
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward the stereochemical assignment of euvesperins A and B: total synthesis of the possible structures of the natural products†
Kenichi
Kobayashi
 *a,
Yusuke
Honma
b,
Kosaku
Tanaka
III
*b,
Momoko
Suzuki
ab,
Kazuhiko
Takatori
b and
Hiroshi
Kogen
b
*a,
Yusuke
Honma
b,
Kosaku
Tanaka
III
*b,
Momoko
Suzuki
ab,
Kazuhiko
Takatori
b and
Hiroshi
Kogen
b
aGraduate School of Pharmaceutical Sciences, Health Sciences University of Hokkaido, 1757 Kanazawa, Tobetsu, Ishikari, Hokkaido 061-0293, Japan. E-mail: kkobayashi@hoku-iryo-u.ac.jp
bGraduate School of Pharmaceutical Sciences, Meiji Pharmaceutical University, 2-522-1 Noshio, Kiyose, Tokyo 204-8588, Japan
First published on 16th August 2024
Abstract
The possible structures of euvesperins A and B were synthesized. The results of our synthesis suggest that euvesperin A may be a mixture of the (2R,3R,4S,7S) and (2S,3S,4R,7S) isomers and euvesperin B may be a mixture of the (2R,3S,4S,7S) and (2S,3R,4R,7S) isomers in consideration of their putative biosynthetic pathways.
In 2016, euvesperins A (1) and B (2) were isolated from Metarhizium sp. FKI-7236 by Ōmura and Shiomi's research group (Fig. 1)1,2 and identified as new circumventors of arbekacin resistance in MRSA as part of their ongoing research.3–5 The structures of compounds 1 and 2 were deduced based on extensive NMR studies, which indicated that both of them were formed as diastereomeric mixtures at the C4 position. However, the relative and absolute configurations of the C2, C3, and C7 stereocenters remained unclear. Thus, determination of the stereochemical configurations of 1 and 2 has been strongly anticipated.
 | ||
| Fig. 1 Reported structures of euvesperins A (1) and B (2). | ||
Previously, we completed the first total synthesis of L-755,807 (3), which has a molecular structure that is very similar to that of euvesperin A (Scheme 1).6–8 In our synthetic approach to 3, a novel highly diastereoselective Darzens reaction between α-silyloxy aldehyde 4 and di-tert-butyl bromomalonate was developed,9,10 and late-stage coupling of ring segment 6 and side-chain segment 7 was accomplished using the Horner–Wadsworth–Emmons (HWE) reaction to effectively produce the desired compound 3.
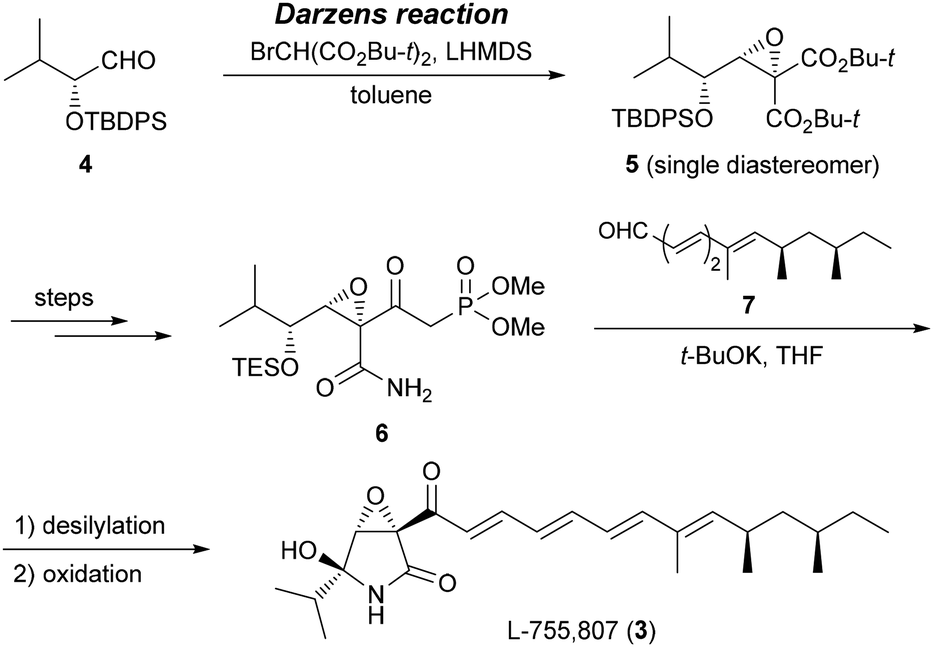 | ||
| Scheme 1 Our previous work on the synthesis of L-755,807 (3). | ||
Comparing the structures of euvesperin A and L-755,807, we presumed that both of them have the same epoxide configuration. Hence, a mixture of (4S,7S)-8 and (4R,7S)-9 or a mixture of (4S,7R)-10 and (4R,7R)-11 were proposed as the putative structures of euvesperin A (Fig. 2), which could be prepared by a synthetic strategy similar to the one we used for L-755,807. Additionally, euvesperin B could be obtained via a biomimetic conversion from euvesperin A as reported in the literature,1 suggesting that its possible structures were a mixture of (4S,7S)-12 and (4R,7S)-13 or a mixture of (4S,7R)-14 and (4R,7R)-15, as depicted in Fig. 2. Thus, we directed our attention toward the total synthesis and stereochemical assignment of these natural products. In this paper, we describe the synthesis of the possible isomers of euvesperins A and B and discuss the stereochemistries of the natural products.
 | ||
| Fig. 2 Putative structures of euvesperins A and B. | ||
Our retrosynthetic analysis of the putative structures of euvesperins A and B is shown in Scheme 2. As mentioned above, euvesperin B can be biomimetically derived from euvesperin A, and then we planned to obtain the four putative structures of euvesperin A 8–11 from enone 17 by a non-stereoselective Mukaiyama hydration, and 17 could be retrosynthetically disconnected via an HWE reaction into n-hexanal (18) and phosphonate 19, which could be prepared from known Weinreb amide 20.6–8
 | ||
| Scheme 2 Retrosynthetic analysis of euvesperins A and B. | ||
Our synthesis commenced with the preparation of enone 17 from the known TES-protected Weinreb amide 206–8 (Scheme 3). According to a procedure from the literature,6–8 amide 20 was similarly converted into phosphonate 19 in 74% yield, which was then subjected to attempted coupling with n-hexanal (18) via an HWE reaction. After some unsuccessful experiments, we realized that phosphonate 19 to be unreactive, presumably owing to the bulkiness around the reactive site.
 | ||
| Scheme 3 Synthetic efforts toward enone 17. | ||
Next, we changed the enone-type intermediate from 17 to aminal 24 or 26, which were synthesized from 216–8via aminal 22 (Scheme 4). Alcohol 21 was initially oxidized with Dess–Martin periodinane, and the resulting hemiaminal was immediately converted into aminal 22 as a single diastereomer. The stereostructure of the aminal portion of 22 was confirmed through NOESY experiments, which revealed a correlation between the methine proton and aminal O-methyl protons, allowing us to determine the stereochemistry. The coupling reaction of aminal 22 with vinyl iodide 2311 afforded enone 24 in 51% yield. With iodide 25,12 which has a terminal double bond, enone 26 was obtained in high yield (88%). Therefore, we elected to use enone 26 for the subsequent hydration step.
 | ||
| Scheme 4 Successful synthesis of enones 24 and 26. | ||
Enone 26 was next subjected to Mukaiyama hydration with cobalt (Co(acac)2) or manganese (Mn(dpm)3) catalysts (Scheme 5).13,14 All conditions afforded a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the desired alcohols (7S)- and (7R)-27, although the yields were only moderate (31–53%) and it proved very difficult to separate the two isomers.
:1 mixture of the desired alcohols (7S)- and (7R)-27, although the yields were only moderate (31–53%) and it proved very difficult to separate the two isomers.
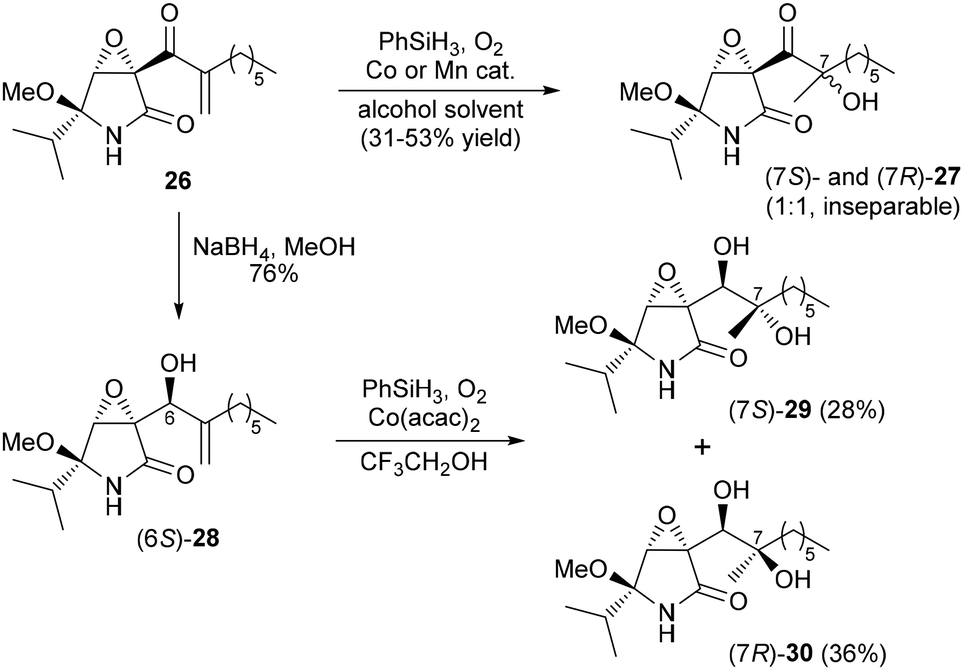 | ||
| Scheme 5 Preparation of the two isomers with respect to the C7 stereocenter. | ||
To overcome these issues, enone 26 was initially reduced with sodium borohydride in methanol to stereoselectively form allyl alcohol (6S)-28 in 76% yield.15 Mukaiyama hydration of (6S)-28 proceeded effectively to yield two diastereomeric diols, which were easily separated to furnish (7S)-29 and (7R)-30 in 28% and 36% yields, respectively.16
Having prepared the two isomers (7S)-29 and (7R)-30, the remaining tasks en route to our goal were oxidation of the C6 alcohol and hydrolysis of the aminal portion (Scheme 6). Accordingly, AZADOL oxidation17,18 of alcohol (7S)-29 was conducted to afford ketone (7S)-31 in 93% yield, which was finally hydrolyzed with aqueous trifluoroacetic acid (TFA) to furnish the 4S isomer of ketone 8 in 40% yield and the 4R isomer of ketone 9 in 5% yield. Similarly, (7R)-30 provided (4S,7R)-10 (43%) and (4R,7R)-11 (8%) in two steps.
 | ||
| Scheme 6 Completion of the synthesis of four possible isomers of euvesperin A. | ||
We performed NOESY experiments to determine the stereochemistry at the C4 position of the four synthetic compounds, and the key correlations are indicated in Scheme 6. As mentioned in the introduction, euvesperin A was reported to be a diastereomeric mixture at the C4 position.1 However, among the four synthetic compounds, (4S,7S)-8 and (4S,7R)-10 exhibited NMR spectra identical to those of the isomers of euvesperin A, which implies that the stereochemistry of euvesperin A was incorrectly assigned. Consequently, four possible combinations were proposed for euvesperin A: a mixture of 8 and 10, a mixture of 8 and the enantiomer (ent) of 10, a mixture of ent-8 and 10, or a mixture of ent-8 and ent-10.19
Natural euvesperin A is anticipated to be biosynthetically produced from natural products PI-09020 with the (7S) configuration via epoxidation (Scheme 7). Therefore, we consider that natural euvesperin A may be a combination of 8 and ent-10.
 | ||
| Scheme 7 Proposed biosynthesis of euvesperin A. | ||
With the proposed structures of euvesperin A in hand, we turned our attention to the synthesis of euvesperin B (Scheme 8). Treatment of a mixture of (4S,7S)-8 and (4R,7S)-9 with sodium methoxide in methanol gave (4S,7S)-12 in 84% yield. Interestingly, (4S,7S)-12 was obtained as a single diastereomer even though the starting materials 8 and 9 were a diastereomeric mixture at the C4 position. Similarly, (4S,7R)-14 was formed as a sole isomer from a diastereomeric mixture of (4S,7R)-10 and (4R,7R)-11. The stereochemistry of the products was deduced from the NOESY correlations shown in Scheme 8. The NMR spectra of synthetic (4S,7S)-12 and (4S,7R)-14 were identical to those of the isomers of euvesperin B. Like euvesperin A, euvesperin B was previously reported to be a diastereomeric mixture at the C4 position; nevertheless, our work has ruled this out.
 | ||
| Scheme 8 Synthesis of proposed structure of euvesperin B. | ||
To determine the correct structure of euvesperin B, we considered which of the combinations could be euvesperin B: a mixture of 12 and 14, a mixture of 12 and ent-14, a mixture of ent-12 and 14, or a mixture of ent-12 and ent-14.19 As described in the literature, euvesperin A is presumed to be a biosynthetic precursor of euvesperin B.1 Thus, euvesperin B should have the (S) configuration at C7, as in the case of euvesperin A. Consequently, we speculate that natural euvesperin B may be a mixture of 12 and ent-14 (Scheme 9).
 | ||
| Scheme 9 Proposed biosynthesis of euvesperin B. | ||
Conclusions
In conclusion, we completed the total synthesis of four possible structures of euvesperin A. By consideration of the putative biosynthetic pathway of this natural product, euvesperin A is proposed to be a mixture of compounds 8 and ent-10. In addition, two possible structures of euvesperin B were synthesized, and we propose that natural euvesperin B may be a mixture of compounds 12 and ent-14.Data availability
Raw data were generated at Meiji Pharmaceutical University and Health Sciences University of Hokkaido. Derived data which supports the results of this study are available from the corresponding authors (Kenichi Kobayashi and Kosaku Tanaka III) upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 20K06951 and The Akiyama Life Science Foundation, and partially supported by a grant from the Dementia Drug Resource Development Center, Project S1511016, the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.Notes and references
- M. Shiina, T. Suga, Y. Asami, K. Nonaka, M. Iwatsuki, S. Ōmura and K. Shiomi, J. Antibiot., 2016, 69, 719–722 CrossRef CAS PubMed.
- H. Iwasaki, T. Tokiwa, M. Shiina, Y. Asami, K. Shiomi, S. Ōmura and K. Nonaka, Mycoscience, 2019, 60, 313–318 CrossRef.
- M. Iwatsuki, T. Ishimori, T. Yamamoto, K. Takata, M. Mori, K. Nonaka, R. Masuma, Y. Hayakawa, H. Hanaki, K. Shiomi and S. Ōmura, Tetrahedron, 2011, 67, 6644–6648 CrossRef CAS.
- K. Takata, M. Iwatsuki, T. Yamamoto, T. Shirahata, K. Nonaka, R. Masuma, Y. Hayakawa, H. Hanaki, Y. Kobayashi, G. A. Petersson, S. Ōmura and K. Shiomi, Org. Lett., 2013, 15, 4678–4681 CrossRef CAS PubMed.
- T. Suga, M. Shiina, Y. Asami, M. Iwatsuki, T. Yamamoto, K. Nonaka, R. Masuma, H. Matsui, H. Hanaki, S. Iwamoto, H. Onodera, K. Shiomi and S. Ōmura, J. Antibiot., 2016, 69, 605–610 CrossRef CAS PubMed.
- K. Tanaka III, K. Kobayashi and H. Kogen, Org. Lett., 2016, 18, 1920–1923 CrossRef PubMed.
- K. Tanaka III, Y. Honma, C. Yamaguchi, L. Aoki, M. Saito, M. Suzuki, K. Arahata, K. Kinoshita, K. Koyama, K. Kobayashi and H. Kogen, Tetrahedron, 2019, 75, 1085–1097 CrossRef.
- K. Tanaka III, K. Kobayashi and H. Kogen, J. Synth. Org. Chem., Jpn., 2019, 77, 673–683 CrossRef.
- K. Tanaka III, K. Kobayashi, K. Takatori and H. Kogen, Tetrahedron, 2017, 73, 2062–2067 CrossRef.
- For other applications of the Darzens reaction to natural product synthesis, see: K. Tanaka III, K. Kobayashi and H. Kogen, Chem. Commun., 2021, 57, 9780–9783 RSC.
- D. H. R. Barton, G. Bashiardes and J. L. Fourrey, Tetrahedron, 1988, 44, 147–162 CrossRef CAS.
- N. Kamiya, Y. Chikami and Y. Ishii, Synlett, 1990, 675–676 CrossRef CAS.
- H. Shigehisa, Y. Suwa, N. Furiya, Y. Nakaya, M. Fukushima, Y. Ichihashi and K. Hiroya, Angew. Chem., Int. Ed., 2013, 52, 3646–3649 CrossRef CAS PubMed.
- P. Magnus, A. H. Payne, M. J. Waring, D. A. Scott and V. Lynch, Tetrahedron Lett., 2000, 41, 9725–9730 CrossRef CAS.
- The absolute configuration of the C6 stereocenter in (6S)-28 was deduced using the modified Mosher's method (please see the ESI†).
- The stereochemistry of the products (7S)-29 and (7R)-30 was established through NOESY experiments after conversion to the corresponding acetonides (please see the ESI†).
- M. Shibuya, Y. Sasano, M. Tomizawa, T. Hamada, M. Kozawa, N. Nagahama and Y. Iwabuchi, Synthesis, 2011, 3418–3425 CAS.
- Y. Iwabuchi, Chem. Pharm. Bull., 2013, 61, 1197–1213 CrossRef CAS PubMed.
- The optical rotation values of euvesperins A and B were not reported in ref. 1 and 2.
- A. Kawashima, Y. Yoshimura, N. Sakai, K. Kamigoori, T. Mizutani and S. Ōmura, Jpn. Pat. Appl, JP02062859A19900302, 1990 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01118j |
| This journal is © The Royal Society of Chemistry 2024 |