PubMed.
- D. A. Butterfield, Antioxidants, 2023, 12, 462 CrossRef CAS PubMed.
- S. Paula, G. I. Lancaster and P. J. Meiklea, Prog. Lipid Res., 2019, 74, 186 CrossRef PubMed.
- M. S. Hossain, S. Mawatari and T. Fujino, Front. Cell Dev. Biol., 2022, 10, 828282 CrossRef PubMed.
- M. Fujino, J. Fukuda, H. Isogai, T. Ogaki, S. Mawatari, A. Takaki, C. Wakana and T. Fujino, Front. Cell Dev. Biol., 2022, 10, 894734 CrossRef PubMed.
- N. E. Braverman and A. B. Moser, Biochim. Biophys. Acta, 2012, 1822, 1442 CrossRef CAS PubMed.
- R. V. Panganamala, L. A. Horrocks, J. C. Geer and D. G. Cornwell, Chem. Phys. Lipids, 1971, 6, 97 CrossRef CAS PubMed.
- T. Fujino, T. Yamada, T. Asada, Y. Tsuboi, C. Wakana, S. Mawatari and S. Kono, EBioMedicine, 2017, 17, 199 CrossRef PubMed.
- Y. Zhou, N. Yu, J. Zhao, Z. Xie, Z. Yang and B. Tian, Front. Cell Dev. Biol., 2020, 8, 765 CrossRef PubMed.
- D. B. Goodenowe, J. Haroon, M. A. Kling, M. Zielinski, K. Mahdavi, B. Habelhah and L. Shtilkind, Front. Cell Dev. Biol., 2022, 10, 864842 CrossRef PubMed.
- J. A. Laszlo, D. L. Compton and K. E. Vermillion, J. Am. Oil Chem. Soc., 2008, 85, 307 CrossRef CAS.
- M. A. G. B. Gomes, A. Bauduin, C. Le Roux, R. Fouinneteau, W. Berthe, M. Berchel, H. Couthon and P.-A. Jaffrès, Beilstein J. Org. Chem., 2023, 19, 1299 CrossRef CAS PubMed.
- W. J. Baumann, T. H. Madson and B. J. Weseman, J. Lipid Res., 1972, 13, 640 CrossRef CAS.
- C. Ferreri, A. Ferocino, G. Batani, C. Chatgilialoglu, V. Randi, M. V. Riontino, F. Vetica and A. Sansone, Biomolecules, 2023, 13, 730 CrossRef CAS PubMed.
- A. J. Slotboom, G. H. De Haas and L. L. M. Van Deenen, Chem. Phys. Lipids, 1967, 1, 192 CrossRef CAS.
- (a) Y. Rui and D. H. Thompson, J. Org. Chem., 1994, 59, 5758 CrossRef CAS; (b) Y. Rui and D. H. Thompson, Chem. – Eur. J., 1996, 2, 1505 CrossRef CAS.
- D. Qin, H. S. Byun and R. Bittman, J. Am. Chem. Soc., 1999, 121, 662 CrossRef CAS.
- J. Shin, O. Gerasimov and D. H. Thompson, J. Org. Chem., 2002, 67, 6503 CrossRef CAS PubMed.
- (a) J. Shin and D. H. Thompson, J. Org. Chem., 2003, 68, 6760 CrossRef CAS PubMed; (b) J. V. Bossche, J. Shin and D. H. Thompson, J. Org. Chem., 2007, 72, 5005 CrossRef PubMed.
- S. Maeda, T. Mohri, T. Inoue, Y. Asano, Y. Otoki, M. Enomoto, K. Nakagawa, S. Kuwahara and Y. Ogura, Biosci. Biotechnol. Biochem., 2021, 85, 1383 CrossRef PubMed.
- M. A. Khan, P. L. Wood and D. Goodenowe, US Patent, 2015, 9,169,280 B2 Search PubMed.
- J. Tromans, B. Zhang and B. T. Golding, Chem. – Eur. J., 2023, e202302708 Search PubMed.
- J. Tromans, B. Zhang and B. T. Golding, Chem. Commun., 2023, 59, 13333 RSC.
- Greene's Protective Groups in Organic Synthesis, ed. P. G. M. Wuts, John Wiley & Sons, Inc., New Jersey, 5th edn, 2014 Search PubMed.
- F. Paltauf and A. Hermetter, Prog. Lipid Res., 1994, 33, 239 CrossRef CAS PubMed.
- S. D. Stamatov and J. Stawinsk, Org. Biomol. Chem., 2010, 8, 463 RSC.
- Z. X. Guistra and K. L. Tan, Chem. Commun., 2013, 49, 4370 RSC.
- P. F. Hudrlik, A. M. Hudrlik and A. K. Kulkarni, Tetrahedron Lett., 1985, 26, 139 CrossRef CAS.
- Q. He, M.-P. Pu, Z. Jiang, H. Wang, X. Feng and X. Liu, J. Am. Chem. Soc., 2023, 145, 15611–15618 CrossRef CAS PubMed.
- M. Lansing, H. Engler, T. M. Leuther, J.-M. Neudörfl and A. Berkessel, ChemCatChem, 2016, 8, 3706 CrossRef CAS.
- C. P. Bold, C. Klaus, B. Pfeiffer, J. Schurmann, R. Lombardi, D. L. Agell, J. F. Diaz and K. H. Altmann, Org. Lett., 2021, 23, 2238 CrossRef CAS PubMed.
- H. Eibl, Chem. Phys. Lipids, 1980, 26, 415 Search PubMed.
- K. Tsubaki, H. Shimooka, M. Kitamura and T. Okauchi, Org. Lett., 2019, 21, 9779 CrossRef CAS PubMed.
- N. S. Chandrakumar and J. Hajdu, J. Org. Chem., 1983, 48, 1197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01233j |
| This journal is © The Royal Society of Chemistry 2024 |