
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of unsymmetrical phosphorus disulfides†
Jeffrey
Ash
and
Jun Yong
Kang
 *
*
Department of Chemistry and Biochemistry, University of Nevada Las Vegas, 4505 S. Maryland Parkway, Las Vegas, Nevada 89154-4003, USA. E-mail: junyong.kang@unlv.edu
First published on 22nd August 2024
Abstract
A sulfur-mediated umpolung strategy employing N-thiosuccinimides and (EtO)2P(O)SH has been developed to synthesize unsymmetrical organophosphorus disulfides (P(O)–S–S motif). A pronucleophile (EtO)2P(O)SH, Brønsted acid and phosphorothioate nucleophile, converts N-thiosuccinimides into unsymmetrical phosphorus disulfides. This protocol achieves catalyst- and additive-free reaction conditions, uses a renewable solvent (EtOH), and avoids harsh reagents.
Introduction
Organophosphorus and organosulfur chemistry have attracted significant research interest due to their widespread applications.1 Especially, organophosphorus disulfides (P(O)–S–S) have received substantial attention in a plethora of fields since they are promising antioxidants and prodrugs as well as pesticides.2 For example, they serve as a key chemical motif of molecular umbrella–nucleoside conjugates (Fig. 1, A).2a The conjugates are amphiphilic molecules interconverting their status in response to environmental changes in cells. This amphiphilic feature of conjugates improves the lipid bilayer transport; thus, they have potential therapeutic applications such as drug delivery. In addition, bisphosphorothioates have shown antioxidant activities (Fig. 1, B).2b They act as metal-ion chelators (Fe, Cu) and radical scavengers. The oligonucleotide phosphorothioates demonstrated improved cellular uptake via a pseudo-disulfide exchange, which has generated a few FDA-approved drugs (Fig. 1, C).3 Furthermore, phosphorus disulfide derivatives have been used for potent pesticides (Fig. 1, D, E).2c These examples highlight several important applications of organophosphorus disulfides.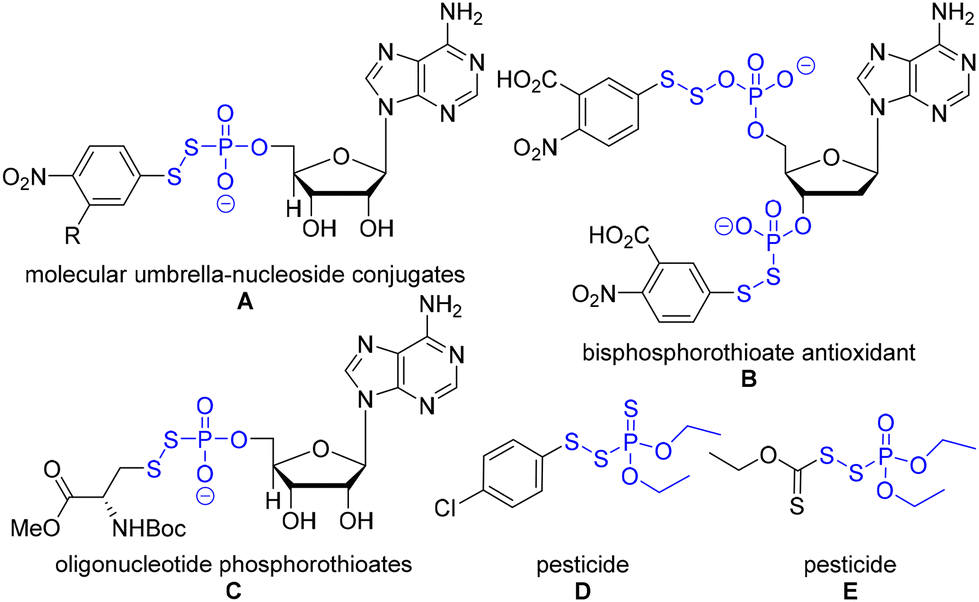 | ||
| Fig. 1 Representative examples of phosphorus disulfide. | ||
The synthesis of unsymmetrical disulfides poses challenges due to the undesired homodimers.4 To access unsymmetrical disulfides, there are three major approaches: an oxidative dehydrogenative coupling reaction, a masked strategy of a coupling reaction, and an umpolung strategy. The oxidative dehydrogenative coupling reaction employs two sterically distinct thiols to utilize the reactivity differences rendered by sterics and kinetics of oxidation.5 A masked strategy of coupling reactions, such as a copper-catalyzed Suzuki and Hiyama-type cross-coupling reaction, affords unsymmetrical disulfides.6,7 This approach demonstrated the late-stage functionalization of biomolecules under borane catalysis.8 Other catalysts such as rhodium, palladium, and N-fluorobenzenesulfonimide catalysts also have been used.9 In addition, the umpolung strategy has been employed to reduce the homodimer by-products. For example, thiol nucleophiles react with preactivated sulfur electrophiles: sulfenamides, thiosulfonate, mercaptobenzotriazole, mercaptobenzothiazole, phosphorothioate bromide, 2-pyridyl disulfide, thiosuccinimide, and Bunte salts.10 By overcoming the homodimer by-product challenges, significant progress on unsymmetrical disulfide synthesis has been made with a rational design of substrates and reactivity differences.
Despite the recent advance in synthesizing unsymmetrical disulfides, the synthetic approach to directly access the phosphorus disulfides is underdeveloped. Recently, the Cao group has reported a mechanochemical disulfur transfer between trisulfide dioxides and secondary phosphine oxides using ball milling to form P(O)–S–S bonds.11 The substrate scope to access the P(O)–S–S bond motif, however, is limited to alkyl dithioperoxoate; when aryl dithioperoxoates were used, an alternative thiophosphate product (P(O)–S) was generated via different reaction pathway. To the best of our knowledge, a synthetic method to access aryl phosphorus disulfides (P(O)–S–S–Ar unit) – a major functionality of the current applications (Scheme 1) – remains elusive.
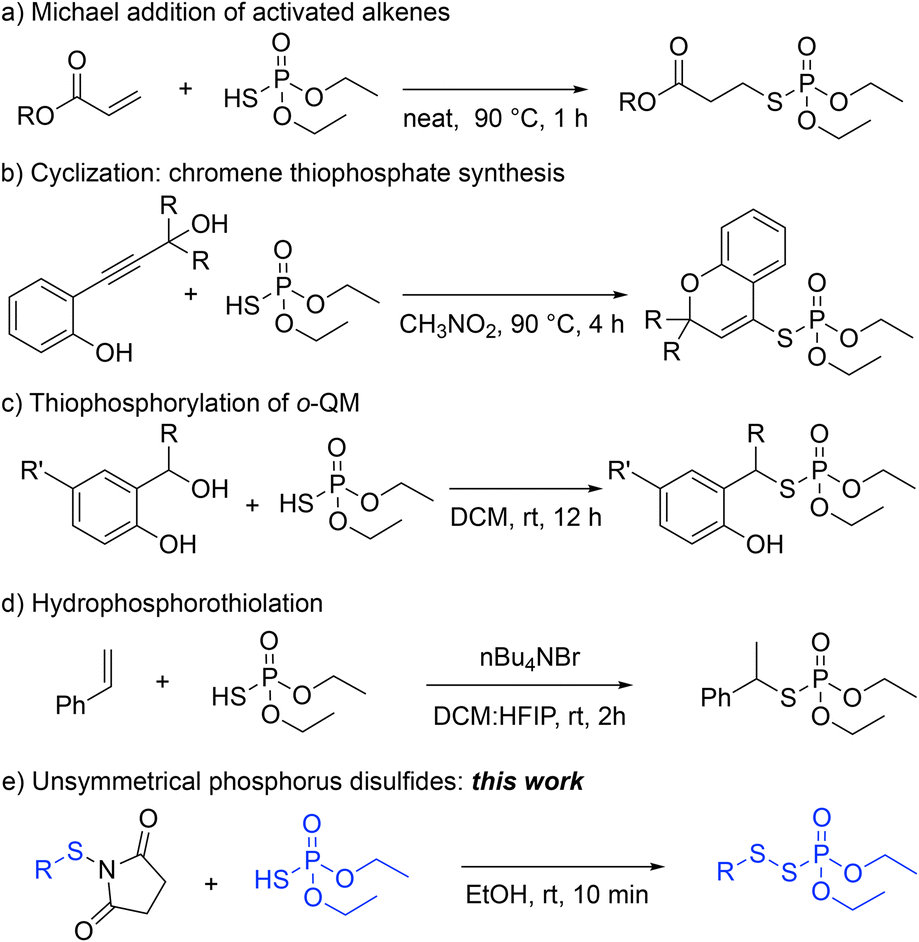 | ||
| Scheme 1 (EtO)2P(O)SH as a pronucleophile. | ||
Harnessing inherent chemical properties and reactivities creates new chemical space and contributes to green synthesis.12 These advantages have been achieved by multifunctional reagents and catalysts.13 Multifunctional reagents can eliminate the need for additives and allow for increased chemoselectivity.13 In this regard, phosphorothioic acid, (EtO)2P(O)SH, has served as a multifunctional reagent – Brønsted acid and phosphorothioate nucleophile – in different transformations (Scheme 1). For example, (EtO)2P(O)SH has been used in a Michael reaction of activated alkenes to synthesize functionalized thiophosphates (Scheme 1, a).14 The Xiao group also reported the utility of (EtO)2P(O)SH by coupling with a propargylic alcohol partner to construct S-(2H-chromen-4-yl) phosphorothioates and polycyclic thiophosphates via a cascade reaction as well as the synthesis of allenyl thiophosphates under elevated temperatures (Scheme 1, b).15 We also demonstrated a bifunctional role of (EtO)2P(O)SH in the thiophosphorylation reaction of in situ formed ortho-quinone methide (o-QM) to synthesize functionalized thiophosphates (Scheme 1, c).16 In addition, the Hajra group used hydrophosphorothiolation of alkenes to form benzyl phosphorothioates employing hexafluoroisopropanol (HFIP) to enhance the acidity of (EtO)2P(O)SH (Scheme 1, d).17 Furthermore, The Wu group used (EtO)2P(O)SH and a Ga(OTf)3 catalyst under photochemical conditions.18,19 Nevertheless, the synthesis of unsymmetrical organophosphorus disulfides (P(O)–S–S) using (EtO)2P(O)SH hasn't been reported (Scheme 1, e).
Since both thiophosphates and disulfides possess useful properties, small molecules containing P(O)–S–S bonds would be of great interest to chemists in academia and industry.2 Based on our previous work on searching for the bifunctionality of (EtO)2P(O)SH,16 serving as both Brønsted acid and phosphorothioate nucleophile, we hypothesized that this reagent would react with N-thiosuccinimides to form P(O)–S–S bond motif (Scheme 1, e).
Results and discussion
To test our hypothesis, we used N-thiosuccinimide 1a and phosphorothioic acid ((EtO)2P(O)SH) 2a as model substrates in ethanol (Table 1). The reaction gave the desired product 3a in a 95% yield (Table 1, entry 1). Other solvents also provided the product 3a in high yields (Table 1, entries 2–7). However, ethanol was selected since it is environmentally friendly and readily available through biomass fermentation.20 Importantly, the reaction conditions address many of the 12 principles of green chemistry as defined by the American Chemical Society.21 For example, the reaction is an atom-economical approach since no additives or excess reagents are required. In addition, the reaction is energy neutral as no heating or cooling is necessary. Furthermore, the use of safer solvents and renewable feedstocks is achieved by ethanol. Lastly, the procedure runs without transition metal catalysts typically used for disulfide synthesis.
With the optimized reaction conditions in hand, the scope of N-thiosuccinimide electrophiles was evaluated to study the steric and electronic effects on the reaction outcome (Scheme 2). First, halogenated aryl N-thiosuccinimides 1b–1d generated the target products 3b–3d in high yields (93–96%). Aryl N-thiosuccinimides bearing electron-donating groups 1e, 1f (4-Me, 4-MeO) yielded the corresponding products 3e, 3f in 88% and 94%, respectively. Next, aryl N-thiosuccinimides 1g and 1h containing sterically demanding groups (2,5-dimethyl, 4-tertbutyl) afforded the desired products 3g, 3h in 90% yields. Alkyl N-thiosuccinimides 1i, 1j (cyclohexyl, n-hexyl) were also well tolerated, providing the target products 3i, 3j in 90% and 79% yields, respectively. Additionally, benzylic N-thiosuccinimide 1k furnished the product 3k in an 89% yield; no competing substitution reactions at the benzylic carbon on the product occurred. Benzylic N-thiosuccinimides containing halogens and electron donating groups 1l–1o (4-fluoro, 4-chloro, 2-chloro, and 4-tertbutyl) also generated the target products 3l–3o in high yields (83–95%). Furthermore, acyl N-thiosuccinimide 1p was tolerated and furnished the product 3p in a high yield of 90%. Heteroaryl N-thiosuccinimides 1q, 1r, however, provided the corresponding products 3q, 3r as inseparable mixtures from succinimide byproduct. Overall, the reaction provided the desired products 3a–3p in high yields and the steric, electronic factors of the N-thiosuccinimides were well tolerated.
 | ||
Scheme 2 Substrate scope of N-thiosuccinimide. Reaction conditions: 1 (0.1 mmol) and 2a (0.1 mmol) in EtOH (0.5 mL) for 10 min. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reported yields are isolated yields. ND: not determined. The reported yields are isolated yields. ND: not determined. | ||
Next, the scope of the phosphorothioic acid nucleophile was examined (Scheme 3). Thioacids with different alkoxy substituents 2b, 2c, and 2d (butyl, isopropyl, and propargyl) were treated with N-thiosuccinimide 1a and they were well tolerated to give the target products 4a, 4b, and 4c in 79%, 71%, and 84% yields, respectively. However, when diphenylthiophosphinic acid was tested, no desired product was formed even at elevated temperatures, presumably due to a weaker acidity than 2a (pKa = −5.14).22
 | ||
| Scheme 3 Substrate scope of phosphorothioic acids. Reaction conditions: 1a (0.1 mmol) and 2 (0.1 mmol) in EtOH (0.5 mL) for 10 min. aThe reported yields are isolated yields. | ||
Having studied an array of substrates, the synthetic utility of this transformation was evaluated (Scheme 4). When beta naphthol 5a was treated with phosphorus disulfide 3a, it was successfully thiolated to give thionaphthol 6a. Imidazopyridine 5b was also effectively thiolated with 3a to give 6b under the catalytic conditions. The imidazopyridine structural motif has shown antibacterial properties.23 In addition, the phosphorus disulfide 3p was used for sulfurization of triphenylphosphine 5c to triphenylphosphine sulfide 6c. These results demonstrated that phosphorus disulfides 3 can serve as electrophilic sulfur sources to functionalize arenols, heteroarenes, and phosphines.
 | ||
| Scheme 4 Synthetic utility. | ||
To further demonstrate the utility of this methodology, we carried out the synthesis of known pesticides 7a, 7b (Scheme 5).2c Pesticide 7a was synthesized by treating N-thiosuccinimide 1d and phosphorodithioic acid 2e under the standard reaction conditions. Pesticide 7b was also readily prepared from N-thiosuccinimide 1q and phosphorothioic acid 2a under elevated thermal conditions. These results provide a direct application of this methodology toward various pesticide syntheses.
 | ||
| Scheme 5 Pesticide synthesis. | ||
To gain insight into the reaction mechanism, control experiments were performed (Scheme 6). The reaction between N-thiosuccinimide 1a and diphenyl phosphoric acid 2f (pKa = −3.95)24 did not yield the target product 4d. This outcome reveals that a stronger acid and more nucleophilic thiolate are necessary for a successful reaction. Next, the role of the proton was tested by using the potassium phosphorothioate 2g. This phosphorothioate, however, generated the desired product 3a in a poor yield (29%) compared to 2a (Scheme 2). This result suggests that a proton donor (Brønsted acid) is necessary to increase the electrophilicity and the reactivity of 1a.
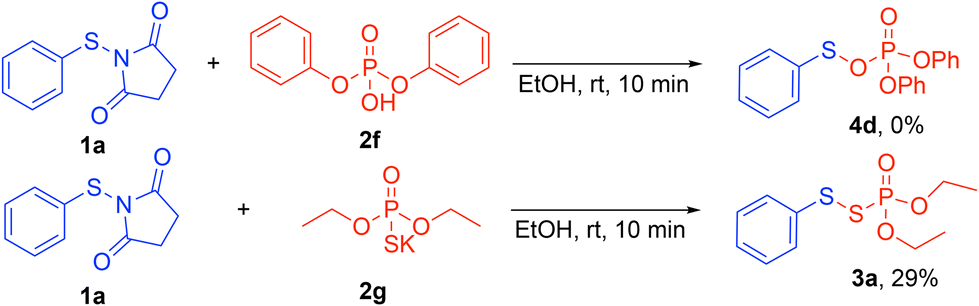 | ||
| Scheme 6 Control experiments. | ||
Based on our previous work and the control experiments,16 a plausible mechanism is proposed in Scheme 7. Concomitantly, the oxygen atom on 1a is protonated by (EtO)2P(O)SH 2a and the resulting thiophosphate attacks the protonated 1a to give succinimide and the target product 3a.
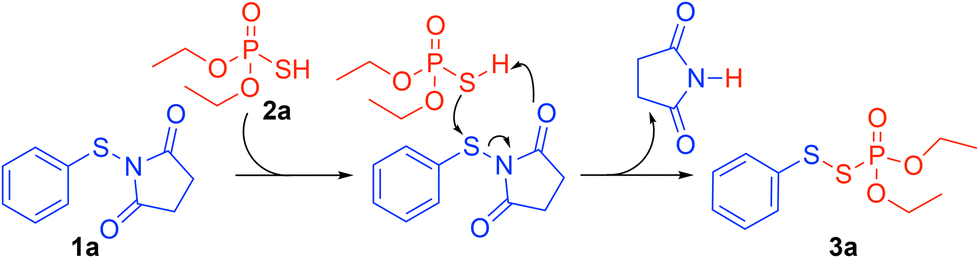 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusion
In summary, we have developed a new mild method to synthesize unsymmetrical organophosphorus disulfides (P(O)–S–S) from (EtO)2P(O)SH. This method addresses the persistent homodimerization issues in unsymmetrical disulfide synthesis. In addition, a wide range of substrate scopes of both N-thiosuccinimide sulfur electrophiles and phosphorothioic acids were well tolerated. Furthermore, the synthetic utility of unsymmetrical organophosphorus disulfides demonstrated an efficient thiolation of arenol, heteroarene, and phosphine as well as the synthesis of several pesticides. Finally, the control experiments support a plausible mechanism. Further studies to understand these novel phosphorus disulfide compounds are under investigation, and they will be reported in due course.Experimental section
General information
All reactions were carried out under air atmosphere in oven-dried glassware with magnetic stirring bar. Dry solvents (THF, toluene, ACN, diethyl ether, and DCM) were obtained by solvent purification system under argon. All commercially available reagents were used as received without further purification. The tubes used for the reaction were showed in Fig. S1.† Purification of reaction products was carried out by flash column chromatography using silica gel 60 (230–400 mesh). Analytical thin layer chromatography was performed on 0.25 mm aluminum-backed silica gel 60-F plates. Visualization was accompanied with UV light and KMnO4 solution. Concentration under reduced pressure refers to the removal of volatiles using a rotary evaporator attached to a dry diaphragm pump (10–15 mm Hg) followed by pumping to a constant weight with an oil pump (<300 mTorr). Infrared (IR) spectra were recorded on an IR spectrometer with KBr wafers or a film on KBr plate. High-resolution mass spectra (HRMS) were recorded on LCMS-IT-TOF mass spectrometer using ESI (electrospray ionization) or APCI (Atmospheric Pressure Chemical Ionization). 1H NMR spectra were recorded in CDCl3 on 400 MHz NMR spectrometer. The 1H chemical shifts are referenced to residual solvent signals at δ 7.26 (CHCl3) or δ 0.00 (TMS). 1H NMR coupling constants (J) are reported in Hertz (Hz) and multiplicities are indicated as follows: s (singlet), bs (broad singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), dt (doublet of triplets), td (triplet of doublets), tt (triplet of triplets). 13C NMR spectra were proton decoupled and recorded in CDCl3 on 100.5 MHz NMR spectrometer. The 13C chemical shifts are referenced to solvent signals at δ 77.16 (CDCl3). 31P NMR spectra were proton decoupled and recorded in CDCl3 on 162 MHz NMR spectrometer. 31P chemical shifts are reported relative to 85% H3PO4 (0.00 ppm) as an external standard.General procedure: synthesis of phosphrous disulfide 3 and 4
To a solution of thiophosphoric acid 2 (0.10 mmol) in ethanol (0.5 mL) was added N-thiosuccinimide 1 (0.10 mmol) at room temperature. The reaction mixture was stirred for 10 minutes. After stirring for 10 minutes at room temperature, the reaction mixture was concentrated under reduced pressure and subjected to column chromatography on silica gel to give the corresponding phosphorus disulfide product 3 or 4.Synthetic utility: synthesis of 6a–6c
To a solution of 3a (0.1 mmol) in DCM (0.5 mL) was added beta naphthol 5a (0.1 mmol), followed by iodine (0.01 mmol). The reaction was stirred at room temperature for 12 hours. After stirring for 12 hours, the reaction mixture was concentrated under reduced pressure, and directly purified by column chromatography (hexane:EtOAc = 8:2) to give 6a (10.8 mg, 43%).
To a solution of 3a (0.1 mmol) in EtOH (0.5 mL) was added imidazopyridine 5b (0.2 mmol), followed by an ammonium iodide (0.01 mmol). The reaction was refluxed for 12 hours. After stirring for 12 hours, the reaction mixture was concentrated under reduced pressure, and directly purified by column chromatography (hexane:EtOAc = 7:3) to give 6b (27.4 mg, 91%).
To a solution of 3p (0.1 mmol) in DCM (0.5 mL) was added triphenylphosphine (0.1 mmol). The reaction was stirred at room temperature for 12 h. After stirring for 12 hours, the reaction mixture was concentrated under reduced pressure, and directly purified by column chromatography (hexane:EtOAc = 95:5) to give 6c (16.7 mg, 95%).
Pesticide synthesis of 7a and 7b
To a solution of dithiophosphoric acid 2e (0.2 mmol) in EtOH (1.0 mL) was added thiosuccinimide 1d (0.2 mmol). The reaction was stirred for 10 minutes. After stirring for 10 minutes, the reaction mixture was concentrated under reduced pressure, and directly purified by column chromatography (hexane:DCM = 8:2) to give 7a (49.3 mg, 75%).
To a solution of thiophosphoric acid 2a (0.1 mmol) in EtOH (0.5 mL) was added thiosuccinimide 1q (0.1 mmol). The reaction was refluxed for 2 hours. After stirring for 2 hours, the reaction mixture was concentrated under reduced pressure, and directly purified by column chromatography (hexane:EtOAc = 7:3) to give 7b (22.6 mg, 78%).
Compound characterization
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Knowledge Fund which is administered by the Nevada Governor's Office of Economic Development (GOED). The Troesh Center Grant and the University of Nevada Las Vegas are acknowledged for their support of this work. Shishir Bhowmik is acknowledged for his support on preparation of some substrates.References
- (a) X. Liu, L. Zhou, R. Yang, X.-R. Song and Q. Xiao, Adv. Synth. Catal., 2023, 365, 2280–2298 CrossRef CAS; (b) S.-W. Rhee, R. P. Iyer, J. E. Coughlin, S. Padmanabhan, J. P. Malerich and M. J. Tanga, J. Labelled Compd. Radiopharm., 2012, 55, 197–200 CrossRef CAS; (c) R. Xie, Q. Zhao, T. Zhang, J. Fang, X. Mei, J. Ning and Y. Tang, Bioorg. Med. Chem., 2013, 21, 278–282 CrossRef CAS PubMed; (d) H. Yan, X. Wang, R. KuoLee and W. Chen, Bioorg. Med. Chem. Lett., 2008, 18, 5631–5634 CrossRef CAS PubMed; (e) T. S. Kumar, T. Yang, S. Mishra, C. Cronin, S. Chakraborty, J.-B. Shen, B. T. Liang and K. A. Jacobson, J. Med. Chem., 2013, 56, 902–914 CrossRef CAS PubMed; (f) Y. Zhang, Y. Cao, Y. Chi, S. Chen, X. Zeng, Y. Liu, G. Tang and Y. Zhao, Adv. Synth. Catal., 2022, 364, 2221–2226 CrossRef CAS; (g) S. Demkowicz, J. Rachon, M. Daśko and W. Kozak, RSC Adv., 2016, 6, 7101–7112 RSC; (h) U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed; (i) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed; (j) C.-S. Jiang, W. E. G. Müller, H. C. Schröder and Y.-W. Guo, Chem. Rev., 2012, 112, 2179–2207 CrossRef CAS PubMed; (k) Z. Wu and D. A. Pratt, Nat. Rev. Chem., 2023, 7, 573–589 CrossRef PubMed.
- (a) V. Janout, B. Jing and S. L. Regen, Bioconjugate Chem., 2002, 13, 351–356 CrossRef CAS; (b) B. Levi Hevroni, A. H. Sayer, E. Blum and B. Fischer, Inorg. Chem., 2014, 53, 1594–1605 CrossRef CAS; (c) G. R. Price, E. N. Walsh and J. T. Hallett, US Pat., 3109770, 1963 Search PubMed.
- (a) Q. Laurent, R. Martinent, D. Moreau, N. Winssinger, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2021, 60, 19102–19106 CrossRef CAS; (b) S. T. Crooke, P. P. Seth, T. A. Vickers and X.-h. Liang, J. Am. Chem. Soc., 2020, 142, 14754–14771 CrossRef CAS.
- (a) O. Schäfer and M. Barz, Chem. – Eur. J., 2018, 24, 12131–12142 CrossRef PubMed; (b) G. Ribeiro Morais and R. A. Falconer, Org. Biomol. Chem., 2021, 19, 82–100 RSC; (c) G. M. Watt and G.-J. Boons, Carbohydr. Res., 2004, 339, 181–193 CrossRef CAS PubMed; (d) N. Stellenboom, R. Hunter, M. R. Caira and L. Szilágyi, Tetrahedron Lett., 2010, 51, 5309–5312 CrossRef CAS.
- (a) X. Qiu, X. Yang, Y. Zhang, S. Song and N. Jiao, Org. Chem. Front., 2019, 6, 2220–2225 RSC; (b) F. Yang, W. Wang, K. Li, W. Zhao and X. Dong, Tetrahedron Lett., 2017, 58, 218–222 CrossRef CAS; (c) B. Dong, Y. Chen, S. Xie, J. Zhang, J. Shen and L.-G. Xie, Org. Biomol. Chem., 2023, 21, 930–934 RSC; (d) P. Huang, P. Wang, S. Tang, Z. Fu and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 8115–8119 CrossRef CAS.
- (a) X. Xiao, M. Feng and X. Jiang, Angew. Chem., Int. Ed., 2016, 55, 14121–14125 CrossRef CAS; (b) Z. Dai, X. Xiao and X. Jiang, Tetrahedron, 2017, 73, 3702–3706 CrossRef CAS.
- (a) S. Chen, S. Cao, C. Liu, B. Wang, X. Ren, H. Huang, Z. Peng and X. Wang, Org. Lett., 2021, 23, 7428–7433 CrossRef CAS PubMed; (b) S. Wu, D. Hu, X. Wan, J. Zhao, Q. He, Z. Su and H. Cao, J. Org. Chem., 2022, 87, 16297–16306 CrossRef CAS.
- (a) X. Xiao, J. Xue and X. Jiang, Nat. Commun., 2018, 9, 2191 CrossRef PubMed; (b) J. Xue and X. Jiang, Nat. Commun., 2020, 11, 4170 CrossRef CAS.
- (a) M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 6624–6625 CrossRef CAS PubMed; (b) J. Guo, J. Zha, T. Zhang, C.-H. Ding, Q. Tan and B. Xu, Org. Lett., 2021, 23, 3167–3172 CrossRef CAS; (c) M. Song, Q. Hu, Z.-Y. Li, X. Sun and K. Yang, Chin. Chem. Lett., 2022, 33, 4269–4272 CrossRef CAS.
- (a) D. N. Harpp and T. G. Back, Tetrahedron Lett., 1972, 13, 1481–1484 CrossRef; (b) D. H. R. Barton, C. Chen and G. Michael Wall, Tetrahedron, 1991, 47, 6127–6138 CrossRef CAS; (c) E. Brzezinska and A. L. Ternay Jr., J. Org. Chem., 1994, 59, 8239–8244 CrossRef CAS; (d) P. Hiver, A. Dicko and D. Paquer, Tetrahedron Lett., 1994, 35, 9569–9572 CrossRef CAS; (e) M. Uragami, Y. Miyake, N. Tokutake, L.-h. Zhang and S. L. Regen, Langmuir, 2000, 16, 8010–8015 CrossRef CAS; (f) R. Hunter, M. Caira and N. Stellenboom, J. Org. Chem., 2006, 71, 8268–8271 CrossRef CAS PubMed; (g) S. Antoniow and D. Witt, Synthesis, 2007, 363–366 CAS; (h) L. Delarue Bizzini, P. Zwick and M. Mayor, Eur. J. Org. Chem., 2019, 6956–6960 CrossRef CAS; (i) M. Bao and M. Shimizu, Tetrahedron, 2003, 59, 9655–9659 CrossRef CAS; (j) Y.-F. Li, Y.-F. Wei, J. Tian, J. Zhang, H.-H. Chang and W.-C. Gao, Org. Lett., 2022, 24, 5736–5740 CrossRef CAS; (k) Z. Bai, S. Zhu, Y. Hu, P. Yang, X. Chu, G. He, H. Wang and G. Chen, Nat. Commun., 2022, 13, 6445 CrossRef CAS.
- Z. Zhang, T. Lao, L. Deng, C. Zhang, J. Liu, M. Fu, Z. Su, Y. Yu and H. Cao, Org. Lett., 2022, 24, 7222–7226 CrossRef CAS.
- (a) B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS; (b) H.-M. Huang, P. Bellotti, J. Ma, T. Dalton and F. Glorius, Nat. Rev. Chem., 2021, 5, 301–321 CrossRef CAS.
- (a) P. S. Fier and S. Kim, J. Am. Chem. Soc., 2024, 146, 6476–6480 CrossRef CAS; (b) P. S. Fier, S. Kim and R. D. Cohen, J. Am. Chem. Soc., 2020, 142, 8614–8618 CrossRef CAS PubMed.
- (a) A. Bacsó, M. Szigeti, S. Varga and T. Soós, Synthesis, 2017, 429–439 Search PubMed; (b) E. Desforges, A. Grysan, N. Oget, M. Sindt and J.-L. Mieloszynski, Tetrahedron Lett., 2003, 44, 6273–6276 CrossRef CAS.
- (a) X.-R. Song, T. Yang, H. Ding and Q. Xiao, Synthesis, 2020, 208–218 Search PubMed; (b) Y. Zhang, S. Du, T. Yang, F. Jin, J. Zhou, B. Cao, Z.-J. Mao, X.-R. Song and Q. Xiao, Org. Chem. Front., 2022, 9, 3156–3162 RSC; (c) S. Jiang, S. Du, J. Bai, X. Chen, M. Liang, S. Lin, M.-J. Luo, X.-R. Song and Q. Xiao, J. Org. Chem., 2023, 88, 14571–14586 CrossRef CAS PubMed; (d) S. Du, S. Jiang, R. Yang, F. Jin, H. Huang, W.-F. Tian, Z.-Z. Zhou, X.-R. Song and Q. Xiao, Org. Lett., 2023, 25, 1263–1267 CrossRef CAS PubMed.
- J. Ash and J. Y. Kang, Org. Biomol. Chem., 2023, 21, 2370–2374 RSC.
- B. Sarkar and A. Hajra, Org. Lett., 2024, 26, 5141–5145 CrossRef CAS PubMed.
- X. Han and J. Wu, Org. Lett., 2010, 12, 5780–5782 CrossRef CAS PubMed.
- X. Han, Y. Zhang and J. Wu, J. Am. Chem. Soc., 2010, 132, 4104–4106 CrossRef CAS PubMed.
- Y. Lin and S. Tanaka, Appl. Microbiol. Biotechnol., 2006, 69, 627–642 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 2000 Search PubMed.
- D. Churchill, J. M. Dust and E. Buncel, Can. J. Chem., 2007, 85, 421–431 CrossRef CAS.
- B. K. R. Sanapalli, A. Ashames, D. K. Sigalapalli, A. B. Shaik, R. R. Bhandare and V. Yele, Antibiotics, 2022, 11, 1680 CrossRef CAS PubMed.
- D. R. Edwards, A. A. Neverov and R. S. Brown, J. Am. Chem. Soc., 2009, 131, 368–377 CrossRef CAS PubMed.
- T. Miyamoto and I. Yamamoto, Agric. Biol. Chem., 1980, 44, 2581–2586 CAS.
- S. K. R. Parumala and R. K. Peddinti, Green Chem., 2015, 17, 4068–4072 RSC.
- X. Huang, S. Wang, B. Li, X. Wang, Z. Ge and R. Li, RSC Adv., 2015, 5, 22654–22657 RSC.
- G. Baccolini, C. Boga and M. Mazzacurati, J. Org. Chem., 2005, 70, 4774–4777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01099j |
| This journal is © The Royal Society of Chemistry 2024 |