
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective Lewis base catalysed allylation of picoline- and quinaldine-based latent pronucleophiles†
Markus
Lange
,
Nikita
Alistratov
and
Ivan
Vilotijevic
 *
*
Institute of Organic Chemistry and Macromolecular Chemistry, Friedrich Schiller University Jena, Humboldtstrasse 10, 07743 Jena, Germany. E-mail: ivan.vilotijevic@uni-jena.de
First published on 3rd August 2024
Abstract
Picolines and quinaldines are valuable building blocks and intermediates in the synthesis of natural products and pharmaceuticals. Functionalization of the methyl group in picolines and quinaldines under mild conditions is challenging. We report that the concept of latent pronucleophiles enables Lewis base catalysed allylation of picolines and quinaldines with allylic fluorides starting from silylated picolines and quinaldines. Reactions afford enantioenriched allylation products when chiral Lewis base catalysts are used. The allylation products can be rapidly transformed to quinolizine-4-ones.
Introduction
Nitrogen containing mono-, bi-, and polycyclic ring systems are omnipresent in biologically active compounds (Scheme 1a).1 Among them, quinolines and pyridines that feature alkyl substituents in position 2, often referred to as picolines and quinaldines, are frequently found as key structural motifs in natural products and pharmaceuticals.2 C–H functionalization of pyridines and their higher homologues to access picoline type structures is challenging due to their low intrinsic reactivity and often requires two-step processes.3 Another strategy toward these structures, functionalization of the methylene carbon in picolines, usually involves deprotonation followed by reaction with an electrophile. However, the formation of picoline and quinaldine anions requires strong bases4 or combinations of a strong Lewis acid and a base which greatly reduces the reaction scope.5 The reported enantioselective alkylations or allylations of methyl pyridines or quinolines require equimolar chiral auxiliaries,4 harsh Lewis acids6 or an intervention by a palladium catalyst7 (Scheme 2b).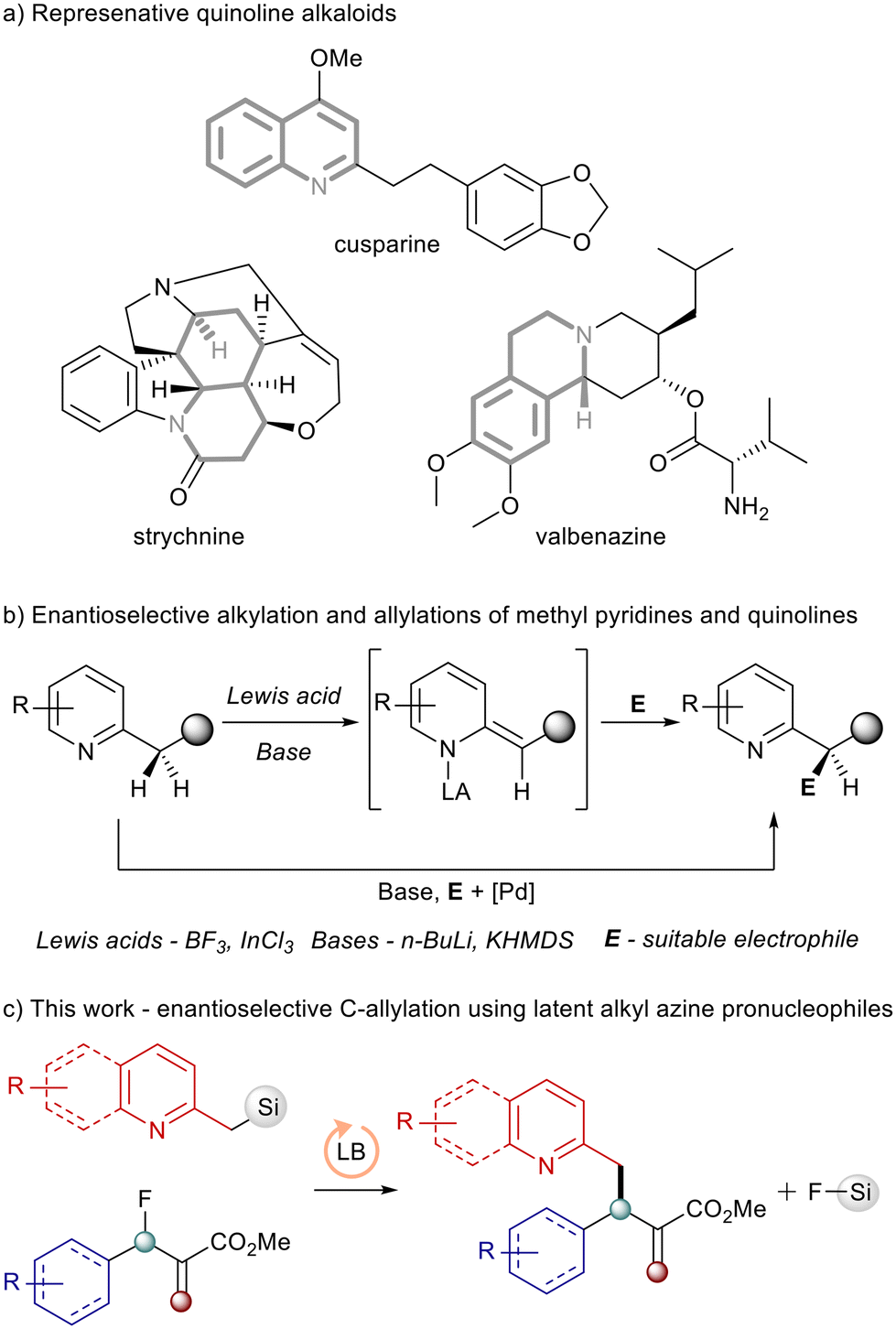 | ||
| Scheme 1 (a) Representative examples of quinoline and tetrahydroquinoline alkaloids; (b) established enantioselective alkylation and allylation procedures for 2-alkyl azines.4,6,7 (c) The enantioselective allylation of alkyl pyridines and quinolines using latent pronucleophiles reported here. | ||
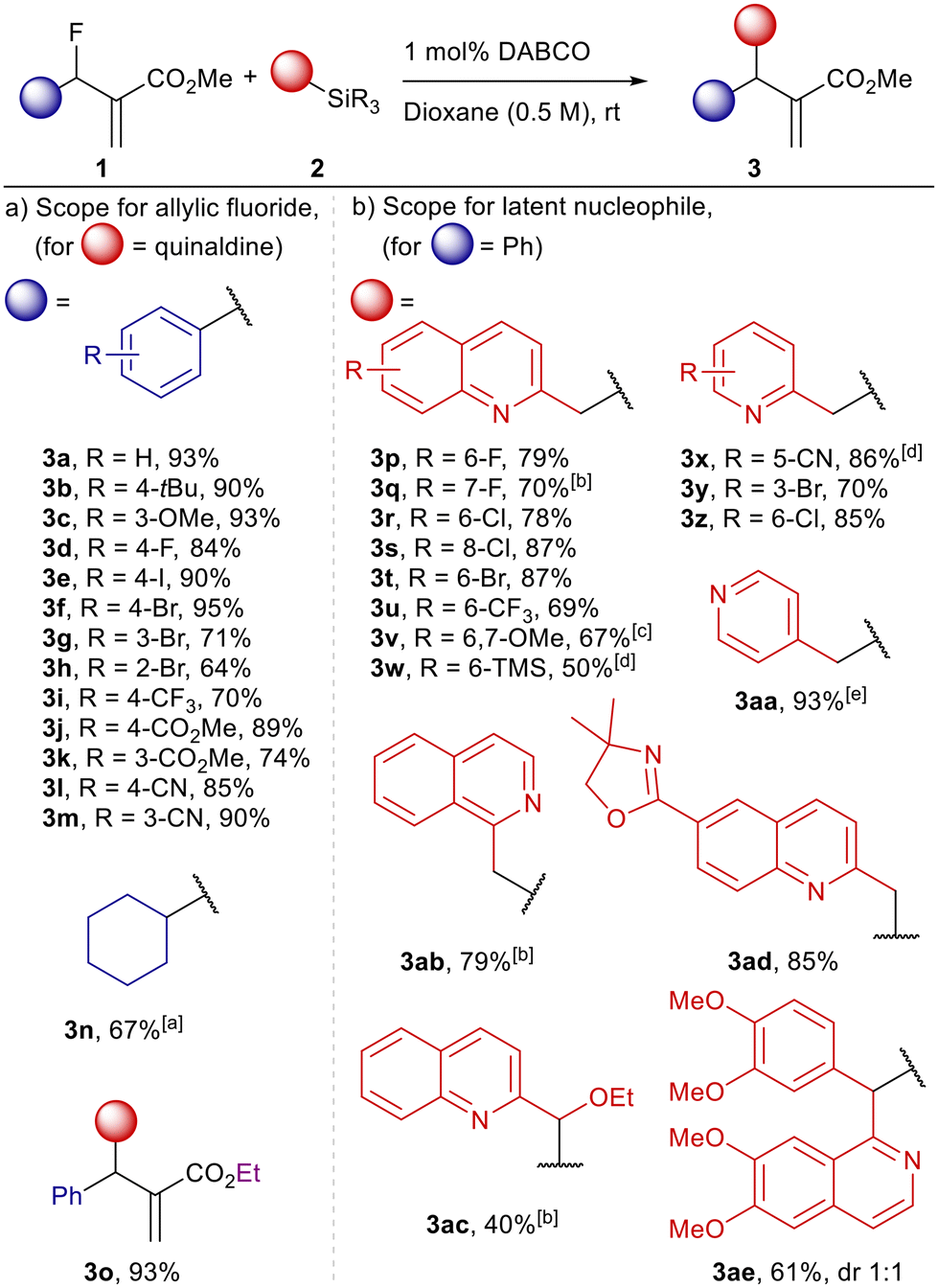 | ||
Scheme 2 Reaction scope for (a) allylic fluorides and (b) latent pronucleophiles. For evaluation of the allylic fluorides trimethyl silyl quinaldine was used. Reaction scope for quinaldines and picolines was evaluated using fluoride 1a and triethyl silyl quinaldines or picolines unless stated otherwise. Conditions: latent pronucleophile (1.1 equiv., 0.11 mmol), DABCO (1 mol%), allylic fluoride (1 equiv., 0.1 mmol), dioxane (0.5 M), N2-atmosphere. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 mol% of DABCO; btrimethylsilyl derivative was used; ccrude trimethylsilyl derivative was used; d10 mol% of DABCO; etrimethylsilyl derivative was used and 10 mol% of DABCO. 5 mol% of DABCO; btrimethylsilyl derivative was used; ccrude trimethylsilyl derivative was used; d10 mol% of DABCO; etrimethylsilyl derivative was used and 10 mol% of DABCO. | ||
Functionalization of alkyl pyridines and quinolines in the presence of Lewis base catalysts is hard to achieve and has not been demonstrated universally.8 A possible reason for this is the nucleophilic character of the nitrogen atom in picolines and quinaldines which may compete with the Lewis base catalyst.9 In enantioselective Lewis base catalysis this can lead to insurmountable limitations as the reactions may proceed autocatalytically without the involvement of the chiral catalyst preventing the catalyst control of the stereochemical outcome of the reaction.
To address such problems in Lewis base catalysed reactions using N-centered nucleophiles, we introduced the concept of latent (pro)nucleophiles in Lewis base catalysis.10 Latent pronucleophiles are species that are themselves not strongly nucleophilic. However, at an opportune point in the catalytic cycle, they can be activated to react as strong anionic nucleophiles. If this activation is dependent on and mediated by the leaving group released during activation of the electrophile by a chiral Lewis base, the reactions exhibit excellent chemo,- regio- and enantioselectivity.10,11 The combination of silylated pronucleophile, where silyl group serves as a locator of the nucleophilic position and a readily available allylic fluoride12 serving as electrophilic coupling partner13 has also proven effective in allylation of C-centered nucleophiles in previous studies by Shibata,14 Companyó15 and our group.10,11 We hypothesized, and here we report, that the concept of latent pronucleophiles can be a blueprint for the enantioselective C-allylation of methyl pyridines and quinolines when such compounds are used as silylated pronucleophiles in combination with fluorinated electrophiles (Scheme 1c).
Results and discussion
We initiated our investigations with optimization of the DABCO-catalysed reactions between allylic fluorides 1, derived from the corresponding Morita–Baylis–Hilmann (MBH) alcohols, and silyl azines 2 produced by direct silylation of quinaldines or pyridines (Scheme 2, for details of optimization studies see ESI†). Trimethylsilyl (TMS), triethylsilyl (TES) and tert-butyldimethylsilyl (TBS) derivatives of quinaldine were investigated as suitable pronucleophiles. TMS quinaldine reached full conversion faster compared to the TES analogue while TBS quinaldine did not show notable reactivity under comparable conditions. When TMS quinaldine 2a was used in combination with 1a in the absence of Lewis base catalyst no reaction occurred, yet only 0.5 mol% of DABCO was sufficient to achieve full conversion in larger scale reactions using 5 mmol of 1a. More polar solvents enabled faster conversion and high product yields with dioxane delivering the best results. The optimized reaction conditions included a slight excess of latent pronucleophile and 1 mol% of DABCO as catalyst in dioxane at room temperature.The reaction scope for allylic fluorides was evaluated first with a set of electron rich allylic fluorides (3b–3c) which gave the corresponding products in excellent yields of 90% and above (Scheme 2a). Allylic fluorides featuring ortho, meta, and para substituted aryl halides were well tolerated and furnished the allylated quinaldines (3d–3g) in good yields (70–95%) though slightly lower yield of 64% was observed for 3h which features a bromo substituent in ortho position adjacent to the reactive centre. Reactions of allylic fluorides featuring electron withdrawing groups (3i–3m) were qualitatively observed to proceed with higher rates and the yields remained high as in the previous cases (70–90%). Allylic fluoride carrying a cyclohexyl group instead of an aryl substituent delivered the desired product 3n with satisfying efficacy of 67%.
The reaction scope for the nucleophilic partner was examined with a structurally diverse set of silylated quinaldines, picolines and related compounds (Scheme 2b). The TES derivatives were preferred as they proved easier to handle than the corresponding TMS derivatives. Halogenated quinaldines with substituents in 6, 7, and 8 position, respectively, underwent the reactions with 1a smoothly with yields for products 3p–3t between 70 and 87%. Quinaldines with electron-withdrawing trifluoromethyl and oxazoline substituents also delivered products in high yields up to 85% (3u and 3ad). Interestingly, the presence of a trimethylaryl silane within the allylic fluoride was also well tolerated in these reactions with 6-TMS-substituted quinaldine 3w obtained in 50% yield though increased catalyst loading was required to drive the reaction to completion. Even more complex quinaldines with additional aryl or alkoxy substituents at the methylene group performed well despite the increased steric crowding at the nucleophilic carbon. When 2-(ethoxy(trimethylsilyl)methyl)quinoline was used as the pronucleophile, the corresponding product 3ac was obtained in 40% yield as a single diastereomer (the crude mixture contained two diastereomers but only one could be isolated in pure form). The silylated pronucleophile derived from papaverine gave the allylation product 3ae in 61% yield as a statistical mixture of diastereomers. The use of the related silylated 1-methylisoquinoline further confirmed the generality of the process giving the product 3ab in 79% yield. Picoline-based pronucleophiles also performed well under the optimized conditions with silylated 5-cyano-, 3-bromo and 6-chloro-2-picolines giving the desired products 3x, 3y and 3z in 86%, 70% and 85% yield, respectively. Strikingly, silylated 4-picoline, which is itself nucleophilic and could compete with some Lewis base catalysts,9 provided the corresponding product 3aa in excellent yield of 93%. In this example, silylation has negligible effect on the nucleophilicity of the pyridine yet the desired reaction pathway appears to kinetically outcompete other possible pathways, i.e. pyridine acting as the nucleophile.
After extensively establishing the reactivity patterns in DABCO catalysed reactions, we focused on the asymmetric variant and examined the reactions in the presence of chiral Lewis base catalysts (Scheme 3). The commercially available cinchona alkaloids with a proven track record in transformations of other MBH adducts have been in focus of this study.10,14e,16 In our previous work, we observed dimeric catalysts such as (DHQD)2PHAL and its pseudoenantiomer (DHQ)2PHAL to catalyse similar reactions exceptionally well. In the current study, (DHQD)2PHAL furnished products with high degrees of enantiocontrol with er up to 95:5 but showed poor regioselectivity for substitution of allylic electrophile. After extensive testing of reaction parameters, we arrived at the final set of conditions which involve 10 mol% (DHQD)2AQN in o-xylene with 1.5 equivalents of latent pronucleophile (for details of optimization studies see ESI†). In contrast to the reactions with DABCO, good conversions were observed only with TMS-quinaldines which was attributed to significantly lower reaction rates in reactions with chiral Lewis base catalysts. We employed a variety of allylic fluorides to test the reaction scope (Scheme 3a). Among electron rich (3b′ and 3c′), electron poor (3i′–3m′), and allylic fluorides featuring aryl halides (3e′–3g′) the enantioselectivities ranged from 14:86 er for 3k′ to 94:6 er for 3b′ while yields varied between 59% (3b′) to 95% (3j′). Yields were adversely influenced by electron donating substituents in allylic fluoride (3c′, 35%). Surprisingly, ethyl ester 3o′ was obtained in comparable yield but with a lower degree of stereocontrol compared to methyl ester 3a′. While testing the scope for quinaldine and 1-methylchinoline based pronucleophiles (Scheme 3b), the degree of enantiocontrol remained consistently around 15:85 (3q′–3af′) and yields ranged from 50% to 69%. The absolute configuration of the preferred product enantiomer when (DHQD)2AQN was used as the catalyst is tentatively assigned as R based on similar transformations using the same catalyst.16d,e
 | ||
| Scheme 3 Scope of enantioselective allylation for (a) allylic fluorides and (b) latent pronucleophiles. Conditions: (DHQD)2AQN (10 mol%), latent pronucleophile (1.5 equiv., 0.15 mmol), allylic fluoride (1 equiv., 0.1 mmol), xylene (0.3 M) N2-atmosphere. The er values represent the order of elution and do not account for the stereochemical outcome being either R or S. | ||
We observed the reactions to be a kinetic resolution. Both (DHQD)2PHAL and (DHQD)2AQN consume the same enantiomer of the allylic fluoride faster, however, the products are delivered as antipodes indicating that stereocontrol does not solely depend on the chiral backbone of the cinchona alkaloid but that it also depends on the linker in the dimeric catalyst (for details see ESI†).17 Similar observations related to the influence of the linker in dimeric cinchona alkaloid catalysts have recently been reported in other transformations.18
Several mechanistic proposals have been put forward for the Lewis base catalysed reactions of allylic fluorides with different nucleophiles including concerted,14c,15 stepwise,10 and silicon-assisted pathways (Scheme 4a).15 We first sought to test if the stepwise mechanism which has been reported in DABCO catalysed allylations of N-silylindoles with allylic fluorides holds in the presence of the less nucleophilic (DHQD)2PHAL. In a crossover experiment, allylic fluoride 1a was reacted with silylated indole 5 and deuterium labelled indole 6 in the presence of (DHQD)2PHAL as the catalyst (Scheme 4b). The isolated products showed incorporation of deuterium slightly over 50% both in xylene and trifluorotoluene. We confirmed that deuterated indole does not react with 1a in the presence of Lewis base catalyst and that there is no silyl transfer between 5 and 6 under the reaction conditions. These results indicate the transient formation of an indole anion and a crossover via acid–base equilibrium of indole anions derived from 5 and 6 and are consistent with a stepwise mechanism. To test if similar crossover can be observed in allylation of quinaldines, an equimolar mixture of 15N enriched quinaldine 8 and trimethylsilyl quinaldine 2a was subjected to allylation with 1a using (DHQD)2AQN in o-xylene or 1,4-dioxane as solvent (Scheme 4c). The HRMS analysis of the reaction product did not show incorporation of the 15N concluding that there was no acid–base equilibrium which would lead to crossover as in the case of N-allylation of indole. While this could speak in favour of a concerted mechanism, formation of a tight ion pair in a solvent cage19 would also be consistent with this observation. The concerted mechanism proposed in allylations of silylenolethers15 would either lead to N-allylation of picolines and quinaldines via a six membered transition state which was not observed or would proceed via a highly ordered ternary four-membered transition state that leads to C-allylation products observed in these reactions. Finally, the fact that (DHQD)2PHAL and (DHQD)2AQN consume the same enantiomer of the allylic fluoride faster but deliver the opposite antipodes of the product also speaks against the concerted mechanism.
 | ||
| Scheme 4 Mechanistic considerations. (a) Potential mechanistic scenarios for the allylic substitution. (b) Crossover experiment of latent nucleophile 5 and 2-D-indole 6 under enantioselective substitution conditions. (c) Crossover experiment of latent pronucleophile 2a and 15N enriched quinaldine 8 under standard enantioselective substitution conditions. | ||
To demonstrate the synthetic utility of the C-allylation products obtained in this study, a short synthetic sequence was developed for synthesis of functionalized quinolizine-4-ones (Scheme 5). To this end, a modified protocol for reductive alkylation20 was employed to obtain reduced quinaldine. A two-step process of saponification followed by PyBop mediated lactam formation gave the desired quinolizine-4-ones. Gratifyingly, both diastereomers of each product could be isolated separately and the combined yields were 57% for 4a/4a′, 54% for 4b/4b′, and 47% for 4f/4f′ over two steps.
 | ||
| Scheme 5 Transformation of allylated quinaldines to functionalized quinolizine-4-ones. (a) Hantzsch ester (2.5 equiv.), m-F3C-C6H4-B(OH)2 (0.25 equiv.), C2H4Cl2, 60 °C; (b) LiOH (4.0 equiv.), THF:EtOH, 40 °C; (c) PyBOP (1.015 equiv.), DIPEA (1.1 equiv.), DCM:DMF. | ||
Conclusions
We have developed a robust tool for regio- and enantioselective allylation of quinaldines and picolines in the presence of Lewis base catalysts. The reactions are enabled by the concept of latent pronucleophiles in Lewis base catalysis and the use of silyl derivatives of quinaldines and picolines in combination with allylic fluorides. They feature broad scope and deliver the products with degrees of enantiocontrol as high as 94:6. The products land themselves to a rapid transformation to quinolizine-4-one frameworks. Interestingly, the stereocontrol in these reactions does not solely depend on the chiral backbone of the cinchona alkaloid but it also depends on the linker in the dimeric catalyst. The mechanistic implications of this observation are currently under investigation.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was a part of a German Science Foundation (DFG) funded project number 445755502. State of Thuringia (Fellowship to M. L.) is gratefully acknowledged. We thank Dr Nico Ueberschaar for help and assistance with the MS analysis in the crossover experiment and Ms Olena Nosovska for critical proofreading of the manuscript. Support of Thüringer Aufbaubank through project NMR-ESU (2022 FGI 0015) is gratefully acknowledged.References
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- (a) A. Fournet, A. A. Barrios, V. Muñoz, R. Hocquemiller, A. Cavé and J. Bruneton, Antimicrob. Agents Chemother., 1993, 37, 859–863 CrossRef CAS PubMed; (b) X.-F. Shang, S. L. Morris-Natschke, Y.-Q. Liu, X. Guo, X.-S. Xu, M. Goto, J.-C. Li, G.-Z. Yang and K.-H. Lee, Med. Res. Rev., 2018, 38, 775–828 CrossRef CAS PubMed.
- S. Maity, A. Bera, A. Bhattacharjya and P. Maity, Org. Biomol. Chem., 2023, 21, 5671–5690 RSC.
- J. J. Gladfelder, S. Ghosh, M. Podunavac, A. W. Cook, Y. Ma, R. A. Woltornist, I. Keresztes, T. W. Hayton, D. B. Collum and A. Zakarian, J. Am. Chem. Soc., 2019, 141, 15024–15028 CrossRef CAS PubMed.
- (a) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092–14093 CrossRef CAS PubMed; (b) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2009, 131, 12056–12057 CrossRef CAS PubMed.
- M. Meazza, F. Tur, N. Hammer and K. A. Jørgensen, Angew. Chem., Int. Ed., 2017, 56, 1634–1638 CrossRef CAS PubMed.
- (a) R. Murakami, K. Sano, T. Iwai, T. Taniguchi, K. Monde and M. Sawamura, Angew. Chem., Int. Ed., 2018, 57, 9465–9469 CrossRef CAS PubMed; (b) S.-C. Sha, J. Zhang, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 17602–17609 CrossRef CAS PubMed; (c) B. M. Trost, D. A. Thaisrivongs and J. Hartwig, J. Am. Chem. Soc., 2011, 133, 12439–12441 CrossRef CAS PubMed.
- (a) N. S. Kumar, L. C. Rao, N. JagadeeshBabu, V. Dileep Kumar, U. S. N. Murthy and H. M. Meshram, Synlett, 2015, 26, 1808–1814 CrossRef CAS; (b) J. Izquierdo, A. Landa, I. Bastida, R. López, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2016, 138, 3282–3285 CrossRef CAS PubMed.
- F. Brotzel, B. Kempf, T. Singer, H. Zipse and H. Mayr, Chem. – Eur. J., 2007, 13, 336–345 CrossRef CAS PubMed.
- Y. Zi, M. Lange, C. Schultz and I. Vilotijevic, Angew. Chem., Int. Ed., 2019, 58, 10727–10731 CrossRef CAS PubMed.
- (a) Y. Zi, M. Lange and I. Vilotijevic, Chem. Commun., 2020, 56, 5689–5692 RSC; (b) M. Lange, Y. Zi and I. Vilotijevic, J. Org. Chem., 2020, 85, 1259–1269 CrossRef CAS PubMed; (c) M. Lange, Y. Zi and I. Vilotijevic, Synlett, 2020, 31, 1237–1243 CrossRef CAS; (d) S. Kumar, M. Lange, Y. Zi, H. Görls and I. Vilotijevic, Chem. – Eur. J., 2023, 29, e202300641 CrossRef CAS PubMed; (e) M. Lange, F. L. Meyer, O. Nosovska and I. Vilotijevic, Org. Lett., 2023, 25, 9097–9102 CrossRef CAS PubMed.
- (a) F. O. Usman, A. R. Gogoi, J. C. Mixdorf, O. Gutierrez and H. M. Nguyen, Angew. Chem., Int. Ed., 2023, 62, e202314843 CrossRef CAS PubMed; (b) J. C. Mixdorf, A. M. Sorlin, Q. Zhang and H. M. Nguyen, ACS Catal., 2018, 8, 790–801 CrossRef CAS.
- (a) K. Balaraman and C. Wolf, Org. Lett., 2021, 23, 8994–8999 CrossRef CAS PubMed; (b) K. Balaraman and C. Wolf, Sci. Adv., 2022, 8, eabn7819 CrossRef CAS PubMed.
- (a) Y. Sumii, T. Nagasaka, J. Wang, H. Uno and N. Shibata, J. Org. Chem., 2020, 85, 15699–15707 CrossRef CAS PubMed; (b) T. Nishimine, H. Taira, S. Mori, O. Matsubara, E. Tokunaga, H. Akiyama, V. A. Soloshonok and N. Shibata, Chem. Commun., 2017, 53, 1128–1131 RSC; (c) S. Okusu, H. Okazaki, E. Tokunaga, V. A. Soloshonok and N. Shibata, Angew. Chem., Int. Ed., 2016, 55, 6744–6748 CrossRef CAS PubMed; (d) T. Nishimine, H. Taira, E. Tokunaga, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2015, 55, 359–363 CrossRef PubMed; (e) T. Nishimine, K. Fukushi, N. Shibata, H. Taira, E. Tokunaga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2013, 53, 517–520 CrossRef PubMed.
- J. Duran, J. Mateos, A. Moyano and X. Companyó, Chem. Sci., 2023, 14, 7147–7153 RSC.
- (a) J.-R. Huang, H.-L. Cui, J. Lei, X.-H. Sun and Y.-C. Chen, Chem. Commun., 2011, 47, 4784–4786 RSC; (b) Z. Li, M. Frings, H. Yu, G. Raabe and C. Bolm, Org. Lett., 2018, 20, 7367–7370 CrossRef CAS PubMed; (c) A. Lin, H. Mao, X. Zhu, H. Ge, R. Tan, C. Zhu and Y. Cheng, Chem. – Eur. J., 2011, 17, 13676–13679 CrossRef CAS PubMed; (d) V. Dočekal, M. Šimek, M. Dračínský and J. Veselý, Chem. – Eur. J., 2018, 24, 13441–13445 CrossRef PubMed; (e) B. Formánek, M. Šimek, M. Kamlar, I. Císařová and J. Veselý, Synthesis, 2019, 51, 907–920 CrossRef; (f) O. Nosovska, P. Liebing and I. Vilotijevic, Chem. – Eur. J., 2024, 30, e202304014 CrossRef CAS PubMed.
- In previous studys on the enantioselective substituion of allylic fluorides, we and other (compare ref. 10) observed that (DHQD)2AQ N and (DHQD)2PH AL produce the same product enantiomere.
- (a) P. J. Boratyński, Mol. Diversity, 2015, 19, 385–422 CrossRef PubMed; (b) S. E. Luderer, B. Masoudi, A. Sarkar, C. Grant, A. Jaganathan, J. E. Jackson and B. Borhan, J. Org. Chem., 2023 DOI:10.1021/acs.joc.3c00084; (c) K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134–8137 CrossRef CAS PubMed; (d) M. Wilking, C. G. Daniliuc and U. Hennecke, Synlett, 2014, 25, 1701–1704 CrossRef.
- (a) L. Herk, M. Feld and M. Szwarc, J. Am. Chem. Soc., 1961, 83, 2998–3005 CrossRef CAS; (b) R. K. Lyon and D. H. Levy, J. Am. Chem. Soc., 1961, 83, 4290–4290 CrossRef CAS; (c) C. Reichardt, J. Org. Chem., 2022, 87, 1616–1629 CrossRef CAS PubMed; (d) C. G. Swain, M. S. Swain, A. L. Powell and S. Alunni, J. Am. Chem. Soc., 1983, 105, 502–513 CrossRef CAS.
- P. Adhikari, D. Bhattacharyya, S. Nandi, P. K. Kancharla and A. Das, Org. Lett., 2021, 23, 2437–2442 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01063a |
| This journal is © The Royal Society of Chemistry 2024 |