
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On resin synthesis of phosphoethanolamine cellulose†
Jhih-Yi
Huang
 ab and
Martina
Delbianco
*a
ab and
Martina
Delbianco
*a
aDepartment of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: martina.delbianco@mpikg.mpg.de
bDepartment of Chemistry and Biochemistry, Freie Universitäte Berlin, Arnimallee 22, 14195 Berlin, Germany
First published on 15th August 2024
Abstract
Phosphoethanoamine (pEtN) cellulose is a chemically modified cellulose present in some bacterial biofilms. To deepen our understanding of this biopolymer and its biological function, access to chemically defined pEtN-cellulose oligosaccharides is desirable. Herein, we report an on resin protocol for the fast synthesis of tailor-made pEtN-celluloses. The cellulose backbone is prepared by automated glycan assembly and then specifically functionalized with pEtN groups, allowing for access to a collection of ten pEtN-cellulose oligomers with different amount and pattern of pEtN.
Phosphoethanoamine (pEtN) cellulose (Fig. 1A) is a chemically modified cellulose found in bacterial biofilms of Escherichia coli and Salmonella enteric, rendering them elastic and adhesive.1–4 The pEtN modification may also provide resistance against cellulase-producing microorganisms.1 During biosynthesis, the pEtN transferase BcsG in the bacterial cellulose synthase (Bcs) complex functionalizes about half of cellulose's glycosyl units with pEtN at the C-6 position.3 Yet, the pEtN substitution pattern has not been identified1,3,5 and it is unclear whether specific patterns could serve as code to trigger a particular response.6,7 To support this hypothesis, we recently showed that the nanocomposite made of synthetic pEtN cellulose oligomers and an amyloidogenic peptide displayed an intriguing modulation of adhesion dependent on the pEtN substitution pattern.8
 | ||
| Fig. 1 Chemical structure of pEtN cellulose (A). On resin approaches to expedite the synthesis of sulfated, phosphorylated, and pEtN-functionalized (this work) glycans (B). | ||
To dissect the role of the pEtN pattern in biological settings, synthetic oligosaccharide standards are valuable tools.9 Previously, we reported the synthesis of well-defined pEtN cellulose oligomers via a combination of solid-phase technology and solution-phase chemistry.8 In this approach, the cellulosic backbone was prepared on resin via automated glycan assembly (AGA),10,11 followed by in-solution post-AGA manipulations to introduce the pEtN groups regiospecifically along the backbone. Upon functionalization, ester hydrolysis and hydrogenolysis of the remaining protecting groups (PGs), afforded the pEtN cellulose carrying an amino linker at the reducing end. Due to the zwitterionic nature of the pEtN moieties, the post-AGA steps (i.e. phosphorylation and PGs removal) required extremely long reaction times and multiple purifications. Moreover, the lability of the pEtN groups mandated cautious manipulations. As a result, individual targets required customized reaction conditions.
The introduction of the pEtN groups directly on solid support has several advantages and could speed up the synthetic procedure.11 On resin protocols were previously demonstrated for sulfated12 as well as phosphorylated13,14 glycans (Fig. 1B), increasing the efficiency of the synthetic process while easing the deprotection and purification steps. Here, we report a general method for the on resin synthesis of pEtN cellulose. The protocol features phosphorylation and hydrolysis of ester PGs to yield the solid-bound semi-protected intermediate. Lengthy purifications are substituted with repetitive washing steps to quickly remove excess reagents. Photo-cleavage followed by hydrogenolysis of the remaining PGs provides access to the target pEtN-functionalized oligosaccharide. The on resin approach also bypasses the requirement of an unnatural group at the reducing end, granting access to native free reducing glycans.
Translating glycan synthesis from solution-phase to solid-phase carries different challenges, such as finding compatible solvents/reagents which can ensure resin swelling and allow for nearly quantitative reaction conversion.11 Reaction monitoring poses additional hurdles. To develop a general on resin pEtN modification protocol, resin-bound glucose 6 bearing a hydroxyl group at the C-6 position was prepared (see ESI†). This model substrate served to systematically optimize the (i) on-resin phosphorylation, (ii) on-resin ester hydrolysis, (iii) photocleavage to release the compound from the solid support, and (iv) hydrogenolysis of the remaining PGs. We analyzed two different approaches based on either H-phosphonate (Fig. 2, Path A) or phosphoramidite (Fig. 2, Path B) chemistry.8,15–17 The choice of P(III) reagents, requiring an additional oxidation step to obtain the target P(V) product, was based on the abundant literature on nucleic acid synthesis.15,18 P(III) reagents are generally more reactive than P(V) reagents, making those attractive for solid phase approaches.18,19
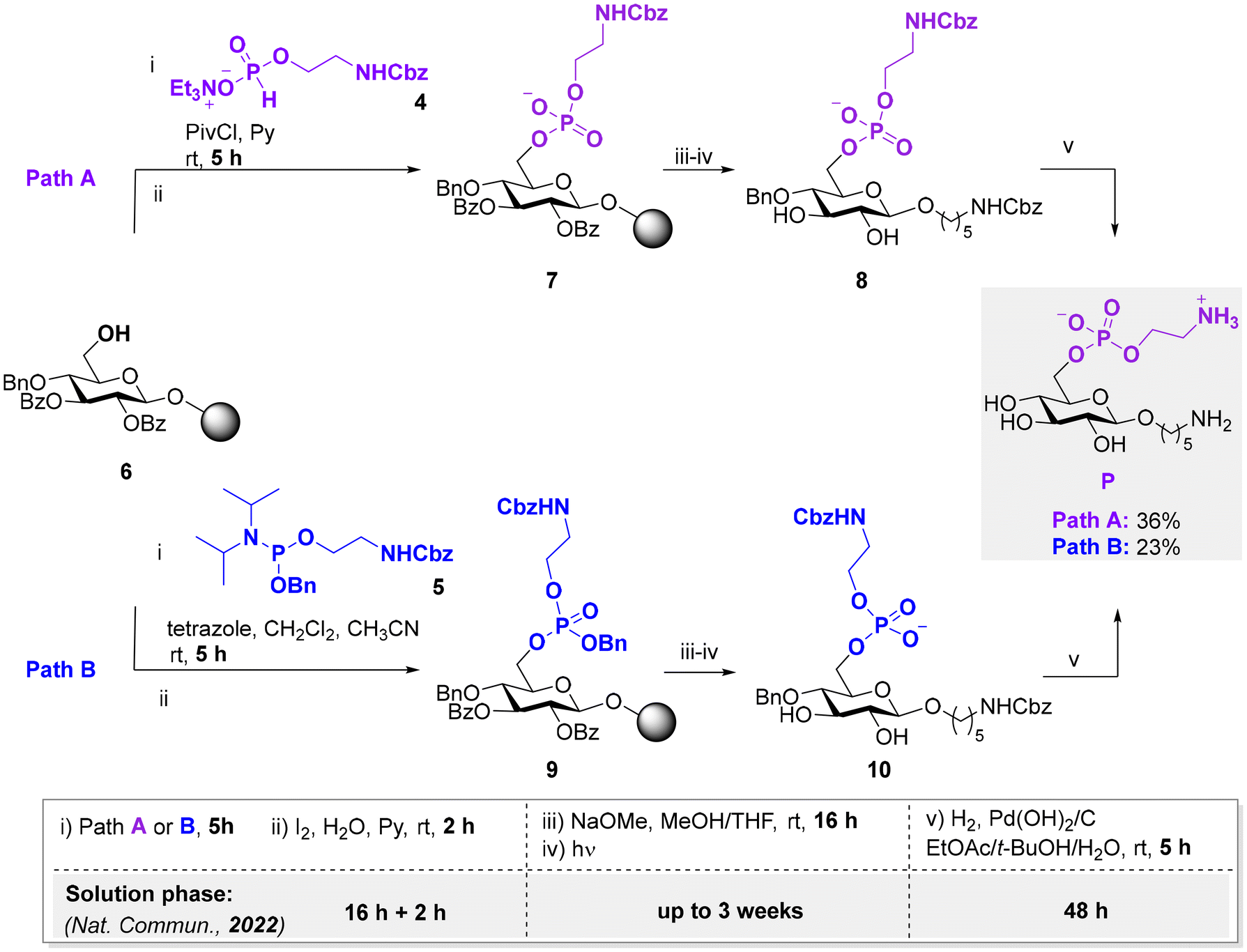 | ||
| Fig. 2 On resin protocols to obtain pEtN-functionalized glucose P using the H-phosphonate (Path A) or the phosphoramidite (Path B) approach. Reaction times are highlighted in comparison to the previously reported solution-phase approach.8 The resin bound starting material 6 was prepared by AGA, for synthetic details we refer to the ESI.† | ||
The formation of the phosphorous–oxygen (P–O) bond via Path A required the condensation of 6 with Cbz-protected ethanolamine H-phosphonate 4, promoted by pivaloyl chloride (PivCl). Aqueous oxidation using iodine (20 equiv.) in a 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pyridine/water mixture was subsequently performed to produce the phosphate diester 7. Reaction optimization (Table S1†) showed complete phosphorylation upon treating the solid bound substrate 6 with 5 equiv. of 4 over 5 h (Table S1, entry 4†). The benzoyl (Bz) esters in anionic compound 7 were hydrolyzed with sodium methoxide, before photocleavage in 1:9 MeOH/CH2Cl2 mixture afforded the semi-deprotected compound 8. The removal of the benzyl (Bn) and carboxybenzyl (Cbz) groups was performed by hydrogenolysis in the presence of Pd(OH)2/C. Upon completion, the reaction mixture was treated with thiourea as palladium scavenger, to ensure the release of the target compound P from the catalyst (Table S1, entry 9†).12 The catalyst was filtered and the product purified by reverse phase preparative HPLC. Phosphorylated compound P was isolated in a 36% overall yield over five steps (including AGA and post-AGA manipulations).
:1 pyridine/water mixture was subsequently performed to produce the phosphate diester 7. Reaction optimization (Table S1†) showed complete phosphorylation upon treating the solid bound substrate 6 with 5 equiv. of 4 over 5 h (Table S1, entry 4†). The benzoyl (Bz) esters in anionic compound 7 were hydrolyzed with sodium methoxide, before photocleavage in 1:9 MeOH/CH2Cl2 mixture afforded the semi-deprotected compound 8. The removal of the benzyl (Bn) and carboxybenzyl (Cbz) groups was performed by hydrogenolysis in the presence of Pd(OH)2/C. Upon completion, the reaction mixture was treated with thiourea as palladium scavenger, to ensure the release of the target compound P from the catalyst (Table S1, entry 9†).12 The catalyst was filtered and the product purified by reverse phase preparative HPLC. Phosphorylated compound P was isolated in a 36% overall yield over five steps (including AGA and post-AGA manipulations).
The phosphoramidite approach (Path B) is another common method to form P–O on solid-phase.18,19 Treatment of compound 6 with phosphoramidite 5 either in the presence of 1H-tetrazole or 5-benzylthio-1H-tetrazole (BTT) as coupling reagent resulted in the P(III) phosphite intermediate, which was subsequently oxidized to afford compound 9 (Table S2, entries 11 and 12†). At this stage, the lability of the phosphate triester in basic conditions13 prompted us to explore milder hydrolysis conditions. However, treatment with lithium hydroxide (LiOH) led to no conversion, even after long reaction times (Table S2, entry 13†). Ester hydrolysis was finally accomplished with sodium methoxide, followed by photocleavage in 1:9 MeOH/CH2Cl2 mixture to afford compound 10. Hydrogenolysis, treatment with thiourea, filtration, and purification furnished P in a 23% yield over five steps. Overall, both methods appeared promising for the synthesis of pEtN-functionalized glycans in solid phase. The entire process (i.e., AGA and post-AGA) required less than two days, in contrast to the long post-AGA reaction times of the solution phase approach8 (Fig. 2, table). Path A resulted in higher isolated yields than Path B and therefore it was chosen for the synthesis of more complex glycans.
To test the applicability of the on resin protocol, nine pEtN cellulose analogues were constructed, with different backbone lengths and pEtN substitution patterns. All glycan backbones, were assembled using AGA from BB1a and BB2a (Fig. 3A, see ESI for complete protocols†). BB2a was strategically introduced within the backbone at the position to be substituted with the pEtN group, permitting the specific liberation of the C-6 hydroxyl group upon Lev deprotection. The optimized phosphorylation procedure (Path A) worked smoothly for all primary alcohols, regardless of the glycan sequence. Increasing the amount of phosphorylating reagent 4 (5 equiv. per hydroxyl group) permitted to install multiple pEtN groups on the same glycan (Fig. 3A). Hydrolysis of the ester groups was completed using sodium methoxide within 16 h for all targets. Reaction progress could be monitored by either ESI-MS or MALDI analysis on a minute reaction sample after microcleavage. A 1:9 MeOH/CH2Cl2 mixture was utilized for the photocleavage of all structures, ensuring good solubility of the cleaved compound while maintaining high resin swelling. LH-20 size exclusion chromatography (1:1 MeOH/CH2Cl2) was conducted to eliminate residual side-products before subjecting the semi-deprotected intermediate to hydrogenolysis and thiourea treatment. Excess thiourea was removed using LH-20 size exclusion chromatography (1:1 MeOH/H2O). A final reverse phase preparative HPLC furnished the target structures in overall yields between 2–20% (Fig. 3B).
 | ||
| Fig. 3 (A) On resin synthesis of pEtN oligosaccharide (PA)3 starting from photolabile linker L2 and BB1-BB2. For synthetic details, we refer to the ESI.† (B) Collections of pEtN oligosaccharides prepared in this study and their isolated yields. The yield of the target is shown in purple and side-products containing α(1,4) in dashed-blue. (C) 1H NMR spectra of APA4 obtained from the thioglycoside BBs containing unwanted α(1,4) linkages (blue) vs APA4 obtained from glycosyl phosphate BBs (purple). | ||
NMR analysis of some sequences showed the presence of trace amounts of oligomers containing the undesired α(1,4) linkages (Fig. 3C). While these side-products could be removed with an additional HPLC purification, they decreased the isolated yields and complicated purifications. These side-products could be connected with the dioxane required for thioglycoside glycosylation during AGA20 (see Module C1 in the ESI†). Thus, we repeated the synthesis using phosphate donors BB1b and BB2b, whose activation is performed in pure CH2Cl2 (see Module C2 in the ESI†). The use of phosphate donors suppressed the formation of the α(1,4)-containing side-products (Fig. 3C), simplifying the isolation of pure pEtN celluloses.
Conclusions
We developed an on-resin approach for the synthesis of zwitterionic pEtN cellulose. On resin phosphorylation could be performed using either H-phosphonate or phosphoramidite reagents (Path A or B, respectively). Path A was showcased in the synthesis of ten oligosaccharides with different amount and pattern of pEtN. The on resin approach shortened the synthetic time and eased the purification steps, allowing for fast access to pEtN celluloses. The evaluation of these pEtN celluloses as substrates for Bcs or as functional modulators of bacterial biofilm is underway.The pEtN moiety is a recurrent modification of bacterial glycans.21 Our method could provide fast access to new synthetic probes to clarify different aspects of glycobiology. Moreover, the zwitterionic nature of pEtN could offer a tool to modulate mechanical properties of cellulosic materials, opening new avenues for tailor-made biomaterials.3
Author contributions
M. D. conceived this project. J. Y. H. developed the protocols and synthesized all the target compounds. M. D. supervised the project. J. Y. H. and M. D. contributed to and discussed the manuscript.Data availability
All experimental and characterisation data are available in the ESI.† Raw data for NMR analysis can be downloaded from https://doi.org/10.17617/3.4G3BOJ, Edmond.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Max Planck Society, the Max Planck Queensland Centre on the Materials Science of Extracellular Matrices, and the German Federal Ministry of Education and Research (BMBF, grant number 13XP5114) for generous financial support. J. Y. H. acknowledges the International Max Planck Research School on Multiscale Bio-Systems for funding. Open Access funding provided by the Max Planck Society.References
- W. Thongsomboon, D. O. Serra, A. Possling, C. Hadjineophytou, R. Hengge and L. Cegelski, Science, 2018, 359, 334–338 CrossRef CAS PubMed.
- E. C. Hollenbeck, A. Antonoplis, C. Chai, W. Thongsomboon, G. G. Fuller and L. Cegelski, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10106–10111 CrossRef CAS PubMed.
- J. Jeffries, G. G. Fuller and L. Cegelski, Microbiol. Insights, 2019, 12, 1–5 Search PubMed.
- J. M. Nguyen, R. E. Moore, S. K. Spicer, J. A. Gaddy and S. D. Townsend, ChemBioChem, 2021, 22, 2540–2545 CrossRef CAS PubMed.
- A. C. Anderson, A. J. N. Burnett, L. Hiscock, K. E. Maly and J. T. Weadge, J. Biol. Chem., 2020, 295, 6225–6235 CrossRef CAS PubMed.
- S. Cord-Landwehr and B. M. Moerschbacher, Fungal Biol. Biotechnol., 2021, 8, 19–26 CrossRef CAS PubMed.
- C. I. Gama, S. E. Tully, N. Sotogaku, P. M. Clark, M. Rawat, N. Vaidehi, W. A. Goddard, A. Nishi and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2006, 2, 467–473 CrossRef CAS PubMed.
- T. Tyrikos-Ergas, S. Gim, J. Huang, S. Pinzón Martín, D. Varón Silva, P. H. Seeberger and M. Delbianco, Nat. Commun., 2022, 13, 3954–3961 CrossRef CAS.
- R. D. Cummings, M. Etzler, M. G. Hahn, A. Darvill, K. Godula, R. J. Woods, L. K. Mahal, L. K. M. Richard, D. Cummings, M. Etzler, M. G. Hahn, A. Darvill, K. Godula, R. J. Woods, R. D. Cummings, M. Etzler, M. G. Hahn, A. Darvill, K. Godula, R. J. Woods and L. K. Mahal, Glycan-Recognizing Probes as Tools, 4th edn, 2022, vol. 130 Search PubMed.
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- J. Huang and M. Delbianco, Synthesis, 2023, 1337–1354 CAS.
- T. Tyrikos-Ergas, E. T. Sletten, J.-Y. Huang, P. H. Seeberger and M. Delbianco, Chem. Sci., 2022, 13, 2115–2120 RSC.
- E. T. Sletten, J. Danglad-Flores, S. Leichnitz, A. Abragam Joseph and P. H. Seeberger, Carbohydr. Res., 2022, 511, 108489–108491 CrossRef CAS PubMed.
- E. T. Sletten, G. Fittolani, N. Hribernik, M. C. S. Dal Colle, P. H. Seeberger and M. Delbianco, ACS Cent. Sci., 2024, 10, 138–142 CrossRef CAS PubMed.
- Y. H. Tsai, S. Götze, I. Vilotijevic, M. Grube, D. V. Silva and P. H. Seeberger, Chem. Sci., 2013, 4, 468–481 RSC.
- K. Puri and S. S. Kulkarni, Org. Lett., 2021, 23, 7083–7087 CrossRef CAS PubMed.
- J. M. Nguyen and S. D. Townsend, Org. Lett., 2021, 23, 5922–5926 CrossRef CAS PubMed.
- S. Roy and M. Caruthers, Molecules, 2013, 18, 14268–14284 CrossRef PubMed.
- A. F. Sandahl, T. J. D. Nguyen, R. A. Hansen, M. B. Johansen, T. Skrydstrup and K. V. Gothelf, Nat. Commun., 2021, 12, 2760–2766 CrossRef CAS PubMed.
- M. Guberman, M. Bräutigam and P. H. Seeberger, Chem. Sci., 2019, 10, 5634–5640 RSC.
- K. Paschinger and I. B. H. Wilson, Glycoconjugate J., 2020, 37, 27–40 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01121j |
| This journal is © The Royal Society of Chemistry 2024 |