
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemistry of installing epitranscriptomic 5-modified cytidines in RNA oligomers
Anna
Kuszczynska†
 ,
Milena
Bors
,
Karolina
Podskoczyj
and
Grazyna
Leszczynska
*
,
Milena
Bors
,
Karolina
Podskoczyj
and
Grazyna
Leszczynska
*
Institute of Organic Chemistry, Faculty of Chemistry, University of Technology, 90-924 Lodz, Zeromskiego 116, Poland. E-mail: grazyna.leszczynska@p.lodz.pl
First published on 16th August 2024
Abstract
Studies of 5-hydroxymethylcytidine (hm5C), 5-formylcytidine (f5C) and 5-carboxycytidine (ca5C) modifications as products of the 5-methylcytidine (m5C) oxidative demethylation pathway in cellular mRNAs constitute an important element of the new epitranscriptomic field of research. The dynamic process of m5C conversion and final turnover to the parent cytidine is considered a post-transcriptional layer of gene-expression regulation. However, the regulatory mechanism associated with epitranscriptomic cytidine modifications remains largely unknown. Therefore, oligonucleotides containing m5C oxidation products are of great value for the next generation of biochemical, biophysical, and structural studies on their function, metabolism, and contribution to human diseases. Herein, we summarize the synthetic strategies developed for the incorporation of hm5C, f5C and ca5C into RNA oligomers by phosphoramidite chemistry, including post-synthetic C5-cytidine functionalization and enzymatic methods.
 Anna Kuszczynska | Anna Kuszczynska received her BSc (2021) and MSc (2022) degrees in chemistry from the Lodz University of Technology, Poland. Currently, she is pursuing her PhD under the supervision of Prof. Leszczynska. Her research interest encompasses the development of new methods for the synthesis of modified ribonucleosides and their incorporation into RNA oligomers. Her latest project focuses on the metabolism of 5-hydroxymethylcytidine (hm5C) in cancer cells. |
 Milena Bors | Milena Bors received her BSc and MSc degrees in chemistry from the Lodz University of Technology, Poland. Currently, she is pursuing her doctoral degree under the supervision of Prof. Leszczynska at the Department of Chemistry, Institute of Organic Chemistry. Her research primarily revolves around the development of innovative methodologies for the synthesis of modified nucleosides and the facile, robust, and orthogonal incorporation of these monomers into RNA oligomers to evaluate the role of epigenetic modifications in the translation process. |
 Karolina Podskoczyj | Karolina Podskoczyj received her PhD in chemistry from the Lodz University of Technology, Poland, in 2023. She is currently an Assistant Professor at the Institute of Organic Chemistry at the Lodz University of Technology, where she joined the research group of Prof. Leszczynska. Her research interests comprise the synthesis of modified nucleosides and RNA oligomers, as well as investigating the impact of modified nucleosides present in the anticodon domain on the structure and properties of tRNA molecules. |
 Grazyna Leszczynska | Grazyna Leszczynska completed her postgraduate studies at the Lodz University of Technology (LUT) in 2000 and the Medical University of Lodz in 2010, specializing in organic chemistry and medical biotechnology, respectively. After receiving her PhD, she joined the group of Prof. A. Malkiewicz and Prof. E. Sochacka as an Assistant Professor. In 2019, she was appointed an Associate Professor and started independent research at LUT, supported by the NCN in Poland. Her research focuses on the chemistry and physicochemical/structural properties of modified ribonucleosides and RNA. |
1. Introduction
In addition to the four canonical nucleosides (A, C, G and U), cellular RNA molecules contain over 160 structurally distinct modified nucleosides.1 Each modification yields specific properties, which can have a crucial impact on the RNA structure, folding, stability, cellular localization and biological function.2 Although most of the modifications have been found in transfer RNAs (tRNAs), recent advances in analytical and next-generation sequencing strategies have revealed an increasing number of modified nucleosides in other non-coding RNAs and coding messenger RNAs (mRNAs).3–7 Among others, N6-methyladenosine (m6A), 5-methylcytidine (m5C) and 5-hydroxymethylcytidine (hm5C) represent post-transcriptional, dynamic and reversible epitranscriptomic-type mRNA modifications linked to the regulation of gene expression.8–105-Methylcytidine (m5C, 1, Fig. 1a) is one of the major RNA modifications identified in mRNA and non-coding RNAs, including tRNA, ribosomal RNA (rRNA), long non-coding RNA, small nuclear RNA, microRNA and enhancer RNA.1,4,11–13 Although m5C modification is widespread in many RNA species in all three domains of life, its increased distribution has been reported in eukaryotic tRNA and mRNA sequences.11,12,14 In tRNA molecules, m5C is known to stabilize the tRNA secondary structure and facilitate codon–anticodon pairing and tRNA aminoacylation.15,16 In addition, tRNA m5C modification regulates the cellular stress response, potentially by controlling the translation rate.17,18 In mRNA, m5C has been reported to regulate the nuclear-cytoplasmic transport,19 mRNA stability,20 splicing21 and translation.22,23 mRNA m5C was also associated with bladder cancer and autoimmune diseases.24–26
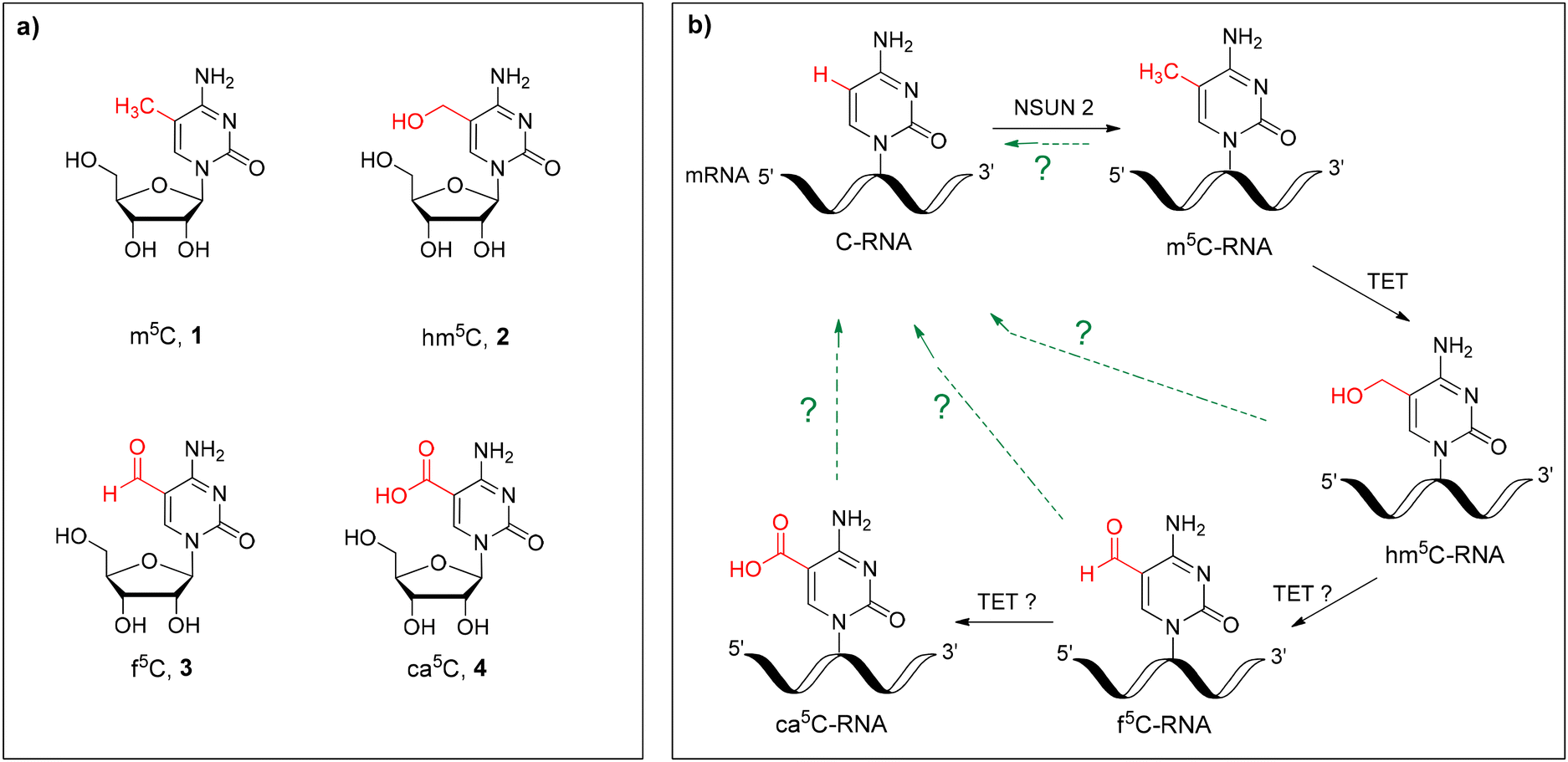 | ||
| Fig. 1 (a) Chemical structures of 5-methylcytidine (m5C, 1), 5-hydroxymethylcytidine (hm5C, 2), 5-formylcytidine (f5C, 3) and 5-carboxycytidine (ca5C, 4); (b) cellular oxidation of m5C-mRNA with the TET and hypothetical pathways of cytidine turnover (green arrows). | ||
Over the last decade, it was demonstrated that m5C undergoes in vivo oxidation to 5-hydroxymethylcytidine (hm5C, 2) and 5-formylcytidine (f5C, 3) (Fig. 1) in total RNA from organisms representing the three domains of life27,28 as well as in mammalian cells,29,30 including human and mouse embryonic stem cells.31,32 Similar to that in DNA,33,34 the ten–eleven translocation enzyme (TET) has been proven to catalyze the in vitro oxidation of m5C-RNA to hm5C as well as f5C-RNA to 5-carboxycytidine (ca5C, 4) (Fig. 1).29,35 Recently, the existence of four cytidines 1–4 has been confirmed in mammalian mRNA, including human colorectal carcinoma and hepatocellular carcinoma tissues.27 The dynamic character of m5C, hm5C, f5C and ca5C epitranscriptomic modifications and the possible but unknown turnover pathway to the parent cytidine (green arrows, Fig. 1b) imply the regulatory roles of these modifications in biological processes.
Originally, hm5C was identified in rRNA isolated from wheat seedlings.36 In mRNA, epitranscriptomic 5-hydroxymethylcytidine was found to increase translation and brain development in Drosophila melanogaster.37 Recently, the regulatory function of hm5C has been discovered related to its contribution to the mRNA flexibility required for mouse embryonic stem cell differentiation.9 In contrast to hm5C, the biological significance of epitranscriptomic f5C and ca5C modifications remains unknown. The stable existence of 5-formylcytidine and 2′-O-methyl-5-formylcytidine (f5Cm) has been confirmed at the wobble position (first anticodon letter) of mammalian mitochondrial tRNAsMet (mt-tRNAMet)38 and cytoplasmic tRNALeu,39 respectively. 5-Formylcytidine increases the flexibility of the mt-tRNAMet anticodon loop domain, facilitating the recognition of both purine-ending codons,40–42 and has been associated with several human diseases.43,44 Recently, f5C has also been detected in chromatin-associate RNA using a new, quantitative f5C-seq method.32
To acquire more insight into the function of hm5C, f5C and ca5C in RNA, multidirectional and interdisciplinary studies are required. In this context, synthetically obtained modified RNA oligomers are valuable tools to elucidate the effect of modified nucleosides on the physicochemical and structural properties of RNA45–48 and to identify the proteins that might recognize and process epitranscriptomic cytidine marks.29,35 Despite the numerous benefits of modified RNA oligomers, e.g. site-specific positioning of modified nucleoside(s) and possibility of a large scale synthesis, incorporation of C5-hydroxymethyl-, formyl- or carboxyl-functionalized cytidines is challenging and troublesome. The major obstacles are selection of appropriate blocking groups for modified building blocks (particularly for electrophilic-type carboxyl and formyl residues49) and appropriate modification of the standard phosphoramidite chemistry protocol.
In this review, we summarize the chemical methods for hm5C-, f5C- and ca5C-incorporation into RNA chains, including the preparation of modified monomer units and methods for C5-substituent installation. We focus on standard and post-synthetic phosphoramidite methods and in vitro enzyme-mediated transformations. In addition, the synthetic approaches to produce the epigenetically-modified DNA fragment are briefly discussed to highlight the difficulties in their implementation in RNA chemistry.
2. Chemical synthesis of modified RNA oligomers
Solid-phase phosphoramidite chemistry is the most common strategy for modified RNA synthesis.50 It involves two approaches: (1) the standard approach via the preparation of a modified phosphoramidite building block and its subsequent incorporation into the RNA chain (Scheme 1a) and (2) the post-synthetic RNA modification approach based on the selective chemical reaction of an easily convertible precursor oligonucleotide prepared by a standard procedure (Scheme 1b).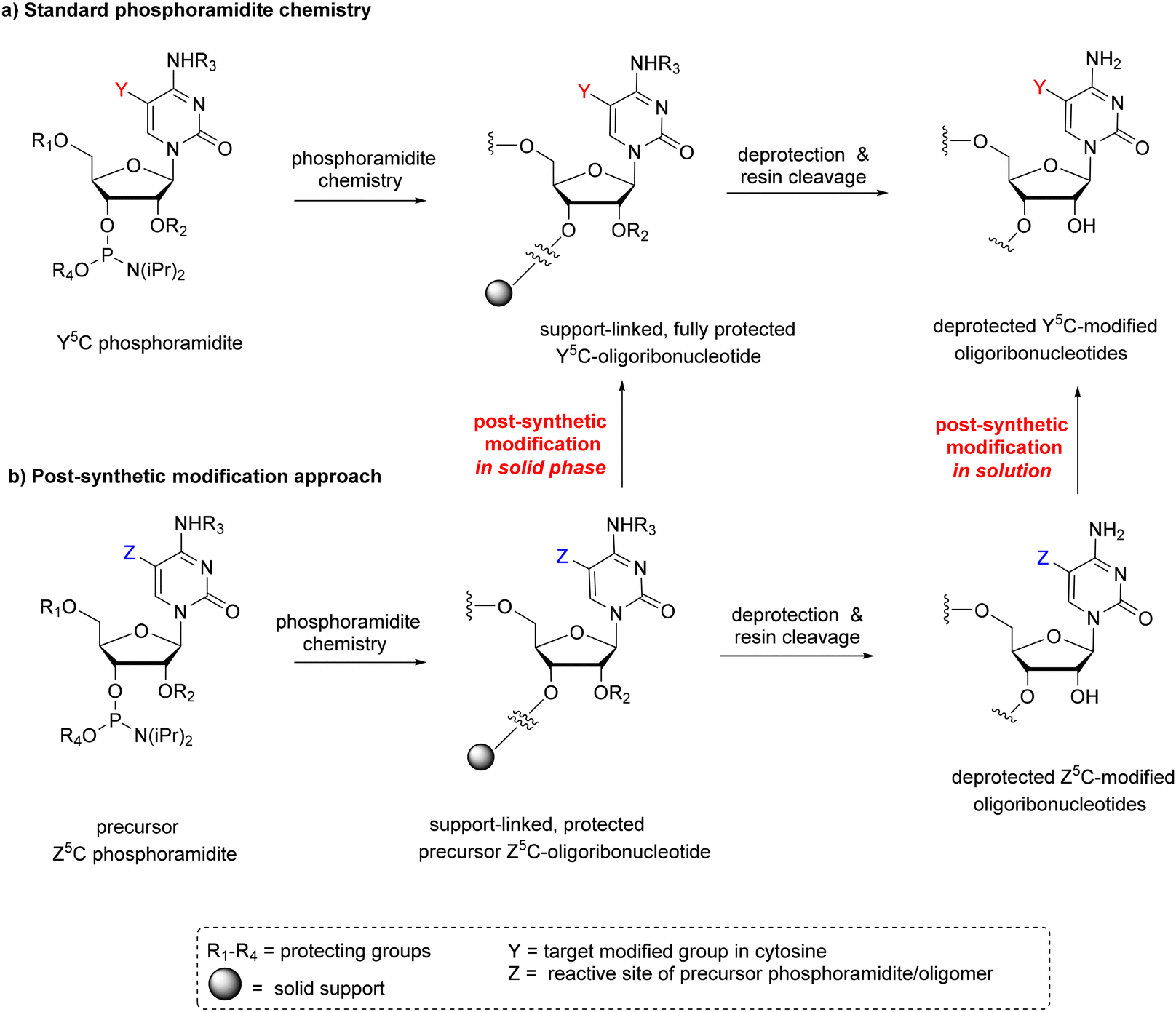 | ||
| Scheme 1 Two approaches in phosphoramidite chemistry; (a) standard method and (b) post-synthetic method of RNA modification divided into two strategies: in the solid phase and in solution. | ||
Post-synthetic conversions of RNA oligomers can be performed in the solid or liquid phase (Scheme 1b). The solid-phase approach involves a fully protected, support-linked oligoribonucleotide as a substrate. After conversion, the oligomer is subjected to a deprotection step and support cleavage. The most typical pattern of RNA deprotection involves a two-step procedure: (1) base deprotection of exoamino functions and phosphate residues with simultaneous support cleavage and (2) removal of 2′-protecting groups, e.g. desilylation when tert-butyldimethylsilyl (TBDMS) or triisopropylsilyloxymethyl (TOM) groups are present. If the Z → Y transformation is processed under alkaline conditions, the simultaneous removal of base-labile protecting groups and support cleavage are often observed. In this case, the post-synthetic reaction is carried out in one conversion–deprotection step.
Alternatively, the post-synthetic RNA modification can be performed in the liquid phase (Scheme 1b). The solution approach requires a fully deprotected oligoribonucleotide as a substrate. The hydrophilic character of RNA and the presence of free 2′-hydroxyl groups restrict the post-synthetic reaction conditions to polar solvents and mild basic conditions. The latter is required to prevent RNA cleavage or phosphate migration. Consequently, the number of organic reactions that can be used for post-synthetic transformation in solution is significantly reduced.
For successful solid-phase RNA synthesis, the phosphoramidite monomeric units must be protected with a combination of orthogonal R1 transient, and R2, R3, and R4 permanent protecting groups (Scheme 1). The standard protection strategy (Fig. 2) involves the R1 acid-labile 4,4′-O-dimethoxytrityl (DMTr) group, the R2 fluorolabile tert-butyldimethylsilyl (TBDMS) or triisopropylsilyloxymethyl (TOM) groups and two base-labile R3 and R4 protecting groups, acyl and β-cyanoethyl, respectively.
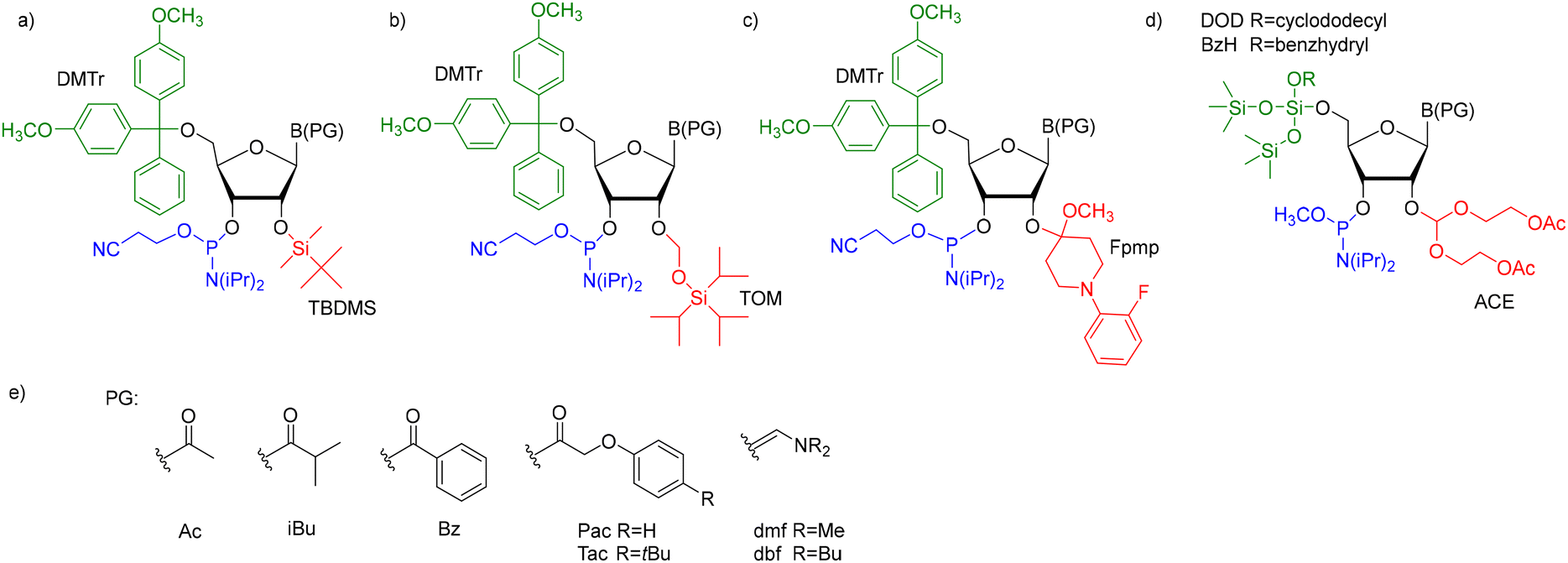 | ||
| Fig. 2 The most common ribonucleoside phosphoramidite building blocks for solid-phase RNA synthesis: (a) 5′-O-DMTr-2′-O-TBDMS-3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite); (b) 5′-O-DMTr-2′-O-TOM-3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite); (c) 5′-O-DMTr-2′-O-Fpmp-3′-O-(2-cyanoethyl-N,N-diisopropylphosphoramidite); (d) 5′-O-DOD(BzH)-2′-O-ACE-3′-O-(methyl-N,N-diisopropyl)phosphoramidite; and (e) commonly used nucleobase protecting groups. | ||
3. 5-Hydroxymethylcytidine (hm5C) modified RNA oligomers
The solid-phase synthesis of hm5C-RNA is challenging because of the pseudobenzylic positioning of the 5-hydroxymethyl group. The choice of the appropriate protection for –CH2OH is crucial to avoid the attack of nucleophiles (e.g. ammonia or methylamine used during oligomer deprotection) on the pseudobenzylic carbon and the formation of undesired side products.51,52 The problem worsens when –CH2OH is protected by an ester type group, for instance, the use of sterically hindered pivaloyl ester (5-CH2OPiv) almost exclusively promotes substitution of the –OPiv group with nitrogen nucleophiles.53,54To this day, two protection concepts of the hm5riboC phosphoramidite have been published (Fig. 3), both using the 5′-O-DMTr-2′-O-TBDMS-type ribose blockage. The first strategy utilizes an acetyl (Ac) protecting group to mask both 5-hydroxymethyl and 4-exoamino functions (I, Fig. 3).45,55–57 In the second strategy, a combination of tert-butyldimethylsilyl (TBDMS) and p-methoxybenzoyl (p-MeOBz) groups was employed for 5-hydroxymethyl and N4-exoamino functions, respectively (II, Fig. 3).58
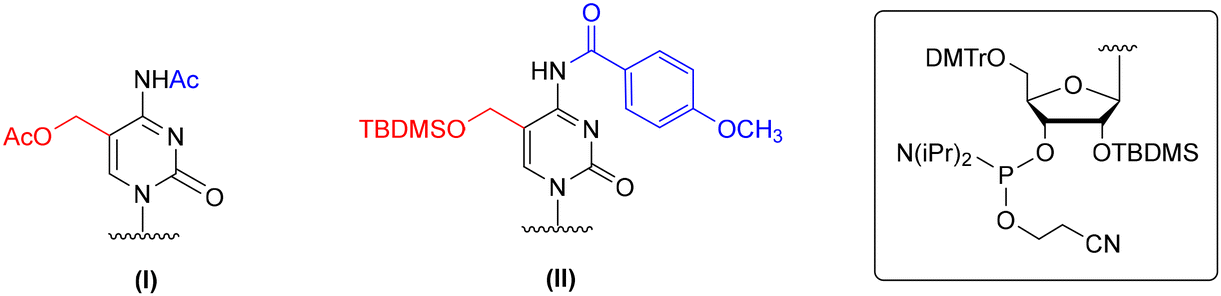 | ||
| Fig. 3 Protection patterns of 5-hydroxymethylcytidine phosphoramidites, as reported in the literature. | ||
Bisacetyl-protected hm5C phosphoramidite 13 was obtained by the Micura and Balasubramanian groups starting with 5-functionalized uridine or cytidine derivatives (Scheme 2a and b).45,55–57 The use of uridine derivatives, 5-hydroxymethyluridine (hm5U, 5)55,56 and 5-methyluridine (m5U, 7),57 as substrates extends the synthetic route, since an additional U → C conversion step is required (Scheme 2a). On the other hand, both uridines are commercially available or easy to synthesize and their use eliminates the problem of N4-amino group reactivity observed in cytidine when the C5-side chain is installed. To synthesize hm5C phosphoramidite 13, the 5-acetoxymethyl group was introduced by selective acetylation of 5-hydroxymethyluridine 5 (easily prepared from uridine) with acetic acid and catalytic amounts of trifluoroacetic acid55,56 or by radical bromination of m5U 7 and subsequent substitution with potassium acetate.57 Although a significant drop in yield is observed when installing acetyl via the 5-CH2Br derivative (42% yield of 8 → 9 conversion), the comparison of overall yields indicates its advantage over the first method starting with uridine due to the shorter synthetic route. 5-Acetylated product 9 was converted into 5-hydroxymethylcytidine 11via substitution of the O4-(2,4,6-triisopropyl)-benzenesulfonyl derivative 10 with aq ammonia. Protection of the N4-exoamino function with the acetyl group, 2′-silylation and phosphitylation furnished bisacetylated hm5C phosphoramidite 13 in an overall yield of 3%55,56 or 6.4%57 in eleven or eight steps, respectively. The relatively low yields of both synthetic routes result from a 50% decrease in the efficiency of the non-regioselective 2′-silylation reaction. This step was improved in the next method by temporarily protecting the ribose with a 3′,5′-di-tert-butylsilylene group.
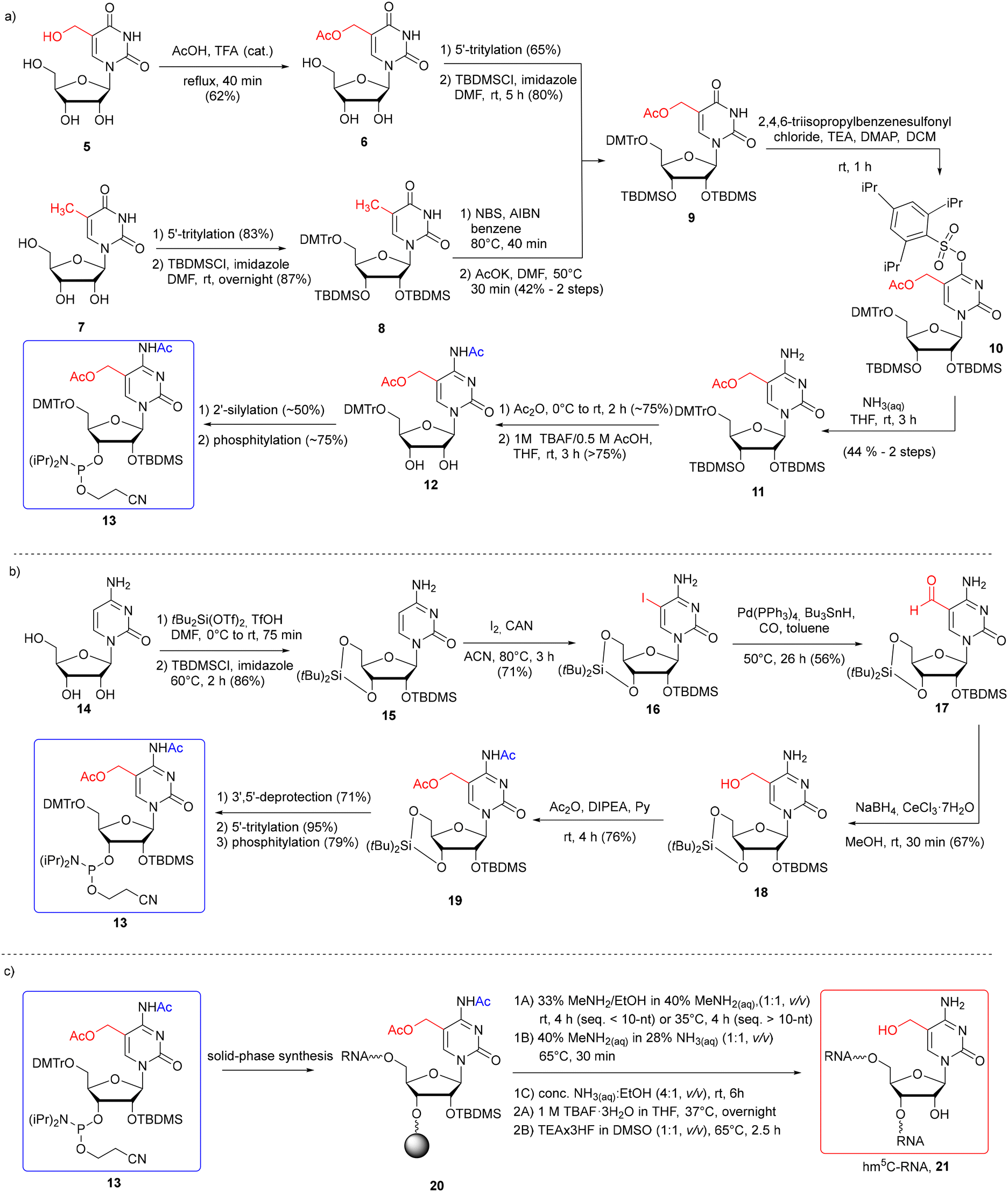 | ||
| Scheme 2 a) Synthesis of hm5C phosphoramidite with 5-CH2OAc and N4-Ac systems of protection with hm5U 5 or m5U 7 as the starting material;55–57 (b) synthesis of hm5C phosphoramidite with 5-CH2OAc and N4-Ac systems of protection with cytidine 14;45 (c) synthesis of hm5C-RNA under different deprotection conditions (1A–1C refers to the alkaline deprotection step; 2A and 2B refer to desilylation).45,55–57 | ||
Therefore, in the second approach (Scheme 2b), bisacetylated hm5C-amidite 13 was synthesized starting with 5-iodination of ribose-protected cytidine 15.45 5-Iodocytidine 16 offers rapid (2 step procedure) access to 5-hydroxymethylated cytidine 18 by Pd-catalyzed reductive carbonylation, followed by the reduction of 5-formylcytidine 17 with NaBH4 under Luche conditions. Simultaneous protection of both the N4-amino and 5-hydroxymethyl groups with acetic anhydride furnished bisacetylated product 19, which was converted into the desired hm5C phosphoramidite 13. This methodology appears more effective and faster than those starting with uridines, providing an hm5C building block 13 in a 9% overall yield in eight steps.
Monomeric unit 13 was successfully incorporated into RNA oligomers 21 (Scheme 2c) as a single modification at one, two, and three positions of RNA oligomers according to the 2′-O-TOM55,56 or 2′-O-TBDMS57 approach. The hm5C building block 13 was coupled within 6–10 min, with 80–98% coupling efficiency, based on a trityl assay. The resulting RNA 20 was subjected to simultaneous support cleavage and base labile group removal under three different conditions. Since it has been previously reported that 5-acetoxy-protected hm5dC-DNA undergoes partial substitution of 5-CH2OAc → 5-CH2NH2 with aq ammonia at 50 °C,59 Riml and colleagues employed more nucleophilic conditions offering fast and clean cleavage of base-labile groups: water–ethanol solution of methylamine (rt for shorter and 35 °C for longer RNA), or alternatively, aq methylamine–ammonia solution at 65 °C.45,55,56 Under both conditions, small amounts of side products (<15%) were detected, resulting from the methylamine attack on the pseudobenzylic carbon. Importantly, the use of the N4-acetyl group in hm5C monomeric unit 13 instead of the N4-benzoyl group prevented transamination at C4 by methylamine.60,61 As the third option, Tanpure and co-workers used a mixture of conc. NH3(aq)–EtOH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, rt, 6 h) for hm5C-RNA 20 deprotection.57 In this case, no comments about side product formation were reported.
:1, v/v, rt, 6 h) for hm5C-RNA 20 deprotection.57 In this case, no comments about side product formation were reported.
The problem of the benzylic nature of hm5C in solid-phase RNA synthesis was overcome by choosing the base-stable 5-CH2OTBDMS protection.58 The N4-exoamino function was protected with the N4-p-methoxybenzoyl (p-MeOBz) group since the standard N4-acetyl or benzoyl groups tend to be cleaved during solid-phase synthesis, yielding branched oligomers (additionally extended at the N4-amino site).58 To synthesize the TBDMS-protected hm5C building block 23 (Scheme 3a), 5-hydroxymethylcytidine 18 (obtained by the reduction of 5-formylcytidine, see Scheme 2b) was treated with TBDMS chloride in DMF, in the presence of imidazole. The subsequent reaction with p-methoxybenzoyl chloride in pyridine furnished fully protected hm5C 22, which was effectively converted into hm5C phosphoramidite 23 (note that the 5′-O-DMTr intermediate product was obtained using freshly prepared 4,4′-dimethoxytrityl triflate instead of the commonly used DMTrCl).
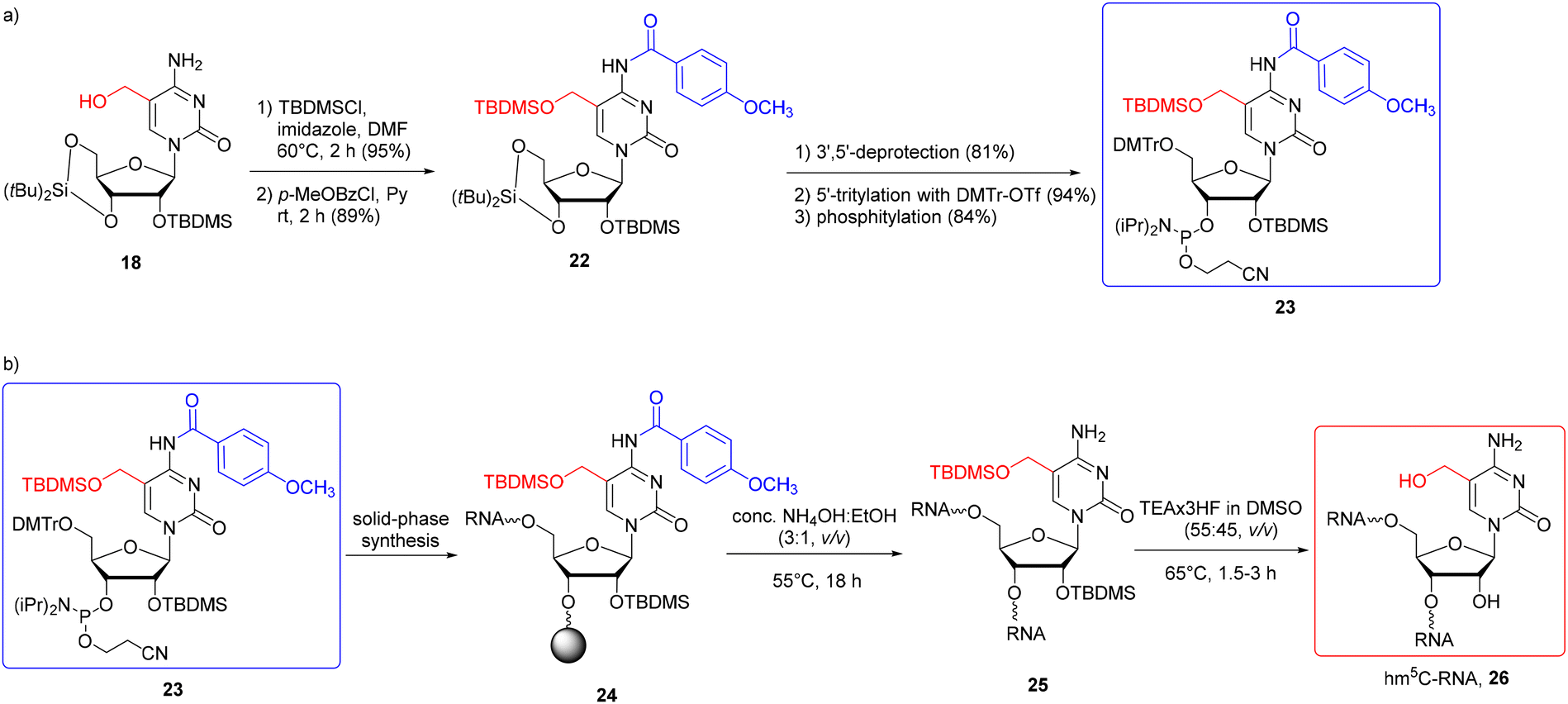 | ||
| Scheme 3 a) Synthesis of hm5C phosphoramidite with 5-CH2OTBDMS and N4-p-MeOBz protection systems; (b) synthesis of hm5C-RNA.58 | ||
The building block 23 has been successfully incorporated into RNA strands (Scheme 3b) with an extended coupling time of 20 min. Support-linked oligomer 24 was treated with conc. NH3(aq)–EtOH (3:1, v/v, 55 °C) to cleave the support and the base-labile protecting groups. Ammonolysis was prolonged to 18 h to achieve complete cleavage of the p-MeOBz group. Subsequent treatment of 25 with TEA·3HF in DMSO (55:45, v/v, 65 °C, 1.5–3 h) caused simultaneous deprotection of both 2′-OH and 5-CH2OH groups.
The hm5C-modified RNA oligomer was also obtained enzymatically, by the TET-mediated oxidation of chemically produced single-stranded m5C-RNA 27 (Scheme 4) under in vitro conditions.29 The recombinant catalytic domain of mouse TET1 protein was incubated with 11-nt m5C-oligomer 27 in the reaction buffer containing Fe2+ and 2-oxoglutarate at 37 °C. The LC-MS monitoring of the TET1-mediated oxidation of m5C-RNA revealed the formation of hm5C-oligomer 28 after 3 min and almost complete conversion of m5C-RNA 27 after 30–40 min.
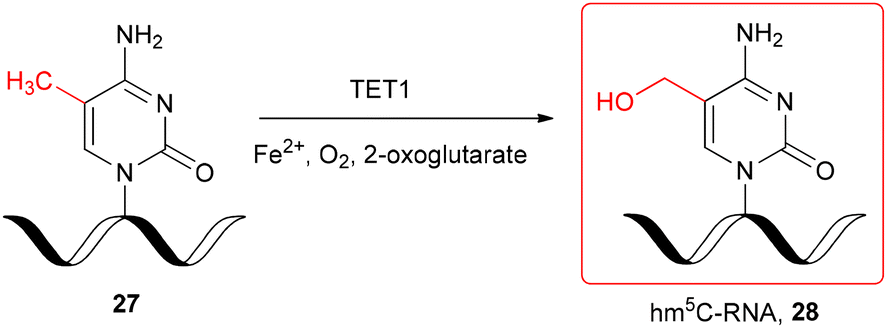 | ||
| Scheme 4 TET1-mediated formation of hm5C-RNA under in vitro conditions.29 | ||
Several protection concepts have been published for incorporation of 5-hydroxymethyl-2′-deoxycytidine (hm5dC) into DNA oligomers (Fig. 4), including 2-cyanoethyl62–65 and TBDMS66,67 protecting groups with N4-benzoyl or dbf (III, IV, and V, Fig. 4). The TBDMS group was also employed in combination with a triazolyl ring at the C4 position (VI, Fig. 4), which constitutes a precursor for post-synthetic 4-triazolyl → 4-NH2 conversion by ammonia treatment.66,67 In addition to the ether-type protecting groups, an acetyl group representing an ester-type protection was employed in combination with N4-benzoyl (VII, Fig. 4).59 Furthermore, an intrinsic cyclic carbamate was used to ensure simultaneous protection of both reactive centers of hm5dC (VIII, Fig. 4).68–71
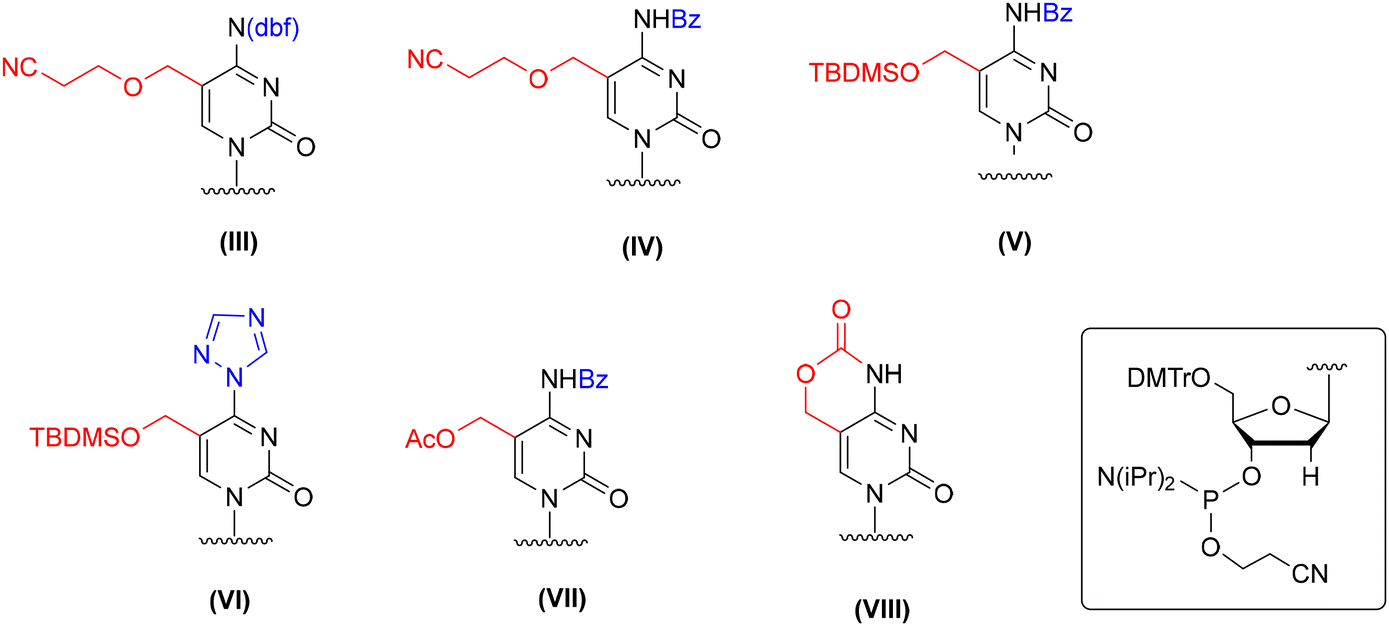 | ||
| Fig. 4 Different protection patterns of 5-hydroxymethyl-2′-deoxycytidine phosphoramidite for application in the solid-phase synthesis of hm5dC-DNA, as reported in the literature. | ||
Although the elaborated protection systems worked well for incorporation of hm5dC amidite into DNA chains, their direct implementation in RNA synthesis is unlikely to succeed. For instance, carbamate68–71 and acetyl59 removal (VII and VIII, Fig. 4) requires DNA incubation with NaOHaq instead of standard aq ammonia to avoid the formation of amide side products. The 5-CH2OCH2CH2CN (III and IV, Fig. 4)62–65,72 and 5-CH2OTBDMS (V and VI, Fig. 4)66,67 protecting groups are superstable and their complete removal is achieved by prolonged DNA incubation with aq ammonia at 65–75 °C. All these conditions are too harsh to keep RNA intact. It is likely that partial deprotection of 2′-O-TBDMS groups will occur, resulting in the cleavage of phosphodiester linkages.73,74
In the past, the hm5dC amidite blockage strategy with 5-CH2OTBDMS and N4-benzoyl groups (V, Fig. 4)66,67 was employed in hm5C-RNA chemistry (see Scheme 3); however, the conditions of post-synthetic hm5C-RNA deprotection were adapted to ensure RNA stability.
An interesting alternative for hm5dC-DNA synthesis is the post-synthetic approach, based on the reduction of support-linked 5-formyl-2'-deoxycytidine-DNA (f5dC-DNA, 29) (Scheme 5) with NaBH4 under Luche conditions.66 Hm5dC-DNA 30 obtained in 15 min was subjected to alkaline deprotection under two mild conditions: K2CO3 in MeOH/H2O (rt, 2 h) or NH3(aq) (rt, 2 h). Both conditions are potentially safe for RNA stability; however, the preparation of support-linked, formyl-unprotected f5C-RNA is still challenging (see the next section).
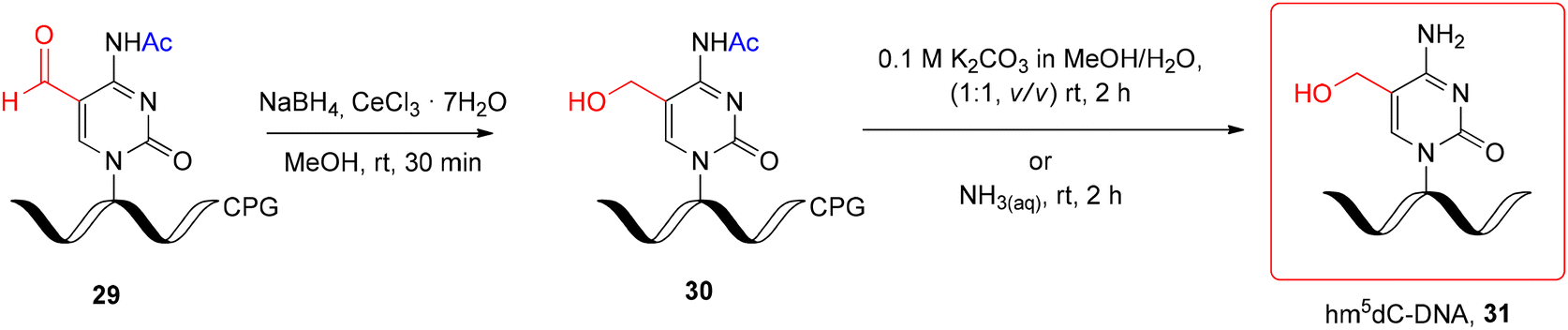 | ||
| Scheme 5 Post-synthetic reduction of support-linked f5dC-DNA and subsequent alkaline deprotection step.66 | ||
4. 5-Formylcytidine (f5C) modified RNA oligomers
The formyl group present in 5-formylcytidine (f5C) is susceptible to some conditions of solid-phase phosphoramidite synthesis, including acidic pH that facilitates N-glycosidic bond cleavage, oxidizers that promote –CHO oxidation and ammonia/amine treatment (commonly used during alkaline oligomer deprotection), which leads to imine formation.62,66,69,75 Due to these restrictions, there is only one example of the successful use of formyl-unprotected f5C-phosphoramidite in f5C-RNA synthesis.42 To keep the formyl group intact, the 5′-O-silyl-2′-O-acetal system of f5C phosphoramidite protection was used (IX, Fig. 5). An alternative concept of f5C-RNA synthesis involved formyl-protected f5C in the 2′-O-TBDMS approach. Since commonly used formyl protecting groups are incompatible with phosphoramidite chemistry, only the cyclic γ-acetal group (X and XI, Fig. 5) has been described so far in the synthesis of f5C-RNA.57,58 A convenient alternative for the standard phosphoramidite method is the post-synthetic strategy approach based on oxidation of a precursor RNA containing 5-(1,2-diacetoxyethyl)cytidine (XII, Fig. 5).76 | ||
| Fig. 5 Different protection patterns of 5-formylcytidine phosphoramidite (or the precursor unit) for the preparation of f5C-RNA oligomers using phosphoramidite chemistry, as reported in the literature. | ||
The first attempt to incorporate f5C into an RNA chain was made by Lusic and co-workers.42 They employed formyl-unprotected f5C phosphoramidite – prepared as methyl diisopropylphosphoramidites, equipped with 5′-O-BzH-2′-O-ACE and N4-dbf protecting groups (IX, Fig. 5). This non-standard system of protection enables to avoid the acidic conditions during RNA synthesis (hazardous for the formyl group, but typical for the 5′-O-DMTr-2′-O-silyl approach). On the other hand, the mild acidic conditions applied in the final step of oligomer deprotection promote the removal of 2′-O-ACE groups but also the hydrolysis of any imine side products that may be formed in the prior reaction of the “free” formyl group with an amine.
To synthesize the monomeric unit 35 (Scheme 6a), 5-hydroxymethylcytidine 32 was selectively oxidized with RuO2 affording 5-formylcytidine 33. Further steps involved the selective protection of N4-amino and 2′-OH functions, followed by phosphitylation.
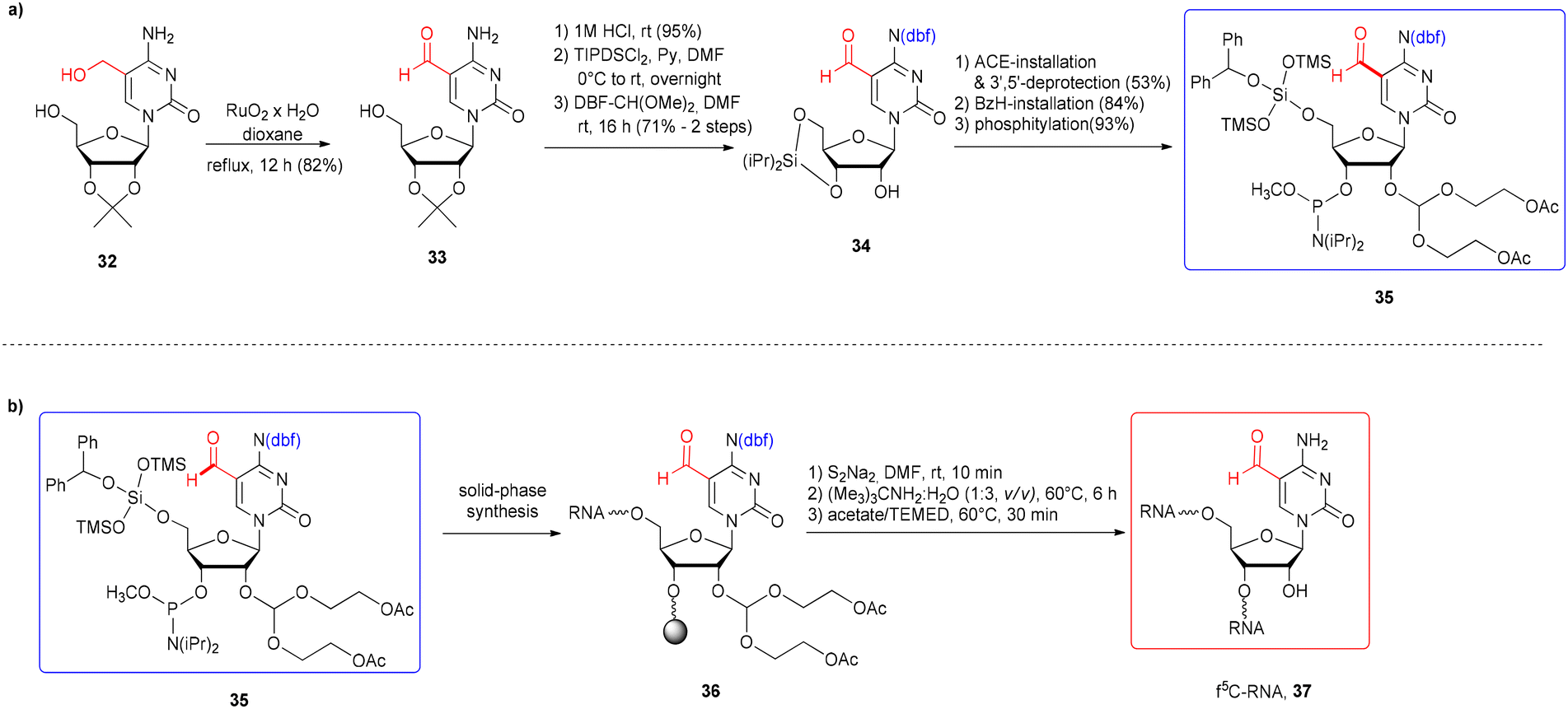 | ||
| Scheme 6 a) Synthesis of formyl-unprotected 5′-O-BzH-2′-O-ACE f5C phosphoramidite; (b) synthesis of f5C-RNA.42 | ||
5-Formylcytidine amidite 35 was introduced into the 17-nt RNA 36 by activation with 5-ethylthio-1H-tetrazole, with a 3.5 min coupling time (Scheme 6b). After 5′-O-BzH removal (TEA·3HF),77 the oligomer 36 was subjected to a three-step deprotection procedure, involving release of phosphate groups by disodium-2-carbamoyl-2-cyanoethylene-1,1-dithiolate (S2Na2), resin cleavage and removal of base-labile protecting groups with tert-butylamine–water solution (including acetyl groups from the 2′-O-ACE blockage) and finally, removal of acid-labile 2′-O-bis(2-hydroxyethoxy)methyl orthoesters with acetate/TEMED. Since the formation of imine adducts with the formyl group was considered, three alkaline deprotection conditions were tested: aq ammonia (rt, 24 h), methylamine (rt, 6 h) and tert-butylamine in water (1:3, v/v, 60 °C, 6 h). Only sterically hindered tert-butylamine provided the desired product with no tert-butylamine adducts.
To avoid problems with the potential reactivity of the aldehyde residue during f5C-RNA synthesis and deprotection, the formyl group was masked with a 1,3-dioxane residue (Scheme 7a).57,58 The cyclic γ-acetal formyl protection is able to survive the standard conditions of solid-phase synthesis in the 5′-O-DMTr-2′-O-TBDMS approach. To eliminate the risk of N4-Ac/Bz cleavage during oligomer synthesis, Michaelides and co-workers used p-MeOBz for the N4-exoamino function.58 The acetal remains intact during the alkaline oligomer deprotection step, preventing the aldehyde from reacting with ammonia/amine. Cyclic acetal is removed under mild acidic conditions in the final deprotection step.
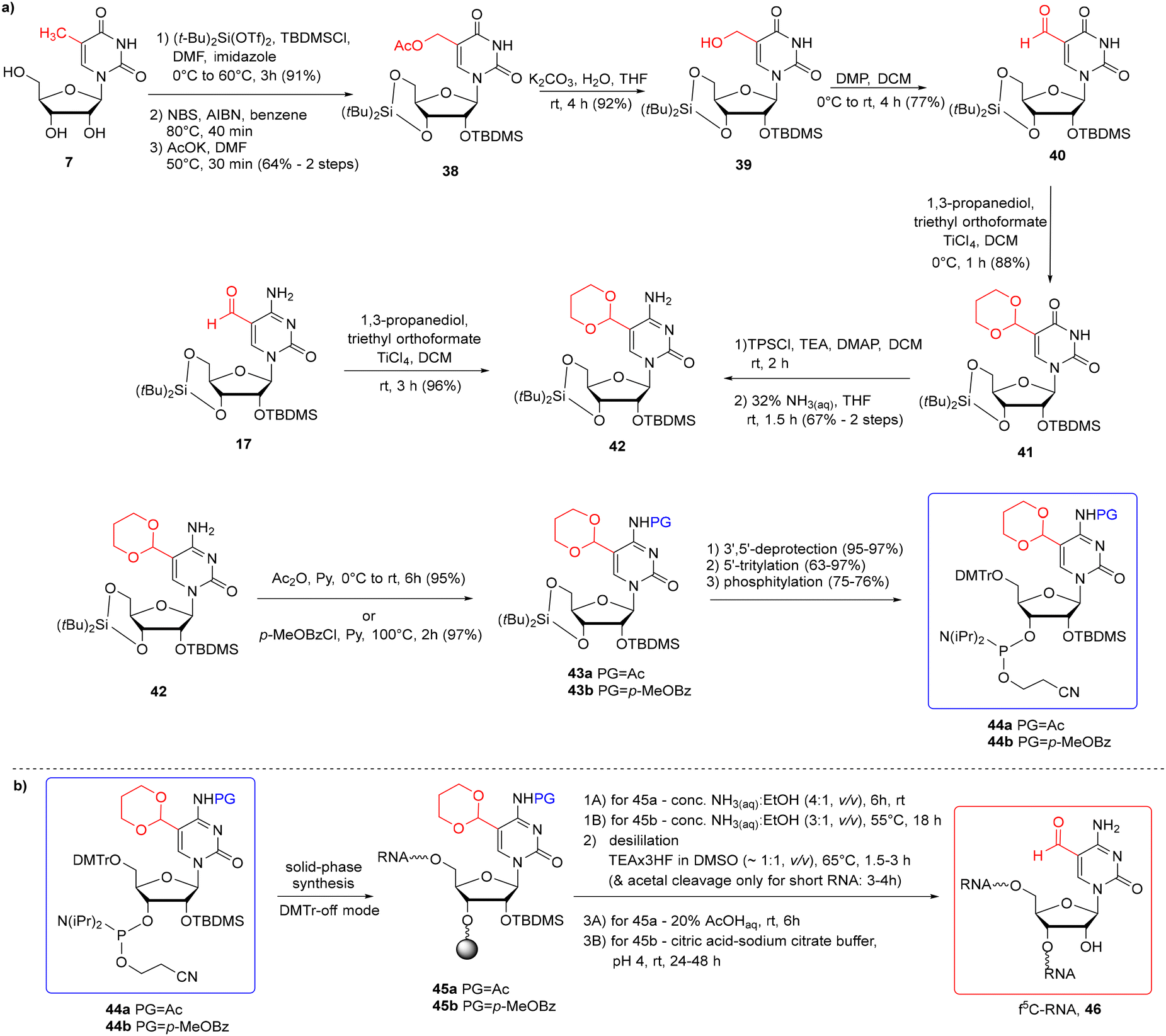 | ||
| Scheme 7 a) Synthesis of f5C phosphoramidites with cyclic acetal and the N4-Ac-57 or the N4-p-MeOBz-58 protection system; (b) synthesis of f5C-RNA. | ||
To introduce the cyclic γ-acetal protection, the 5-formyl-modified uridine 40 (Scheme 7a) or cytidine 17 was treated with propane-1,3-diol, in the presence of triethyl orthoformate and TiCl4 Lewis acid as an activator, affording 4157 and 42,58 respectively. 5-Formylcytidine 17 was obtained by Pd-catalyzed reductive carbonylation of 5-iodocytidine (see Scheme 2a). To prepare the 5-formyluridine derivative 40, 5-methyluridine 7 was converted to 5-hydroxymethylcytidine 39 through a three-step procedure: 5-bromination, nucleophilic substitution with potassium acetate and subsequent acetate ester hydrolysis. Next, 5-hydroxymethyluridine 39 was selectively oxidized to aldehyde 40 with Dess–Martin periodinane (DMP). Following the installation of the acetal group, protected uridine 41 was converted into cytidine 42via the O4-(2,4,6-triisopropyl)benzenesulfonyl intermediate. Acetal-protected f5C 42 was then blocked at the N4-amino function by an acetyl57 or p-MeOBz58 group and further converted into 5′-O-DMTr-2′-O-TBDMS-protected phosphoramidite 44a/44b.
Acetal-masked f5C phosphoramidite 44a/44b was incorporated into RNA (Scheme 7b) with a coupling time of 10 min (ref. 57) or 20 min (ref. 58) and 80–90% coupling efficiency. The first two steps of oligonucleotide deprotection were performed under standard conditions: conc. NH3(aq)–EtOH solution to cleave the support and the base-labile protecting groups, and TEA·3HF to remove the TBDMS groups. Since the desilylation solution is mildly acidic, prolonged incubation of short oligomers at 65 °C cleaved both 2′-O-TBDMS and acetal groups. For longer oligomers, an additional step of acetal deprotection was required by RNA incubation with citric acid–sodium citrate buffer at pH 4 (rt, 24–48 h)58 or 20% AcOHaq (rt, 6 h).57 The orthogonal protection systems in f5C 44a and hm5C 13 building blocks enabled the incorporation of both epitranscriptomic modifications into a single RNA strand according to the above-described protocol.57
5-Formylcytidine-RNA oligomers were also prepared by the post-synthetic approach based on the oxidation of 5-diol-containing cytidine with sodium periodate (Scheme 8).76 The 5′-O-DMTr-2′-O-TBDMS-protected precursor phosphoramidite 49 offers a fully orthogonal system of protection as the C5-linked diol residue is protected with the acetyl groups.
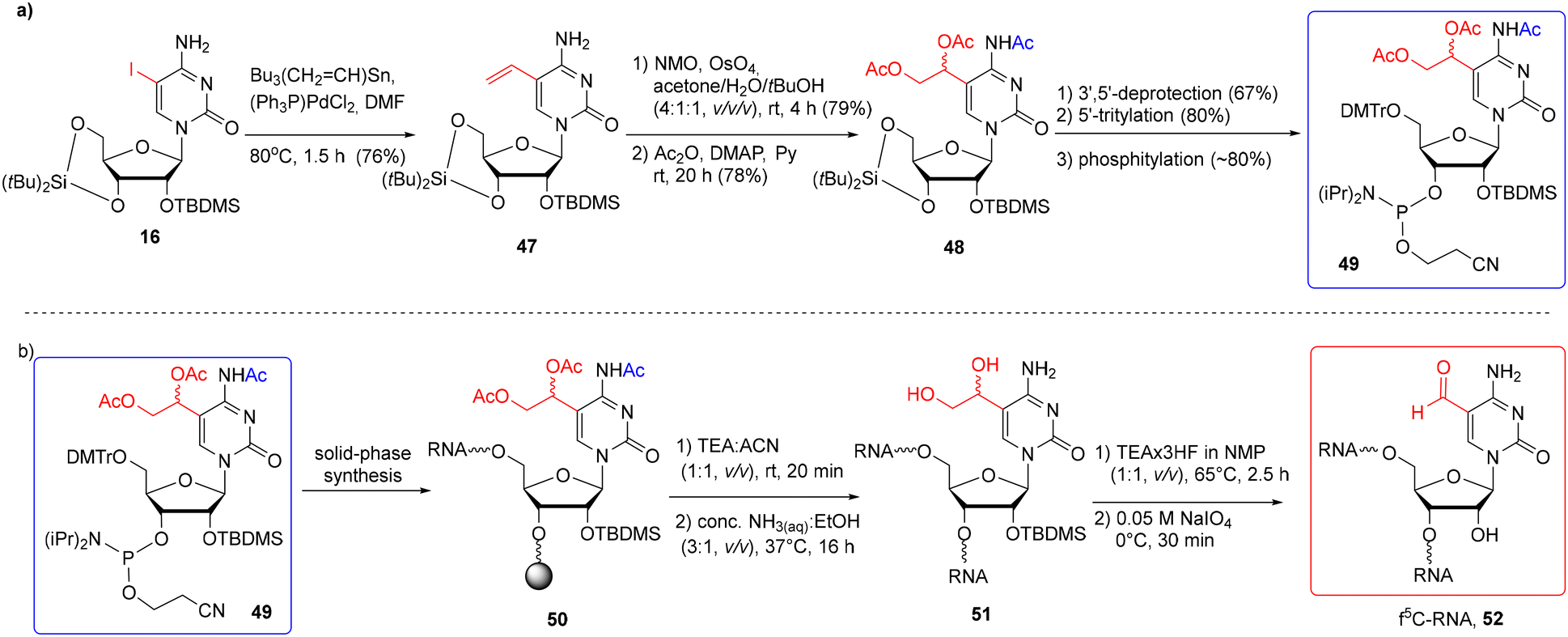 | ||
| Scheme 8 a) Synthesis of f5C-precursor phosphoramidite; (b) synthesis of f5C-RNA via the post-synthetic modification approach.76 | ||
The precursor building block 49 was synthesized as shown in Scheme 8a. 5-Iodocytidine 16 was subjected to a Pd-catalyzed Stille reaction, affording 5-vinylcytidine 47. Treatment of 47 with OsO4 in the presence of N-methylmorpholine-N-oxide, followed by acylation, gave the triacetylated product 48. After selective removal of the di-tert-butylsilylene protection, the resulting nucleoside was tritylated and phosphitylated to furnish the 3′-O-phosphoramidite of 5-(1,2-diacetoxyethyl)-N4-acetylcytidine 49. Phosphoramidite 49 was coupled twice with a 10 min coupling time, using the phosphoramidite chemistry on the CPG-Am(Pac) support (the use of a 2′-O-methylated nucleoside was necessary to preserve the 3′-terminal nucleoside against the C2′–C3′ bond cleavage). After the synthesis, precursor RNA 50 (Scheme 8b) was subjected to alkaline deprotection consisting of the selective release of phosphate groups under mild conditions (TEA-ACN, 1:1, v/v, rt, 20 min), followed by treatment with conc. NH3(aq)–EtOH to remove the support and the remaining base-labile groups. Diol-containing oligomer 51 was desilylated with TEA·3HF/NMP (1:1, v/v, 65 °C, 2.5 h) and purified by IE HPLC. The fully deprotected oligomer was quantitatively oxidized to f5C-RNA 52 with 0.05 M NaIO4 used in a 50-fold molar excess at 0 °C for 30 minutes. The excess of the oxidizing reagent was rapidly removed using a Sep-Pak cartridge.
The synthesis of f5dC-containing DNA was performed in several ways, including the incorporation of formyl-unprotected f5dC phosphoramidites (XIII and XIV, Fig. 6),64,66,69 acetal-protected cytidine (XV)70,71 and a C5-diol-functionalized precursor building block (XVI).78 The latter two monomeric units were successfully adopted in f5C-RNA synthesis, although the protocols of alkaline deprotection were modified to avoid the risk of RNA degradation, e.g. prolonged incubation of DNA with conc. NH3(aq) at 55 °C was replaced with rapid incubation of RNA with conc. NH3(aq)–EtOH (3:1, v/v) at 37–55 °C.58,76 To introduce the formyl-unprotected f5dC phosphoramidites (XIII and XIV, Fig. 6), a 5′-O-DMTr approach was employed with N4-benzoyl or N4-acetyl protecting groups. The benzoyl protection required the use of more harsh conditions in the deprotection step (unfavorable in RNA chemistry): prolonged incubation with aq ammonia64,69 or 0.4 M NaOH in water–methanol.69 The N4-acetyl group is more labile under basic conditions; its cleavage was performed by treatment of f5dC-DNA with 0.1 M K2CO3 in MeOH:H2O (1:1, v/v, rt, 2 h) or conc. NH3(aq) (rt, 2 h).66 In all cases, where ammonia was used, the authors did not observe imine formation. It is likely that the traces of the imine side products were cleanly hydrolyzed into aldehydes under the acidic conditions during the final 5′-detritylation. Overall, the latter two deprotection methods, utilizing the N4-acetyl group, could be implemented in f5C-RNA chemistry; however, no trials have been done to employ the formyl-unprotected f5C phosphoramidite in the 5′-O-DMTr approach.
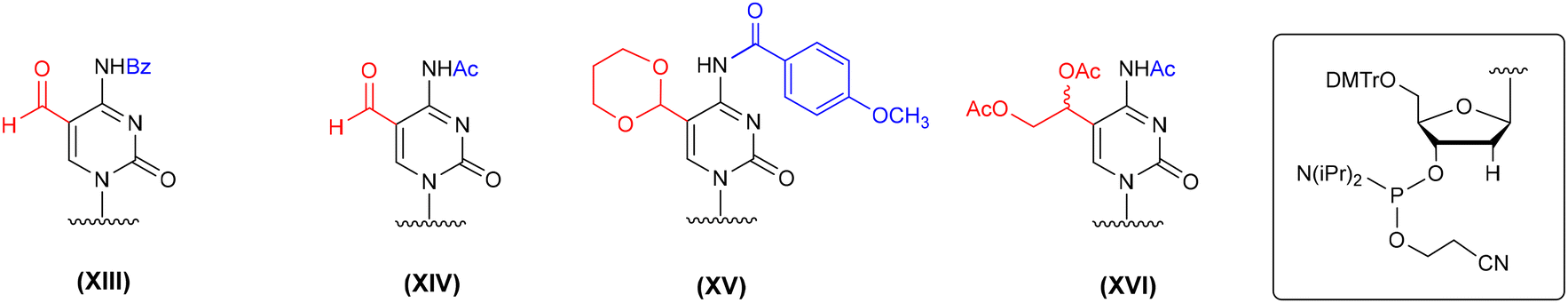 | ||
| Fig. 6 Different protection patterns of f5dC phosphoramidites employed in the solid-phase synthesis of f5dC-DNA, as reported in the literature. | ||
5. 5-Carboxycytidine (ca5C) modified RNA oligomers
In general, only two carboxyl protecting groups, 2-(p-nitrophenyl)ethyl (NPE) and 2-(trimethylsilyl)ethyl (TMSE), have been employed so far in the synthesis of modified RNA oligomers via phosphoramidite chemistry.79–84 The benefit of both ester-type protections is that they can be removed under conditions that prevent ester ammonolysis and the formation of amide side products. For instance, the NPE group is removed under anhydrous DBU in THF through a β-elimination mechanism prior to the ammonolysis.Carell's group reported the first synthesis of a 5-carboxycytidine-modified RNA oligomer (ca5C-RNA) using the standard 5′-O-DMTr-2′-O-TBDMS approach.58 The p-nitrophenylethyl group was selected to protect the carboxyl function in the ca5C building block 55 (Scheme 9) in combination with N4-p-MeOBz.
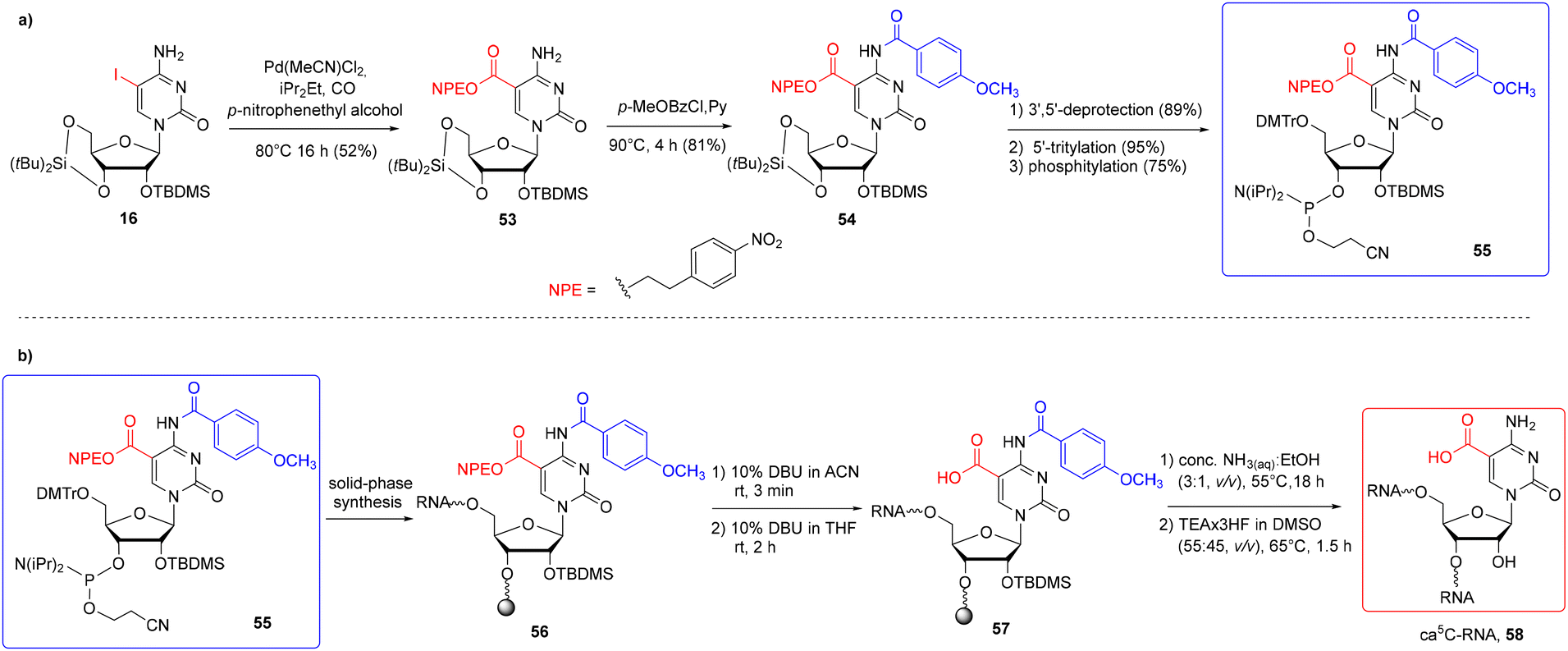 | ||
| Scheme 9 a) Synthesis of ca5C phosphoramidite 55; (b) synthesis of ca5C-RNA 58.58 | ||
The NPE-protected ca5C phosphoramidite 55 was synthesized via Pd-mediated esterification of silyl-protected iodocytidine 16 using molten p-nitrophenylethyl alcohol as the solvent and reagent (Scheme 9a). The carboxyl-protected cytidine 53 was treated with p-methoxybenzoyl chloride to protect the N4-exoamino function and then converted to ca5C phosphoramidite 55. A standard phosphoramidite chemistry protocol was used for ca5C-phosphoramidite incorporation, with the coupling time extended to 20 min. To avoid the formation of amide side products, the support-linked ca5C-RNA 56 (Scheme 9b) was subjected to a three-step alkaline deprotection: a short treatment with 10% DBU in acetonitrile for selective removal of β-cyanoethyl groups from phosphate residues, selective deprotection of the NPE group with 10% DBU in THF (rt, 2 h) and incubation of carboxyl-unprotected ca5C-RNA 57 with conc. NH3(aq)–EtOH (3:1, v/v, 55 °C) to deprotect the exoamino functions and cleave the oligomer from the support. Oligomer desilylation finished the deprotection protocol providing the desired ca5C-RNA 58.
A slight modification of the above RNA synthesis method allowed the insertion of two other phosphoramidites hm5C 23 and f5C 44b into a single RNA strand.58 Since f5C-phosphoramidite 44b was protected with an acidic-labile acetal group, the aforementioned protocol of oligomer deprotection was extended with an additional step of RNA incubation with citric acid–sodium citrate buffer at pH 4 (rt, 24 h).
In 2017, the catalytic activity of mammalian ten–eleven translocation methylcytosine dioxygenase 1 (TET1) was discovered for in vitro oxidation of 5-formylcytidine-modified RNA 37 to the 5-carboxycytidine derivative 59 (Scheme 10).35 For this purpose, a synthetically prepared f5C-RNA oligomer 37 (see Scheme 6) was incubated with the catalytic domain of the TET1 enzyme (expressed in insect cells) in a buffer containing 100 mM HEPES (pH 7.0), 50 mM NaCl, 1 mM 2-oxoglutarate, 2 mM ascorbic acid, 0.1 mM Fe(NH4)2(SO4)2, 1 mM DTT, 1 mM ATP, and 100 μg ml−1 bovine serum albumin at 37 °C for 0.5 hours. The oligonucleotide was isolated by ultrafiltration and subjected to enzymatic digestion. In addition to the canonical nucleosides A, C, G and U, both f5C and ca5C were identified by UHPLC-MS analysis, demonstrating that the TET1 enzyme catalyzes the oxidative formation of ca5C from f5C in single stranded, double stranded and hairpin forms of RNA, albeit with low overall efficiency.
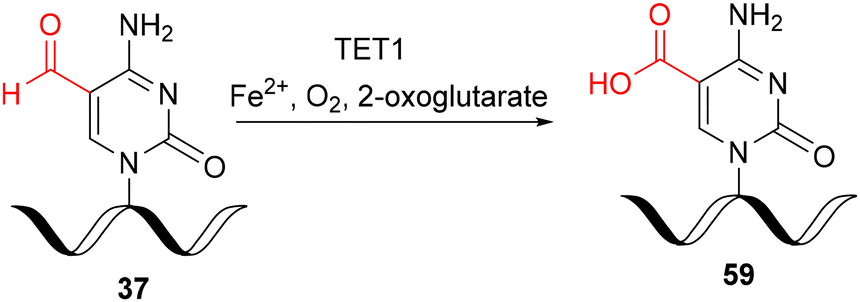 | ||
| Scheme 10 Preparation of ca5C-RNA via TET1-mediated oxidation of f5C-RNA under in vitro conditions.35 | ||
In the literature, several protection concepts for the incorporation of 5-carboxy-2′-deoxycytidine (ca5dC) phosphoramidite into DNA oligomers (Fig. 7) have been published. In most cases, methyl (XVII and XVIII, Fig. 7)66,69,85 or ethyl (XIX, Fig. 7)71 esters were utilized to protect the carboxyl function of ca5dC in combination with N4-Bz71,85 or N4-Ac groups.66,69 The alkyl esters were removed under hydrolytic conditions with an aq solution of NaOH or K2CO3, since standard ammonolysis causes the formation of amide side products. Both hydrolytic conditions are harsh for RNA handling and could lead to the removal of the 2′-O-silyl protecting group and subsequent RNA degradation. The 5-trifluoroethyl ester of ca5dC-DNA (XX, Fig. 7) undergoes effective ammonolysis and aminolysis reactions,86 but does not offer complete ester conversion to 5-carboxylic acid.66
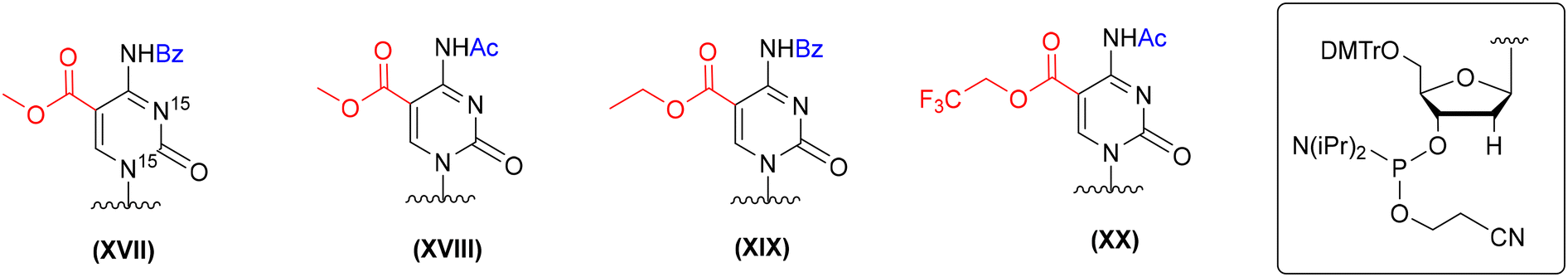 | ||
| Fig. 7 Different protection patterns of ca5dC phosphoramidite for the preparation of ca5dC-DNA oligomers, as reported in the literature. | ||
In addition, the post-synthetic strategy for ca5dC-DNA synthesis was reported via hydrolysis of 5-trifluoromethylcytidine-modified DNA 60 (CF3dC-DNA, Scheme 11) as a precursor oligomer. Hydrolysis was performed in solution by prolonged incubation of fully deprotected, support-free oligonucleotide 60 with NaOHaq at 60 °C.87,88 Although the 5-trifluoromethylcytidine ribonucleoside has been proven to react efficiently with various nucleophilic reagents, including NaOHaq,89,90 its direct implementation in RNA chemistry will lead to phosphodiester cleavage.
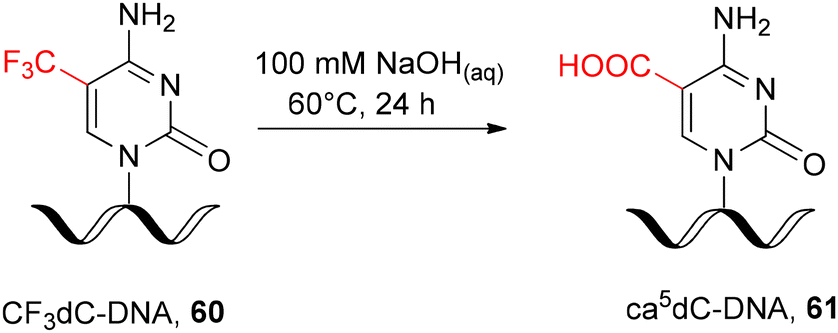 | ||
| Scheme 11 Post-synthetic DNA modification with ca5dC.87,88 | ||
6. Summary
Since 5-hydroxymethyl-, 5-formyl- and 5-carboxycytidine are related to the poorly understood field of epitranscriptome science, future research needs to address the functional significance of these modifications. In this context, RNA solid-phase synthesis has become a key technology to produce RNAs with the target modification at any desired sequence position.Numerous methods for the synthesis of oligonucleotides containing nucleophilic modifying groups, e.g. the hydroxymethyl group in hm5C/hm5dC, are known; their incorporation is usually achieved using standard protocols of phosphoramidite solid-phase synthesis. Therefore, no further efforts in this area should be expected.
In contrast, the preparation of oligonucleotides containing reactive electrophilic groups, e.g. carboxyl or aldehyde groups, is problematic because the commonly used protecting groups are incompatible with solid-phase conditions. The main problem concerns the alkaline deprotection conditions, which are difficult to change. An alternative methodology for installing troublesome modifying groups is the post-synthetic RNA modification approach. It seems that future work will focus on the selection of promising precursor monomeric units and post-synthetic reaction conditions that yield the desired RNAs.
Abbreviations
| A | Adenosine |
| Ac | Acetyl |
| ACE | Bis(2-acetoxyethoxy)methyl |
| AIBN | Azobisisobutyronitrile |
| aq | Aqueous |
| ATP | Adenosine triphosphate |
| B | Base |
| Bz | Benzoyl |
| BzH | Benzhydryloxybis-(trimethylsiloxy)silyl |
| C | Cytidine |
| ca5C | 5-Carboxycytidine |
| ca5dC | 5-Carboxy-2′-deoxycytidine |
| CAN | Ceric ammonium nitrate |
| conc. | Concentrated |
| CPG | Controlled pore glass |
| dbf | Dibutylaminomethylene |
| DBF-CH(OMe)2 | N,N-Diisobutylformamidinedimethyl acetal |
| DBU | Diazabicyclo[5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DIPEA | N,N-Diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| dmf | Dimethylaminomethylene |
| DMF | Dimethylformamide |
| DMP | Dess–Martin periodinane |
| DMSO | Dimethyl sulfoxide |
| DMTr | 4,4′-O-Dimetoxytrityl |
| DNA | Deoxyribonucleic acid |
| DOD | Bis(trimethylsiloxy)cyclododecyloxysilyl |
| f5C | 5-Formylcytidine |
| Fpmp | 1-(2-Fluorophenyl)-4-methoxypiperidin-4-yl |
| G | Guanosine |
| HEPES | 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| hm5C | 5-Hydroxymethylcytidine |
| hm5dC | 5-Hydroxymethyl-2′-deoxycytidine |
| hm5U | 5-Hydroxymethyluridine |
| HPLC | High-performance liquid chromatography |
| iBu | Isobutyryl |
| IE HPLC | Ion-exchange high-performance liquid chromatography |
| iPr | Isopropyl |
| LC | Liquid chromatography |
| Leu | Leucine |
| m5C | 5-Methylcytidine |
| m5U | 5-Methyluridine |
| m6A | N6-Methyladenosine |
| Me | Methyl |
| Met | Methionine |
| mRNA | Messenger ribonucleic acid |
| MS | Mass spectrometry |
| mt | Mitochondrial |
| NBS | N-Bromosuccinimide |
| NMO | N-Methylmorpholine N-oxide |
| NMP | N-Methyl-2-pyrrolidone |
| NPE | 2-(p-Nitrophenyl)ethyl |
| nt | Nucleotide |
| Pac | Phenoxyacetyl |
| PG | Protecting group |
| Py | Pyridine |
| RNA | Ribonucleic acid |
| rt | Room temperature |
| S2Na2 | Disodium-2-carbamoyl-2-cyanoethylene-1,1-dithiolate |
| seq | Sequence |
| Tac | 4-(tert-Butylphenoxy)acetyl |
| TBAF | Tetrabutylammonium fluoride |
| TBDMS | tert-Butyldimethylsilyl |
| tBu | tert-Butyl |
| TEA | Triethylamine |
| TEA·3HF | Triethylamine trihydrofluoride |
| TEMED | N,N,N′,N′-Tetramethylethylene diamine |
| TET | Ten–eleven translocation enzyme |
| Tf | Trifluoromethanesulfonate |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TIPDSCl2 | 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane |
| TMS | Trimethylsilyl |
| TMSE | 2-(Trimethylsilyl)ethyl |
| TOM | Triisopropylsilyloxymethyl |
| TPS | (2,4,6-Triisopropyl)benzenesulfonyl |
| tRNA | Transfer RNA |
| U | Uridine |
| UHPLC | Ultra high-performance liquid chromatography |
| v/v | Volume per volume |
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the National Science Center, Poland, UMO-2021/43/B/ST4/01570 to GL.This paper has been completed while the first authors (AK and MB) were Doctoral Candidates in the Interdisciplinary Doctoral School at the Lodz University of Technology, Poland.
References
- A. Cappannini, A. Ray, E. Purta, S. Mukherjee, P. Boccaletto, S. N. Moafinejad, A. Lechner, C. Barchet, B. P. Klaholz, F. Stefaniak and J. M. Bujnicki, Nucleic Acids Res., 2024, 52, D239–D244 CrossRef PubMed.
- M. Duechler, G. Leszczyńska, E. Sochacka and B. Nawrot, Cell. Mol. Life Sci., 2016, 73, 3075–3095 CAS.
- W. V. Gilbert, T. A. Bell and C. Schaening, Science, 2016, 352, 1408–1412 CAS.
- J. Mathlin, L. Le Pera and T. Colombo, Int. J. Mol. Sci., 2020, 21, 4684 CAS.
- L. Trixl and A. Lusser, Wiley Interdiscip. Rev.: RNA, 2019, 10, e1510 CrossRef PubMed.
- G. Guo, K. Pan, S. Fang, L. Ye, X. Tong, Z. Wang, X. Xue and H. Zhang, Mol. Ther.–Nucleic Acids, 2021, 26, 575–593 CrossRef CAS PubMed.
- E. Destefanis, G. Avşar, P. Groza, A. Romitelli, S. Torrini, P. Pir, S. G. Conticello, F. Aguilo and E. Dassi, RNA, 2021, 27, 367–389 CrossRef CAS PubMed.
- L. Shen, C.-X. Song, C. He and Y. Zhang, Annu. Rev. Biochem., 2014, 83, 585–614 CrossRef CAS PubMed.
- J. Lan, N. Rajan, M. Bizet, A. Penning, N. K. Singh, D. Guallar, E. Calonne, A. Li Greci, E. Bonvin, R. Deplus, P. J. Hsu, S. Nachtergaele, C. Ma, R. Song, A. Fuentes-Iglesias, B. Hassabi, P. Putmans, F. Mies, G. Menschaert, J. J. L. Wong, J. Wang, M. Fidalgo, B. Yuan and F. Fuks, Nat. Commun., 2020, 11, 4956 CAS.
- I. A. Roundtree, M. E. Evans, T. Pan and C. He, Cell, 2017, 169, 1187–1200 CrossRef CAS PubMed.
- J. E. Squires, H. R. Patel, M. Nousch, T. Sibbritt, D. T. Humphreys, B. J. Parker, C. M. Suter and T. Preiss, Nucleic Acids Res., 2012, 40, 5023–5033 CAS.
- S. Edelheit, S. Schwartz, M. R. Mumbach, O. Wurtzel and R. Sorek, PLoS Genet., 2013, 9, e1003602 CAS.
- Y. Motorin, F. Lyko and M. Helm, Nucleic Acids Res., 2010, 38, 1415–1430 CAS.
- S. Shinoda, S. Kitagawa, S. Nakagawa, F.-Y. Wei, K. Tomizawa, K. Araki, M. Araki, T. Suzuki and T. Suzuki, Nucleic Acids Res., 2019, 47, 8734–8745 CrossRef CAS PubMed.
- Y. Motorin and M. Helm, Biochemistry, 2010, 49, 4934–4944 CrossRef CAS PubMed.
- J. E. Squires and T. Preiss, Epigenomics, 2010, 2, 709–715 CAS.
- C. T. Y. Chan, M. Dyavaiah, M. S. DeMott, K. Taghizadeh, P. C. Dedon and T. J. Begley, PLoS Genet., 2010, 6, e1001247 CrossRef CAS PubMed.
- M. Schaefer, T. Pollex, K. Hanna, F. Tuorto, M. Meusburger, M. Helm and F. Lyko, Genes Dev., 2010, 24, 1590–1595 CAS.
- X. Yang, Y. Yang, B.-F. Sun, Y.-S. Chen, J.-W. Xu, W.-Y. Lai, A. Li, X. Wang, D. P. Bhattarai, W. Xiao, H.-Y. Sun, Q. Zhu, H.-L. Ma, S. Adhikari, M. Sun, Y.-J. Hao, B. Zhang, C.-M. Huang, N. Huang, G.-B. Jiang, Y.-L. Zhao, H.-L. Wang, Y.-P. Sun and Y.-G. Yang, Cell Res., 2017, 27, 606–625 CAS.
- Y. Yang, L. Wang, X. Han, W.-L. Yang, M. Zhang, H.-L. Ma, B.-F. Sun, A. Li, J. Xia, J. Chen, J. Heng, B. Wu, Y.-S. Chen, J.-W. Xu, X. Yang, H. Yao, J. Sun, C. Lyu, H.-L. Wang, Y. Huang, Y.-P. Sun, Y.-L. Zhao, A. Meng, J. Ma, F. Liu and Y.-G. Yang, Mol. Cell, 2019, 75, 1188–1202 CAS.
- J. I. Young, E. P. Hong, J. C. Castle, J. Crespo-Barreto, A. B. Bowman, M. F. Rose, D. Kang, R. Richman, J. M. Johnson, S. Berget and H. Y. Zoghbi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17551–17558 CAS.
- T. P. Hoernes, N. Clementi, K. Faserl, H. Glasner, K. Breuker, H. Lindner, A. Hüttenhofer and M. D. Erlacher, Nucleic Acids Res., 2016, 44, 852–862 CAS.
- Q. Li, X. Li, H. Tang, B. Jiang, Y. Dou, M. Gorospe and W. Wang, J. Cell. Biochem., 2017, 118, 2587–2598 CAS.
- G. Guo, H. Wang, X. Shi, L. Ye, K. Yan, Z. Chen, H. Zhang, Z. Jin and X. Xue, Front. Cell Dev. Biol., 2020, 8, 430 CrossRef PubMed.
- X. Chen, A. Li, B.-F. Sun, Y. Yang, Y.-N. Han, X. Yuan, R.-X. Chen, W.-S. Wei, Y. Liu, C.-C. Gao, Y.-S. Chen, M. Zhang, X.-D. Ma, Z.-W. Liu, J.-H. Luo, C. Lyu, H.-L. Wang, J. Ma, Y.-L. Zhao, F.-J. Zhou, Y. Huang, D. Xie and Y.-G. Yang, Nat. Cell Biol., 2019, 21, 978–990 CAS.
- H. Song, J. Zhang, B. Liu, J. Xu, B. Cai, H. Yang, J. Straube, X. Yu and T. Ma, Biomark. Res., 2022, 10, 15 Search PubMed.
- W. Huang, M.-D. Lan, C.-B. Qi, S.-J. Zheng, S.-Z. Wei, B.-F. Yuan and Y.-Q. Feng, Chem. Sci., 2016, 7, 5495–5502 CAS.
- S. M. Huber, P. van Delft, L. Mendil, M. Bachman, K. Smollett, F. Werner, E. A. Miska and S. Balasubramanian, ChemBioChem, 2015, 16, 752–755 CAS.
- L. Fu, C. R. Guerrero, N. Zhong, N. J. Amato, Y. Liu, S. Liu, Q. Cai, D. Ji, S.-G. Jin, L. J. Niedernhofer, G. P. Pfeifer, G.-L. Xu and Y. Wang, J. Am. Chem. Soc., 2014, 136, 11582–11585 CrossRef CAS PubMed.
- H.-Y. Zhang, J. Xiong, B.-L. Qi, Y.-Q. Feng and B.-F. Yuan, Chem. Commun., 2016, 52, 737–740 CAS.
- M. Yu, G. C. Hon, K. E. Szulwach, C.-X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen, B. Park, J.-H. Min, P. Jin, B. Ren and C. He, Cell, 2012, 149, 1368–1380 CAS.
- R. Lyu, K. Pajdzik, H. Sun, L. Zhang, L. Zhang, T. Wu, L. Yang, T. Pan, C. He and Q. Dai, Isr. J. Chem., 2024, 64, e202300111 CrossRef.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CAS.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CAS.
- M. Basanta-Sanchez, R. Wang, Z. Liu, X. Ye, M. Li, X. Shi, P. F. Agris, Y. Zhou, Y. Huang and J. Sheng, ChemBioChem, 2017, 18, 72–76 CAS.
- I. Rácz, I. Király and D. Lásztily, Planta, 1978, 142, 263–267 Search PubMed.
- B. Delatte, F. Wang, L. V. Ngoc, E. Collignon, E. Bonvin, R. Deplus, E. Calonne, B. Hassabi, P. Putmans, S. Awe, C. Wetzel, J. Kreher, R. Soin, C. Creppe, P. A. Limbach, C. Gueydan, V. Kruys, A. Brehm, S. Minakhina, M. Defrance, R. Steward and F. Fuks, Science, 2016, 351, 282–285 CAS.
- J. Moriya, T. Yokogawa, K. Wakita, T. Ueda, K. Nishikawa, P. F. Crain, T. Hashizume, S. C. Pomerantz, J. A. McCloskey, G. Kawai, N. Hayashi, S. Yokoyama and K. Watanabe, Biochemistry, 1994, 33, 2234–2239 CrossRef CAS PubMed.
- J. P. P. de Barros, G. Ketih, C. E. Adlouni, A. L. Glasser, G. Mack, G. Dirheimer and J. Desgrès, Nucleic Acids Res., 1996, 24, 1489–1496 CrossRef PubMed.
- C. Takemoto, T. Ueda, K.-i. Miura and K. Watanabe, Nucleic Acids Symp. Ser., 1999, 42, 77–78 Search PubMed.
- C. Takemoto, L. L. Spremulli, L. A. Benkowski, T. Ueda, T. Yokogawa and K. Watanabe, Nucleic Acids Res., 2009, 37, 1616–1627 CAS.
- H. Lusic, E. M. Gustilo, F. A. P. Vendeix, R. Kaiser, M. O. Delaney, W. D. Graham, V. A. Moye, W. A. Cantara, P. F. Agris and A. Deiters, Nucleic Acids Res., 2008, 36, 6548–6557 CrossRef CAS PubMed.
- N. Jonkhout, J. Tran, M. A. Smith, N. Schonrock, J. S. Mattick and E. M. Novoa, RNA, 2017, 23, 1754–1769 CAS.
- X. Wang, X. Jin and L. Cheng, Isr. J. Chem., 2024, 64, e202300178 CrossRef.
- C. Riml, A. Lusser, E. Ennifar and R. Micura, J. Org. Chem., 2017, 82, 7939–7945 CrossRef CAS PubMed.
- M. Sierant, G. Leszczynska, K. Sadowska, A. Dziergowska, M. Rozanski, E. Sochacka and B. Nawrot, Nucleic Acids Res., 2016, 44, 10986–10998 CAS.
- H. Sun, J. Sheng, A. E. A. Hassan, S. Jiang, J. Gan and Z. Huang, Nucleic Acids Res., 2012, 40, 5171–5179 CrossRef CAS PubMed.
- P. Szczupak, M. Sierant, E. Wielgus, E. Radzikowska-Cieciura, K. Kulik, A. Krakowiak, P. Kuwerska, G. Leszczynska and B. Nawrot, Cells, 2022, 11, 1522 CAS.
- A. V. Kachalova, E. M. Zubin and T. S. Oretskaya, Russ. Chem. Rev., 2002, 71, 1041–1059 CrossRef CAS.
- L. Flemmich, R. Bereiter and R. Micura, Angew. Chem., Int. Ed., 2024, 63, e202403063 CAS.
- L. C. Sowers and G. P. Beardsley, J. Org. Chem., 1993, 58, 1664–1665 CAS.
- N. Shakya, N. C. Srivastav, S. Bhavanam, C. Tse, N. Desroches, B. Agrawal, D. Y. Kunimoto and R. Kumar, Bioorg. Med. Chem., 2012, 20, 4088–4097 CAS.
- K. Bartosik and G. Leszczynska, Tetrahedron Lett., 2015, 56, 6593–6597 CrossRef CAS.
- K. Bartosik, E. Sochacka and G. Leszczynska, Org. Biomol. Chem., 2017, 15, 2097–2103 CAS.
- C. Riml and R. Micura, RNA Methylation: Methods and Protocols, in Methods Mol. Biol, 2017, vol. 1562, pp. 295–302 Search PubMed.
- C. Riml and R. Micura, Synthesis, 2016, 48, 1108–1116 CrossRef CAS PubMed.
- A. A. Tanpure and S. Balasubramanian, ChemBioChem, 2017, 18, 2236–2241 CrossRef CAS PubMed.
- I. N. Michaelides, N. Tago, B. Viverge and T. Carell, Chem. – Eur. J., 2017, 23, 15894–15898 CrossRef CAS PubMed.
- M. de Kort, P. C. de Visser, J. Kurzeck, N. J. Meeuwenoord, G. A. van der Marel, W. Rüger and J. H. van Boom, Eur. J. Org. Chem., 2001, 2075–2082 CAS.
- R. A. Johnsson, J. J. Bogojeski and M. J. Damha, Bioorg. Med. Chem. Lett., 2014, 24, 2146–2149 CrossRef CAS PubMed.
- M. P. Reddy, N. B. Hanna and F. Farooqui, Tetrahedron Lett., 1994, 35, 4311–4314 CAS.
- S. Tardy-Planechaud, J. Fujimoto, S. S. Lin and L. C. Sowers, Nucleic Acids Res., 1997, 25, 553–558 CAS.
- A. S. Hansen, A. Thalhammer, A. H. El-Sagheer, T. Brown and C. J. Schofield, Bioorg. Med. Chem. Lett., 2011, 21, 1181–1184 CAS.
- S. Xuan, Q. Wu, L. Cui, D. Zhang and F. Shao, Bioorg. Med. Chem. Lett., 2015, 25, 1186–1191 CAS.
- K. Sugizaki, S. Ikeda, H. Yanagisawa and A. Okamoto, Org. Biomol. Chem., 2011, 9, 4176–4181 RSC.
- Q. Dai and C. He, Org. Lett., 2011, 13, 3446–3449 CAS.
- Q. Dai, C.-X. Song, T. Pan and C. He, J. Org. Chem., 2011, 76, 4182–4188 CAS.
- M. Münzel, D. Globisch, C. Trindler and T. Carell, Org. Lett., 2010, 12, 5671–5673 CrossRef PubMed.
- M. Münzel, U. Lischke, D. Stathis, T. Pfaffeneder, F. A. Gnerlich, C. A. Deiml, S. C. Koch, K. Karaghiosoff and T. Carell, Chem. – Eur. J., 2011, 17, 13782–13788 CrossRef PubMed.
- A. S. Schröder, O. Kotljarova, E. Parsa, K. Iwan, N. Raddaoui and T. Carell, Org. Lett., 2016, 18, 4368–4371 Search PubMed.
- A. S. Schröder, J. Steinbacher, B. Steigenberger, F. A. Gnerlich, S. Schiesser, T. Pfaffeneder and T. Carell, Angew. Chem., Int. Ed., 2014, 53, 315–318 CrossRef PubMed.
- D.-Z. Yang, Z.-Z. Chen, M. Chi, Y.-Y. Dong, S.-Z. Pu and Q. Sun, Molecules, 2022, 27, 749 CAS.
- S. A. Scaringe, C. Francklyn and N. Usman, Nucleic Acids Res., 1990, 18, 5433–5441 CAS.
- T. Wu, K. K. Ogilvie and R. T. Pon, Nucleic Acids Res., 1989, 17, 3501–3517 CAS.
- S. Schiesser, T. Pfaffeneder, K. Sadeghian, B. Hackner, B. Steigenberger, A. S. Schröder, J. Steinbacher, G. Kashiwazaki, G. Höfner, K. T. Wanner, C. Ochsenfeld and T. Carell, J. Am. Chem. Soc., 2013, 135, 14593–14599 CrossRef CAS PubMed.
- K. Podskoczyj, K. Kulik, J. Wasko, B. Nawrot, T. Suzuki and G. Leszczynska, Chem. Commun., 2021, 57, 12540–12543 CAS.
- S. A. Hartsel, D. E. Kitchen, S. A. Scaringe and W. S. Marshall, Oligonucleotide Synthesis, in Methods Mol. Biol, 2005, vol. 288, pp. 33–49 Search PubMed.
- N. Karino, Y. Ueno and A. Matsuda, Nucleic Acids Res., 2001, 29, 2456–2463 CAS.
- M. Sundaram, P. F. Crain and D. R. Davis, J. Org. Chem., 2000, 65, 5609–5614 CrossRef CAS PubMed.
- A. C. Bajji, M. Sundaram, D. G. Myszka and D. R. Davis, J. Am. Chem. Soc., 2002, 124, 14302–14303 CAS.
- A. C. Bajji and D. R. Davis, J. Org. Chem., 2002, 67, 5352–5358 CAS.
- M. Eshete, M. T. Marchbank, S. L. Deutscher, B. Sproat, G. Leszczynska, A. Malkiewicz and P. F. Agris, Protein J., 2007, 26, 61–73 CAS.
- G. Leszczynska, J. Pięta, K. Wozniak and A. Malkiewicz, Org. Biomol. Chem., 2014, 12, 1052–1056 RSC.
- G. Leszczynska, P. Leonczak, A. Dziergowska and A. Malkiewicz, Nucleosides, Nucleotides Nucleic Acids, 2013, 32, 599–616 Search PubMed.
- S. Schiesser, B. Hackner, T. Pfaffeneder, M. Müller, C. Hagemeier, M. Truss and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 6516–6520 Search PubMed.
- Y. Nomura, N. Haginoya, Y. Ueno and A. Matsuda, Bioorg. Med. Chem. Lett., 1996, 6, 2811–2816 CrossRef CAS.
- Y. Ito, M. Matsuo, K. Yamamoto, W. Yamashita, T. Osawa and Y. Hari, Tetrahedron, 2018, 74, 6854–6860 CrossRef CAS.
- Y. Ito, K. Yamamoto and Y. Hari, Curr. Protoc. Nucleic Acid Chem., 2019, 78, e91 Search PubMed.
- K. Podskoczyj, A. Klos, S. Drewniak and G. Leszczynska, Org. Biomol. Chem., 2023, 21, 2809–2815 RSC.
- K. Podskoczyj, A. Kuszczynska, A. Dziergowska and G. Leszczynska, Curr. Protoc., 2024, 4(2), e984 CAS.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |