DOI:
10.1039/D4OB01044B
(Review Article)
Org. Biomol. Chem., 2024,
22, 6860-6904
Transition metal-catalyzed C(sp2/sp3)–H α-fluoroalkenylation from gem-(bromo/di)fluoroalkenes to monofluoroalkenes: scope, mechanisms, and synthetic applications
Received
24th June 2024
, Accepted 31st July 2024
First published on 1st August 2024
Abstract
Organofluorines have a broad range of industrial applications, such as pharmaceuticals, liquid crystal displays (LCDs), solar cells, textiles, and construction coatings, and are used in peptidomimetics, surfactants, refrigerants, anesthetics, and agrochemicals. Among them are versatile monofluoroalkenes that play a crucial role in medicinal and synthetic chemistry. The synthetic strategies for this class of molecules are limited, and prior efforts frequently suffered from poor atom- and step-economies. As a surrogate pathway for traditional cross-coupling transformations, transition metal (TM)-catalyzed C–H direct α-fluoroalkenylation overcomes these obstacles and provides straightforward techniques to access monofluoroalkenes. Nevertheless, substrate scope is still a challenge for catalysis, where gem-bromofluoroalkene synthons are applicable with electronically biased substrates such as azoles, while gem-difluoroalkene-based strategies are limited to substrates containing N-based directing groups. Herein, we review the cutting-edge fluoroalkenylation research for direct synthesis of monofluoroalkenes achieved during the last decade (2013–2023). This review is divided into two main parts: the first part discusses TM-catalyzed direct α-fluoroalkenylation via the merging of C–H activation and C(sp2)–Br cleavage strategies using gem-bromofluoroalkenes, and the second part describes the same reaction, albeit with C(sp2)–F cleavage of highly explored gem-difluoroolefins. Our review surveys all previously reported monofluoroalkenes in this research area, including their preparation techniques, stereoselectivity, and yield percentages. Furthermore, optimal conditions, reactant scope, mechanistic investigations, synthetic applications, benefits, and drawbacks of each presented methodology are critically discussed.
 Essam M. Eliwa | Essam M. Eliwa is a post-doctoral researcher in the Djukic's group at the University of Strasbourg, France (Nov 2023–Apr 2024). He received his B.Sc. degree in 2006 in Special Chemistry with a grade of excellent and an honors degree. He obtained his M.Sc. and Ph.D. degrees in organic chemistry from Al-Azhar University in 2013 and 2017, respectively. During his Ph.D., he spent one year in Sewald's group at the University of Bielefeld, Germany (2015–2016). His current research interests include C–H fluoroalkenylation reactions of heterocyclic compounds using earth-abundant TM catalysts with the intention of applying green chemistry principles for the design and synthesis of valuable bioactive molecules. Dr Essam M. Eliwa: E-mail: essameliwa85@azhar.edu.eg; https://orcid.org/0000-0001-5570-1427. |
 Ahmed H. Bedair | Ahmed H. Bedair received a B.Sc. in chemistry from Ain Shams University in 1970. He obtained his M.Sc. and Ph.D. degrees in organic chemistry from Al-Azhar University in 1975 and 1977, respectively. As a post-doctoral researcher, he spent four months in Kroeker's group at the University of Winnipeg, Canada (2003). His current research interests include the chemistry of coumarins and dyes and the synthesis of new fluorescent materials. Prof. Dr. Ahmed H. Bedair: E-mail: bedair48azhar@gmail.com; https://orcid.org/0009-0008-0796-6437. |
 Jean-Pierre Djukic | Jean-Pierre Djukic obtained his Ph.D. from University Pierre & Marie Curie, Paris (France), under the guidance of Eric Rose in 1992. He held a post-doctoral research assistant position at the Iowa State University (USA) in the group of L. Keith Woo from 1993 to 1994 and was hired by the CNRS at the University of Strasbourg. He is a fellow of the A. von Humboldt foundation and spent a research period in the group of Karl Heinz Dötz at the University of Bonn (Germany) from 1996 to 1997. As a Director of Research, since 2012 he has been focusing on reaction intermediate trapping, reaction calorimetry, mechanisms of catalyzed C–H bond functionalization and hydrosilylation, and theoretical assessments of noncovalent interactions in various organic and organometallic systems. Dr Jean-Pierre Djukic: E-mail: djukic@unistra.fr; https://orcid.org/0000-0003-3196-4921; Scopus ID: 6603745567, Laboratoire de Chimie et Systémique Organométallique, UMR 7177 Institut de Chimie – CNRS/Université de Strasbourg, 4 rue Blaise Pascal, F-67000 Strasbourg, France. |
1. Introduction
The realm of transition metal (TM)-based catalysts is of extensive interest in academic and industrial research laboratories as an ultimate solution to the synthesis of high-value organic compounds. TM-based catalysts are applied as supported or not on a solid. These might be a wide range of coordination complexes with significant catalytic activity, organometallic complexes, metal oxides, simple metal salts, or even pure metals.1–4
In the organic synthesis community, C–H bond activation and functionalization chemistry has acquired considerable momentum as a powerful tool to access diverse organic frameworks through extraordinary bond cleavage methodologies.5–12
Among organohalogens, organofluorine chemicals are relevant to several aspects of daily life and technology. They are essential reagents for the materials science, pharmaceutical,13 and agrochemical industries,14 among other fields. Organofluorines are thought to make up about 53% of agrochemicals and 20% of medicines, including some of the most popular drugs. As ligands, they have also been studied in coordination and organometallic chemistry.15 Fluorine atoms (or C–F bond formation) can be introduced to the organic molecule skeleton through C–H fluorination, fluoroalkylation, or fluoroalkenylation reactions.16–24
Monofluoroalkenes are a versatile class of organic molecules; they can be used as fluorinated synthons in synthetic organofluorine chemistry.17,23,24 They also play a crucial role in chemical biology as precursors of bioactive molecules and impact the medicinal chemistry domain as a bioisostere for peptide linkage because they can impart desirable electronic and steric characteristics as well as enhance lipophilicity, improve recognition, and increase metabolic and conformational stability.13 As a result, several strategies have been deployed to obtain various monofluoroalkene classes. In the last decade (2013–2023), TM-catalyzed C–H fluoroalkenylation has proven to be a successful and promising strategy.17,23,24 It is endowed with the general advantages of being atom- and step-economical25 as well as being specifically oxidant-free (redox-neutral), stereoselective and regioselective, with good functional group tolerance, no alkynylated side-product, and a scalable reaction.
Despite the fact that C–H α-fluoroalkenylation has attracted widespread attention in the last decade as a major methodology to access monofluoroalkenes, it is still facing significant challenges such as substrate scope (general applicability) and harsh conditions. Regarding the limitations of substrates, they are either electronically biased or pre-directed substrates. We find that the reported protocols that utilized gem-bromofluoroalkene synthons are applicable with “electronically biased” substrates such as azoles,26–28 and their gem-difluoroalkene counterparts are limited to substrates containing N-based directing groups (DG),29–46 whereas other substrates are still unsuitable, such as ketones, esters, acids, etc.
Furthermore, as we will see below, all the reported competition experiments confirmed the superiority of substrates and fluorinated reagents that contain electron-donating groups (EDGs) over the analogues carrying electron-withdrawing groups (EWGs). Though some mild and cost-effective α-fluoroalkenylation procedures (room temperature and abundant TM catalysis) are reported,28,40–46 harsh conditions are still a significant obstacle that needs to be addressed. According to several experimental and theoretical studies,30,39,41,43 the α-fluoroalkenylation mechanistic pathway employing gem-difluoroalkene synthons proceeds via four main steps, including C–H activation, olefin π-coordination, regioselective migratory olefin insertion, and β-fluoride elimination. In contrast with TM-mediated oxidative addition to cleavage C–F bonds, the β-F elimination route proceeds under milder conditions via β-fluoroorganometallic intermediates to install C–C coupling products (monofluoroalkenes).23,24 Additionally, kinetic isotope effect (KIE) tests indicated that either C–H dissociation or alkene migratory insertion is a rate-determining step.30,37,39
Given the importance of monofluoroalkenes and the synthetic obstacles of C–H fluoroalkenylation reactions, there was an imperious need to survey recent achievements in this field. Thus, this review summarizes with mechanistic and synthetic details the newly discovered C–H α-fluoroalkenylation approaches for accessing distinct monofluoroalkenes categorized according to their synthetic protocols, which are based on the types of gem-halofluoroalkene building blocks and TM catalysts.
This review is divided into two parts: the first one focuses on relatively less explored direct α-fluoroalkenylation via the merging of C–H activation and C(sp2)–Br cleavage strategies using gem-bromofluoroalkenes, and the second part describes the same reaction, albeit with C(sp2)–F cleavage of easily accessible and commercially available gem-difluoroolefins. Each part is subdivided into subsections according to the type of TM-based catalysis. This review covers all the reported monofluoroalkenes in this research area from 2013 up to 2024 with the specific procedures, stereoselectivity, and yields. Moreover, this review reports on the optimal conditions, reactant scope, mechanistic studies, synthetic applications, advantages, and shortcomings of each reported protocol. Furthermore, some related out-of-scope examples are also considered for comparison. Finally, this review critically outlines the achievements in the field of C–H direct α-fluoroalkenylation and draws some perspectives for future advances.
2. Direct α-fluoroalkenylation with gem-bromofluoroalkenes
2.1. C–H α-fluoroalkenylation using Pd(0)/Cu(I) bimetallic catalytic system
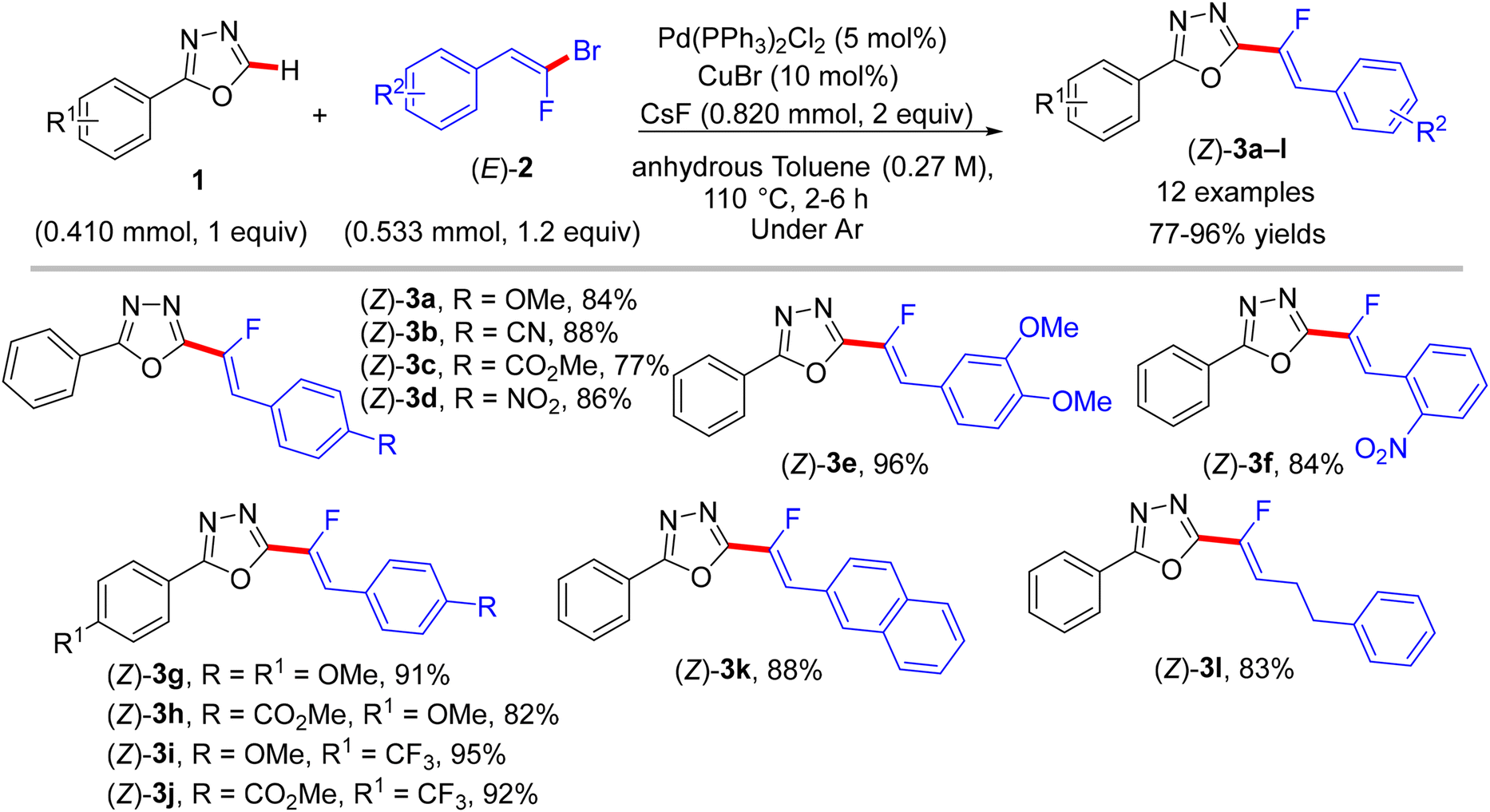
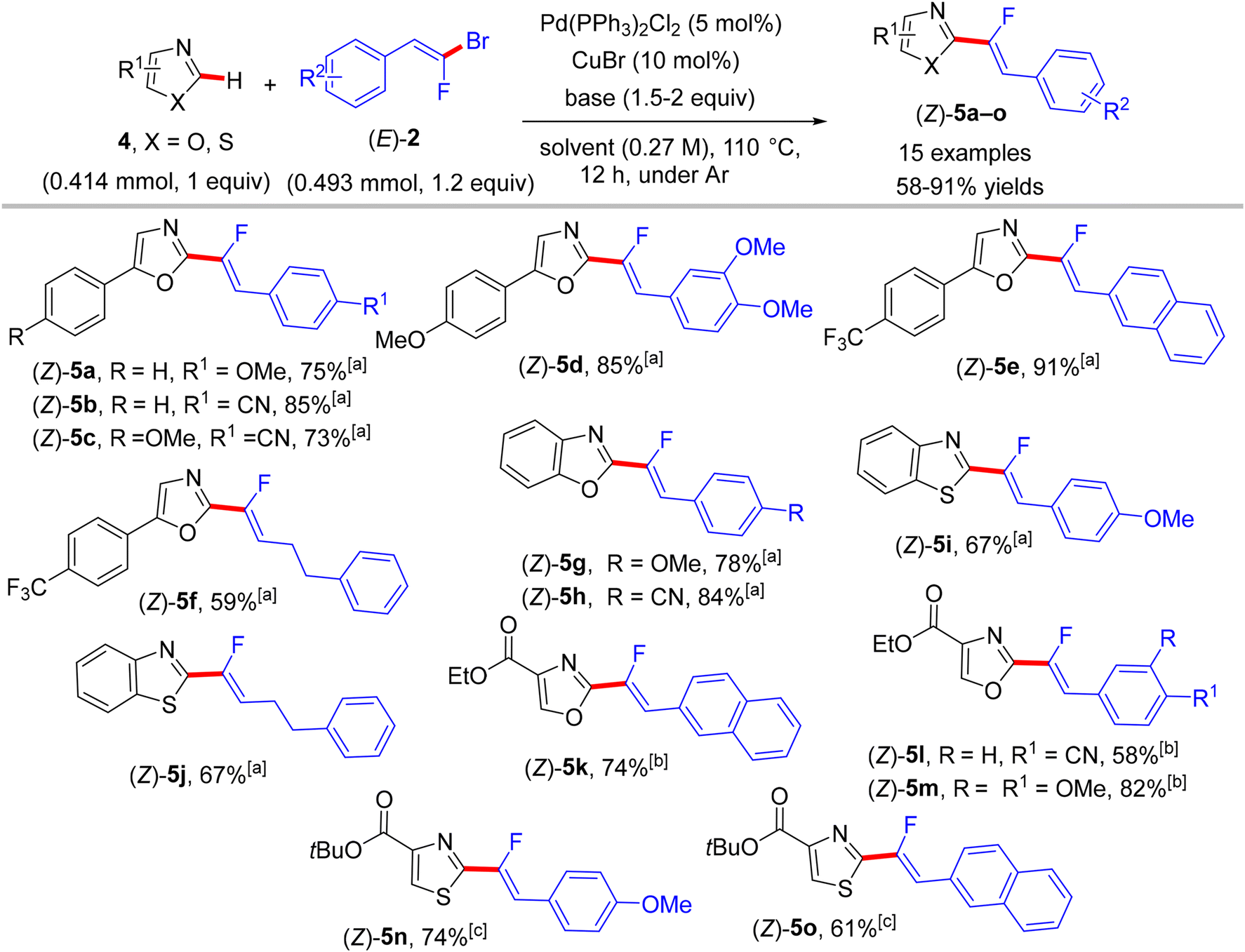
As mixed 1,1-dihalo-1-alkenes, gem-bromofluoroolefins are significant fluorinated substructures and useful building blocks.19,20 They can be prepared as a mixture of stereoisomers by several methods, such as the Wittig reaction, but some separation protocol-dependent chemoselectivity is reported.47,48 They were successfully employed as synthons in organic synthesis to access monofluoroalkenes.49 The paper by Hoarau et al. (2013) documented the first use of trisubstituted (E)-gem-bromofluoroalkenes (2) as electrophilic coupling partners in the direct C(sp2)–H α-fluoroalkenylation of various electronically biased azoles (1 and 4) to access the corresponding (Z)-monofluoroalkenes (3a–l and 5a–o) in fair to excellent yields (Schemes 1 and 2).26
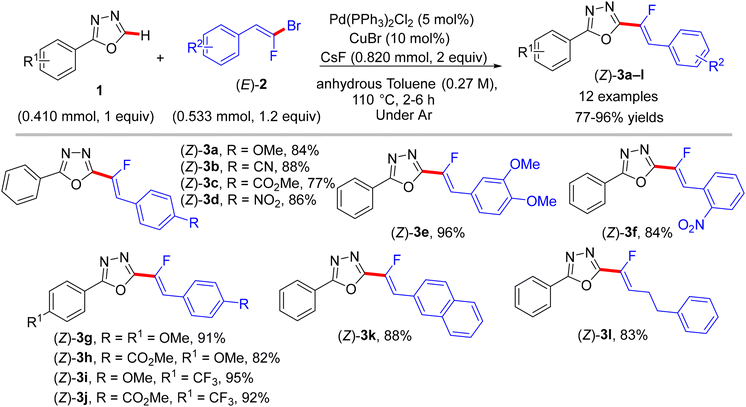 |
| | Scheme 1 Direct C(sp2)–H α-fluoroalkenylation of 1,3,4-oxadiazoles 1 by Pd(0)/Cu(I) catalytic system. | |
 |
| | Scheme 2 Direct C(sp2)–H α-fluoroalkenylation of various 1,3-diazoles 4 by Pd(0)/Cu(I) catalytic system. [a] LiOtBu (1.5–2 equiv.), 1,4-dioxane. [b] Cs2CO3 (2 equiv.), 1,4-dioxane. [c] Cs2CO3 (2 equiv.), dimethylformamide (DMF). | |
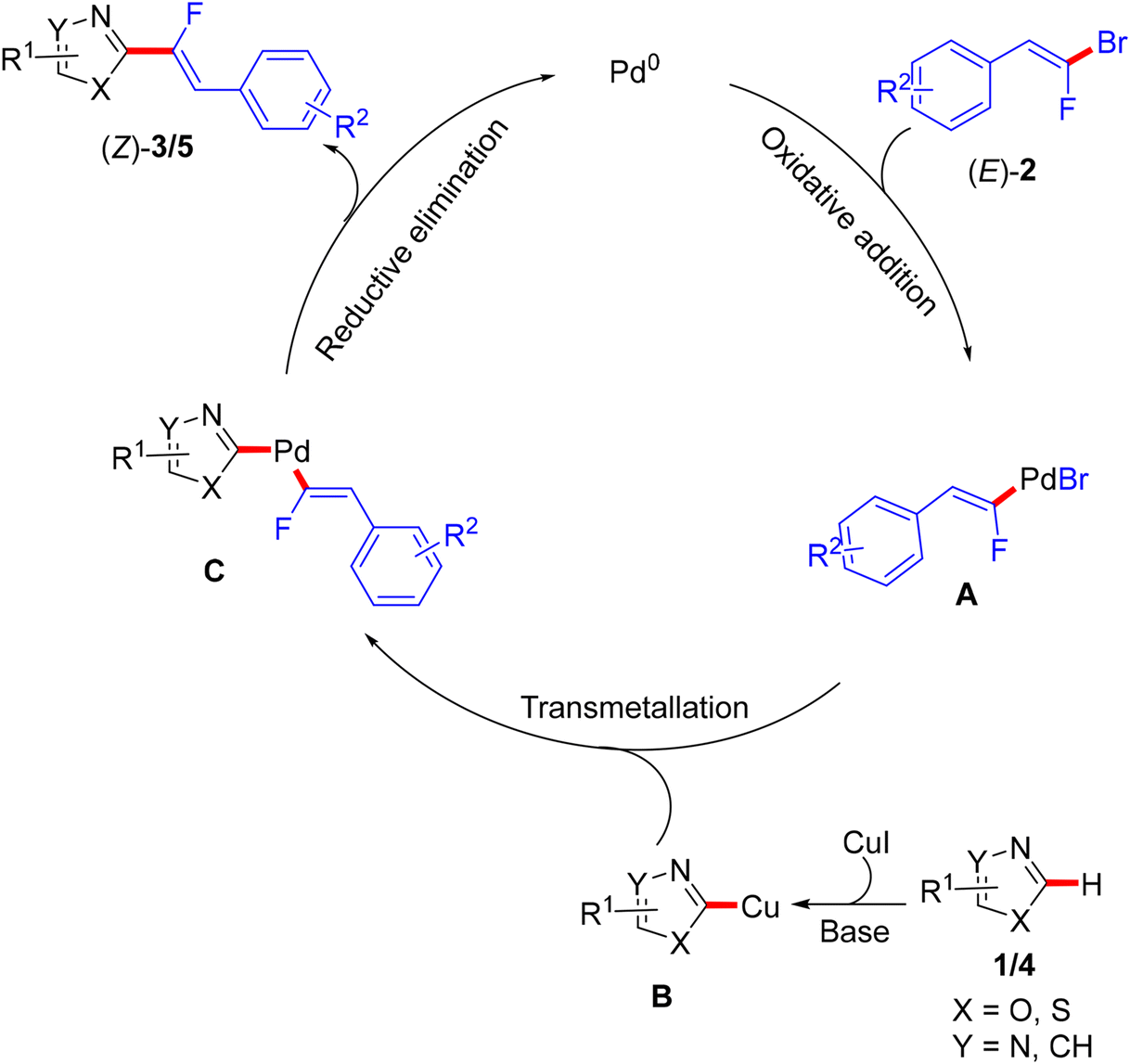
This step-economical reaction proceeds via a base-assisted Pd(0)/Cu(I) bimetallic catalytic system at 110 °C under complete control of stereochemistry with functional group tolerance and without alkynylated (dehydrofluorinated) side-products. This protocol can also be scaled up from 0.41 to 2.05 mmol without losing efficiency. C(sp2)–H α-fluoroalkenylation of oxadiazoles 1 was conducted using CsF as a mild base, while the less acidic 1,3-diazoles 4 needed more basic conditions offered by Cs2CO3 or LiOtBu. The PdCl2(PPh3)2 pre-catalyst was more effective in this transformation than Pd(OAc)2, and the CuBr co-catalyst dramatically influenced the success of the reaction. Although the authors did not perform any mechanistic studies, according to the reported Pd(0)/Cu(I) catalytic cycle,2,50 the oxidative addition afforded organopalladium intermediate A, while the base and Cu(I) mediated the deprotonative metalation of azoles to a 2-azolylcopper intermediate B that was combined with Avia transmetalation, yielding C followed by reductive elimination to furnish the desired monofluoroalkenes (Scheme 3).
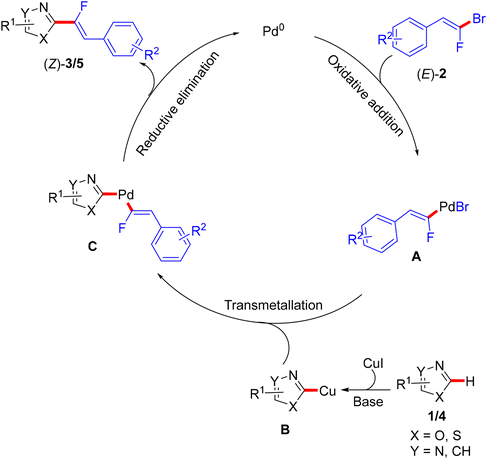 |
| | Scheme 3 Proposed catalytic cycle of Pd(0)/Cu(I)-catalyzed direct C(sp2)–H α-fluoroalkenylation of various azoles 1/4 with 2. | |
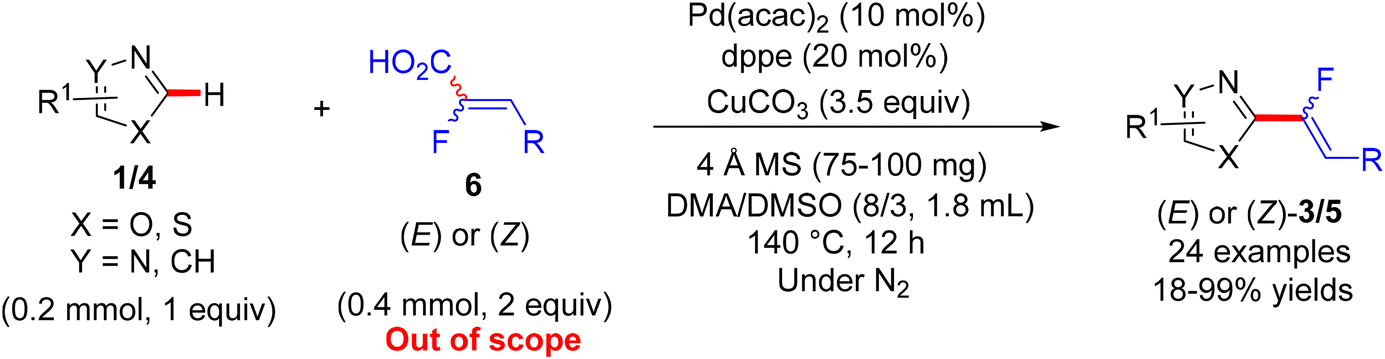
In 2014, the same group reported the synthesis of monofluoroalkenes 3/5via decarboxylative/direct C(sp2)–H α-fluoroalkenylation of various azoles 1/4 using the same catalytic system (Pd/Cu), albeit with α-fluoroacrylic acid coupling partner 6 that is out of scope of our review. Importantly, this transformation proceeds with complete retention of stereochemistry at the double bond, where the E reactant of 6 gives the (E)-monofluoroalkenes (Scheme 4).27
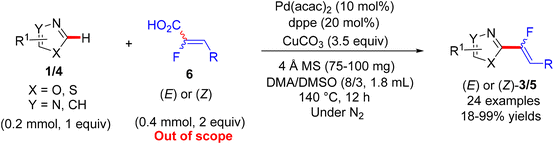 |
| | Scheme 4 Direct C(sp2)–H α-fluoroalkenylation of various azoles 1/4 by Pd(0)/Cu(I) catalytic system using 6 as a coupling partner. | |
2.2. C–H α-Fluoroalkenylation using Cu(I)
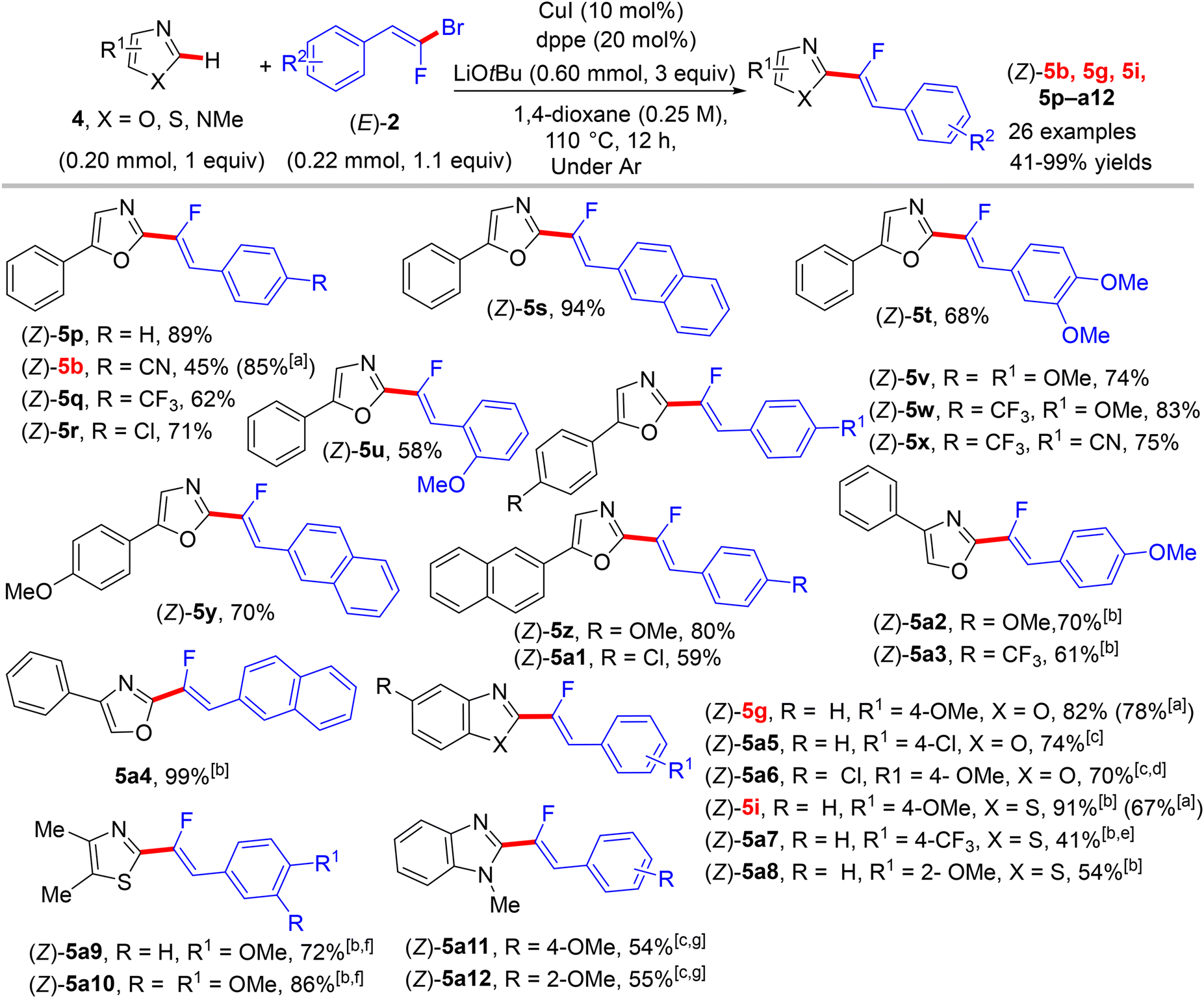
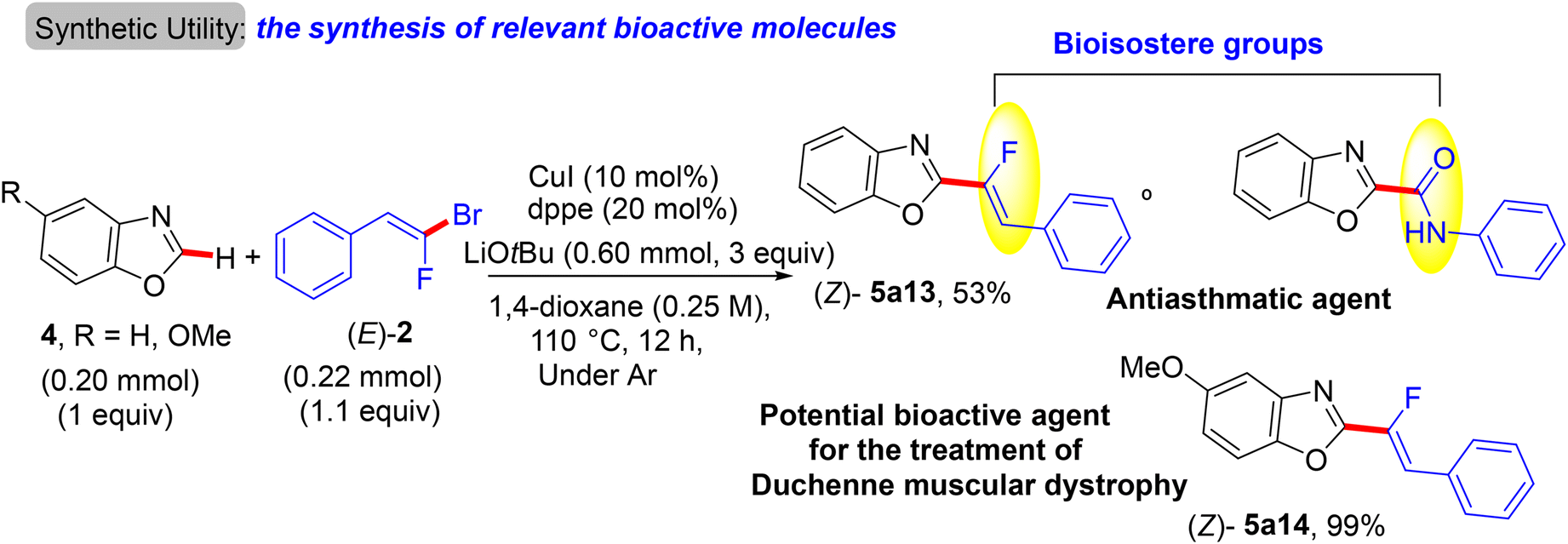
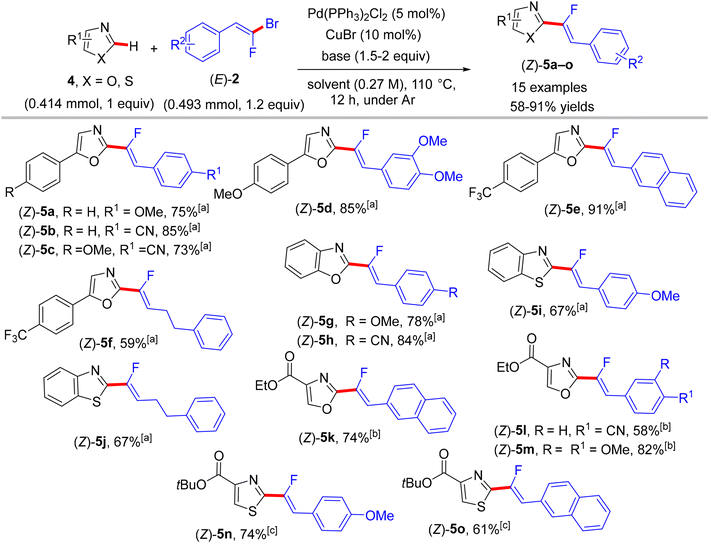
Two years later (2016), the prior research group published an article defining a low-cost protocol for the direct C(sp2)–H α-fluoroalkenylation of various 1,3-diazoles.26,27 This pioneering study describes for the first time the use of Cu(I) alone (Pd-free) to catalyze this type of reaction.28 5-Phenyloxazole (4) was used as a model substrate with (E)-1-(2-bromo-2-fluorovinyl)-4-methoxybenzene (2) to obtain the optimum conditions. The above conditions without Pd were tested,26 but the best performance was generated by CuI, 1,2-bis(diphenylphosphino)ethane (dppe) ligand, and sublimated LiOtBu in refluxing anhydrous 1,4-dioxane under argon at 110 °C for 12 hours. Under these conditions, a variety of 1,3-diazoles 4 were coupled with 2 to afford the corresponding (Z)-monofluoroalkenes 5a–o in moderate to excellent yields (Scheme 5).28 Importantly, in the absence of Cu, the reaction's outcome was negative and faintly sensitive to the Cu precursor types [Cu(I) or Cu(II)]. This procedure had the same advantages as the prior one as well as being less expensive. Unfortunately, in comparison with the above protocol,26 the alkylated or tetrasubstituted gem-bromofluoroalkenes were unreactive, and the ortho-substituted counterparts with an EWG were unstable. A drawback was that the substrate scope in these protocols was limited to azoles, and there was no suggested interpretation for why the activation occurred on the C–H bond that is flanked by two heteroatoms rather than the C–H bond of the phenyl ring (4/5-phenyl azoles) via some metallocyclic intermediates. This protocol was employed to synthesize bioactive compounds, as shown in Scheme 6.28 One of them, 5a13, is an analogue of a known amide-based antiasthmatic reagent, and the second, 5a14, is a potential bioactive agent for the treatment of Duchenne muscular dystrophy.28
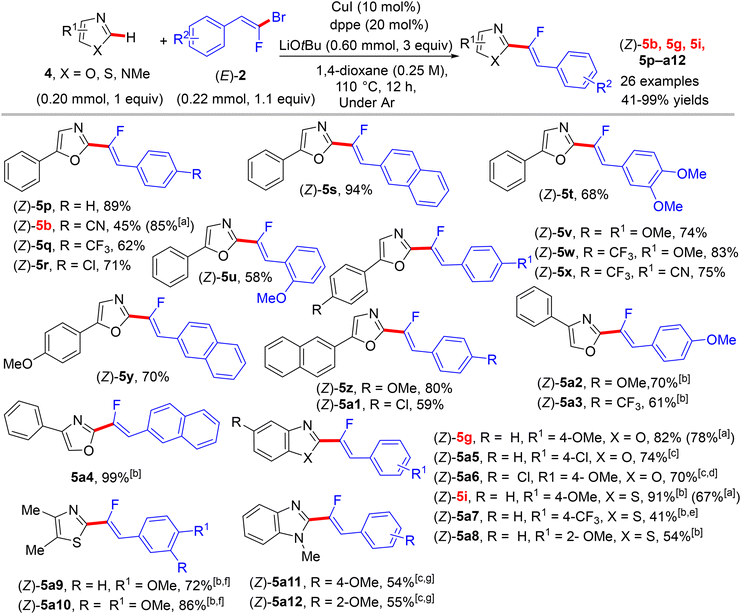 |
| | Scheme 5 Direct C(sp2)–H α-fluoroalkenylation of various 1,3-diazoles 4 by CuI. [a] Yield produced by bimetallic Pd(0)/CuBr catalysis26 for product numbers colored in red. [b] 1,10-Phenanthroline (phen) (20 mol%) was used as a ligand instead of dppe. [c] Reaction was performed at 130 °C. [d] Reaction was performed on a 1 mmol scale. [e] Reaction was performed at 90 °C. [f] CuI (20 mol%) and Phen (40 mol%) were used. [g] Ligand = trans-N,N′-dimethylcyclohexane-1,2-diamine. | |
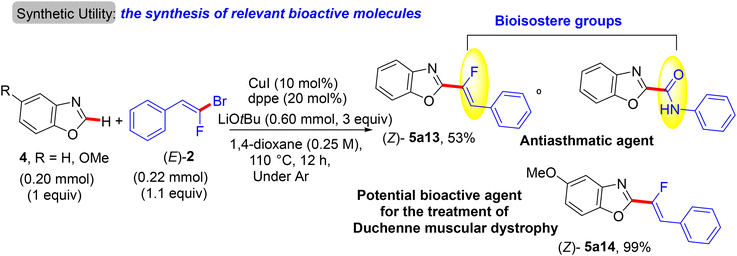 |
| | Scheme 6 Direct C(sp2)–H α-fluoroalkenylation of benzoxazoles by CuI to access relevant bioactive molecules 5a13 and 5a14. | |
3. Direct α-fluoroalkenylation with gem-difluoroalkenes
3.1. Metal-free, base-mediated direct C(N)–H α-fluoroalkenylation at room temperature
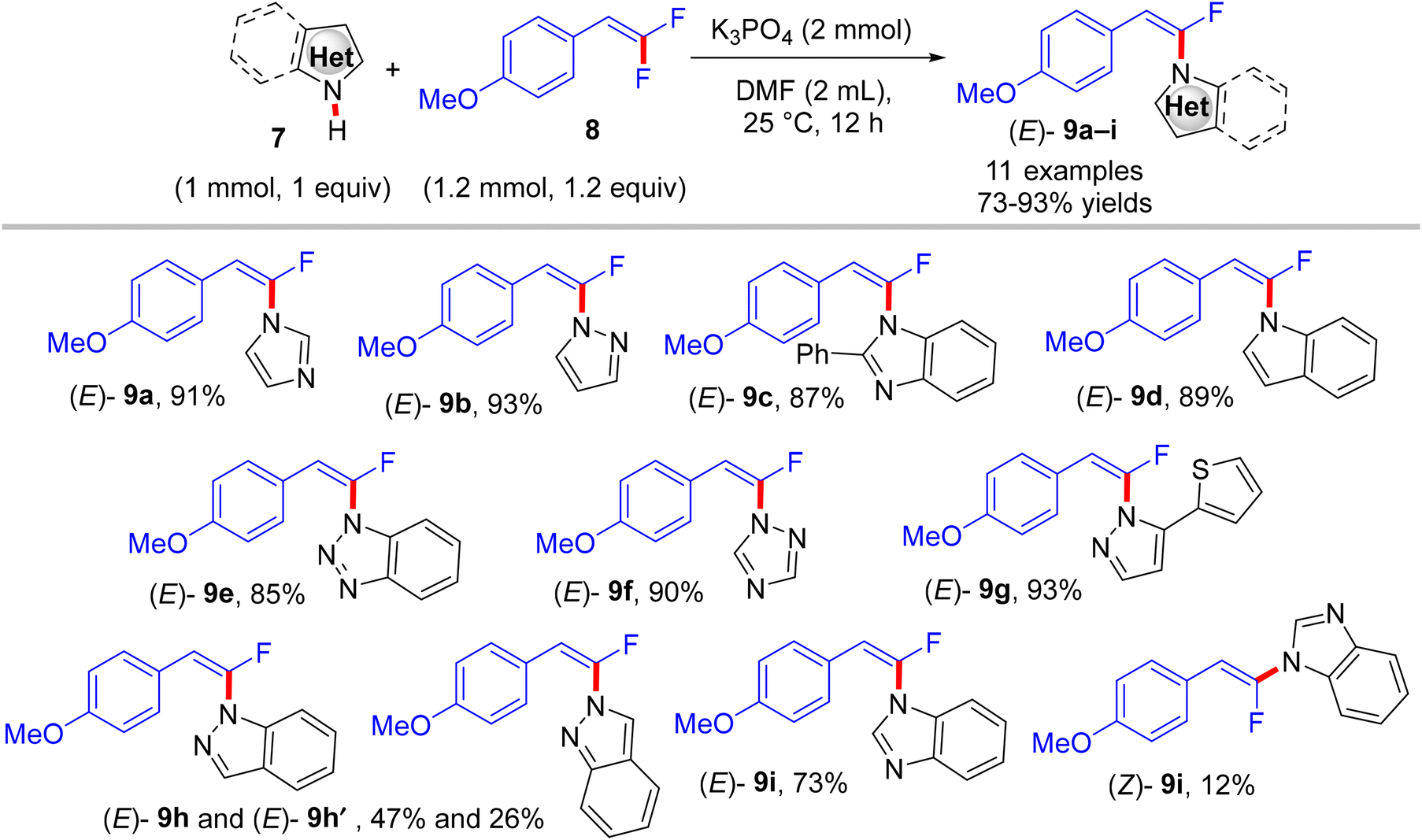
gem-Difluoroalkenes are readily accessible and valuable synthetic building blocks that may be functionalized to produce a diverse range of fluorinated products.17–24 In addition, they are significant alkenyl bidentate electrophiles for organometallic reagents; their oxidative insertion in cross-coupling reactions is rather easy.23,24 Biologically, they have been employed as irreversible electrophilic inhibitors of numerous enzymes.51 As examples for comparison, Cao et al.'s study (2014) effectively detailed a high stereoselective, metal-free, and straightforward C–N coupling approach giving access to a variety of (E)-N-(α-fluorovinyl)azoles 9a–i in good to excellent yields through a direct N–H α-fluoroalkenylation reaction between NH-containing azoles 7 and trisubstituted gem-difluoroalkenes 8.52
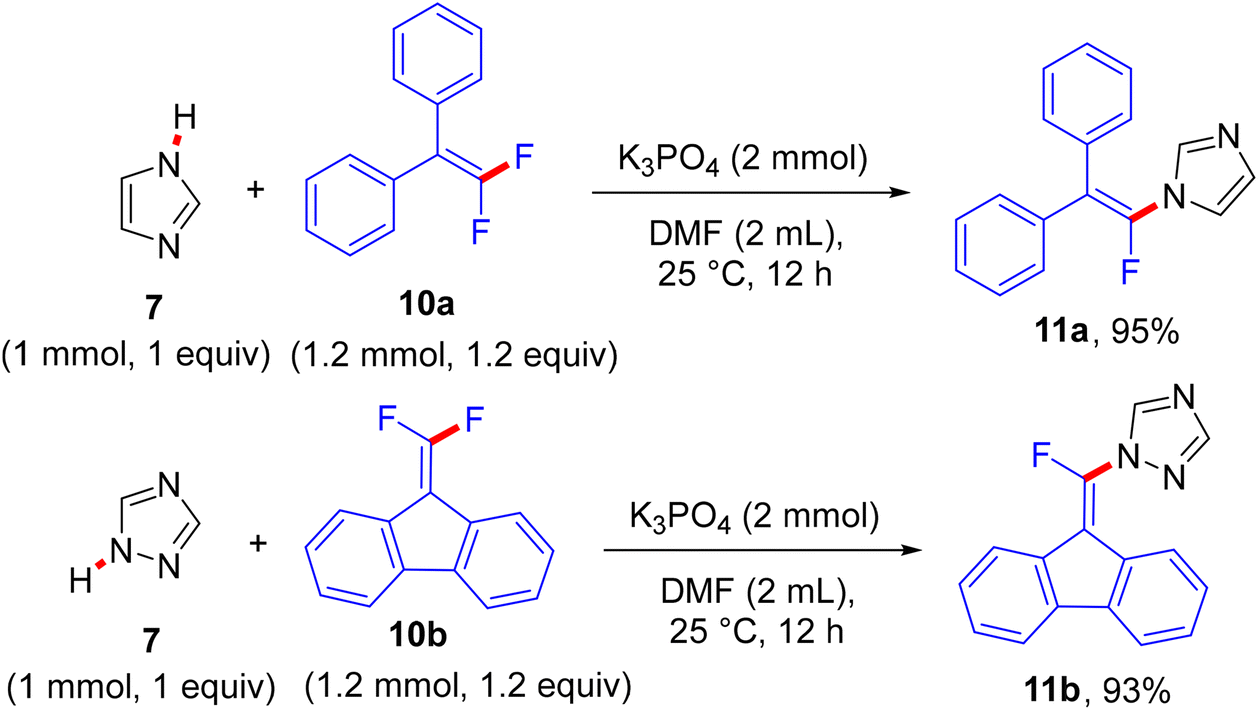
The reaction proceeds via a K3PO4-mediated vinylic nucleophilic substitution (SNV) mechanism53 under rather mild conditions without the formation of alkynylated side-products. Optimal conditions were reported as follows: azole (1 mmol), gem-difluoroalkene (1.2 equiv.), K3PO4 (2 equiv.), and dried dimethylformamide (DMF, 2 mL), stirring for 12 h at 25 °C (Scheme 7).52 Under these conditions, the authors succeeded in achieving the same reaction with tetrasubstituted symmetrical gem-difluoroalkenes 10a,b to obtain the corresponding N-vinylation products 11a,b in excellent yields (Scheme 8).52 In a tunable stereoselective pathway, the produced monofluoroalkenes (e.g., 9a,b) can be converted to N,N′-α,α′-dihetaryl-substituted olefins 12a–c in excellent yields via two-fold C–F cleavages by treatment with another equivalent of 7 at 80 °C, establishing the difficult cleavage of the second C–F bond. Worthy of note, 1,1′-(2-(4-methoxyphenyl)ethene-1,1-diyl)bis(1H-imidazole) (12a) was obtained via this one-step method in a nearly quantitative yield (95%) (Scheme 9).52 In contrast with the aforementioned studies,26–28 the azoles considered here contain a more acidic proton (N–H group) that a suitable base can abstract. Worthy of note, the optimal base K3PO4 did not affect the gem-difluoroalkenes through the formation of the alkynylated products.
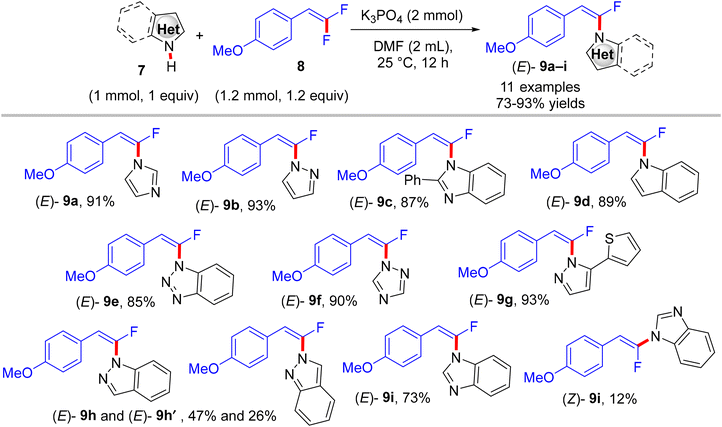 |
| | Scheme 7 Metal-free K3PO4-mediated direct N–H α-fluoroalkenylation of various NH-containing azoles 7. | |
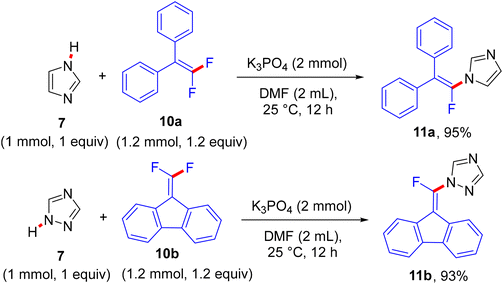 |
| | Scheme 8 Metal-free K3PO4-mediated direct N–H α-fluoroalkenylation of NH-containing azoles 7 with 10a,b. | |
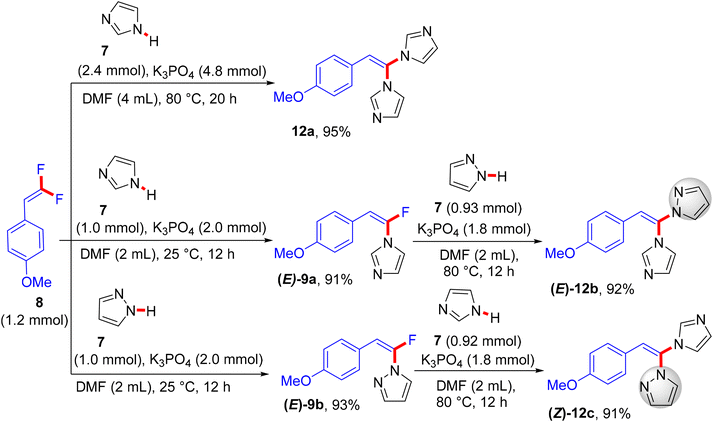 |
| | Scheme 9 Synthesis of N,N′-α,α′-dihetaryl-substituted olefins 12a–c at 80 °C. | |
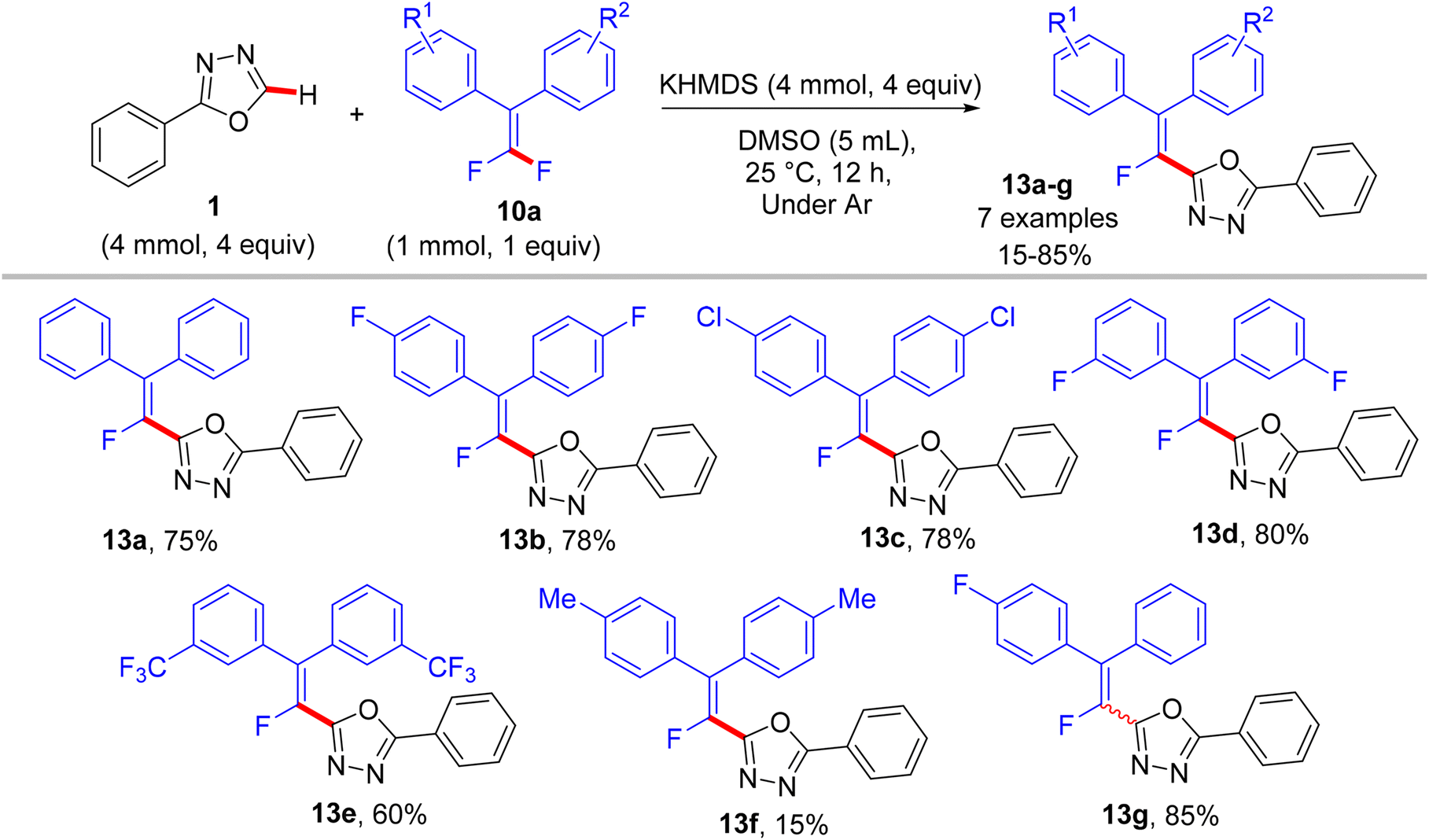
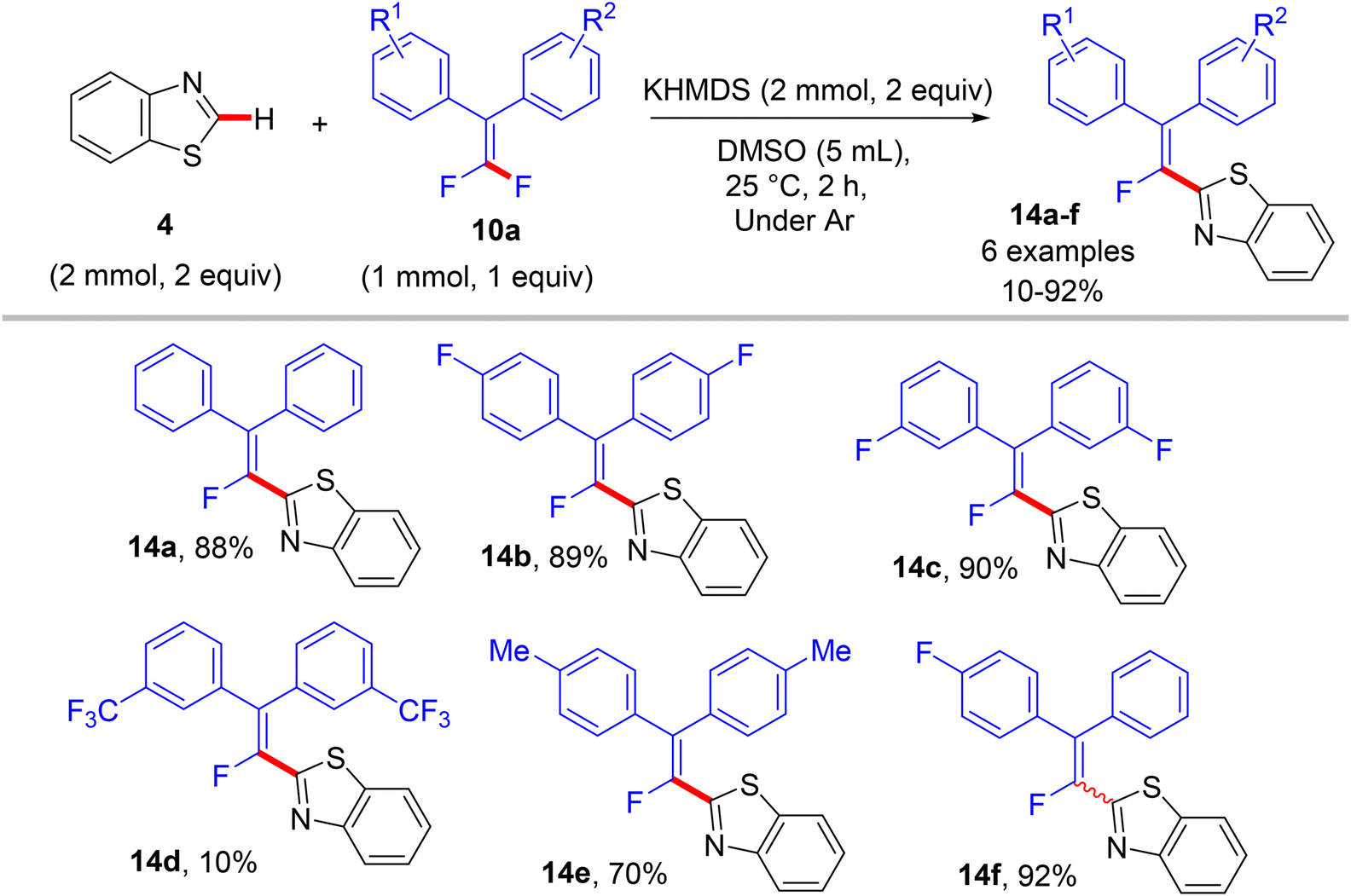
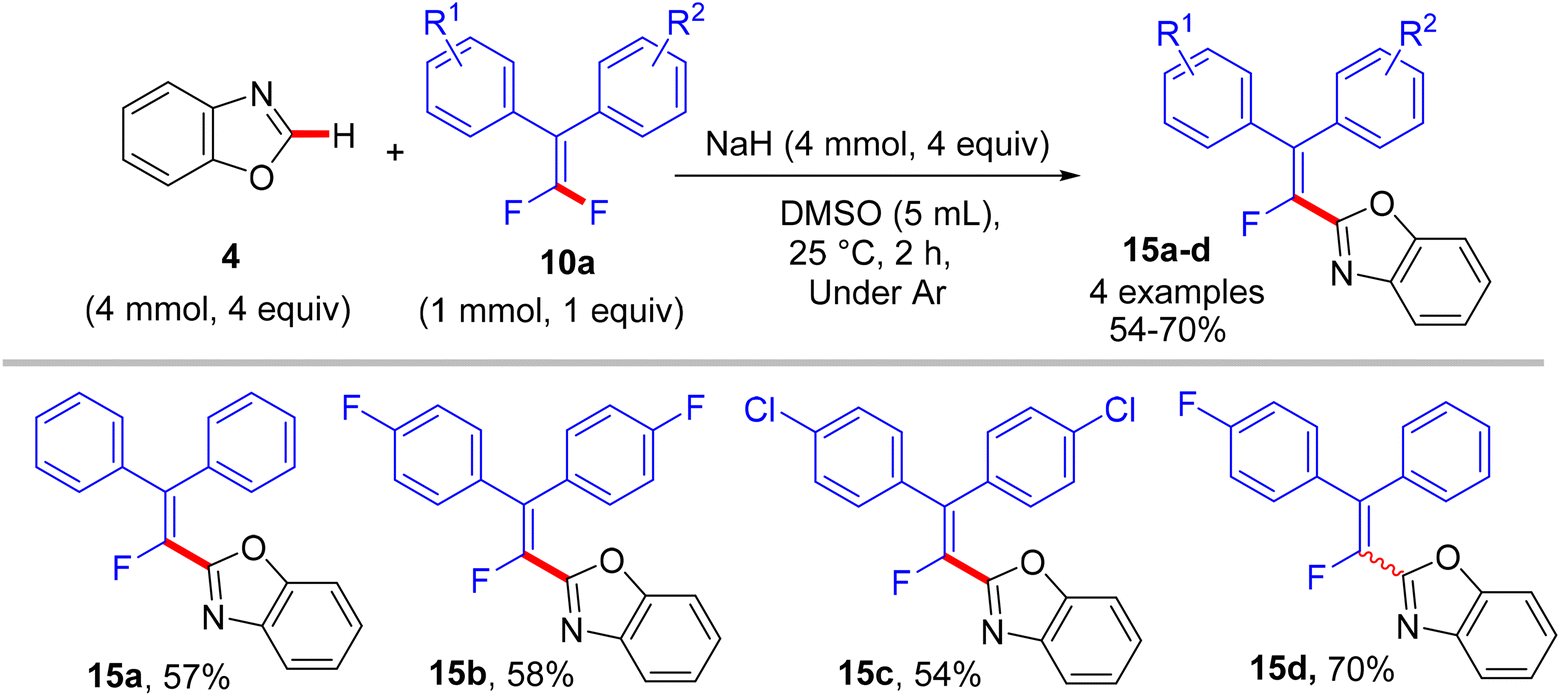
In a similar synthetic vein, Cao's lab (2015) also reported a metal-free, base-mediated direct C(sp2)–H α-fluoroalkenylation of non-containing NH-azoles (1 and 4) with tetrasubstituted symmetrical and unsymmetrical gem-difluoroalkenes 10a to generate the corresponding fluoroalkenylated azoles 13–15.54 As in the prior study,52 the reaction was achieved via the SNV mechanism,53 which included base-assisted formation of nucleophilic sp2 carbanion of azoles (transition state) and subsequent addition–elimination steps with electrophilic 10a to furnish the desired products. After several investigations, the optimum conditions were established as follows: dry dimethyl sulfoxide (DMSO) as a solvent and non-nucleophilic bases such as potassium bis(trimethylsilyl)amide (KHMDS) or NaH were the best choices at ambient temperature (Schemes 10–12).54
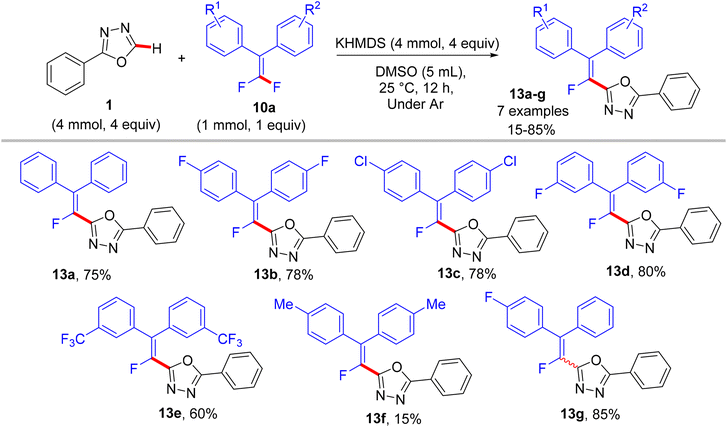 |
| | Scheme 10 Metal-free KHMDS-mediated direct C(sp2)–H α-fluoroalkenylation of 1 with 10a. | |
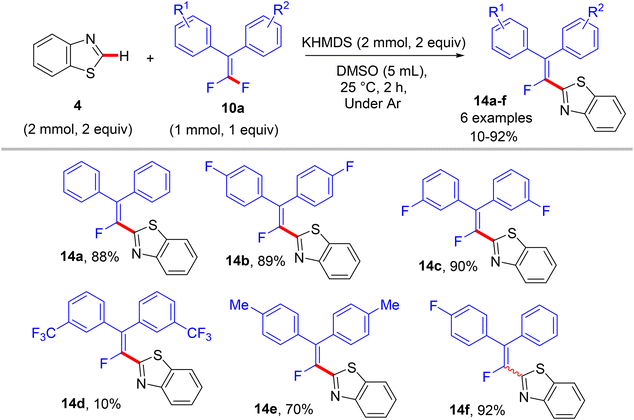 |
| | Scheme 11 Metal-free KHMDS-mediated direct C(sp2)–H α-fluoroalkenylation of benzothiazole (4) with 10a. | |
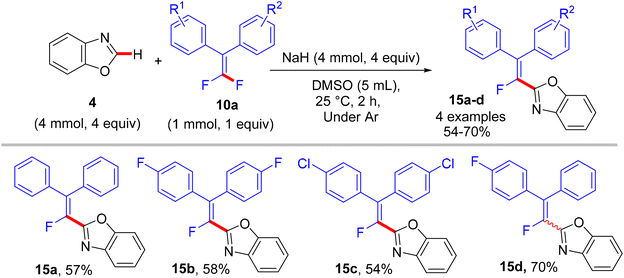 |
| | Scheme 12 Metal-free NaH-mediated direct C(sp2)–H α-fluoroalkenylation of benzoxazole (4) with 10a. | |
In comparison with the above metal-based studies,26,28 the substrate scope in this study is limited as a drawback owing to the absence of a metal catalyst that assisted the cleavage of C–H bonds in heteroarenes. For instance, trisubstituted gem-difluoroalkenes 8 were unsuitable (though the reason was not mentioned, but they might form alkynylated side products), and tetrasubstituted counterparts 10a bearing strong EWG such as CF3 or EDG such as CH3 were unfavorable, resulting in a low conversion. Regarding azole partners, the presence of the benzene ring as a substituent such as 2-phenyl-1,3,4-oxadiazole (1) or fused as in benzothiazole and benzoxazole (4) was essential for driving the reaction smoothly, limiting the azole scope in this reaction.
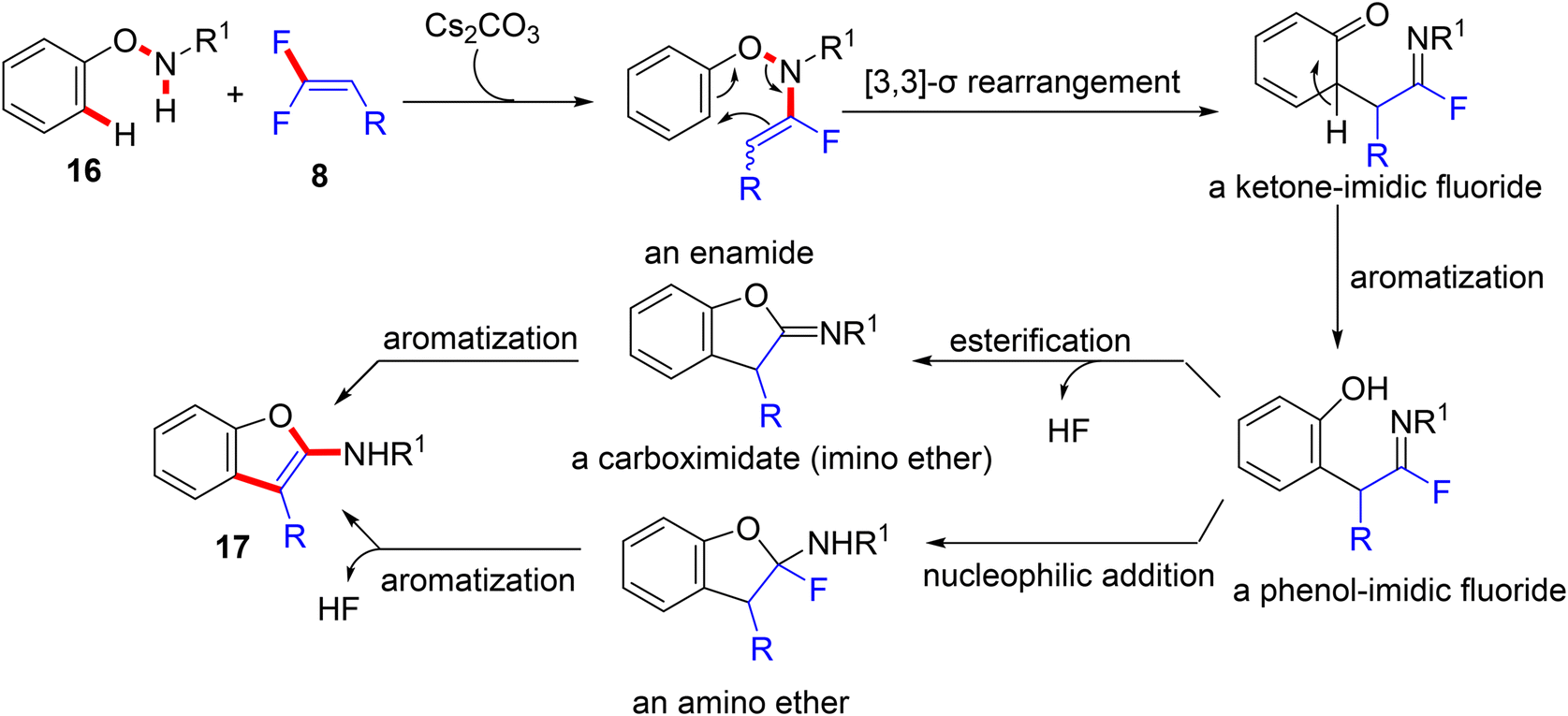
According to the Yi et al. study (2021), when N-phenoxy secondary amides 16 are employed instead of azoles under metal-free conditions, Cs2CO3-assisted domino N–H fluoroalkenylation-[3,3]-sigmatropic rearrangement ensures, which is used to directly access a variety of 2-aminobenzofurans (17, out of scope). The amide-NH fluoroalkenylation was suggested to occur transiently, and the trial with t-amide resulted in no reaction, proving the importance of the N–H bond in this annulation. After that, several cleavages of multiple bonds were performed to deliver the final products (Scheme 13).55 The assumed mechanism is illustrated in Scheme 14. Cs2CO3 promoted the nucleophilic substitution reaction of N-phenoxy secondary amides 16 with 8 to yield the corresponding enamide intermediate. After that, the [3,3]-σ rearrangement afforded a ketone-imidic fluoride that converted to the phenolic analog through aromatization. The latter may be transformed either to the imino ether (carboximidate) or the amino ether via the esterification process or nucleophilic addition. Finally, the aromatization delivered the 2-aminobenzofuran product 17. It is worth noting that the use of Cs2CO3 did not deliver the alkynylated side products.
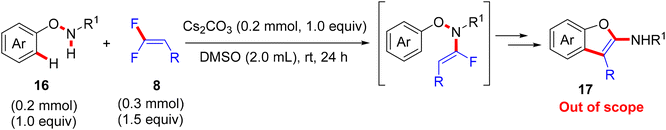 |
| | Scheme 13 Metal-free Cs2CO3-mediated direct synthesis of 2-aminobenzofurans 17via N–H α-fluoroalkenylation of N-phenoxy secondary amides 16 with 8. | |
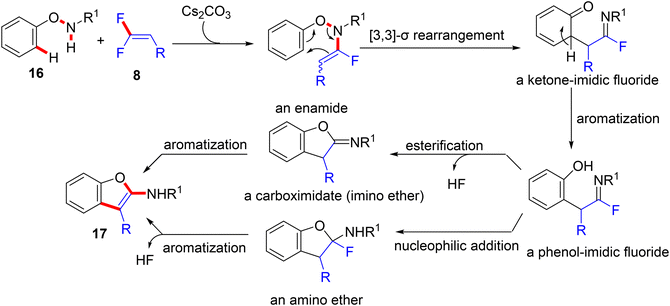 |
| | Scheme 14 Proposed mechanism of Cs2CO3-mediated direct synthesis of 2-aminobenzofurans 17via N–H α-fluoroalkenylation of N-phenoxy secondary amides 16 with 8. | |
3.2. C–H α-Fluoroalkenylation using Rh(III)Cp*
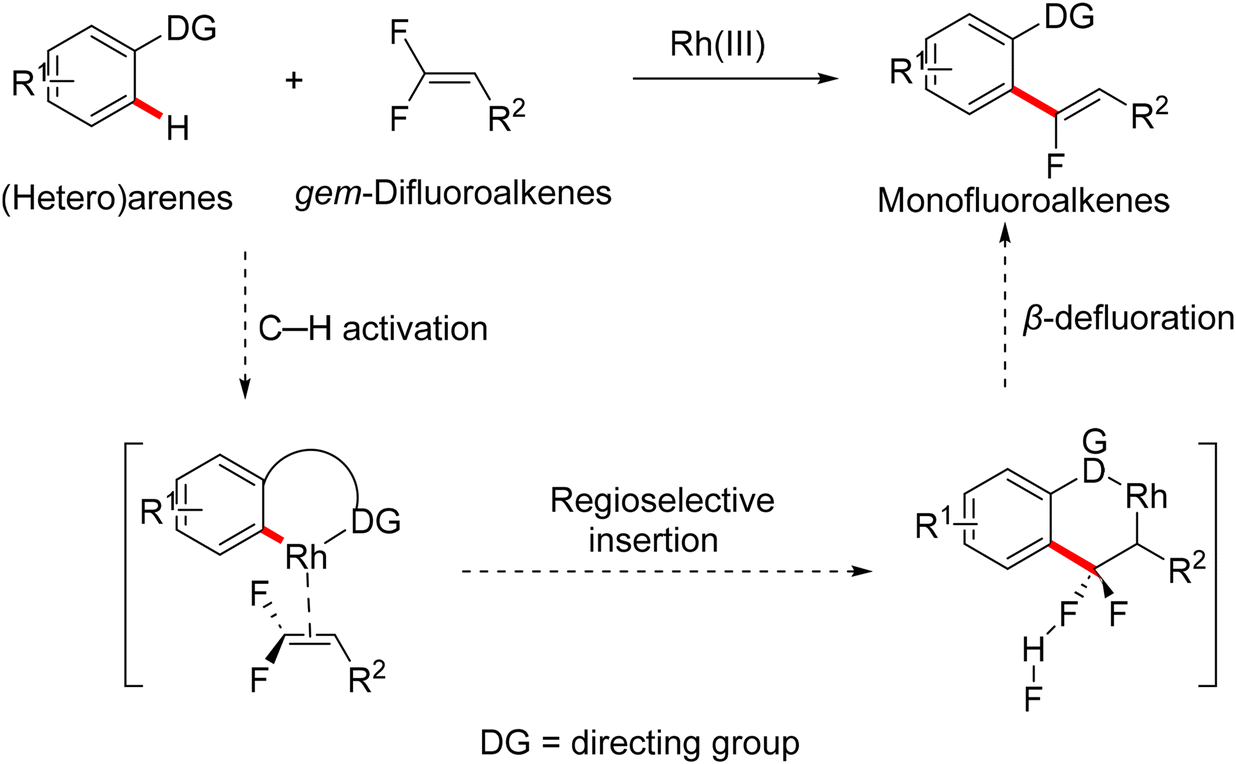
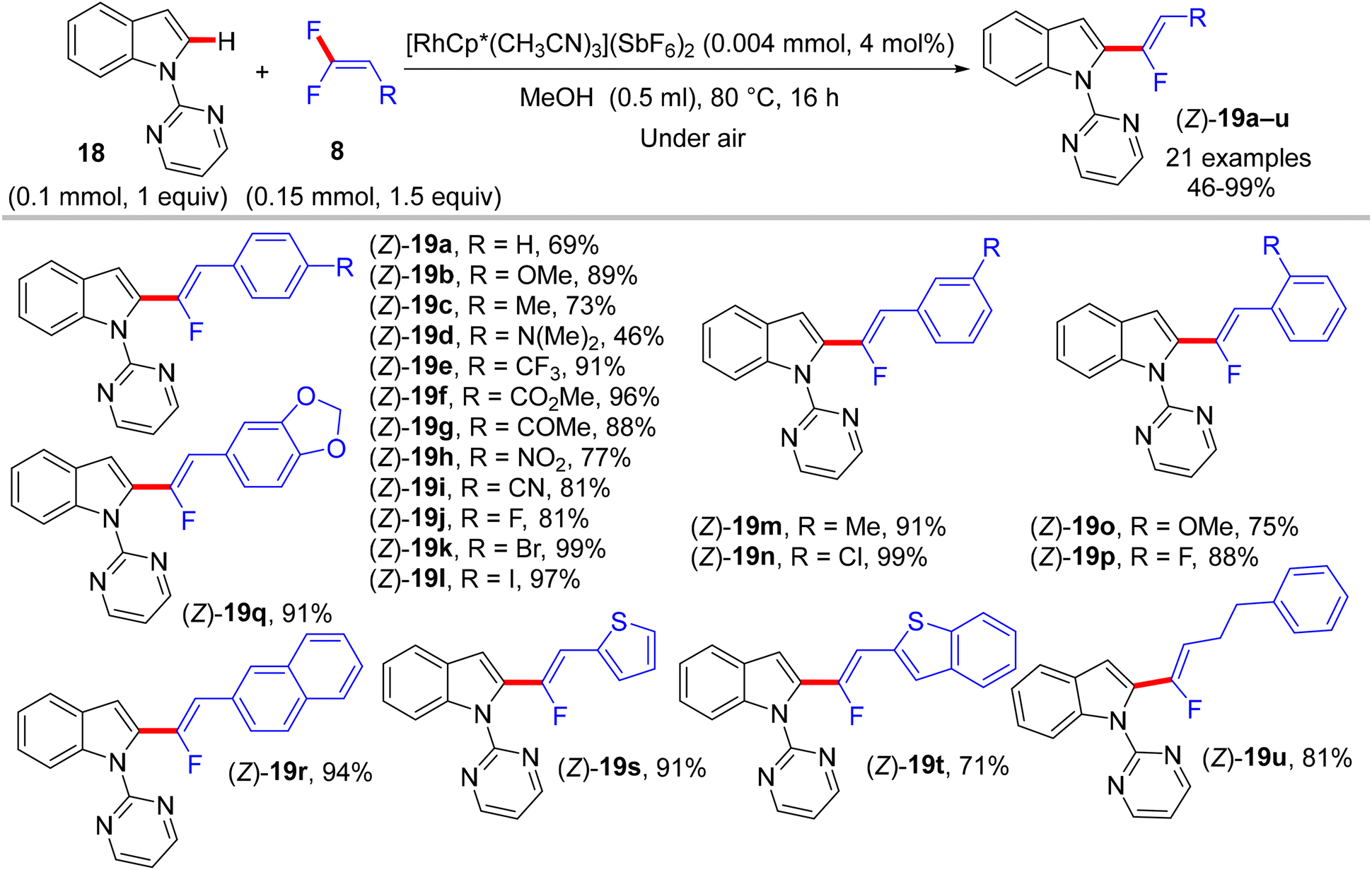
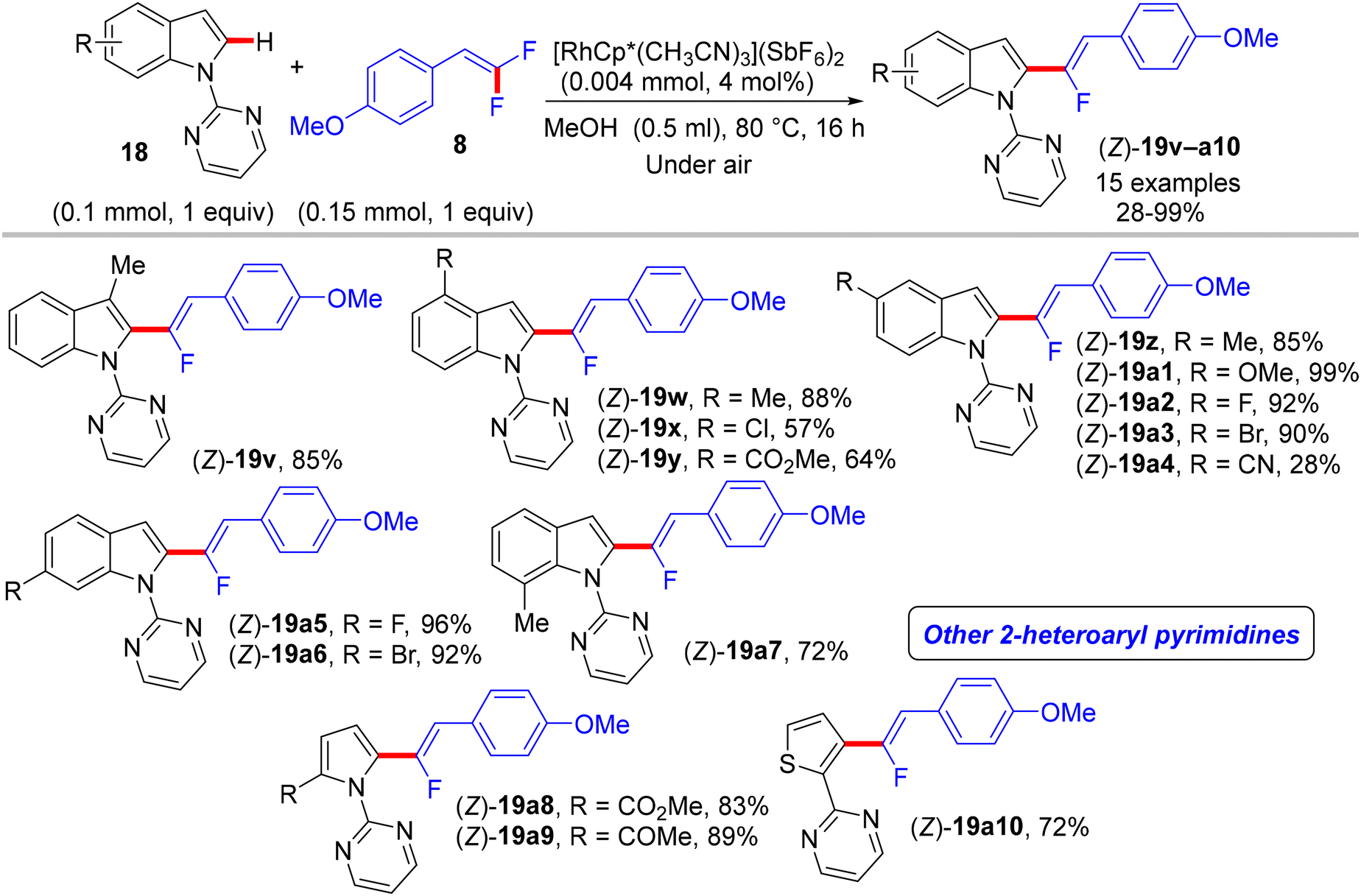
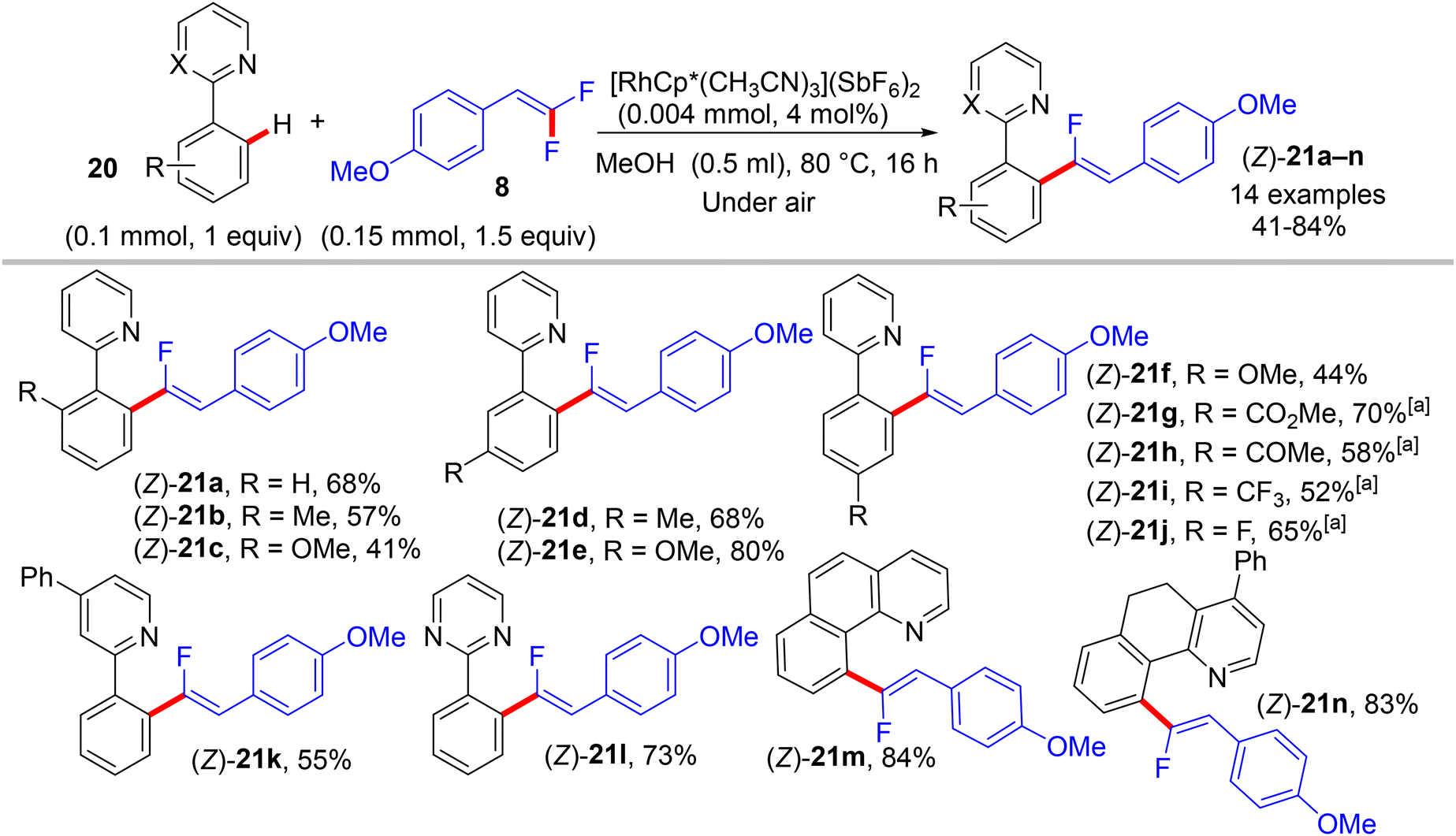
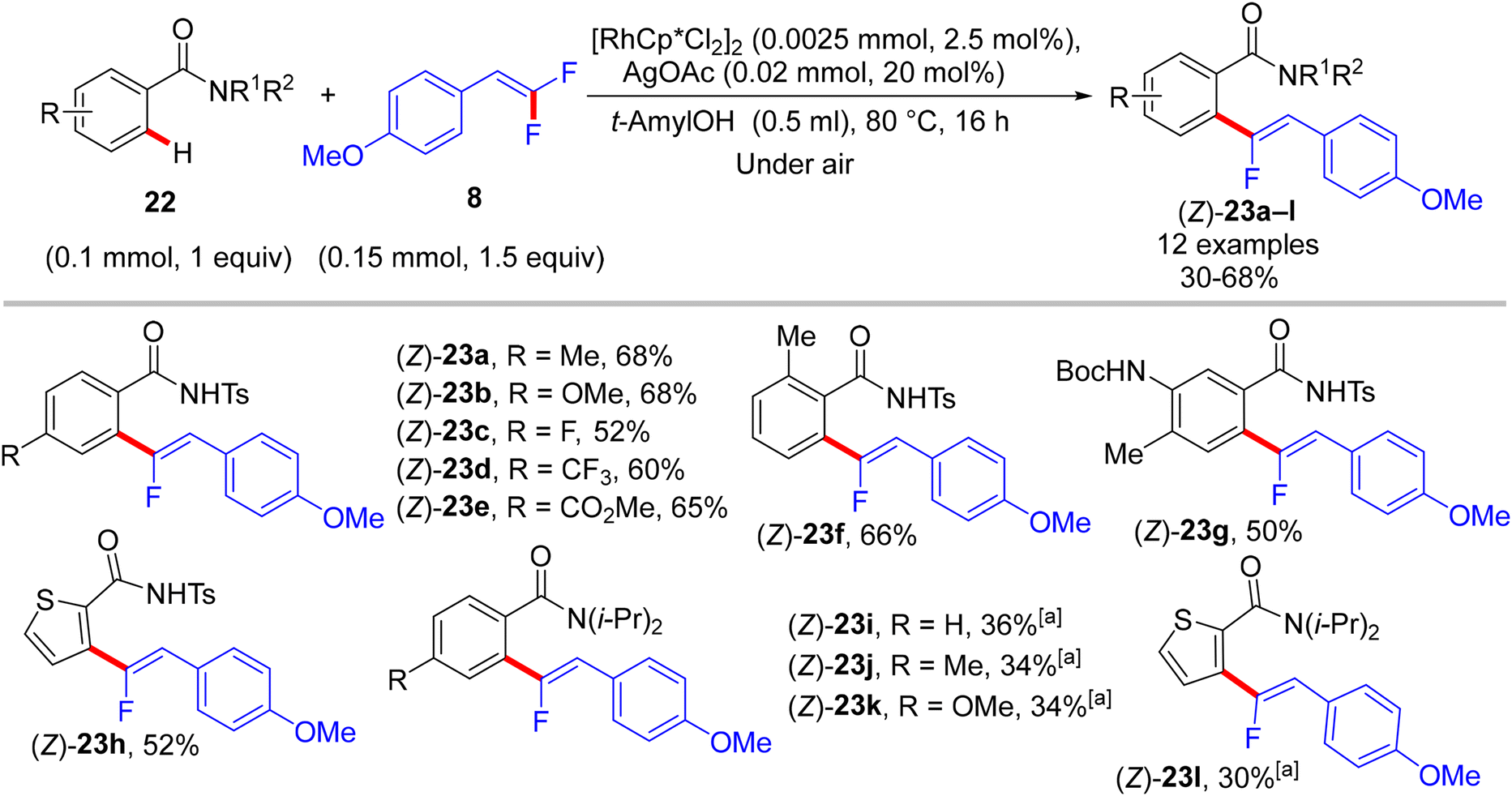
In 2015, Loh and his co-workers pioneering study described a base- and oxidant-free (redox neutral), highly efficient, and chelation-assisted direct C(sp2)–H α-fluoroalkenylation of (hetero)arenes using Rh(III)Cp*-based catalysis for the first time. As a hypothesis postulated by the authors, chelation-assisted C(sp2)–H activation of (hetero)arenes via five-membered rhodacycle formation was followed by C–F activation of gem-difluoroalkenes through H-bond interaction with the in situ generated HF, and hence β-F elimination delivered the final products (Scheme 15). Starting with highly polarized and easily accessible 8 and readily available (hetero)arenes-containing N-based DG (18, 20, and 22), the transformation proceeds smoothly with a regio- and stereoselective manner, wide-scope, and broad functionality tolerance, affording the corresponding (Z)-(hetero)arylated monofluoroalkenes (19a–a7, 21a–n, and 23a–l) in fair to quantitative yields (Schemes 16–19).29
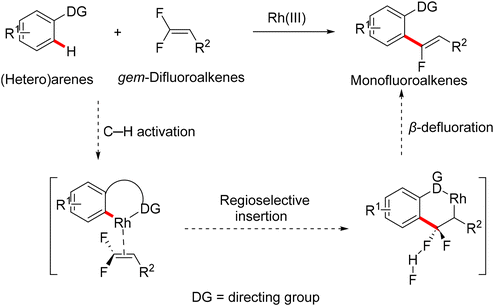 |
| | Scheme 15 Chelation-assisted direct C(sp2)–H α-fluoroalkenylation of (hetero)arenes using Rh(III)-based catalysis. | |
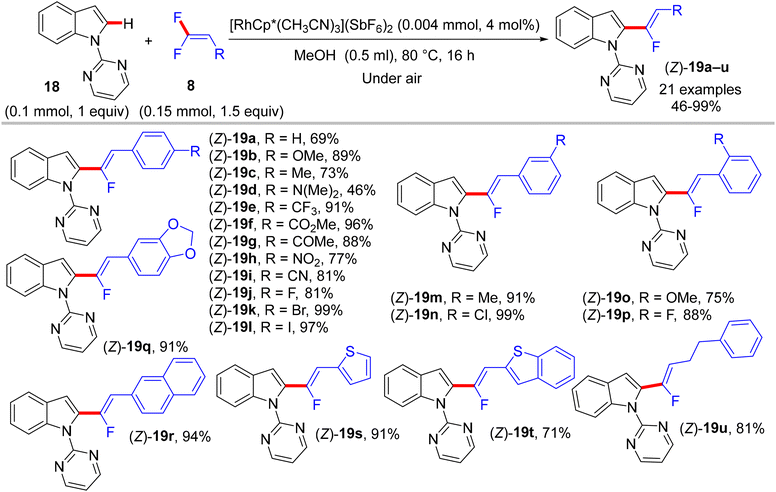 |
| | Scheme 16 Scope of 8 in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18. | |
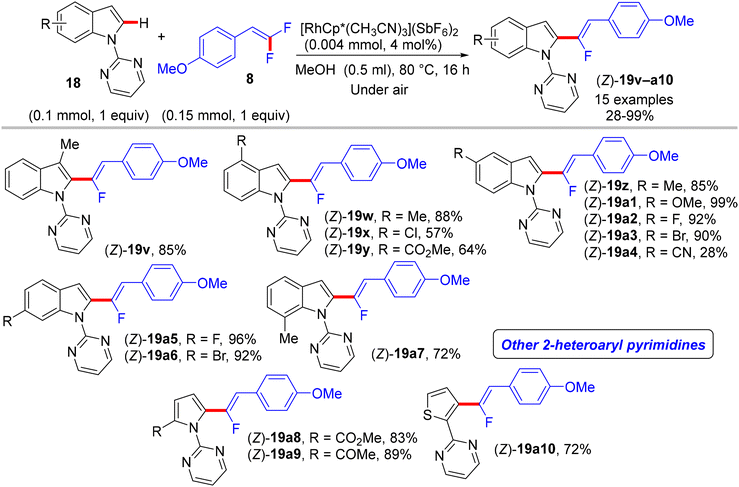 |
| | Scheme 17 Scope of 18 in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation by 8. | |
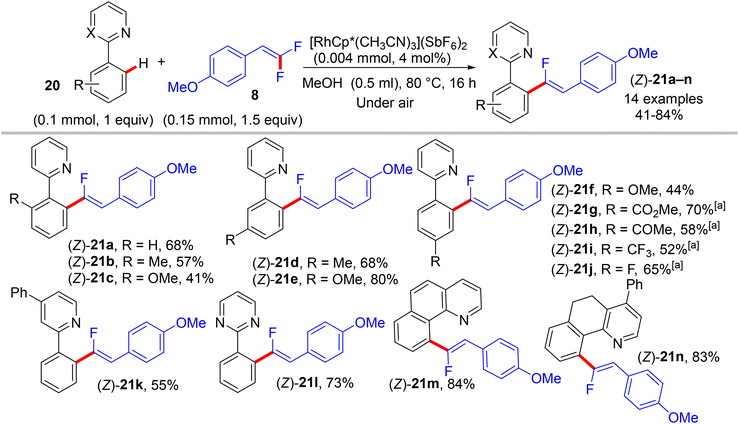 |
| | Scheme 18 Scope of 2-arylpyridines 20 in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation by 8. [a] Use trifluoroethanol (TFE) as reaction solvent instead of MeOH. | |
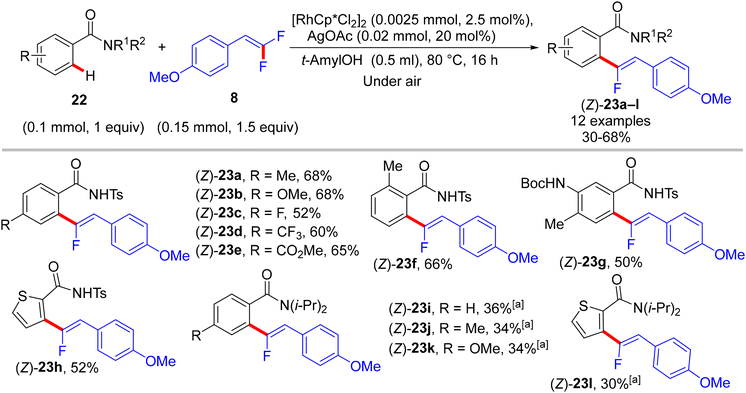 |
| | Scheme 19 Scope of benzamides 22 in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation by 8. [a] Use [RhCp*(MeCN)3](SbF6)2 (0.004 mmol) as catalyst, 1,2-dichloroethane (DCE) as reaction solvent. | |
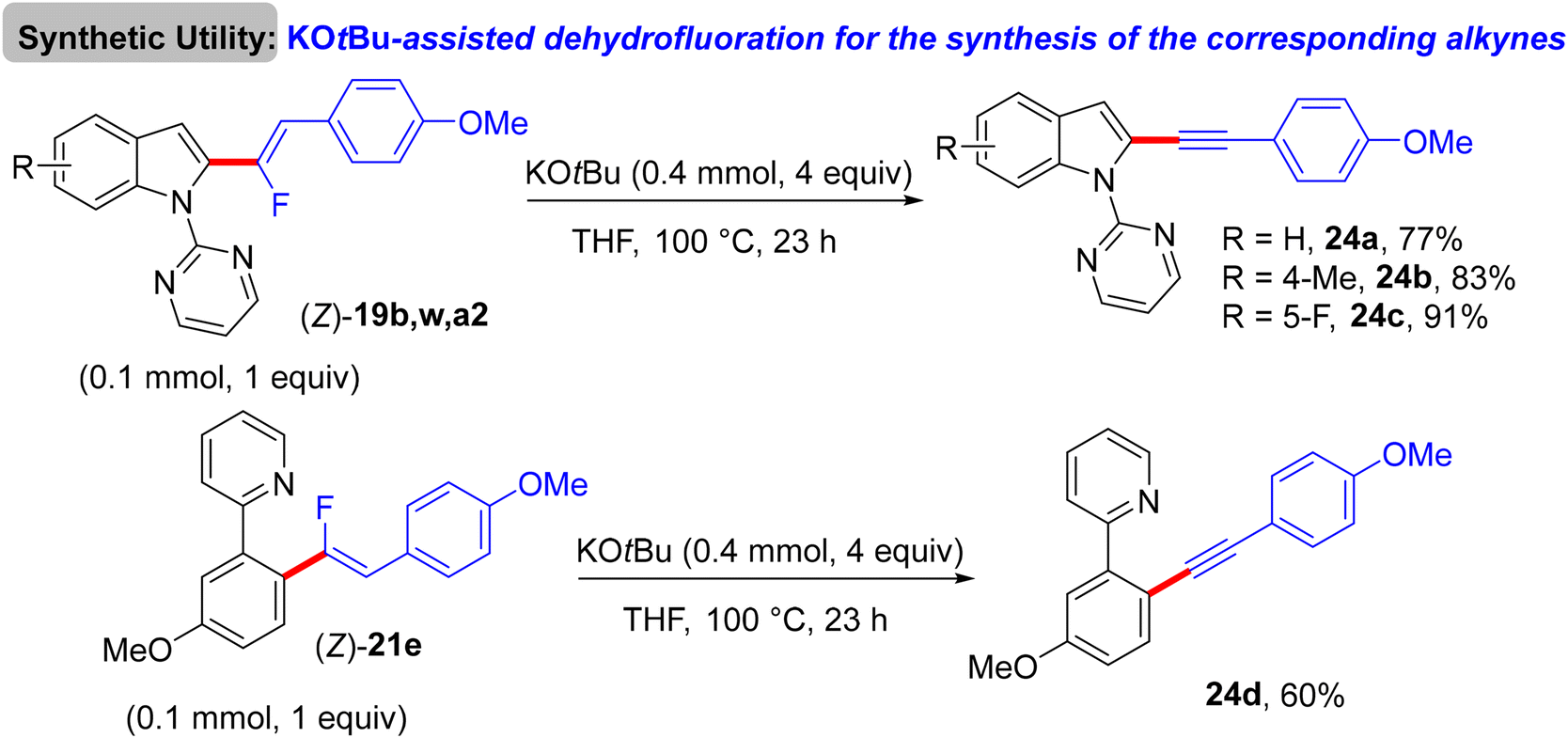
The preliminary investigation was commenced with 1-(2,2-difluorovinyl)-4-methoxybenzene (8) and 1-(pyrimidin-2-yl)-1H-indole (18) starting materials, and the best isolated yield (89%) of the expected monofluoroalkene 19a was achieved under these optimal conditions: cationic [RhCp*(CH3CN)3](SbF6)2 as the catalyst and methanol (MeOH) as solvent, and at 80 °C for 16 h under air. The substrate scope with EWG or EDG substituents was extended, including 1-pyrimidin-2-yl indoles 18, 2-arylpyridines 20, and benzamides 22, as well as a variety of 8. Regarding benzamides 22, Ts-amide derivatives were tested with slight modification [[RhCp*Cl2]2 (2.5 mol%), AgOAc (20 mol%) as catalytic system, solvent was tert-amyl alcohol (t-AmylOH)] and proved to be more effective (23a–h, moderate yields) than N,N-diisopropylbenzamides (23i–l, low yields), which was attributed to their low conversion in this transformation (Scheme 19).29 As a limitation of this simple protocol, chelation-directing groups such as imine, anilide, and ketone proved unsuitable as coupling partners. Preparation of the corresponding alkynes 24a–d as a synthetic utility for this approach was accomplished via the KOtBu-assisted dehydrofluoration process in THF at 100 °C for 23 h (Scheme 20),29 even though the counterpart base, LiOtBu, did not promote this type of process in the (E)-gem-bromofluoroalkene 2 Cu(I)-based protocol as mentioned above.28
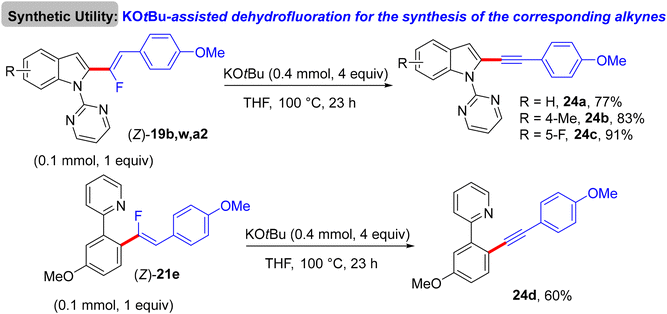 |
| | Scheme 20 KOtBu-assisted dehydrofluoration of 19b, w, a2, and 21e for the synthesis of the corresponding alkynes 24a–d, respectively. | |
Surprisingly, when Loh and co-workers used trisubstituted gem-difluorovinyl tosylate 25 instead of 8 as a coupling partner with 18 under approximately the same conditions, C(sp2)–H alkylation products 26 (out of scope) were generated rather than monofluoroalkenes (Scheme 21).56 The other heteroarenes (20 and 22) in the prior study were unsuccessful substrates.29 In this transformation, the tosyl difluorinated enol ether functioned as an valuable substitute for an acetate building block.17,23
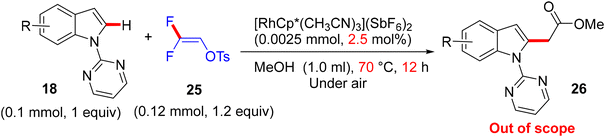 |
| | Scheme 21 Rh(III)-catalyzed direct C(sp2)–H alkylation of 18 with 25. | |
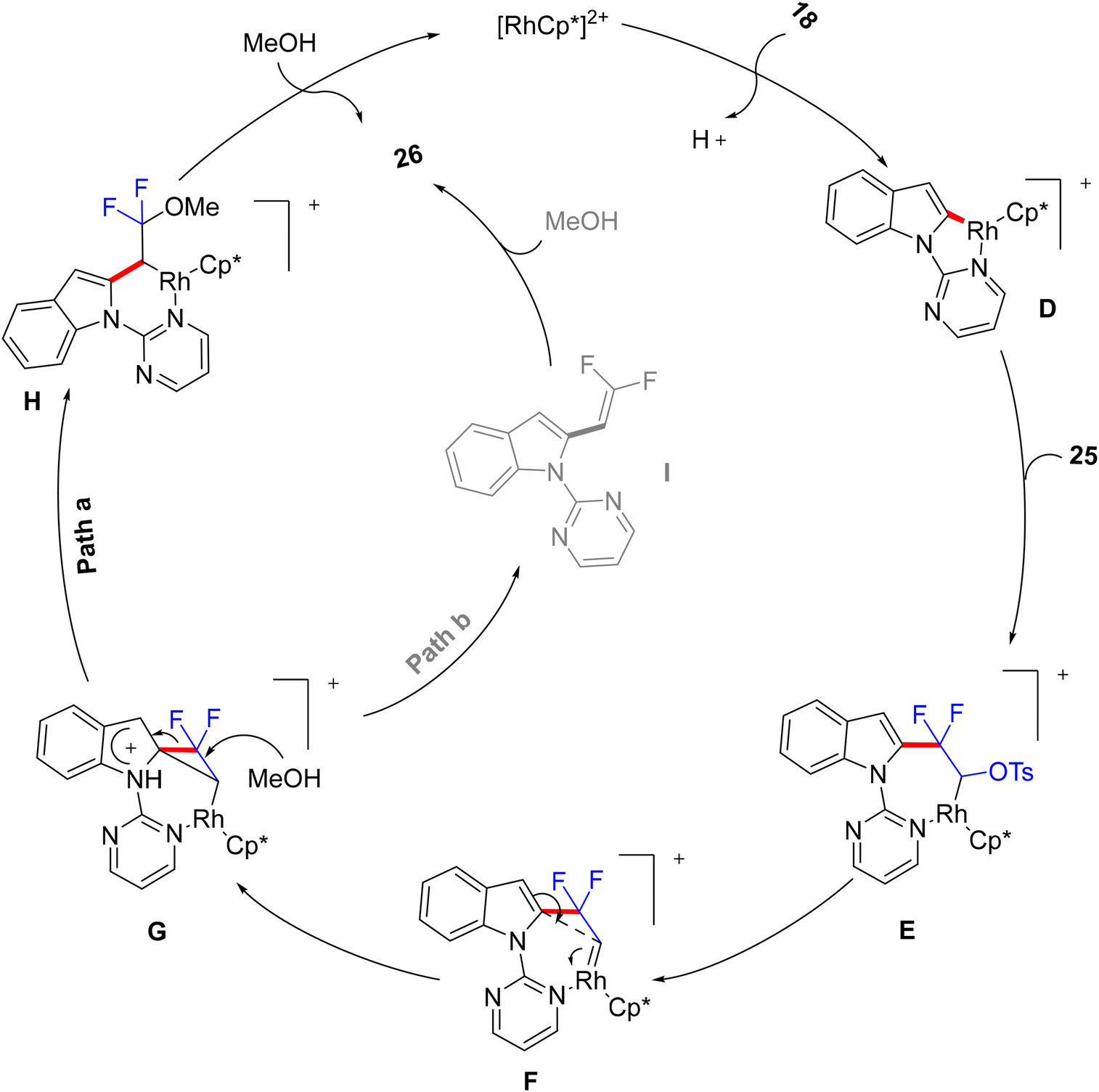
The authors conducted some control experiments to postulate a mechanism (Scheme 22) that was not further theoretically and experimentally consolidated so far. Firstly, pyrimidinyl DG-promoted C(sp2)–H activation of 18 by Rh(III)Cp* catalyst to produce the five-membered rhodacycle complex D. Next, the addition of C(sp2)–Rh to the double bond of 25 produced the seven-membered rhodacycle intermediate E. Subsequently, E underwent a tosylate α-elimination to install the Rh(V)-carbenoid intermediate F.57 Then, an intramolecular migration of the indolyl core delivered a strained difluorocyclopropane transition state G, which produced a six-membered rhodacycle intermediate H by the nucleophilic attack of MeOH. Later, the alcoholysis of H produced the final product 26 and regenerated the catalyst (Path a). In another possible pathway (which still cannot be ruled out at this stage), a nucleophilic attack of MeOH on G might generate an indolyl gem-difluoroalkene intermediate I that might further convert to 26 (Path b).
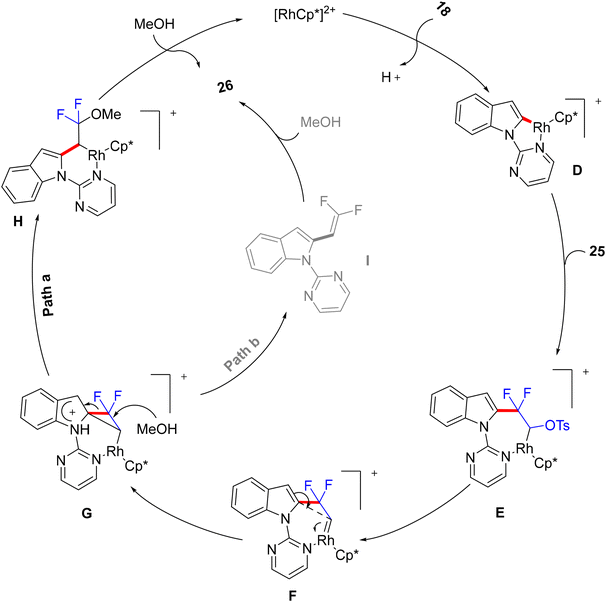 |
| | Scheme 22 Proposed catalytic cycle of Rh(III)-catalyzed C(sp2)–H alkylation of 18. | |
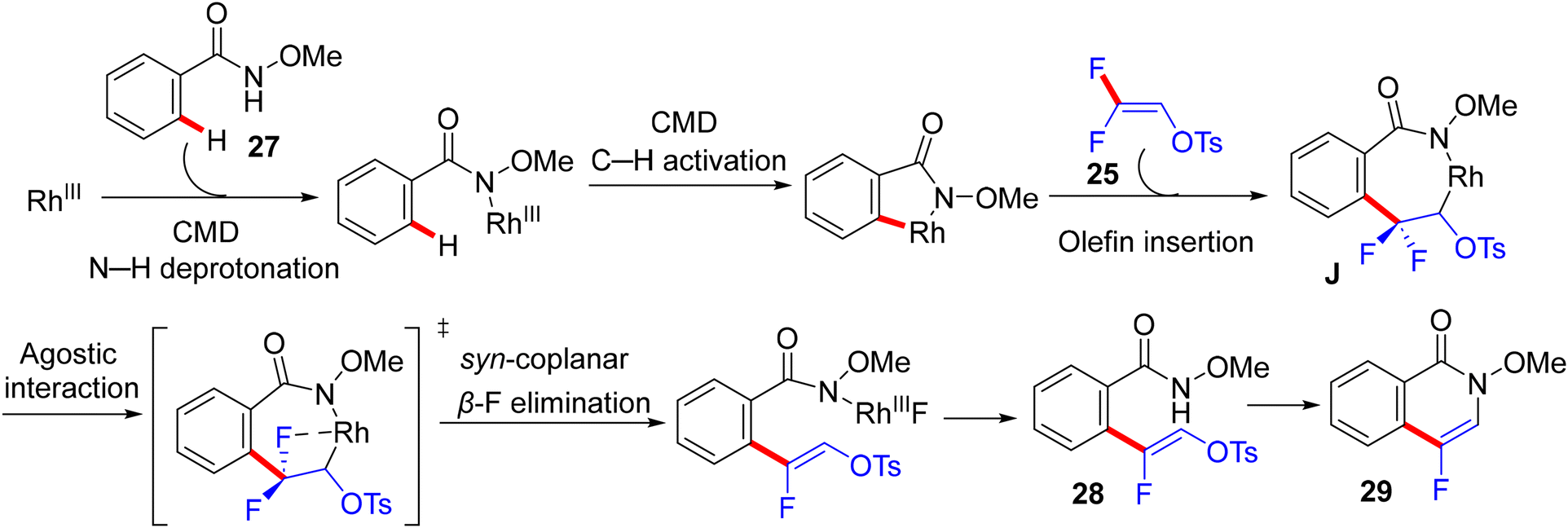
Two years later (2017), Honggen Wang, Xingwei Li et al. described pioneering experimental and supporting theoretical investigations for the direct C(sp2)–H α-fluoroalkenylation of N-OMe secondary amides 27via the doubly bridged dimer [RhCp*Cl2]2 catalyst, using 25 synthon as a versatile coupling partner. After extensive trials of the reaction parameters under inert atmosphere, optimum conditions to produce the expected (Z)-monofluoroalkenes 28 were obtained as follows: [RhCp*Cl2]2 (2.5 mol%), AgBF4 (10.0 mol%), CsOPiv (1.0 equiv.), and Ca(OH)2 (0.2 M 1.0 equiv., as HF scavenger) in hexafluoroisopropanol (HFIP) at 40 °C for 4–8 h. After that, these conditions showed wide N-OMe amide substrate scope and compatibility of functional groups. As a good leaving group, the tosylate unit proved to be a good handle for an intramolecular cyclization upon treatment with concentrated aqueous H2SO4 at ambient temperature to generate the corresponding 4-fluoroisoquinolin-1(2H)-ones 29 that were also obtained via a one-pot reaction, skipping the isolation of acyclic monofluoroalkenes (Scheme 23).30 As a synthetic application for this strategy, a tumor necrosis factor 31 was obtained in 77% yield via multi-step chemical synthesis, including the N-OMe bond cleavage of fluorinated isoquinolinone 29a by NaH/DMF, followed by the alkylation of the NH product (30, 1.0 equiv.) with 4-bromobutyrate (1.5 equiv.) in the presence of K2CO3 (2.0 equiv.). As an analogy, this approach was also extended to include isoquinolone substrates 32a–c, and the monofluoroalkenes 33a–c were isolated in 35–53% yields and afforded the corresponding cyclized 5-fluoro-8H-isoquinolino[3,2-a]isoquinolin-8-ones (34a–c) upon treatment with 2N NaOH solution (Scheme 24).30
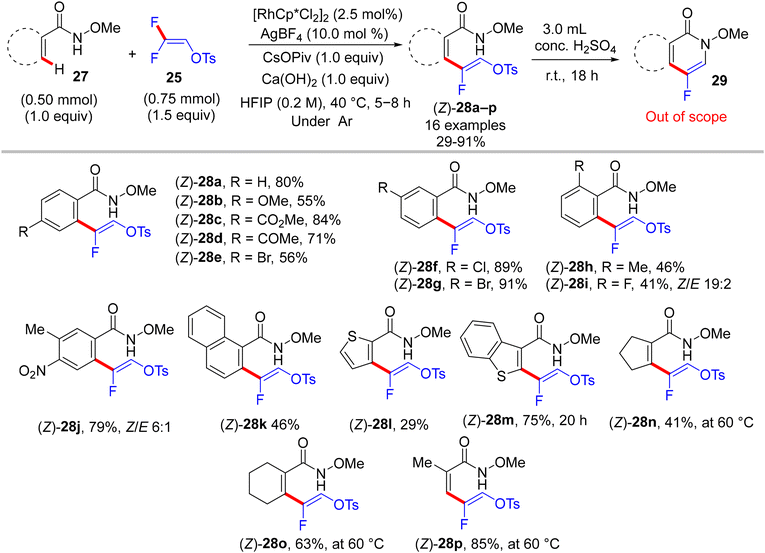 |
| | Scheme 23 Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 27 with 25. | |
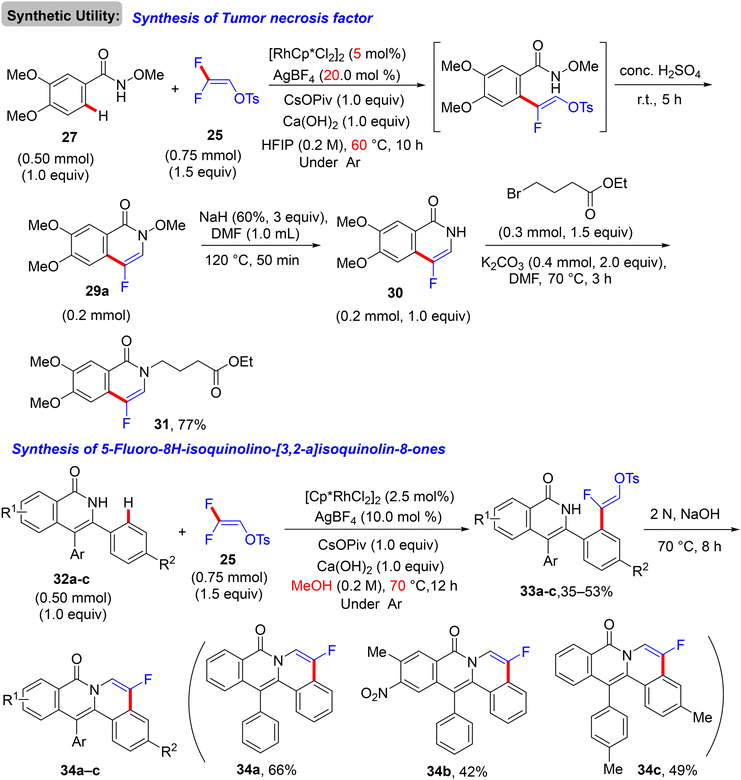 |
| | Scheme 24 Synthesis of the tumor necrosis factor 31 and 5-fluoro-8H-isoquinolino[3,2-a]isoquinolin-8-ones (34a–c). | |
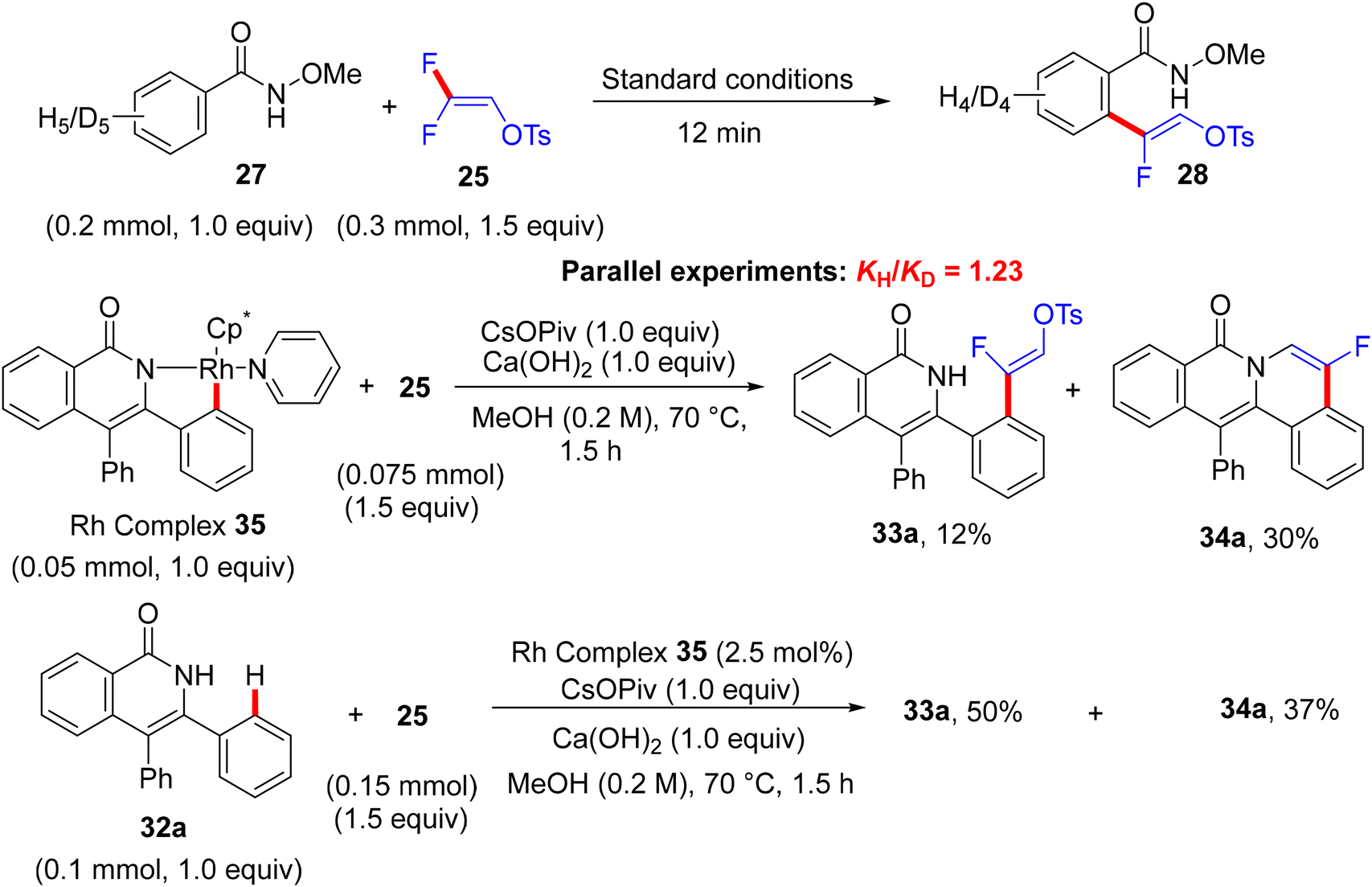
From the mechanistic viewpoint, the authors conducted some experimental work combined with computational studies. KIE studies showed that C–H dissociation is not likely the turnover-limiting step. Attempts to isolate the rhodacycle intermediate from the reaction between the Rh(III) catalyst and N-OMe benzamide failed, but were successful with the isoquinolinone derivative via Wang's protocol.58 The stoichiometric transformation of Rh complex 35 with 25 delivered the alkenylation product 33a in 12% quantitative 1H NMR yield, while the catalytic pathway furnished the same product with 50% quantitative 1H NMR yield (Scheme 25). Regarding the theoretical calculations executed at the density functional theory (DFT) level (B3LYP and M11L),59 the formation of a seven-membered rhodacycle intermediate J was achieved after three steps, including N–H deprotonation, C–H activation, and olefin insertion (Scheme 26). Both N–H deprotonation and C–H activation proceeded through a concerted metalation-deprotonation (CMD) pathway.60 Next, the olefin insertion occurred via a transition state that has the highest activation barrier. Natural Bond Orbital (NBO) analysis61 supported the regioselectivity in the migratory insertion of 25via the β-carbon, which is highly positively charged owing to the strong inductive effect(I) of F atoms.
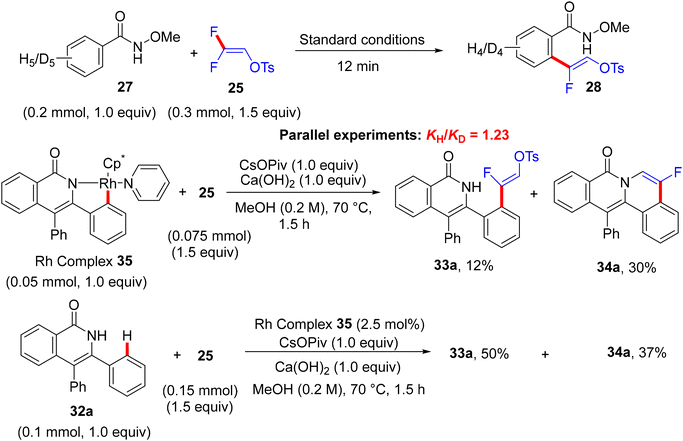 |
| | Scheme 25 Control experiments in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 27 with 25. | |
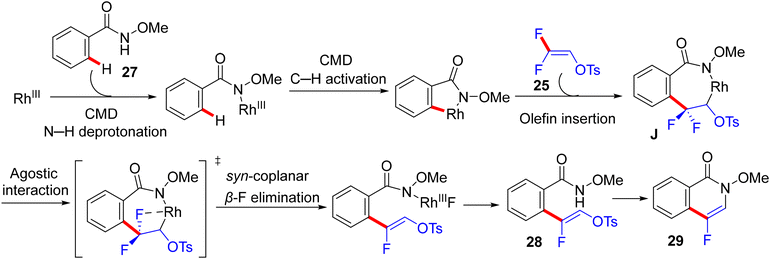 |
| | Scheme 26 Summary of mechanistic proposal of Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 27 with 25. | |
Hence, the computational work points to the establishment of an “agostic” interaction between the C–F bond and Rh center, followed by a syn-coplanar β-F elimination via a transition state with an activation Gibbs energy of ΔG‡ ~7 kcal mol−1 and subsequently the formation of (Z)-monofluoroalkenes 28 as observed experimentally. While the anti-coplanar H-bonding-assisted β-F elimination that was suggested by the prior study29 of Loh was accomplished via a transition state with an activation barrier of ΔG‡ = 19 kcal mol−1, by 12 kcal mol−1, a higher energy barrier than that of the cis-coplanar one, thus giving support to the proposal of a dominant syn-coplanar route for this type of transformation. Overall, the migratory olefin insertion carries the highest activation barrier (ΔG‡ = 26 kcal mol−1) and is the rate-determining step in agreement with experimental results. The authors summarized the mechanistic proposal as displayed in Scheme 26.
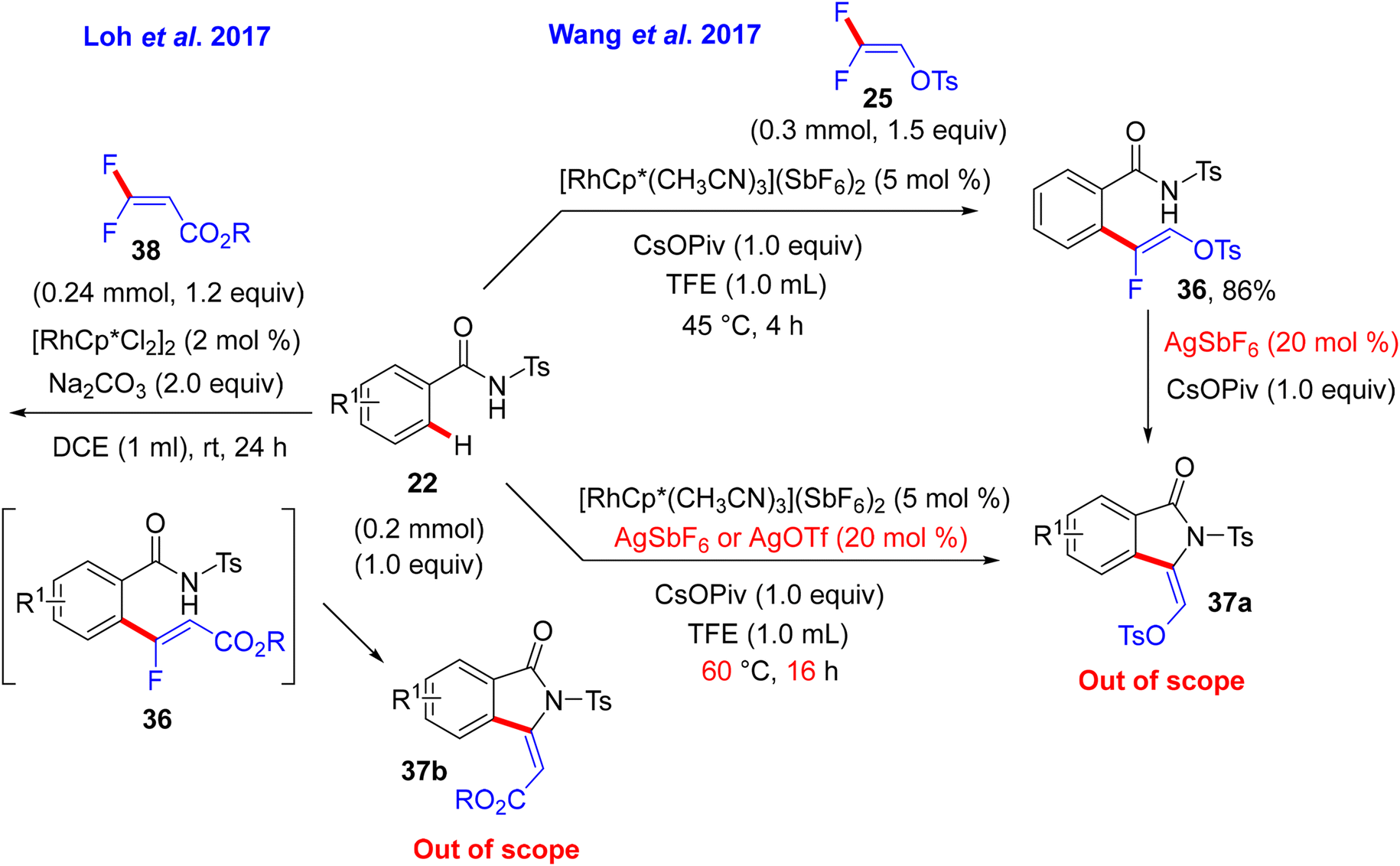
After that, in May 2017, Wang and co-researchers used the same starting materials, but N-tosylbenzamide (22) instead of the prior one N-methoxybenzamide (27), to install the corresponding (Z)-monofluoroalkene 36via a redox-neutral Rh(III)-based catalysis approach. Their study aimed to obtain the well-known bioactive 3-alkylidene isoindolinone 37a (out of scope), and this was achieved by applying a bimetallic Rh(III)/Ag(I) relay catalytic system. The Rh(III) catalyst catalyzed direct C(sp2)–H α-fluoroalkenylation, whereas the Ag(I) salt (AgSbF6 or AgOTf) activated the subsequent cyclization process (Scheme 27).31 In addition, Loh's group in the same year conducted a similar reaction under very mild conditions: [4 + 1] annulation of 22 and gem-difluoroacrylate 38 at room temperature using the [RhCp*Cl2]2/Na2CO3 catalytic system without Ag salts to obtain 37b, which contains CO2R instead of the OTs group. The reaction proceeds via a monofluoroalkene intermediate, followed by a cyclization step to afford the annulation product (Scheme 27).32
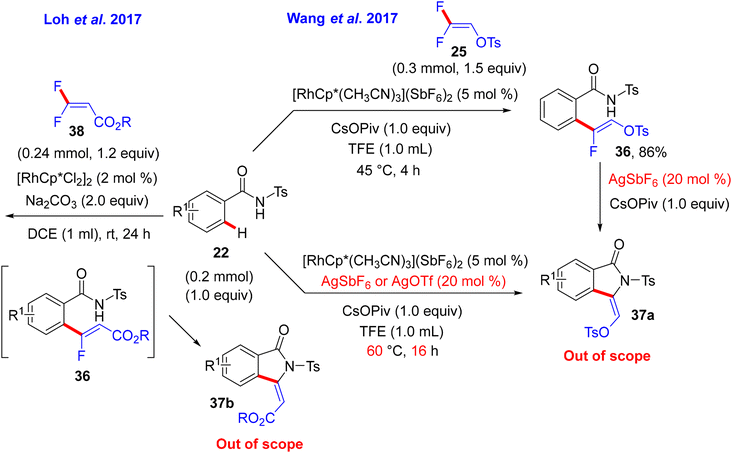 |
| | Scheme 27 Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 22 with 25 and 38. | |
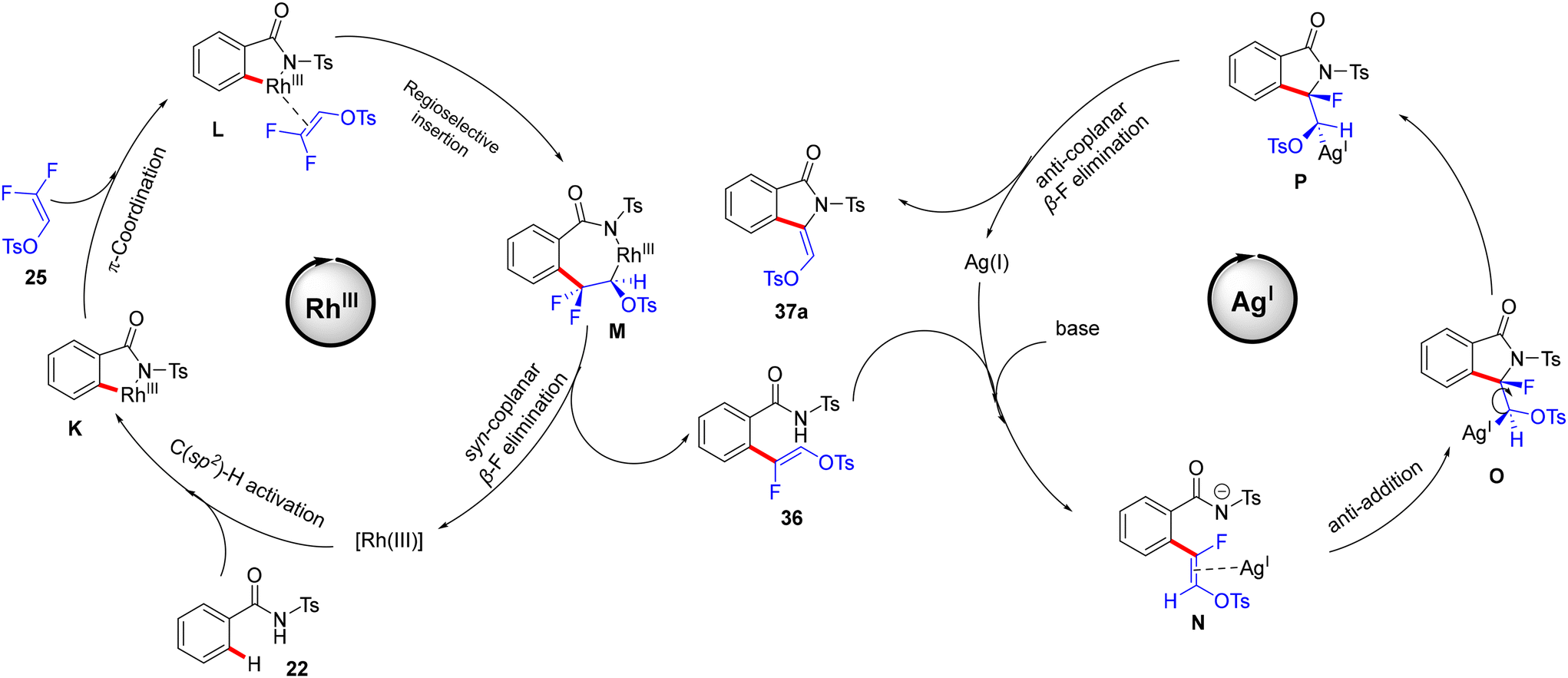
Wang's group proposed a reaction mechanism according to the previous studies30 and the transformation outcomes. In the Rh cycle, the amide DG-assisted Rh(III) species catalyzes the C(sp2)–H activation to afford the five-membered rhodacycle complex K. The π-coordination of alkene 25 generates L, followed by a regioselective migratory alkene insertion to create the seven-membered rhodacycle intermediate M. According to the abovementioned study,30 a syn-coplanar β-F elimination provides the expected (Z)-monofluoroalkene 36 with good stereoselectivity. Hence, while the N–H group is deprotonated by a base, the Ag(I) salt acts as a π acid, which activates the olefin to produce the N intermediate. Next, the anti-addition to the activated double bond results in a 5-exo cyclization to yield O and then P. Finally, an anti-coplanar β-F elimination affords the cyclized (Z)-stereospecific 3-alkylidene isoindolidone product 37a (Scheme 28).
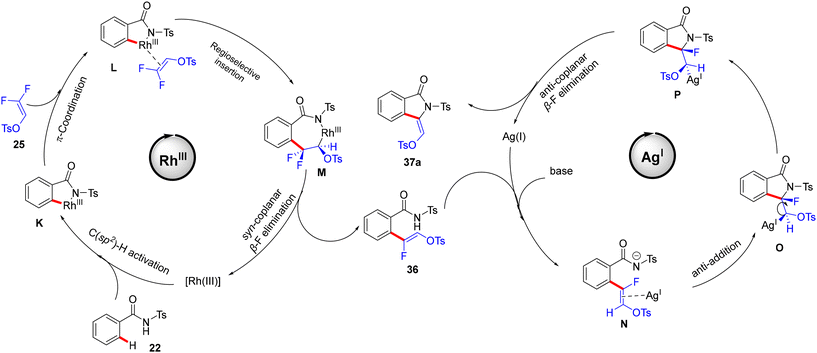 |
| | Scheme 28 Proposed mechanism of a bimetallic Rh(III)/Ag(I) relay catalytic system on 22 with 25. | |
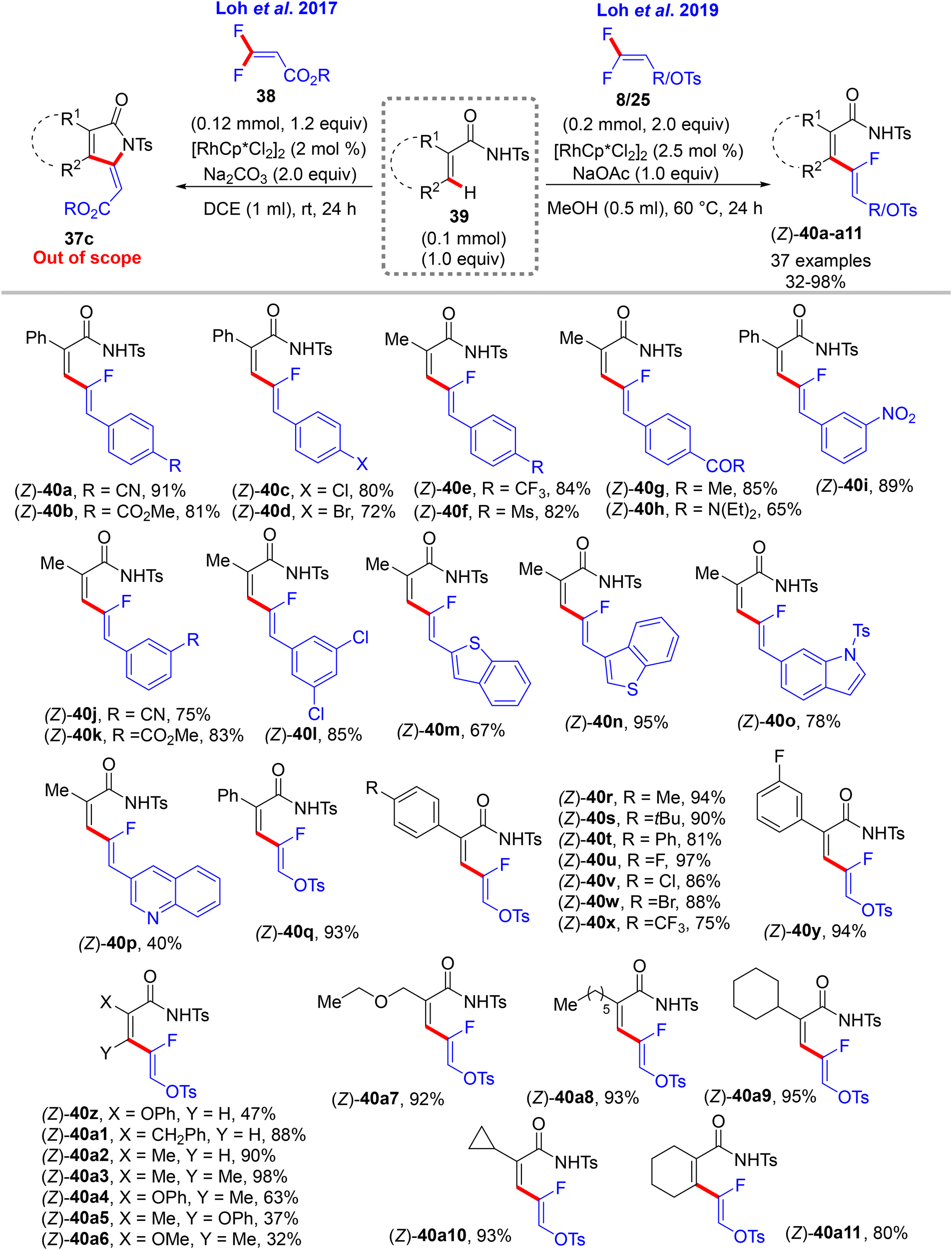
Still with Ts-secondary amide substrates, Loh's research group also introduced acrylamides 39 for C(sp2)–H α-fluoroalkenylation with different gem-difluoroalkenes (8, 25, and 38). Under the aforementioned conditions in Scheme 23,32 such as Ts-benzamides, 39 with 38 form [4 + 1] cyclization product 37cvia Rh(III)-catalyzed alkenyl C(sp2)–H activation and a two-fold C(sp2)–F cleavage strategy. With 8 and 25, slight modifications in the mild conditions lead to the formation of (1Z,3Z)-2-fluoro-1,3-dienes as (Z)-monofluoroalkenes 40a–a11 in poor to excellent yields (32–98%) with complete stereoselectivity (Scheme 29).33
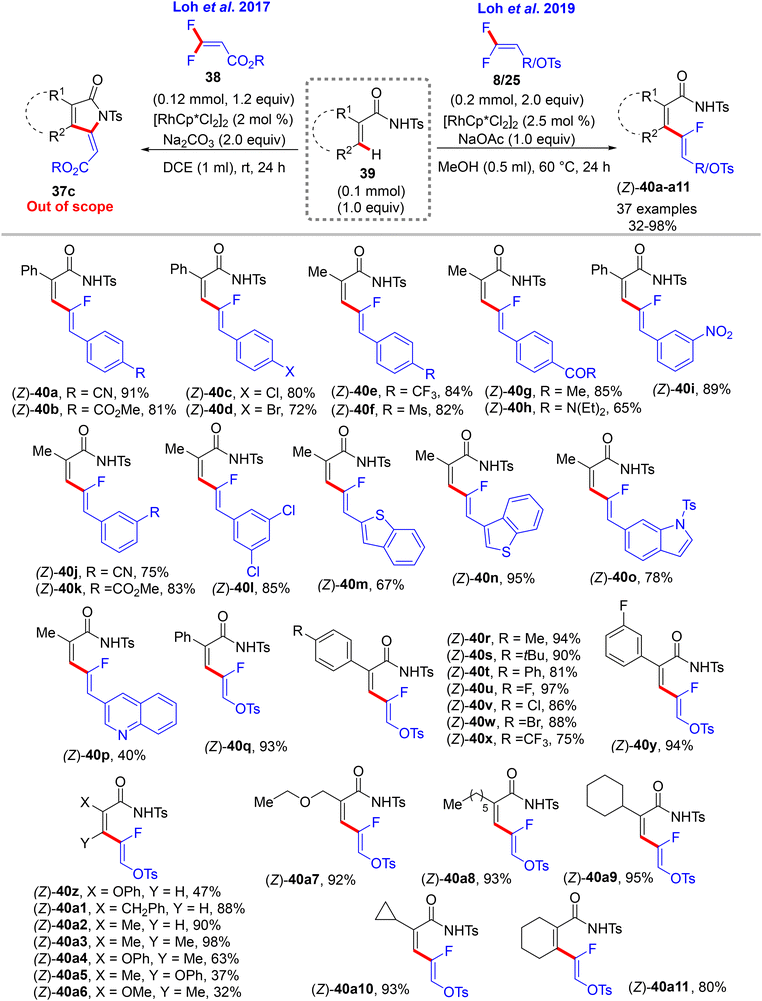 |
| | Scheme 29 Scope investigation in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of acrylamides 39 with 8 to create 2-fluoro-1,3-dienes 40a–a11. | |
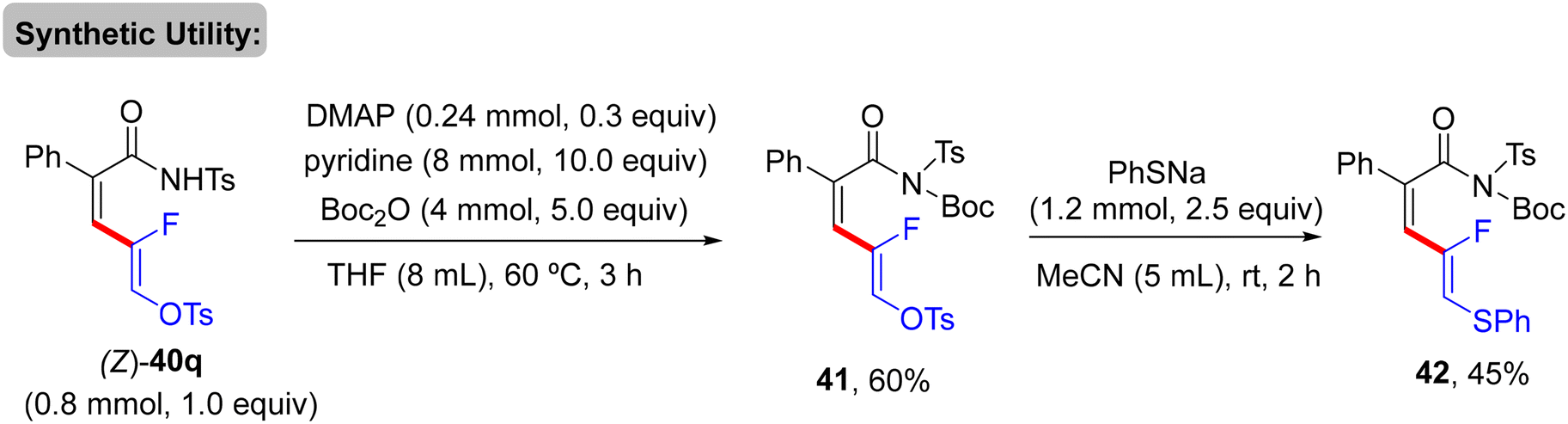
As a synthetic extension of this protocol, the OTs group was replaced by the SPh unit to form the corresponding thioether 42 in 45% isolated yield. Initially, base-promoted NH group protection of 40q by di-t-butyl dicarbonate (Boc2O) generated 41 in 60% yield. Thereafter, treatment of 41 with sodium benzenethiolate at room temperature furnished 42 (Scheme 30).33 Mechanistically, the authors experimentally confirmed that gem-difluoroalkenes are essential for this transformation, where the analogues containing heavier halides (gem-dibromo-, gem-dichloro-, and gem-bromofluoroalkenes 2) were unsuitable, though the specific reason was not mentioned. The plausible mechanism matched those proposed in previous reports.30,31 Namely (Scheme 28), N-tosylamide-assisted C(sp2)–H activation by Rh(III) to form rhodacycle intermediate Q, which was followed by π-coordination to afford R. Subsequent olefin insertion forms the S species, which undergoes a syn-coplanar β-F elimination to deliver 40 as a (Z)-isomer (Scheme 31).
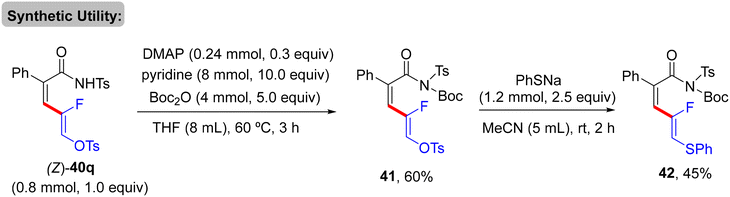 |
| | Scheme 30 Synthesis of thioether 42. | |
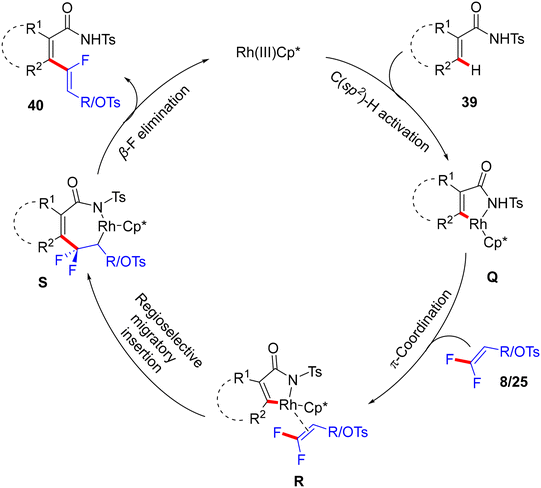 |
| | Scheme 31 Proposed catalytic cycle of Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 39 with 8/25. | |
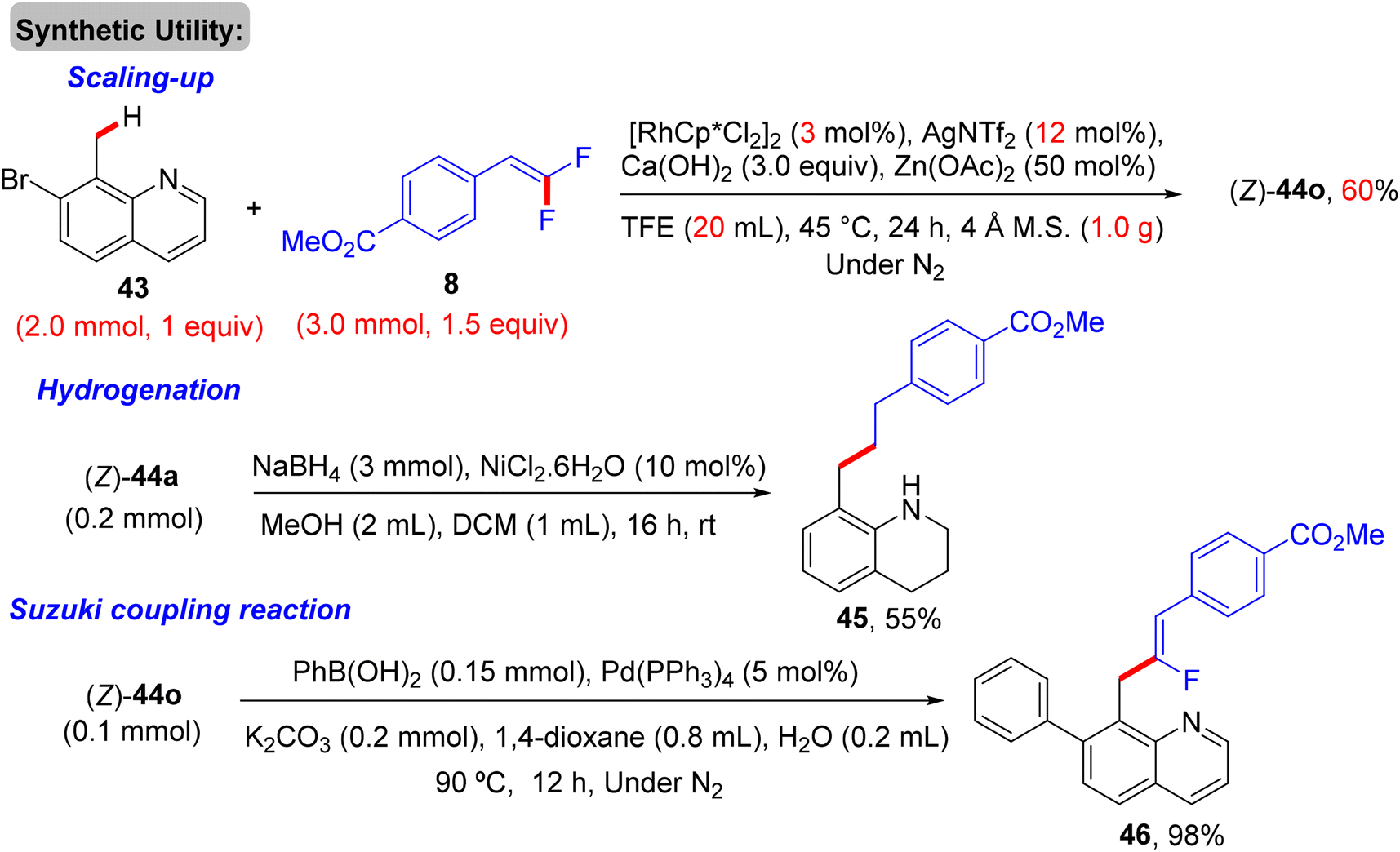
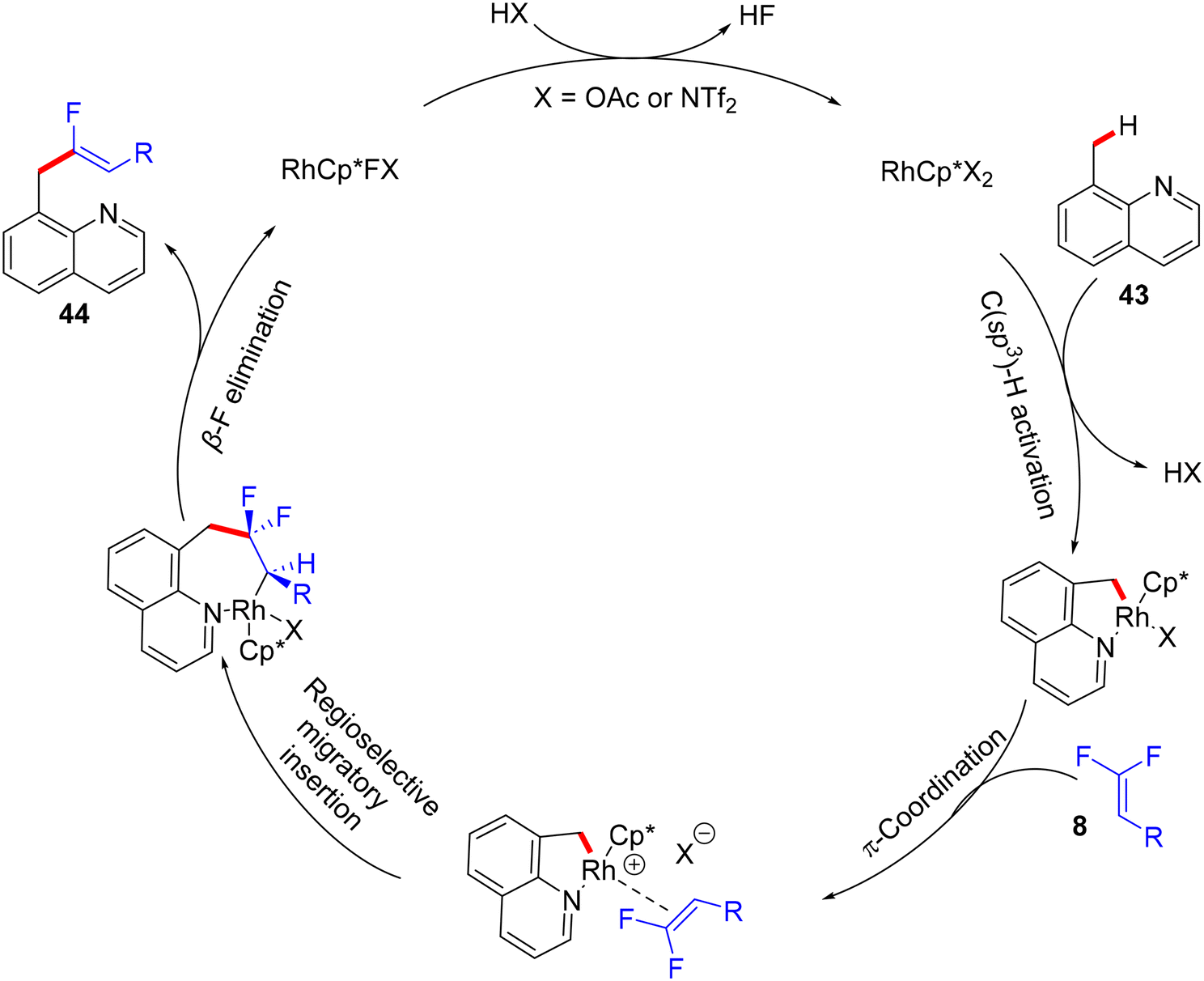
Thereafter, Xingwei Li, Fen Wang, and their co-workers reported the first benzylic α-fluoroalkenylation also by Rh(III)Cp*-based catalysis. This seldom direct C(sp3)–H activation was conducted between 8-methylquinolines 43 and 8via regio- and stereoselective route to produce the corresponding (Z)-monofluoroalkenes 44a–a10 in poor to excellent yields (30–98%) under mild and redox-neutral circumstances (Scheme 32).34 To achieve this transformation type, the best effective conditions were [RhCp*Cl2]2 (5 mol%), AgNTf2 (20 mol%), Ca(OH)2 (3.0 equiv.), and Zn(OAc)2 (50 mol%) in TFE (2 mL) at 45 °C for 24 h using 100 mg of 4 Å molecular sieves. Regarding the reaction scope, coupling between 43 and 25 or perfluoroalkyl alkene partners was successful and exhibited the desired products in 61% (44a9, 100 °C, DCE) and 94% (44a10, 80 °C) isolated yields, respectively (Scheme 32). As a scope limitation, 2-methylquinoline and 8-ethylquinoline were found to be unsuitable substrates for this transformation. Furthermore, the authors demonstrated some synthetic applications, such as a scaling up (2.0 mmol) of the 44o (60%) derivative and catalytic hydrogenation (NaBH4/NiCl2·6H2O) of 44a to produce the corresponding 1,2,3,4-tetrahydroquinoline derivative 45 (55%). Moreover, the Pd-catalyzed Suzuki–Miyaura coupling reaction of organohalide 44o as a late-stage functionalization (LSF) to install the biphenyl product 46 in excellent yield (98%) (Scheme 33).34Scheme 34 shows the catalytic cycle that was proposed according to the previous studies.30,31
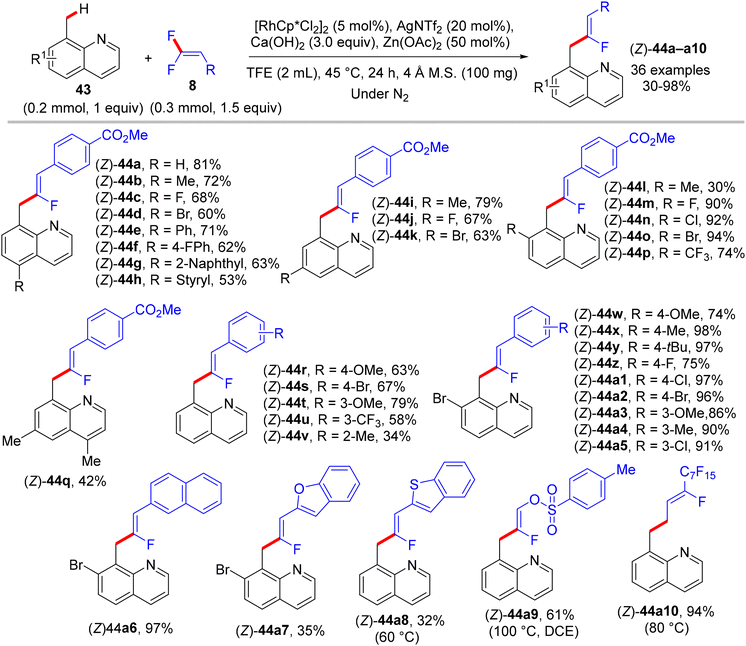 |
| | Scheme 32 Scope investigation in Rh(III)-catalyzed direct C(sp3)–H α-fluoroalkenylation of 43 with 8. | |
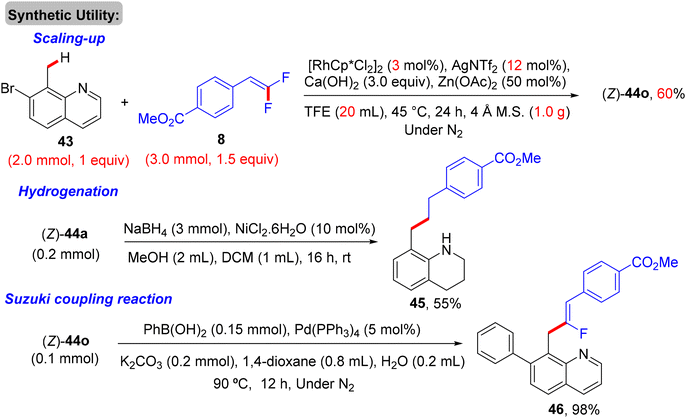 |
| | Scheme 33 Synthetic applications of Rh(III)-catalyzed direct C(sp3)–H α-fluoroalkenylation of 43 to access 45 and 46. | |
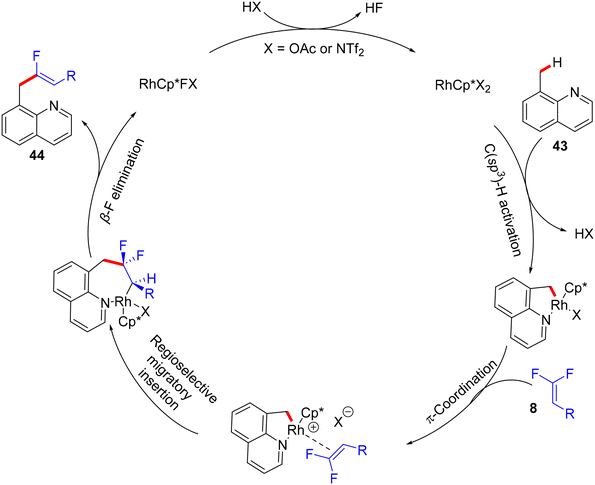 |
| | Scheme 34 Proposed catalytic cycle of Rh(III)-catalyzed direct C(sp3)–H α-fluoroalkenylation of 43. | |
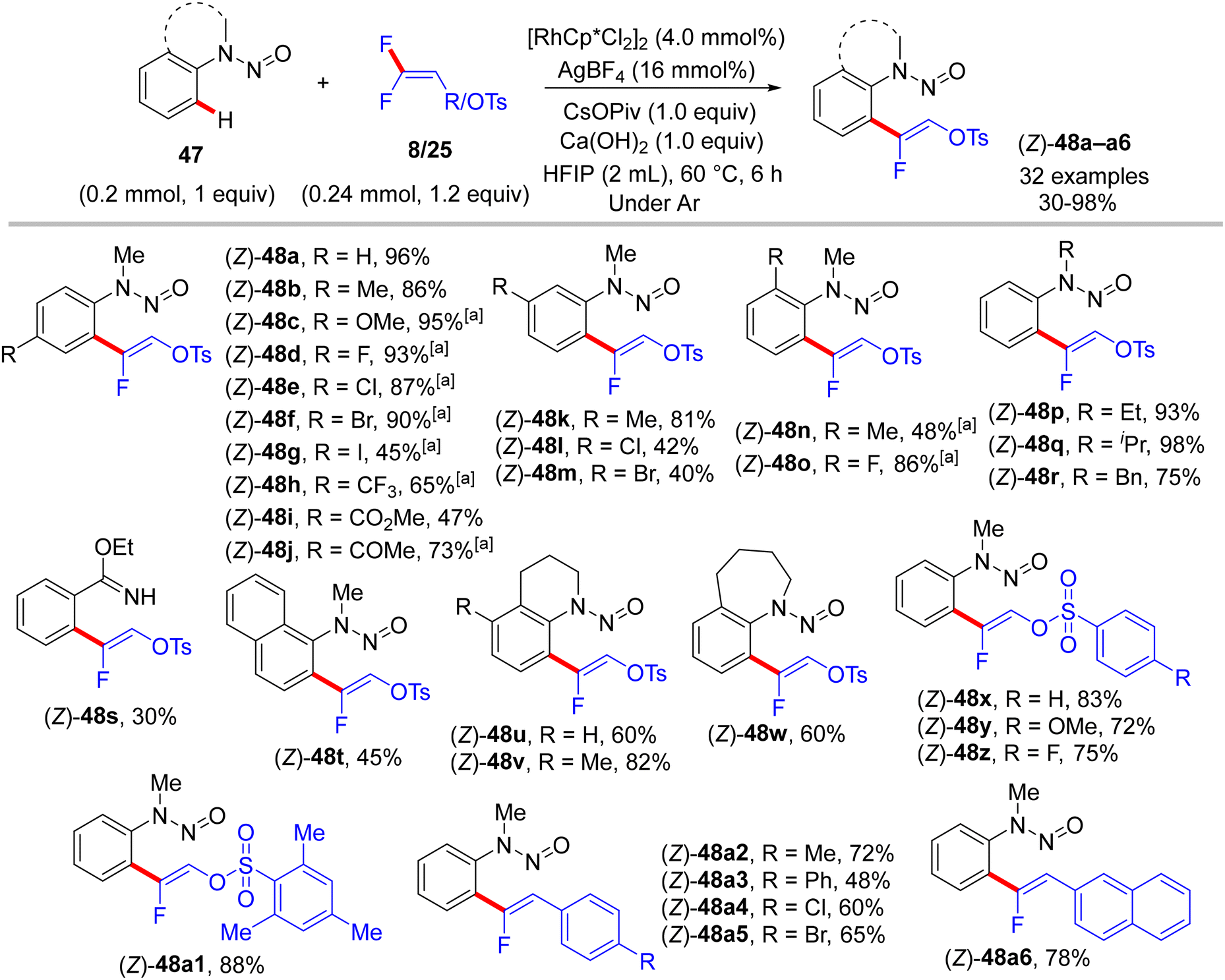
Furthermore, the research work of the Xingwei Li group (2018) described the first catalytic synthesis of (Z)-monofluoroalkenes 48a–a6 from the nitrogenous-directed N-nitrosoaniline substrates 47 and 8/25. This transformation is accomplished under mild and redox-neutral conditions via the aforementioned steps, including chelation-assisted rhodacycle formation, alkene regioselective insertion, and syn-coplanar β-F elimination (Scheme 35). Optimal conditions were identified as follows: [RhCp*Cl2]2 (4.0 mol%), AgBF4 (16.0 mol%), CsOPiv (1.0 equiv.), Ca(OH)2 (1.0 equiv.), HFIP (2 mL) at 60 °C for 6 h under argon. The scope of both reactants was screened (Scheme 36).35
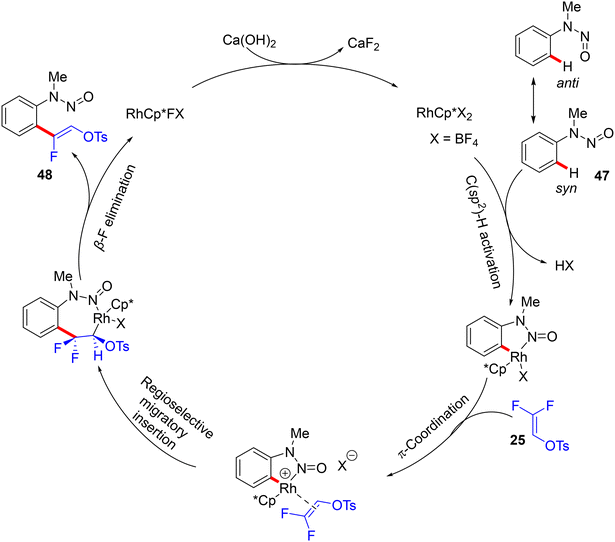 |
| | Scheme 35 Proposed catalytic cycle of Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 47. | |
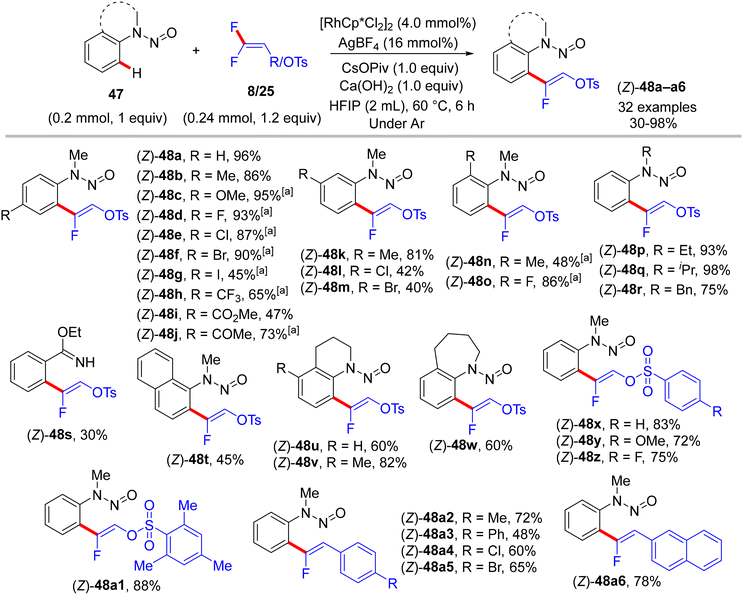 |
| | Scheme 36 Scope investigation in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 47 with 8/25. [a] CsOAc (1.0 equiv.) was used instead of CsOPiv. | |
The synthetic usefulness of the final product (48) was investigated. As a removal directing group (RDG) type,62 the N-nitroso unit was reductively removed using either pathways, that is, Zn (2 equiv.)/NH4Cl (2 equiv.) in a mixed solvent of MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (3:1) at 45 °C or NiCl2·6 H2O (2 equiv.)/NaBH4 (6 equiv.) at room temperature in THF, producing the corresponding N-methylaniline derivative 49 in 68 and 75%, respectively. Next, treatment of 49 with 2N NaOH (6 equiv.) in MeOH at 70 °C provided N-methylisatin (50) in a 70% isolated yield. Further, the versatile pseudohalide tosylate group (OTs) of 49 could be coupled with phenylboronic acid via the Suzuki–Miyaura reaction to produce the cross-coupled product 51 in 78% isolated yield (Scheme 37).35
:H2O (3:1) at 45 °C or NiCl2·6 H2O (2 equiv.)/NaBH4 (6 equiv.) at room temperature in THF, producing the corresponding N-methylaniline derivative 49 in 68 and 75%, respectively. Next, treatment of 49 with 2N NaOH (6 equiv.) in MeOH at 70 °C provided N-methylisatin (50) in a 70% isolated yield. Further, the versatile pseudohalide tosylate group (OTs) of 49 could be coupled with phenylboronic acid via the Suzuki–Miyaura reaction to produce the cross-coupled product 51 in 78% isolated yield (Scheme 37).35
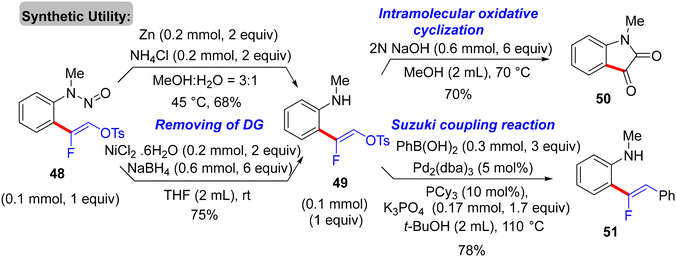 |
| | Scheme 37 Synthetic applications of Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 47 to synthesize 49–51. | |
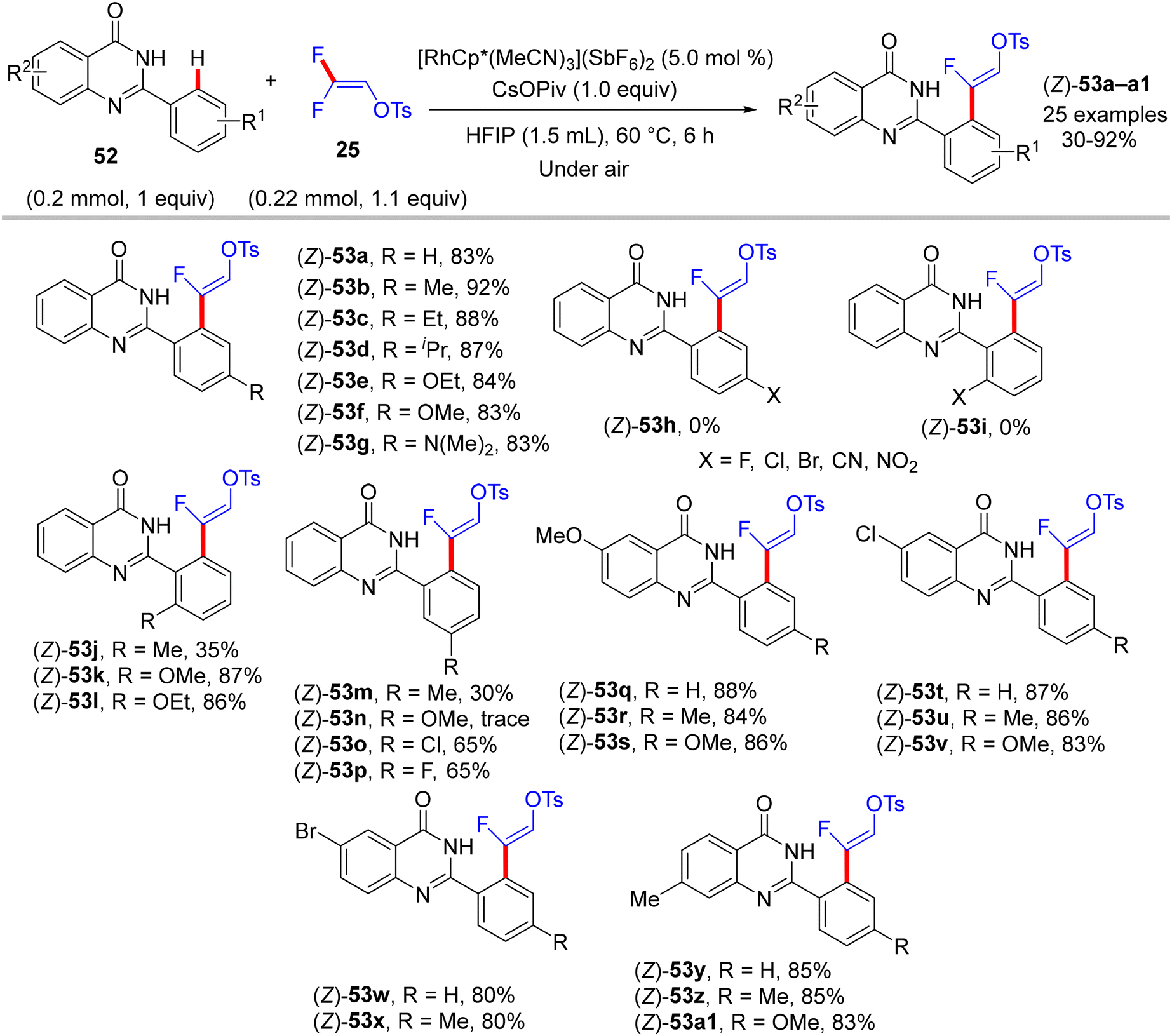
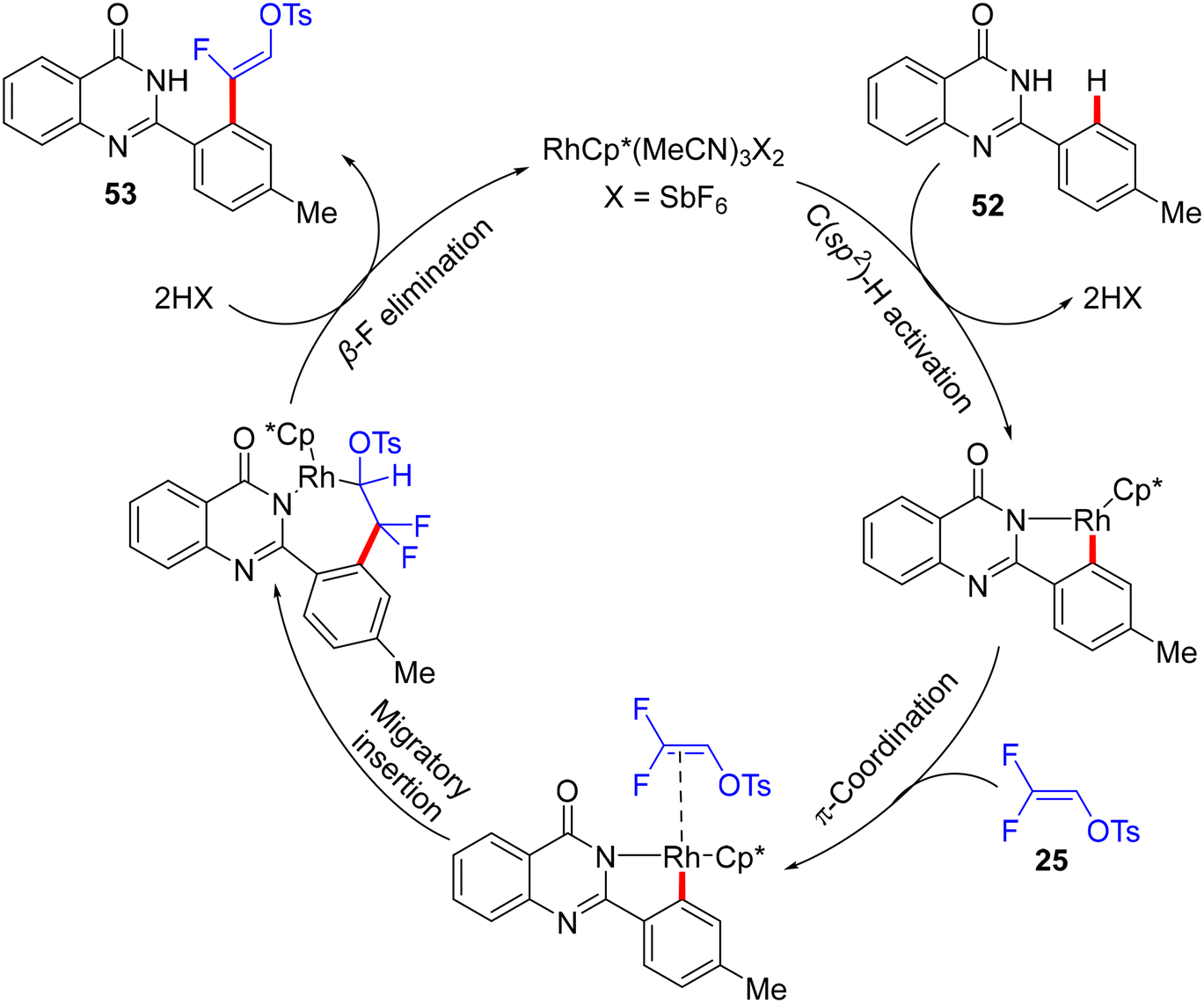
In 2020, five years after Loh's study,29 Peng's lab used the same cationic Rh catalyst [RhCp*(MeCN)3](SbF6)2 to catalyze the direct C(sp2)–H α-fluoroalkenylation of 2-arylquinazolinones 52 using 25 synthon. 2-Arylquinazolinone contains an amide-imine unit that served as a permanent DG (PDG) to assist C(sp2)–H activation via rhodacycle key intermediate formation. Following Honggen Wang's study findings,30 which confirmed the syn-coplanar β-F elimination pathway other than anti-coplanar H-bonding-assisted β-F elimination (Loh's study29) and the impact of the oxygenated bases, Peng and co-workers used CsOPiv, and optimal conditions were obtained after the initial investigation as follows: [RhCp*(MeCN)3](SbF6)2 (5.0 mol%), CsOPiv (1.0 equiv.), HFIP (1.5 mL), at 60 °C for 6 h under atmospheric oxygen.36
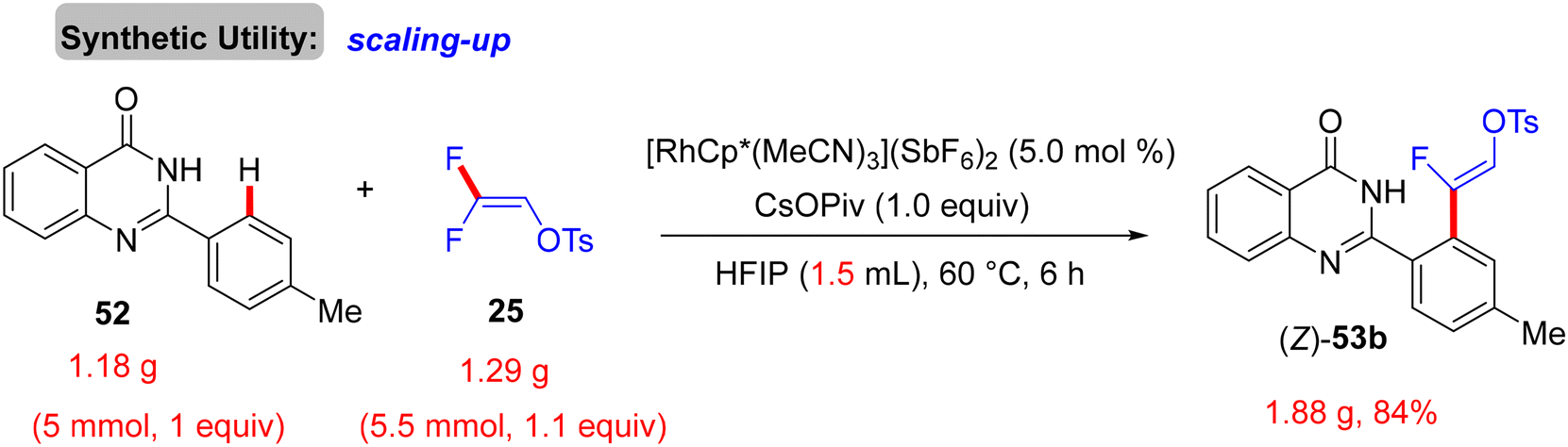
Scheme 38 displays the synthesis of (Z)-53a–a1, the generality, and substrate scope of this protocol. When the R1 group at the para or ortho positions of the 2-aryl group is EDG, the (Z)-monofluoroalkenes are generated in good to excellent yields (83–92%). When the R1 group is an EWG at the same positions, no reaction occurs, but the reverse is correct with the meta position (53m–p). Surprisingly, whatever the electronic feature of the R2 group of the quinazolinone ring, that is either EDG or EWG, the desired monofluoroalkenes (53q–a1) are isolated in high yields (80–88%). Of note, no cyclization products are obtained owing to the absence of either Ag salts (removal of AgF)31 or acidic (H+)/basic (OH−) conditions (removal of TsOH),30 as indicated above. Moreover, the authors succeeded in achieving the gram-scale synthesis of (Z)-53b (Scheme 39). As shown in Scheme 40, according to the findings of the previous studies,30–35 the catalytic cycle of this transformation was established.
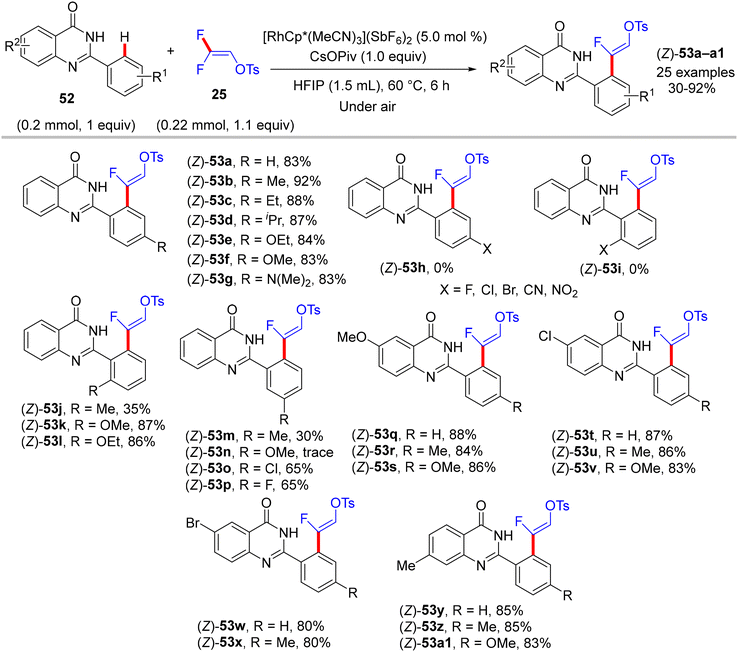 |
| | Scheme 38 Scope investigation in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 52 with 25. | |
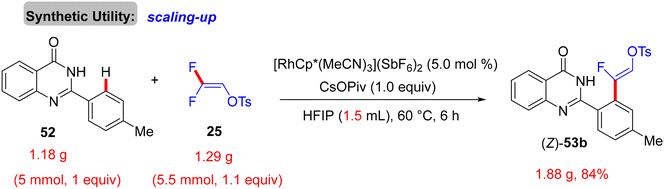 |
| | Scheme 39 Gram-scale synthesis of (Z)-53b. | |
 |
| | Scheme 40 Proposed catalytic cycle of Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 52. | |
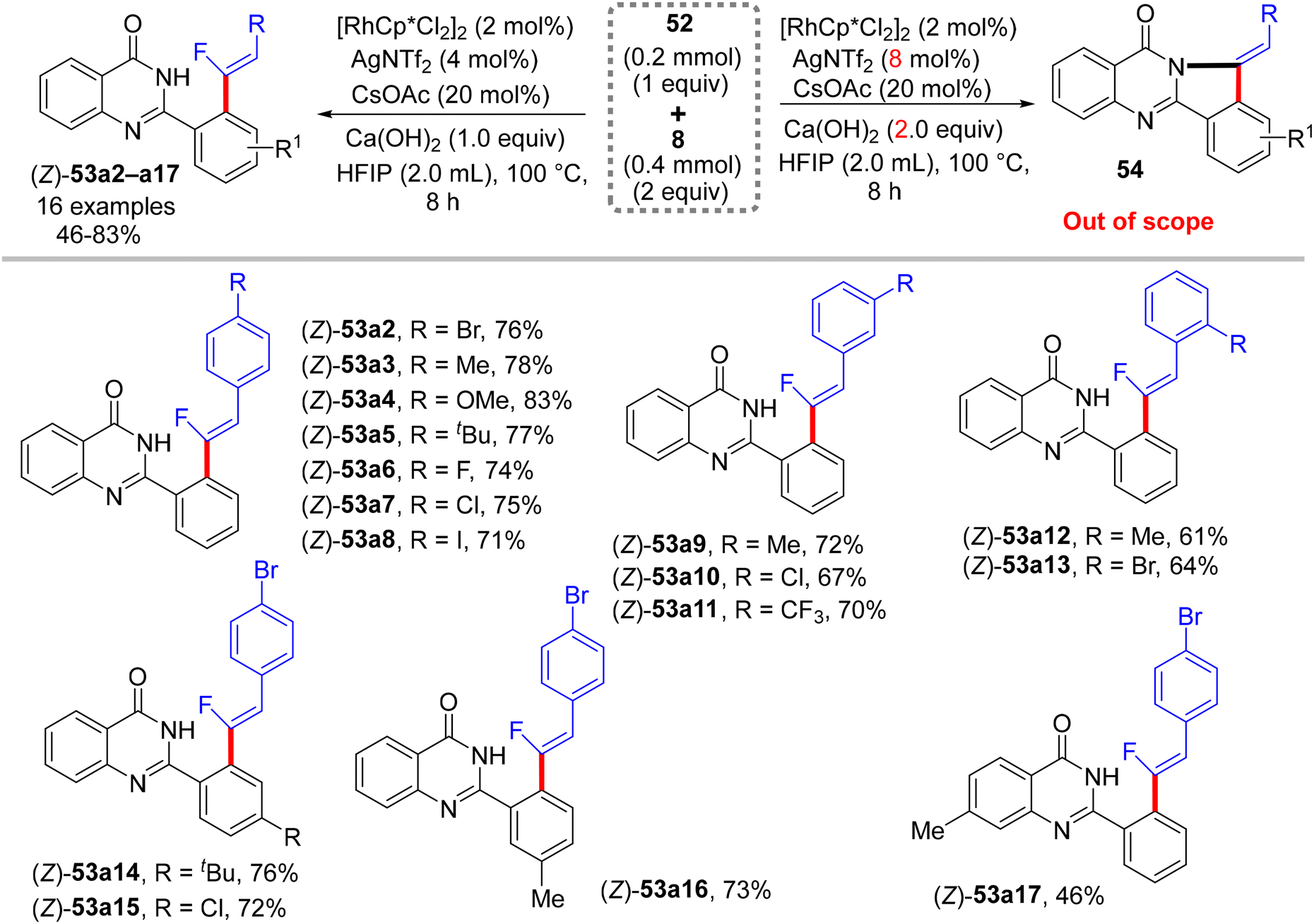
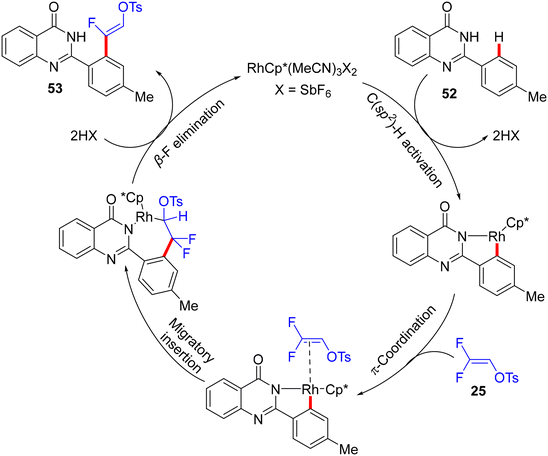
Recently (2023), Ji and co-workers employed the same prior substrate 52 for the direct C(sp2)–H α-fluoroalkenylation with 8 by [RhCp*Cl2]2 catalyst.37 Like in the previous occurrences, this stereoselective transformation was also aided by PDG (amide-imine) and afforded the desired (Z)-monofluoroalkenes (53a2−ax) in fair to very good, isolated yields (46–83%), especially with EDGs of para- and meta-trisubstituted gem-difluoroalkenes (8), as well as featuring good functional group compatibility. After several optimization trials, the optimum conditions were settled as follows: [RhCp*Cl2]2 (2 mol%), Ca(OH)2 (1.0 equiv.), CsOAc (20 mol%), and AgNTf2 (4 mol%) in HFIP (2.0 mL) at 100 °C for 8 h (Scheme 41). Doubling the amount of Ca(OH)2 (2.0 equiv.) and AgNTf2 (8 mol%) led to domino (tandem) C(sp2)–H and N–H activation (two-fold C–F cleavage/[4 + 1] cyclization31) that furnished the corresponding isoindolo[1,2-b]quinazolin-10(12H)-ones 54, which is beyond the scope of this review. It is worthy of note that this catalytic procedure (C(sp2)–H activation) is similar to the prior one (C(sp3)–H activation),34 with the replacement of Zn(OAc)2 by CsOAc.
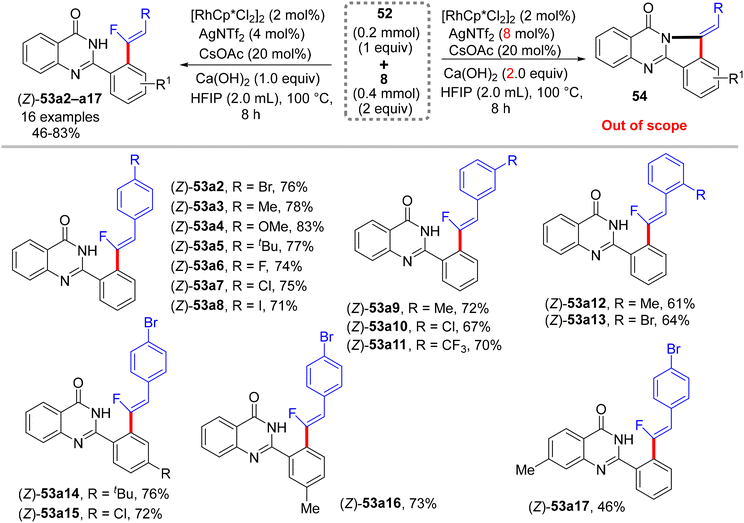 |
| | Scheme 41 Scope investigation in Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 52 with 8. | |
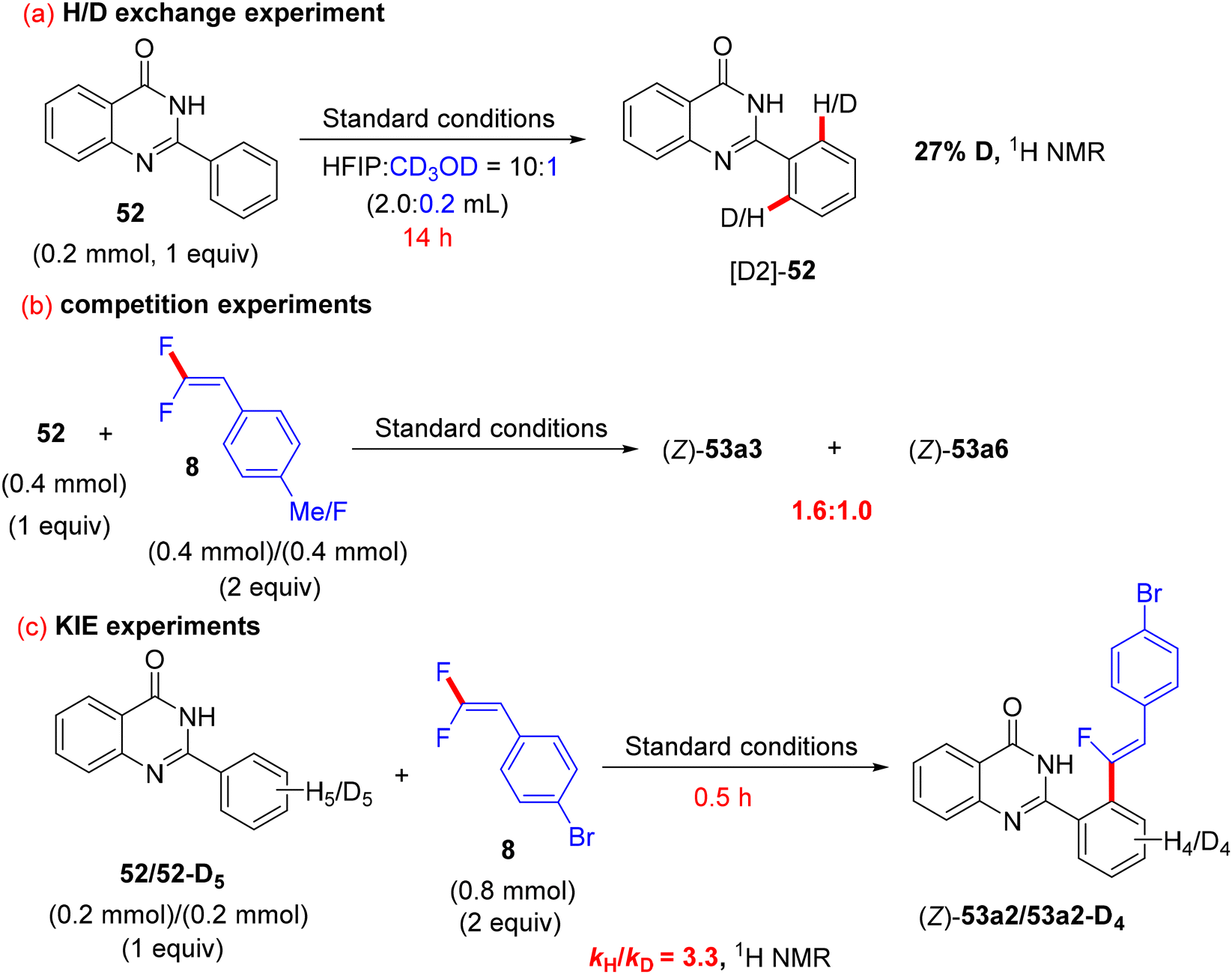
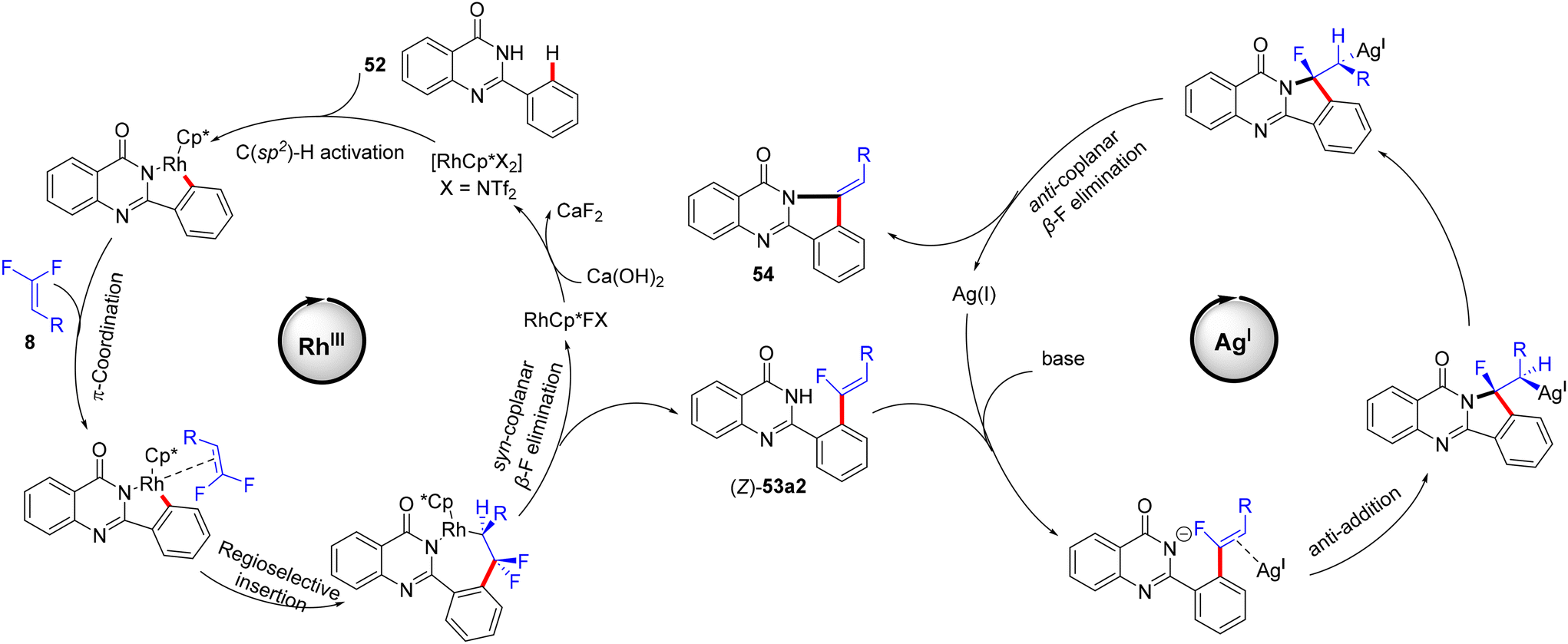
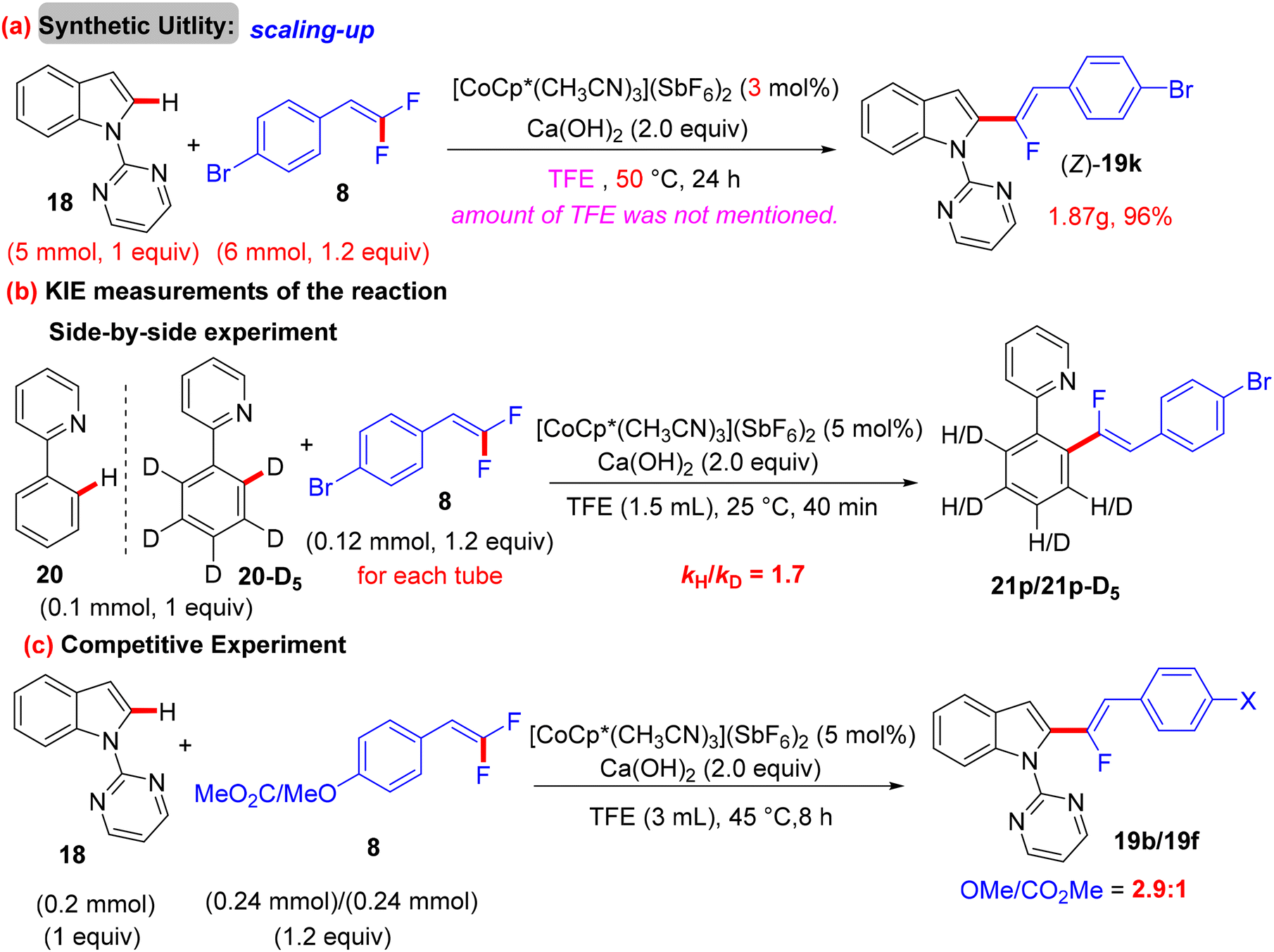
The authors conducted some control mechanistic experiments. An H/D exchange by CD3OD was observed at the two ortho-positions ([D2]-52, 27% D) of 52, demonstrating that the C(sp2)–H activation was reversible. Like the previous studies, the competition experiments proved that 8 with EDGs were more efficient than their EWG counterparts (1.6:1.0). The KIE (kH/kD, 1H NMR) value equals 3.3, indicating that the C(sp2)–H cleavage might participate in the rate-determining step (Scheme 42).37 Besides these observations and the literature findings,30 a catalytic cycle similar to the above-mentioned one in Wang's study31 was proposed and is depicted in Scheme 43.
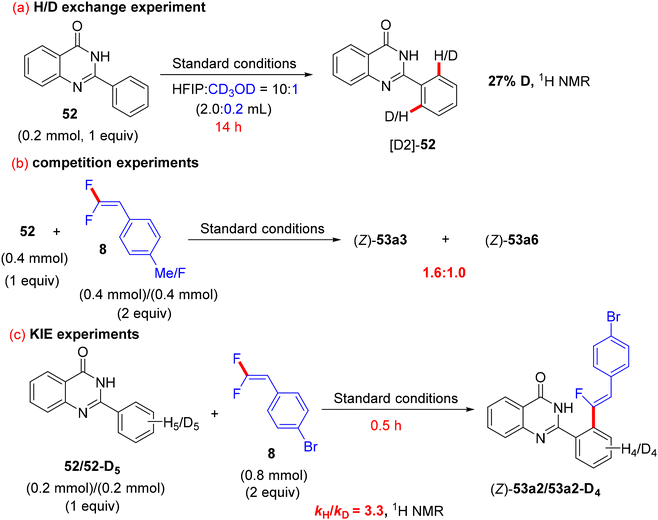 |
| | Scheme 42 Control mechanistic experiments of Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 52 with 8. | |
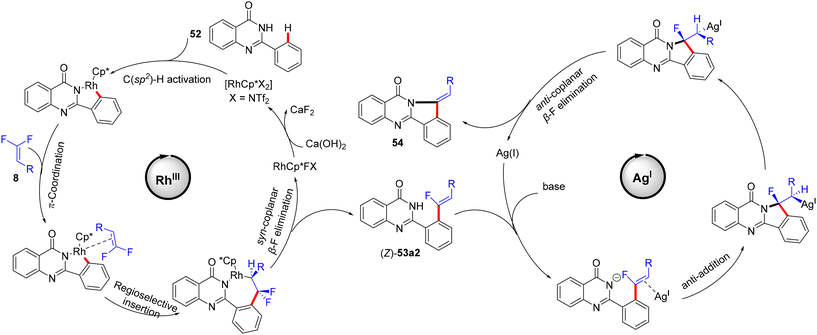 |
| | Scheme 43 Proposed catalytic cycle of Rh(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 52 with 8. | |
3.3. C–H α-Fluoroalkenylation using Ru(II)
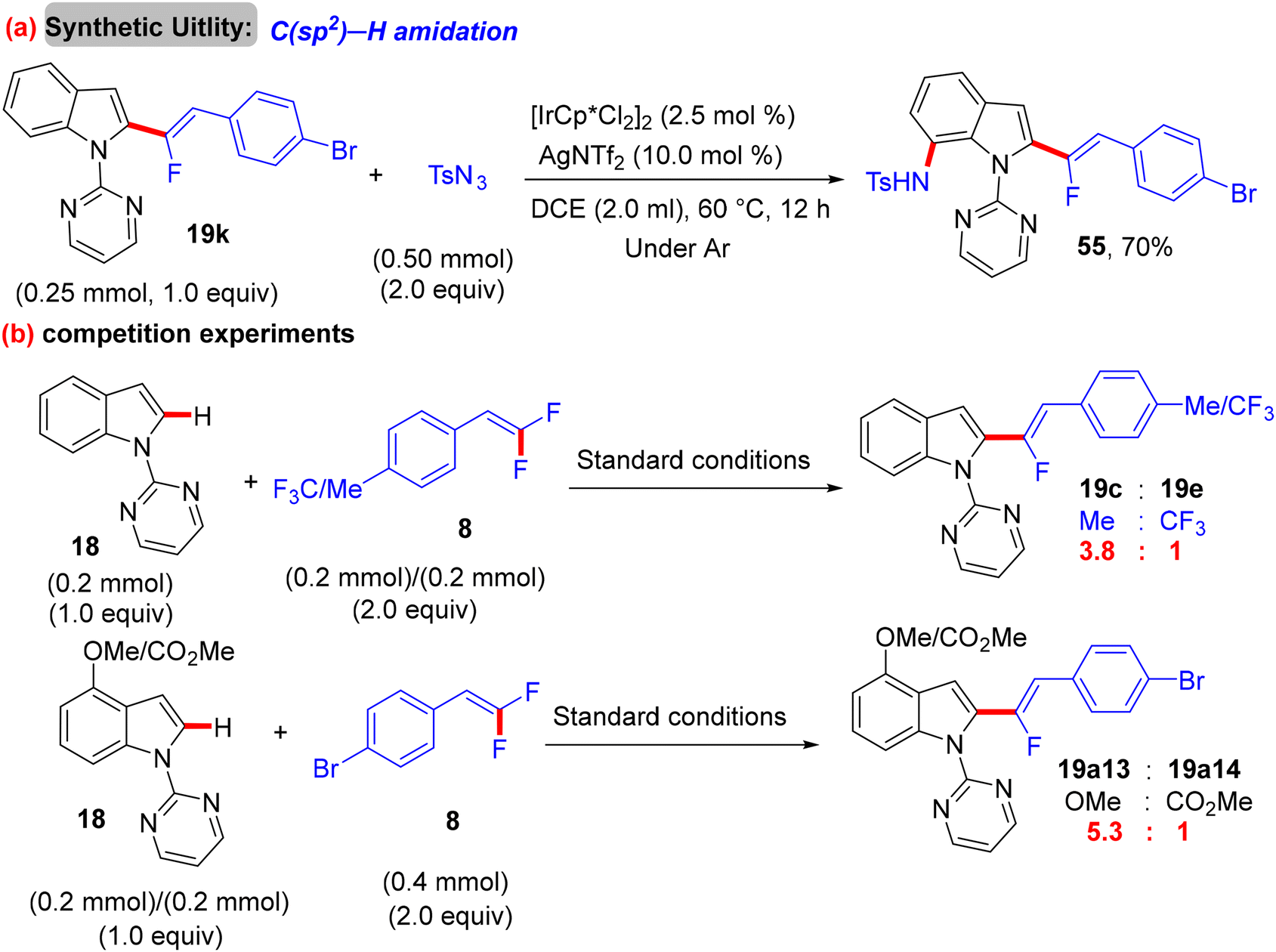
After Loh's study in 2015 on α-fluoroalkenylation of N-pyrimidinylindoles 18 by Rh(III)Cp* catalyst,29 Li's group conducted the same work with a “Ru(II) catalysis” for the first time ever.38 With the [Ru(p-cymene)Cl2]2 (5 mol%)/AgSbF6 (10 mol%)/Ca(OH)2 (2 equiv.) catalytic system, the desired product was not obtained, and after several trials, the optimum Ag-free conditions were demonstrated as follows: [Ru(p-cymene)Cl2]2 (5 mol%), Ca(OH)2 (2 equiv.), TFE (1 mL), stirring under argon at 100 °C for 12 h. To examine the applicability of this Ru(II) protocol, the scopes of 18 and olefinic reagent 8 were investigated, and the expected products (19a11–a32 and 21o–q) were isolated with good to excellent regio- and stereoselectivity (Scheme 44). Overall, the yield and the stereoselectivity (Z/E ratio) are lower than those in Loh's procedure,29 demonstrating the high efficiency of Rh(III)Cp* catalysis. As a synthetic application, compound 19k was subjected to LSF by treatment with sulfonyl azide (TsN3) in the presence of the [IrCp*Cl2]2 (2.5 mol%)/AgNTf2 (10.0 mol%) catalytic system to furnish the corresponding C(sp2)–H amidation product 55 (70% isolated yield, Scheme 45a). As in previous studies, competition experiments indicated that electron-rich substrates (18 and 8) are preferred and most effective (Scheme 45b). Moreover, the mechanistic pathway was proposed based on the aforementioned outcomes,30 and the Ru(II) mode is similar to the Rh(III) one (Scheme 46).
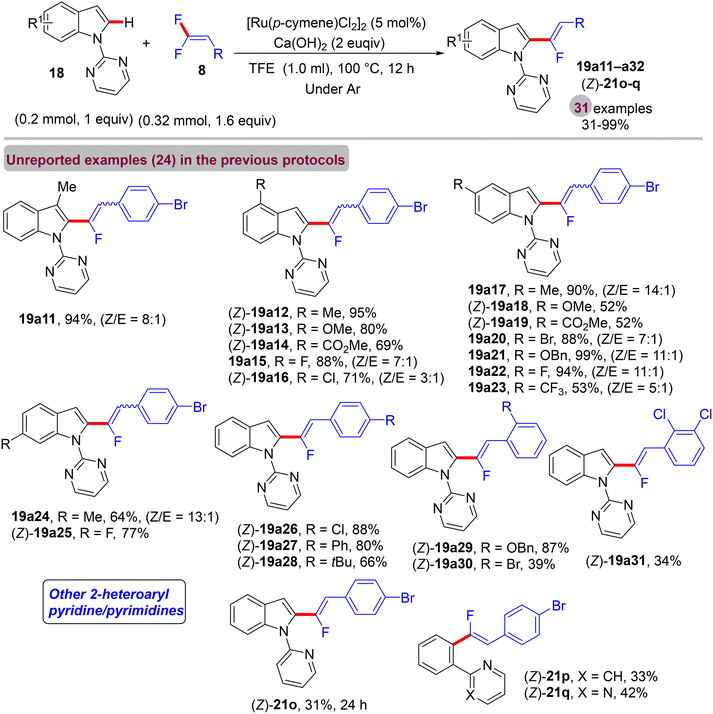 |
| | Scheme 44 Scope investigation in Ru(II)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18 with 8. | |
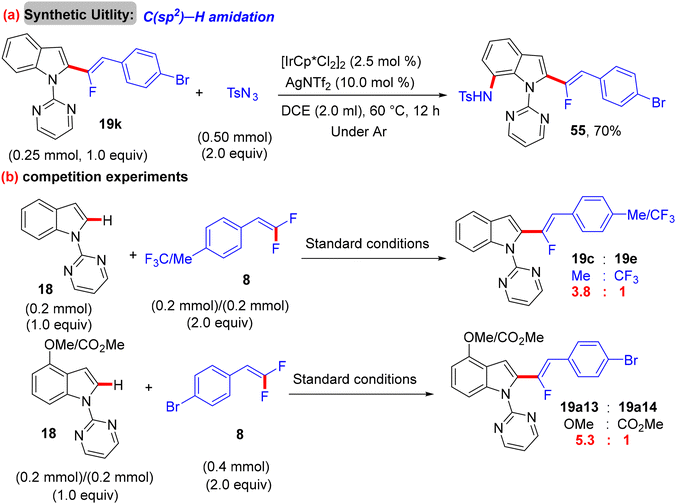 |
| | Scheme 45 Synthetic application and competition experiments of Ru(II)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18 with 8. | |
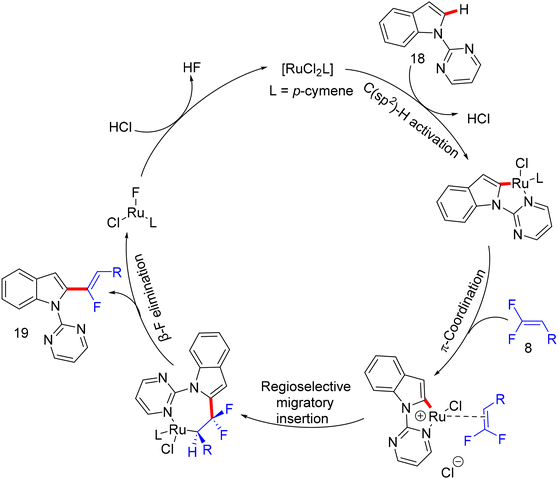 |
| | Scheme 46 Proposed catalytic cycle of Ru(II)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18 with 8. | |
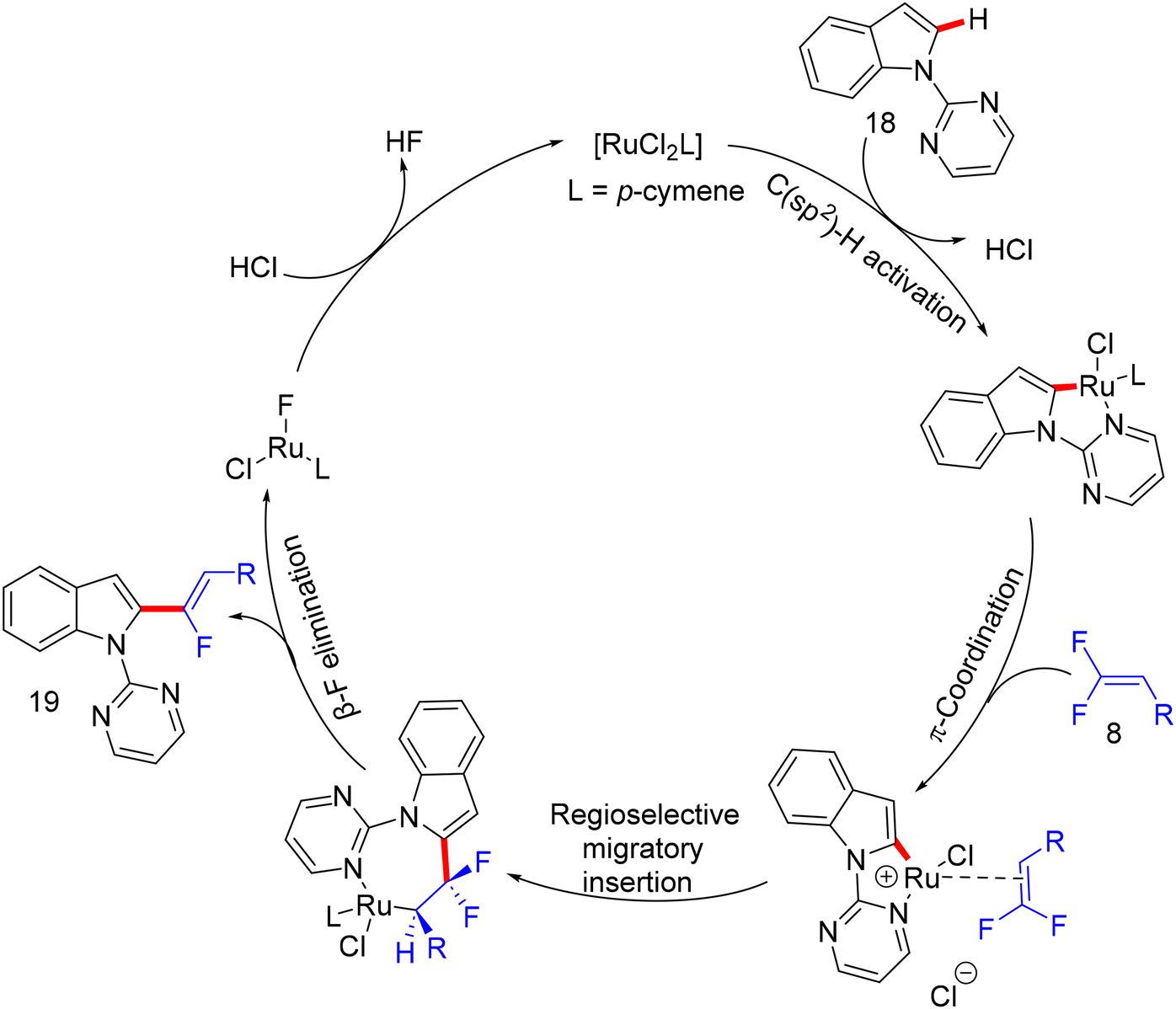
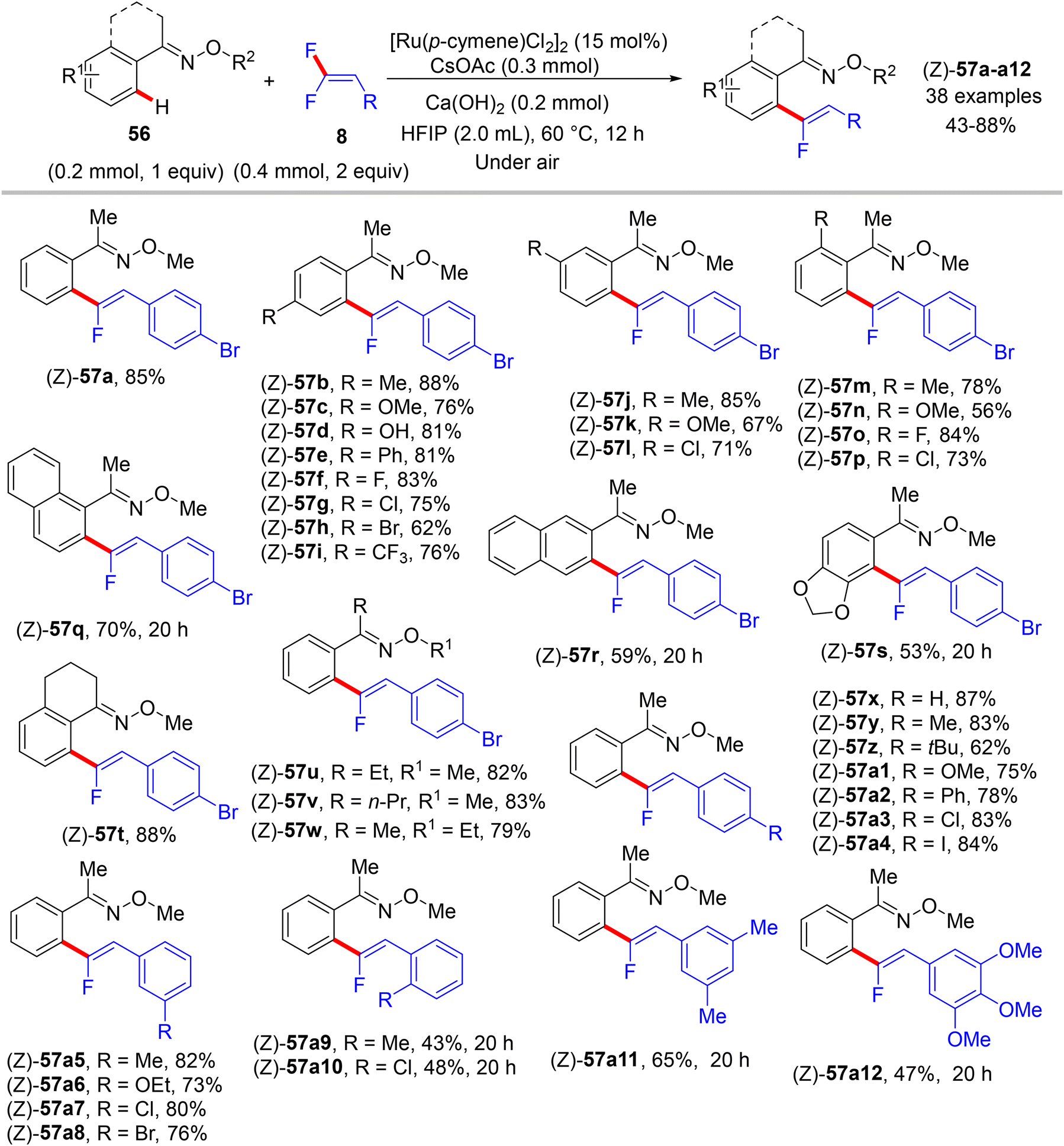
In 2020, an article also from Ji's lab illustrates the usage of the same prior Ru(II) catalyst for direct C(sp2)–H α-fluoroalkenylation of arene-containing oxime ethers 56 (as a removal ortho-directing group, RDG) with 8 to exhibit the expected monofluoroalkenes 57 and subsequent α-fluoroalkenylated acetophenones 58 after removing the oximyl ether DG.39 Additionally, this approach proceeds via C(sp2)–H functionalization and alkenyl C(sp2)–F cleavage in a (Z)-stereoselective manner with good functional group tolerance. The authors attempted to use [RhCp*Cl2]2 and Pd(OAc)2 as catalysts, respectively, but the expected product did not form. Next, they used the diamagnetic dimer [Ru(p-cymene)Cl2]2, and after several tests, they reached the optimal conditions as follows: catalyst (15 mol%), Ca(OH)2 (0.2 mmol), and CsOAc (0.3 mmol) in HFIP (2.0 mL) at 60 °C for 12 h. Under these conditions, the substrate scopes of 56 and 8 were investigated (Scheme 47). Like the previous protocols, para- and meta-substituted substrates are more favorable than their ortho counterparts, proving the importance of the substituent steric effect. Changes in the alkyl group of the oximyl ether are also tolerated in this transformation, and their products were isolated in good yields (57t–w, 79–88%).
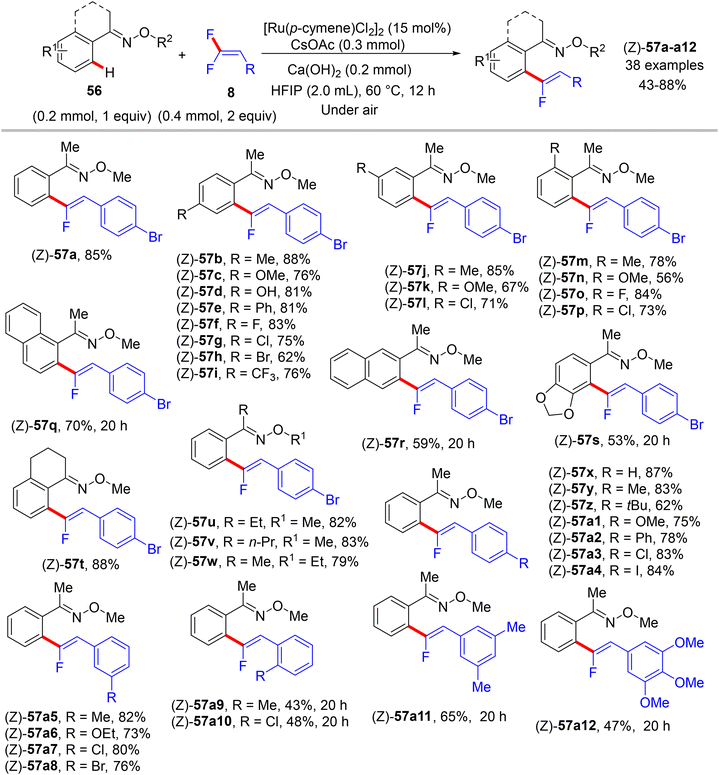 |
| | Scheme 47 Scope investigation in Ru(II)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 56 with 8. | |
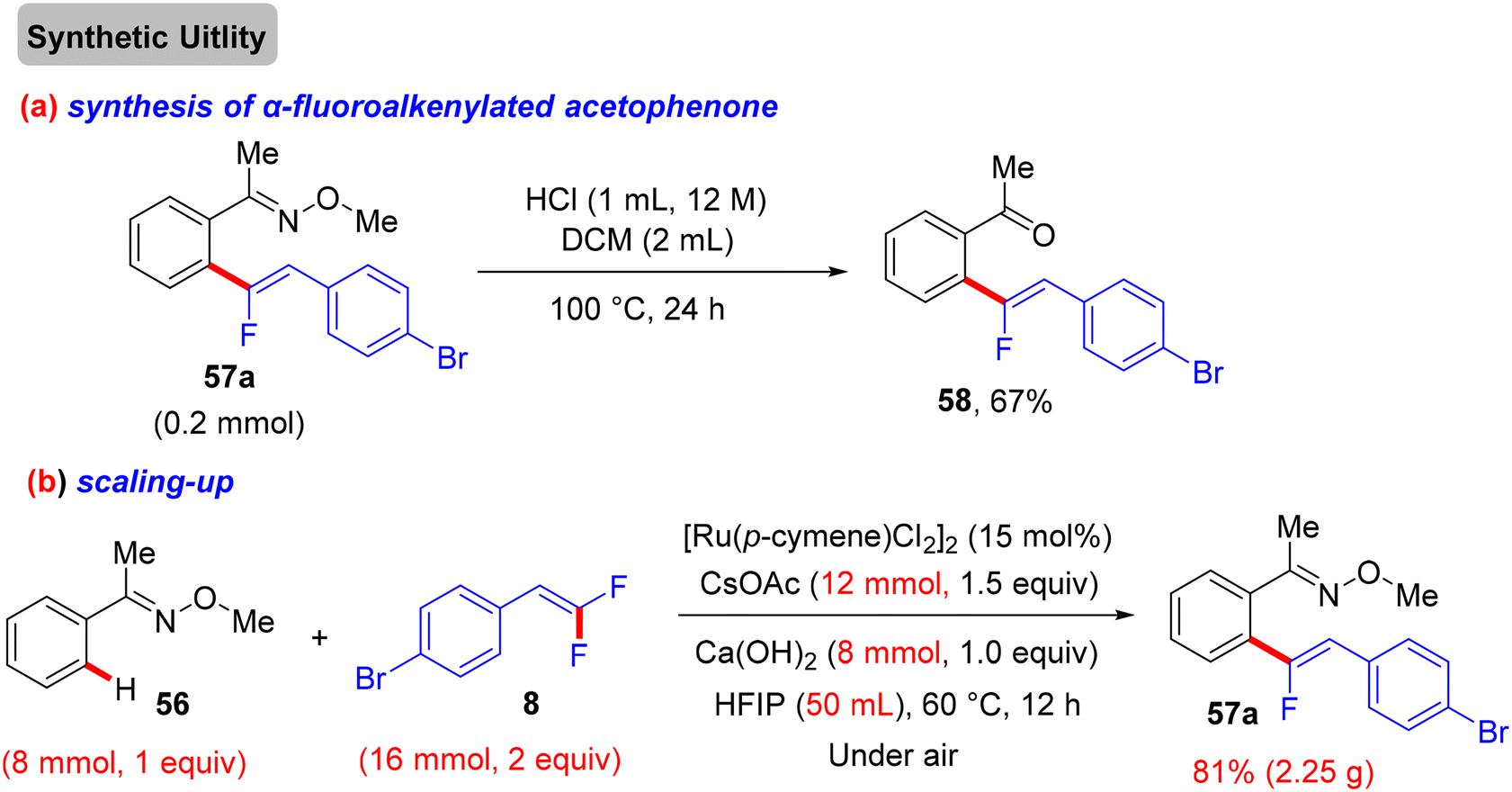
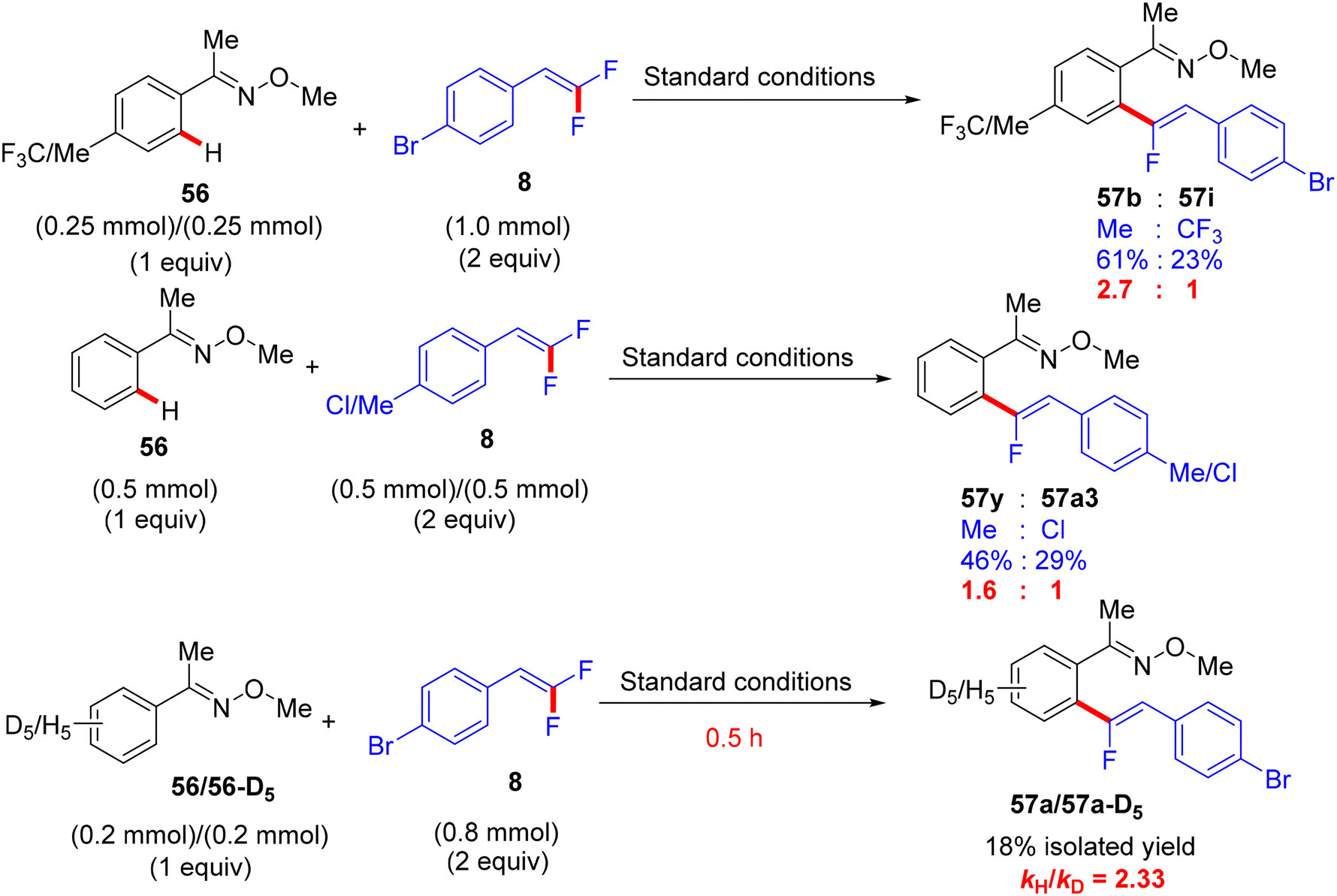
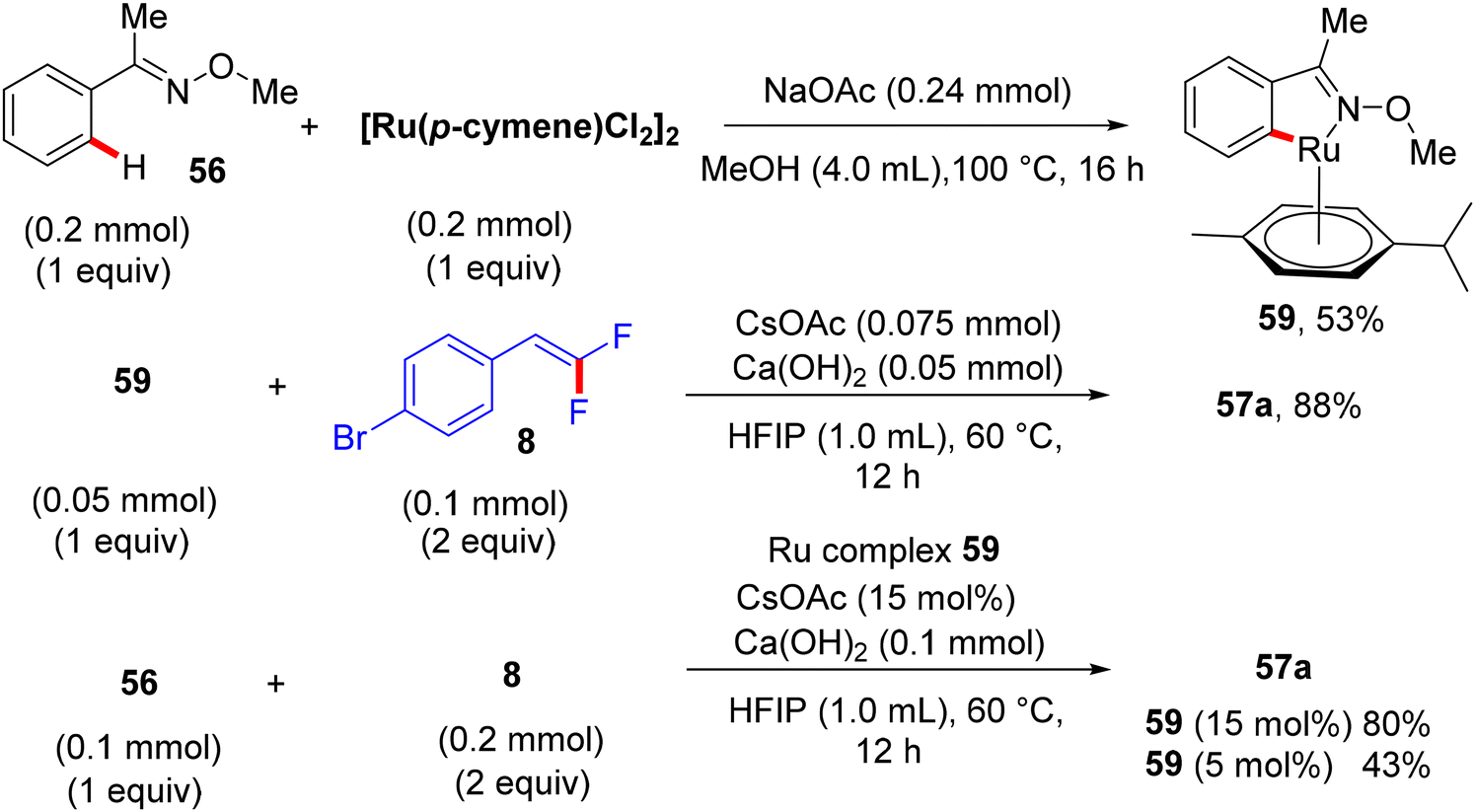
Synthetic applications of this protocol were demonstrated by the authors, where acetophenone derivative 58 was obtained in 67% isolated yield via the removal of the DG by acidic hydrolysis with HCl (12 M) in DCM at 100 °C. In addition, gram-scale synthesis of 57a was achieved, as illustrated in Scheme 48. Most importantly, several control experiments were performed to support the proposed mechanism (Scheme 49). Firstly, the intermolecular kinetic competition experiments of electronically biased 56 and 8 indicated that the EDG on both reactants facilitated the transformation. Secondly, KIE experiments of 56 and 56-D5 with two equivalents of 8 afforded 57a/57a-D5 with a kH/kD value of 2.33 (1H NMR), suggesting that the activation of the C(sp2)–H bond could be rate-determining. Finally, the isolation of cyclometallated Ru complex 59 (Scheme 50) and its successful usage in stoichiometric and catalytic reactions confirm its active role in the catalytic cycle (Scheme 51), which is similar to the previous one.38
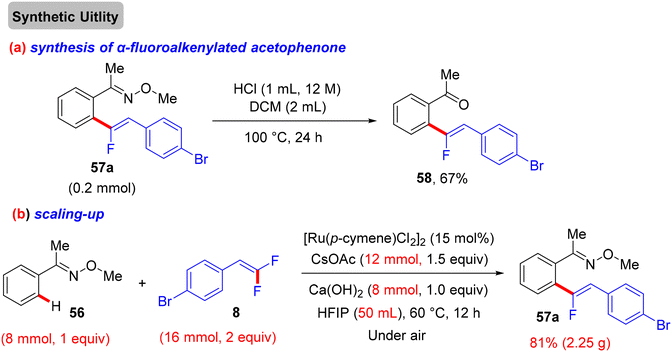 |
| | Scheme 48 Chemical synthesis of acetophenone derivative 58 and gram-scale formation of 57a. | |
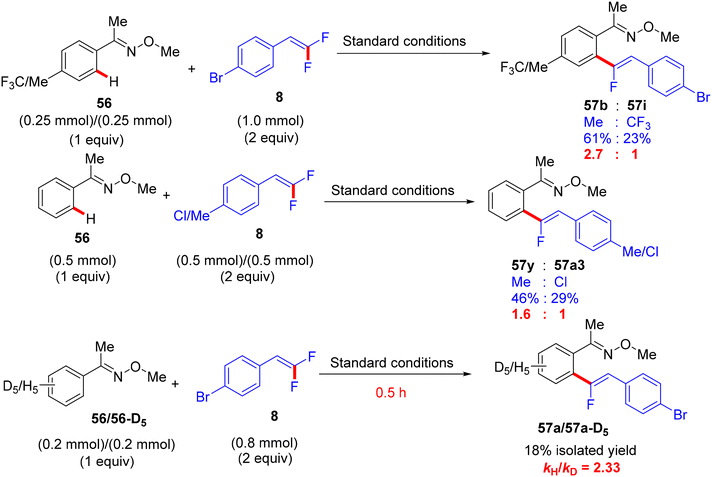 |
| | Scheme 49 Competition experiments of Ru(II)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 56 with 8. | |
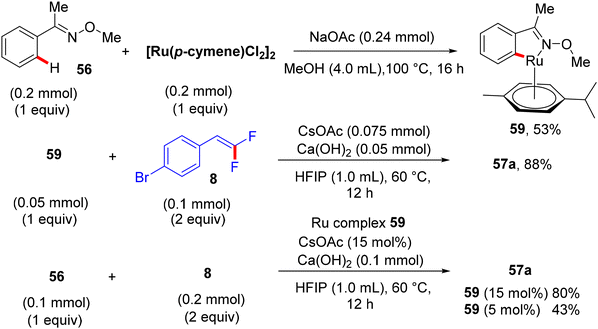 |
| | Scheme 50 Synthesis and α-fluoroalkenylation of intermediate Ru complex 59. | |
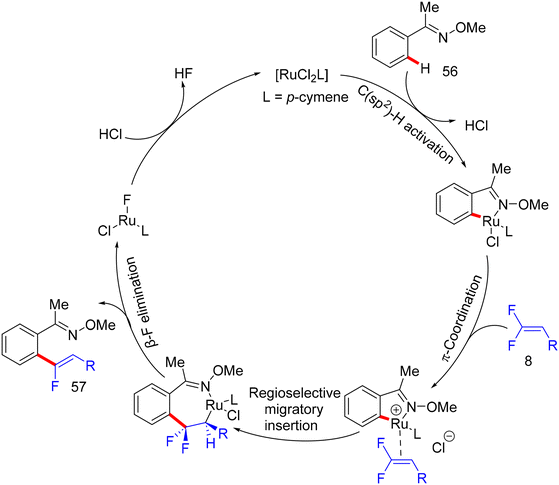 |
| | Scheme 51 Proposed catalytic cycle of Ru(II)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 56. | |
3.4. C–H α-Fluoroalkenylation using Co(III)Cp*
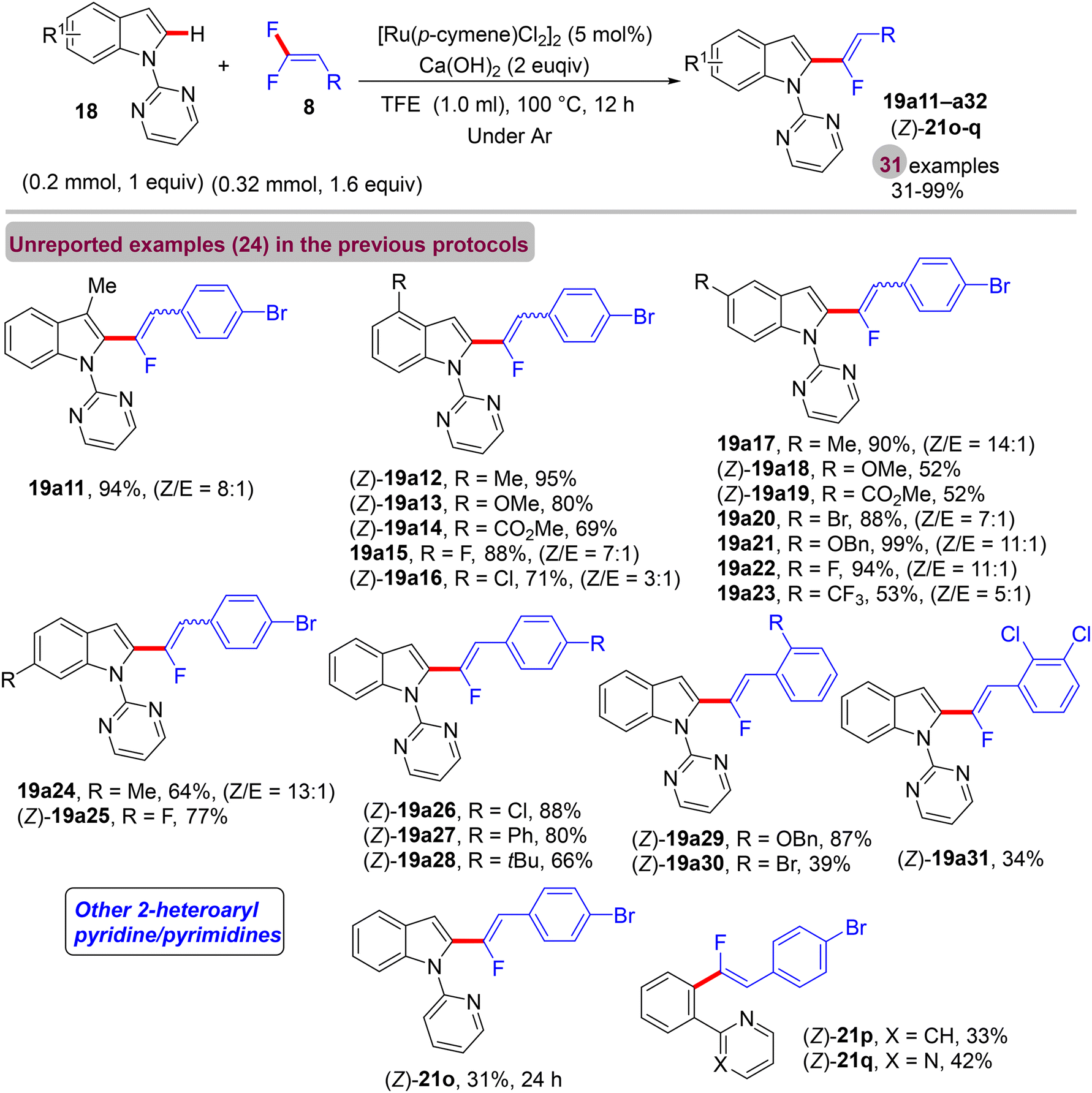
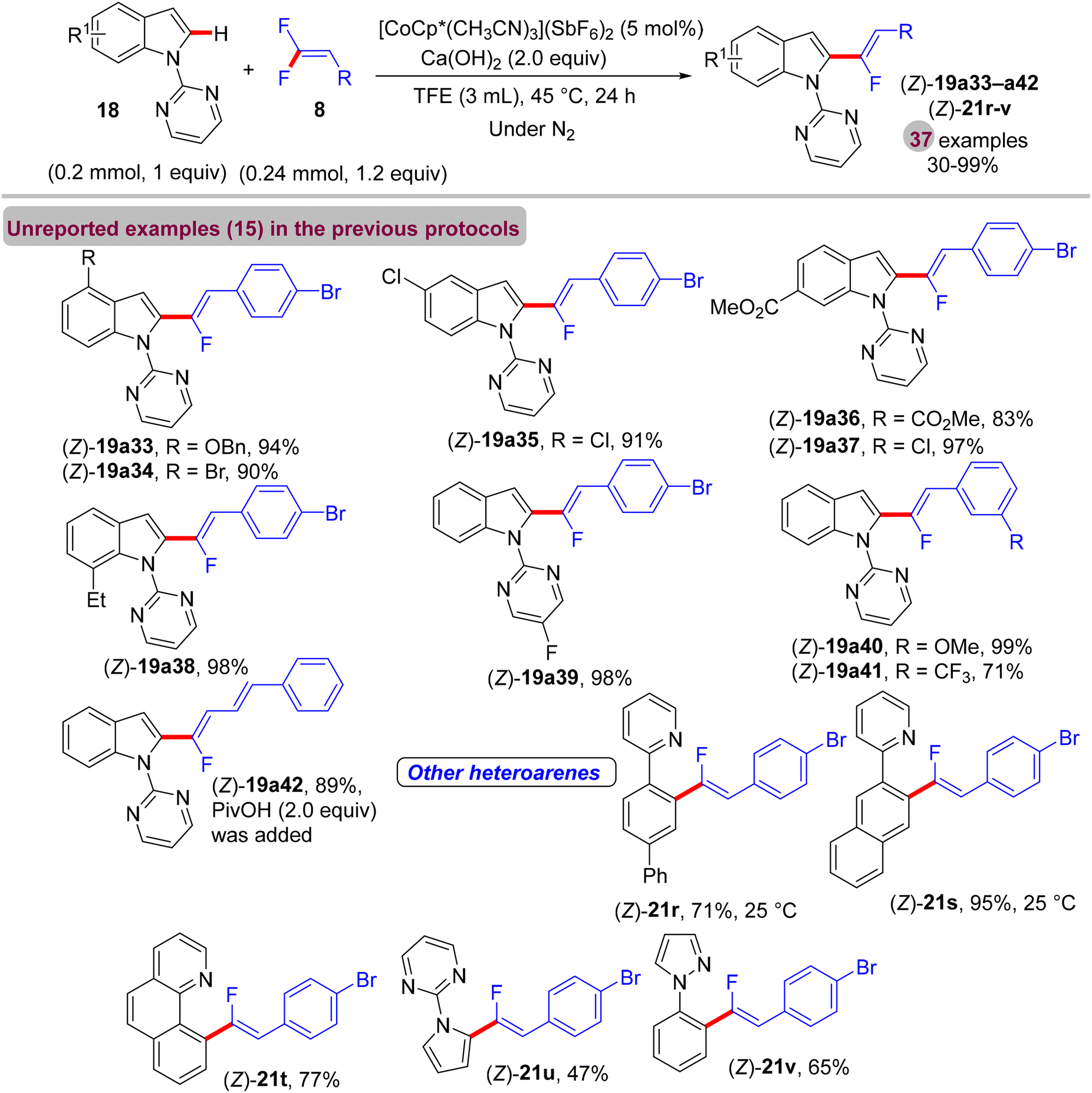
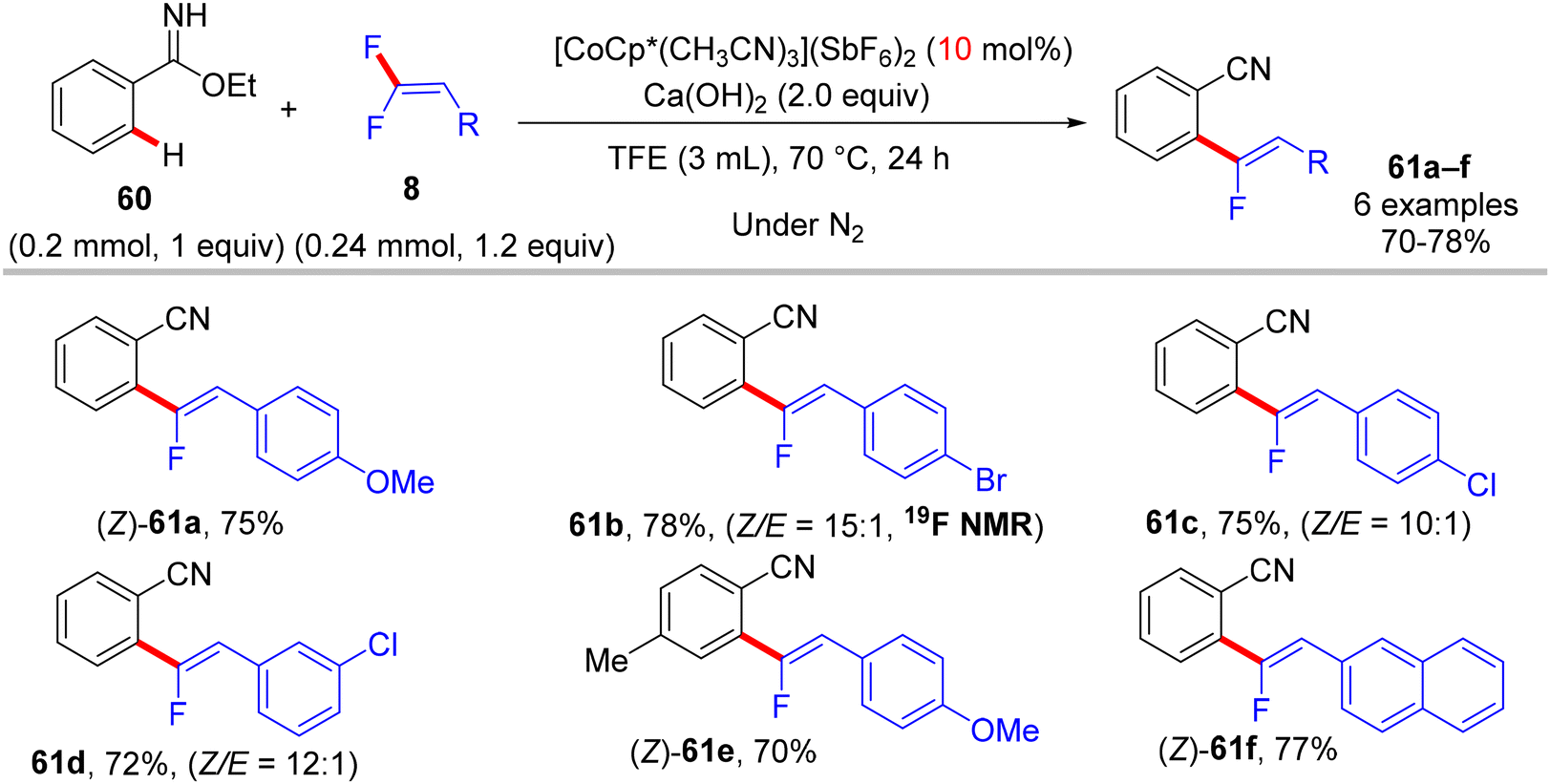
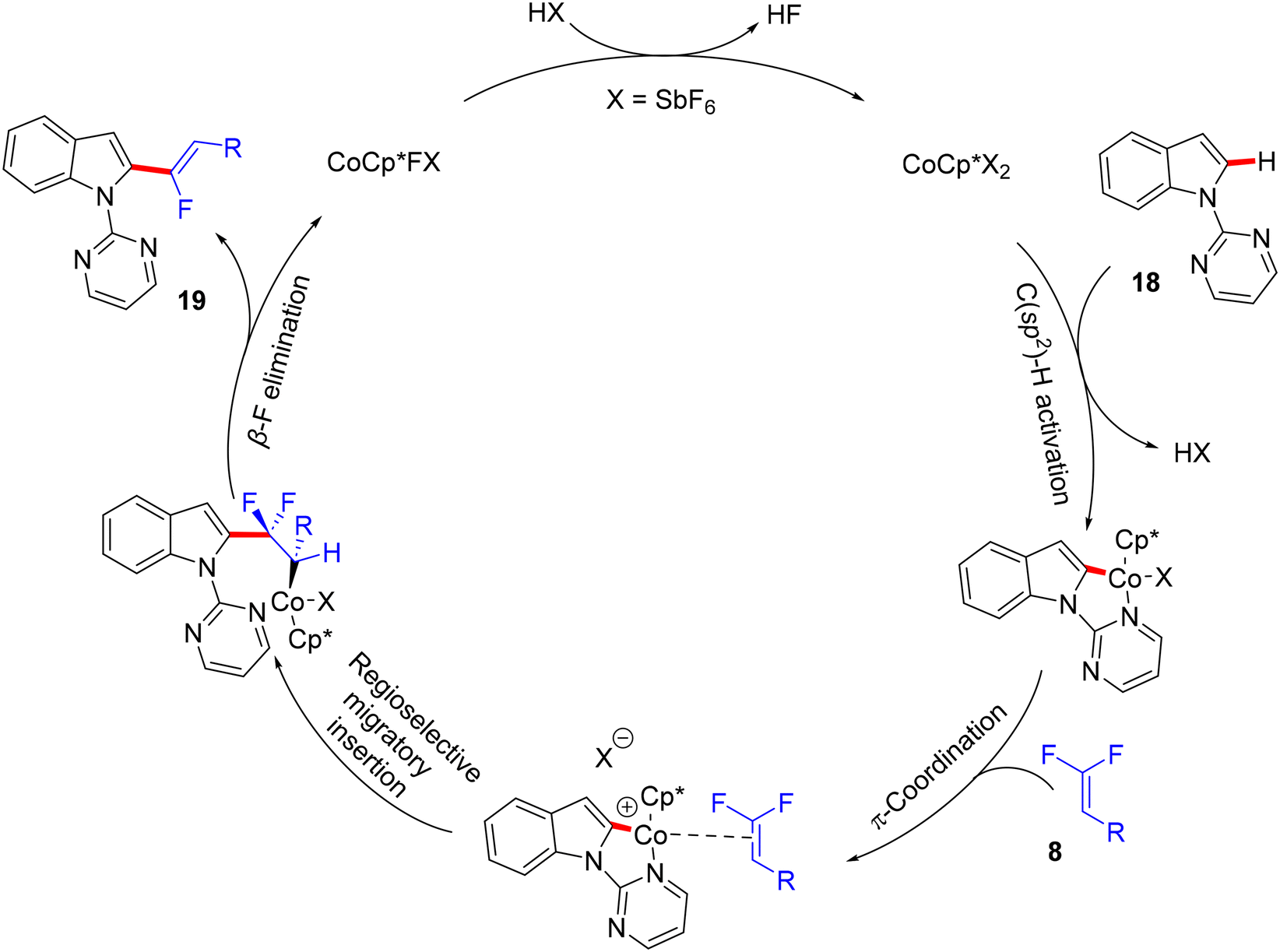
Before their work on the direct C(sp2/3)–H α-fluoroalkenylation of heteroarenes by Rh(III)Cp* and Ru(II) catalysts in 2017 and 2018,30,34,35,38 in 2016 the Li research group40 tried to use Co(III)Cp*-based catalysis for conducting the same reaction as a low-cost alternative to Loh's procedure with Rh(III) in 2015.29 In their preliminary investigation, they used 10 mol% of [CoCp*(CO)I2]63,64 and 20 mol% of AgNTf in TFE solvent (1.5 mL) at 80 °C in the presence of an additive, mostly Ca salts. The desired monofluoroalkenes were obtained in 61–95% isolated yield, but with poor stereoselectivity (Z/E ratio = 1.6–3.7/1, 19F NMR). Among the employed HF scavenger Ca salts, Ca(OH)2 was the best choice and was mostly used after that in similar studies as shown above. Moreover, they switched to the cobalt analogue of [RhCp*(CH3CN)3](SbF6)2, which was utilized in Loh's study.29 The optimal conditions were settled as follows: [CoCp*(CH3CN)3](SbF6)2 (5 mol%) and Ca(OH)2 (2.0 equiv.) in TFE (3 mL) at 45 °C for 24 h, improving the isolated yields and enhancing the stereoselectivity (Z/E > 20/1). As in Loh's protocol,2918 was chosen as a model substrate, and the study was also extended to other nitrogenous-directed substrates. The scope of 18 and 8 to access the corresponding (Z)-monofluoroalkenes (19a33–a42) was investigated (Scheme 52). The scope of the other heteroarenes (20 afforded 21r–v) that are assisted by pyridine, pyrimidine, or pyrazole is also demonstrated. Under the conditions of this procedure, substrate-based ethyl benzimidates 6065 afforded the corresponding benzonitrile monofluoroalkenes 61a–f in good yields (Scheme 53), demonstrating the carboximidate unit as a transformable transient directing group (TDG).62,66 As shown in Scheme 54, the authors achieved the gram-scale synthesis of 19k and the KIE experiment (kH/kD = 1.7 1H NMR, 20/20-D5 gave 21p/21p-D5), which indicated the C–H activation step is not the rate-determining step in agreement with their aforementioned experimental and theoretical studies on gem-difluorovinyl tosylate (25) and N-OMe benzamides 27.30 The Co(III)Cp* catalytic cycle is also similar to the previous Rh(III)Cp* and Ru(II) ones (Scheme 55).
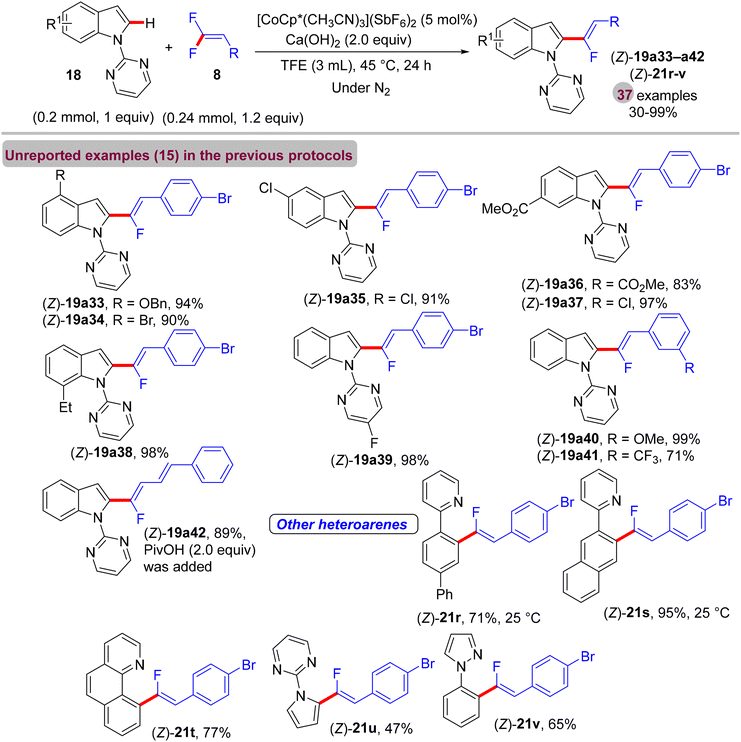 |
| | Scheme 52 Scope investigation in Co(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18/21 by 8. | |
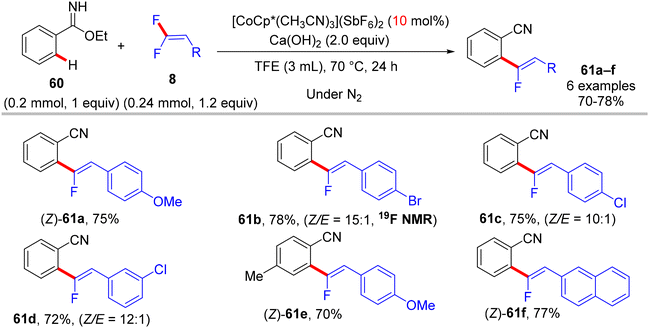 |
| | Scheme 53 Scope of 60 in Co(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation with 8. | |
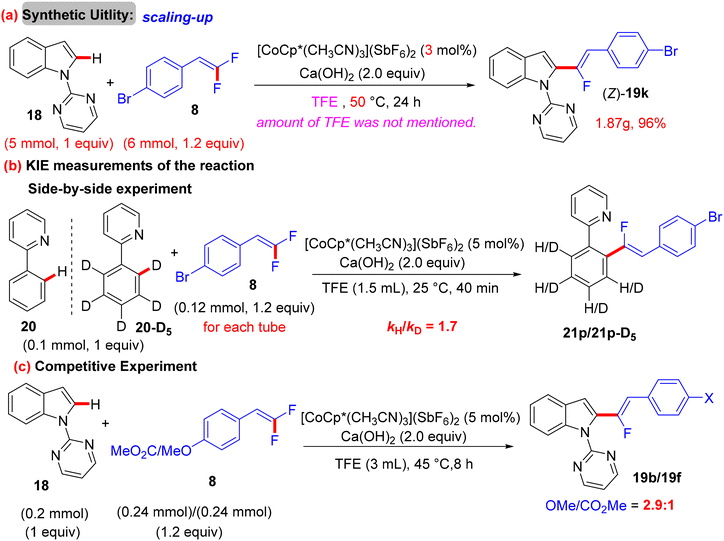 |
| | Scheme 54 Gram-scale synthesis of 19k, KIE, and competitive experiments of Co(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18/20 with 8. | |
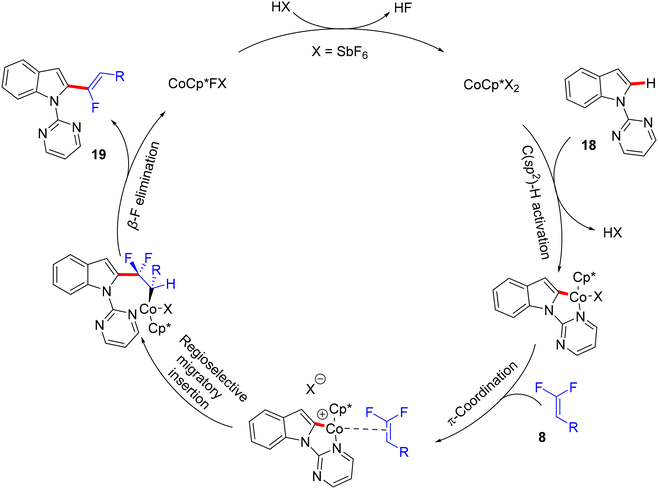 |
| | Scheme 55 Proposed catalytic cycle of Co(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18. | |
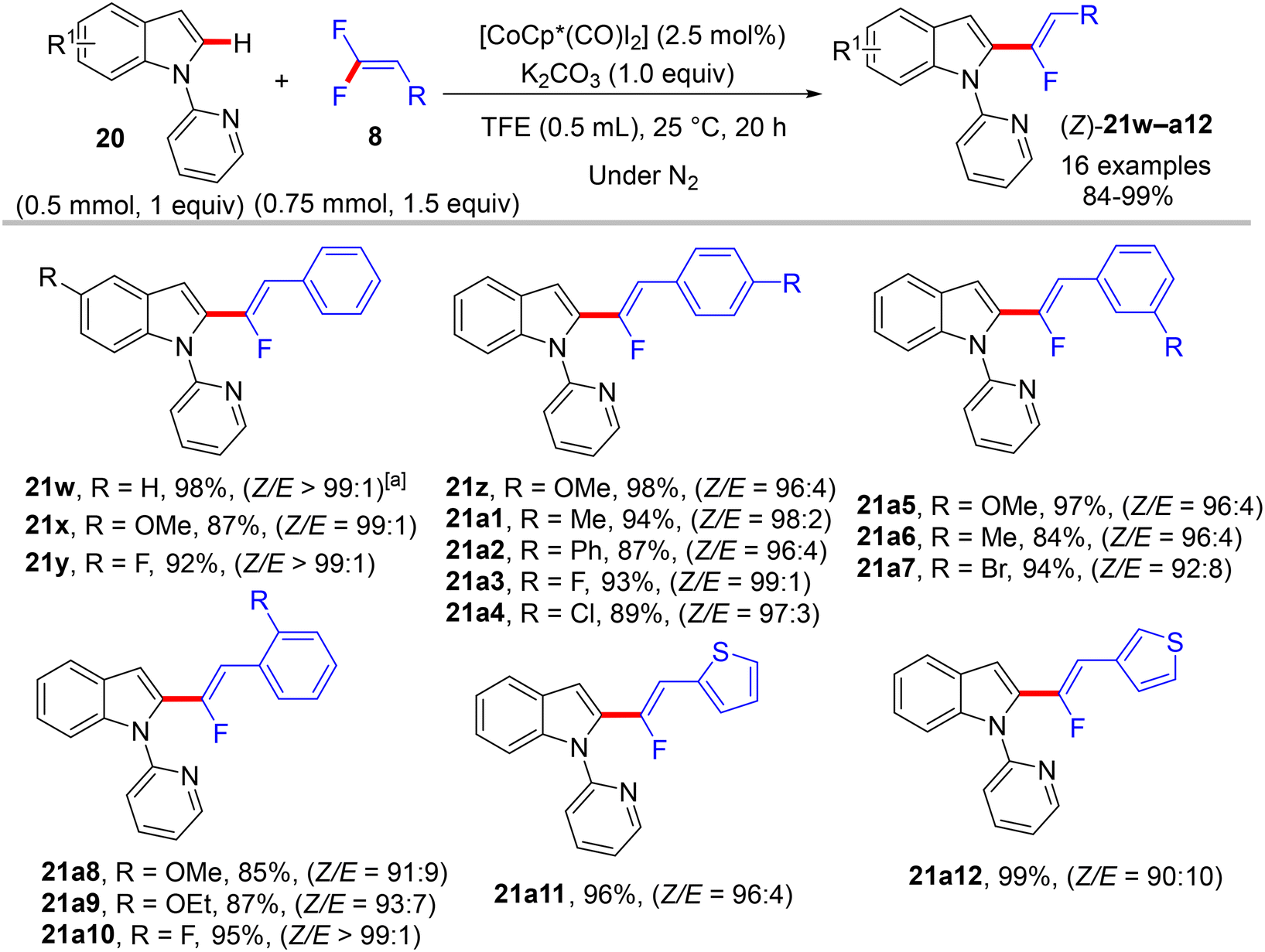
One year later, Ackermann et al. reported the first sustainable room-temperature direct C(sp2)–H α-fluoroalkenylation of N-pyridinylindoles 20 (as 2-heteroaryl pyridines) by 8, employing a low catalyst loading of [CoCp*(CO)I2] and mild base (K2CO3). Optimization studies of this silver-free transformation generated the optimum conditions as follows: [Co(III)] (2.5 mol%), K2CO3 (1.0 equiv.), and TFE solvent (0.5 mL) at 25 °C for 20 h under nitrogen atmosphere to afford the expected monofluoroalkenes 21w–a12 in high to excellent yields (Scheme 56) with a Z/E ratio between 90:10 and 99:1. The stereoselectivity findings were confirmed by computational studies that indicated the (Z)-selective route is energetically favored owing to steric repulsion in the E counterpart.41
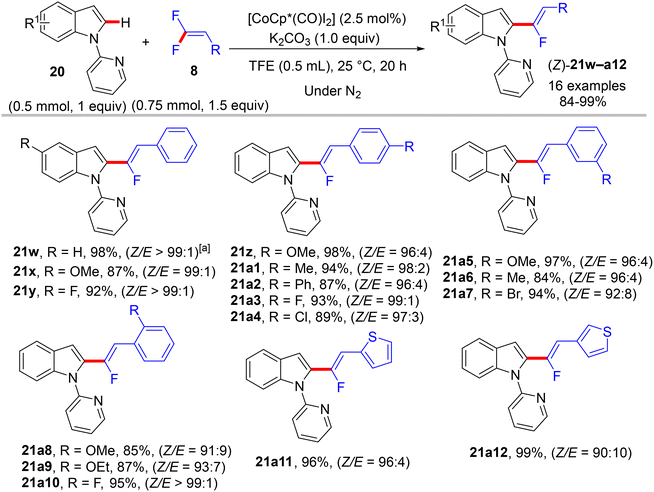 |
| | Scheme 56 Scope investigation in Co(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of N-pyridinylindoles (20) with 8. | |
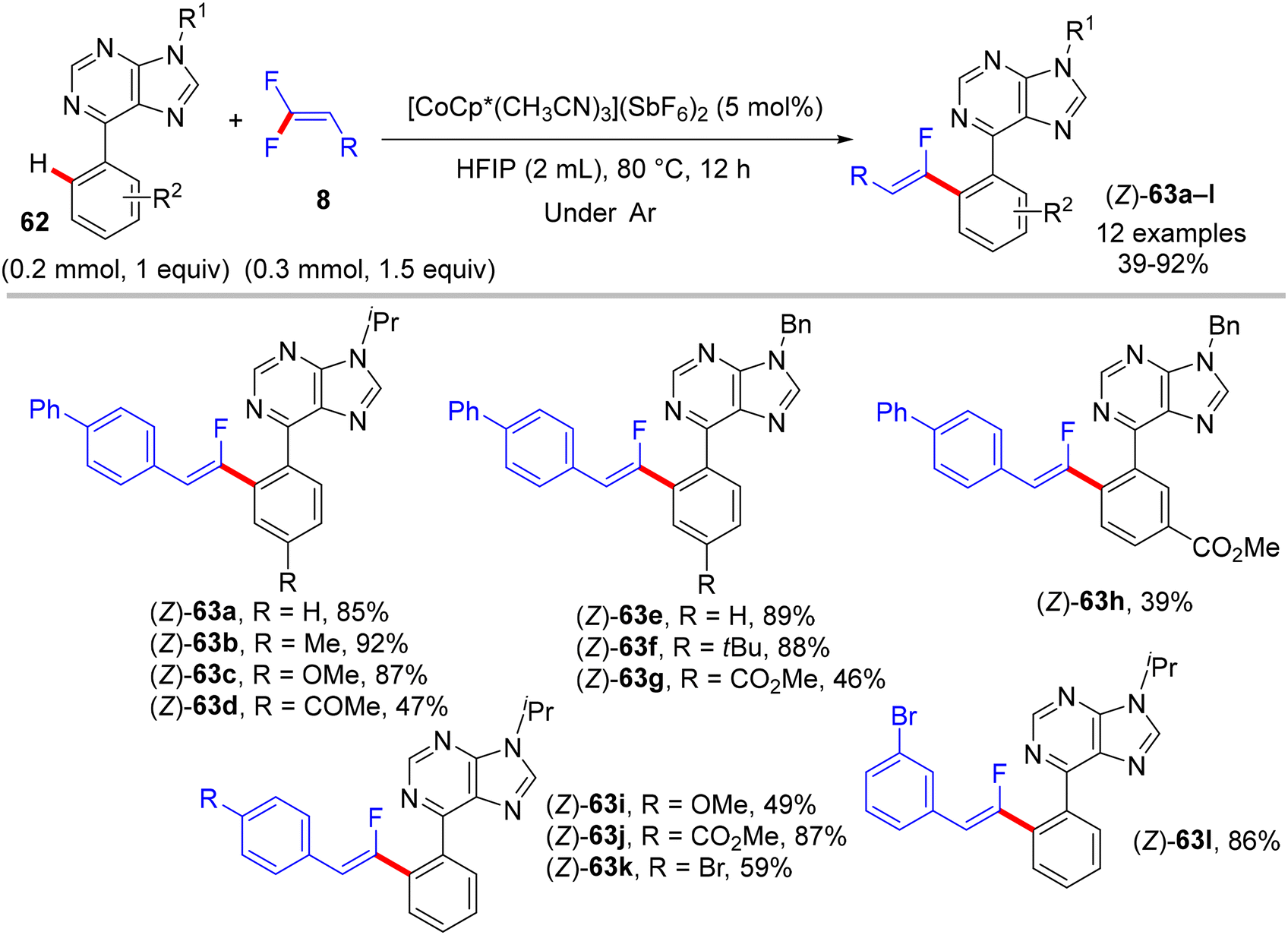
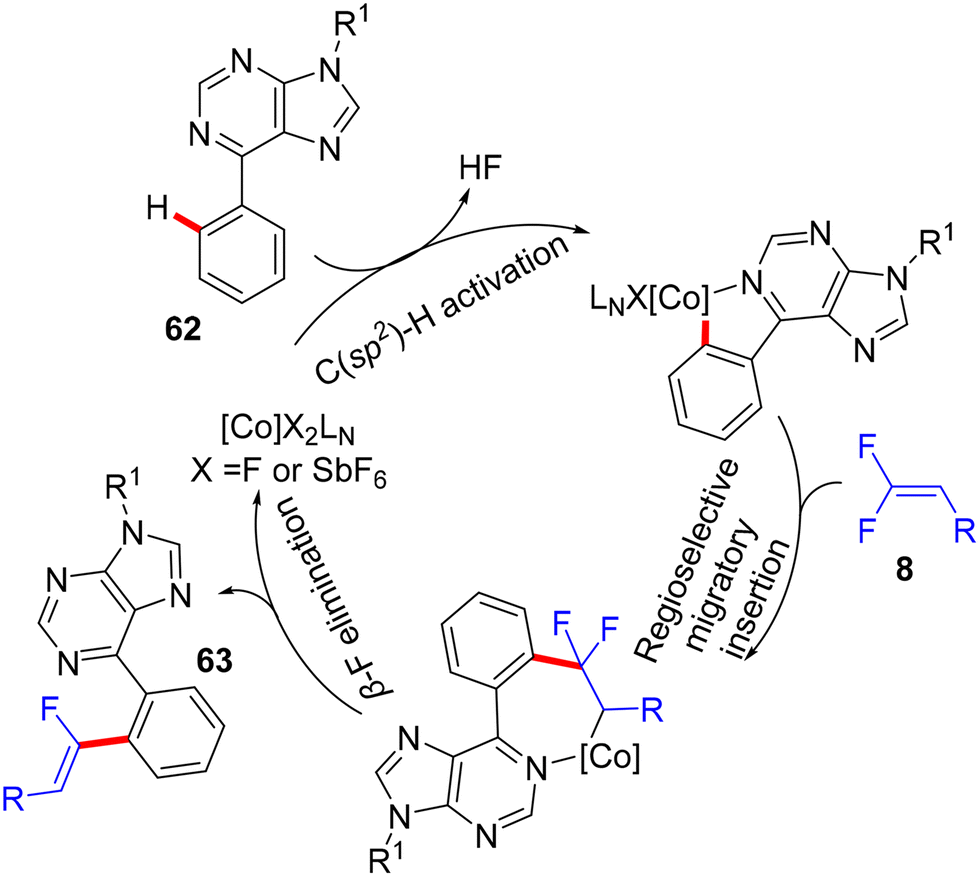
In 2018, fluoroalkenylation this time came from Hokkaido University, Japan, where the Matsunaga research group42 used the same Co(III) catalyst as the Xingwei Li study,40 but via Loh's methodology,29 base- and additive-free. They performed direct C(sp2)–H α-fluoroalkenylation of bioactive67 and versatile substrate-containing PDG62 6-arylpurines 62 using 8 in a high (Z)-stereoselective manner. The optimization stage was conducted on the model N-isopropyl-6-phenylpurine 62 with 8, and the result showed that the single-component [CoCp*(CH3CN)3](SbF6)2 (5 mol%, 95%) was better than the CoCp*(CO)I2 (5 mol%)/AgSbF6 (10 mol%, 81%) catalytic system in the optimal alcoholic solvent HFIP (0.5 mL) at 80 °C. Scheme 57 shows the obtained products 63a–l in moderate to good yields, and Scheme 58 displays the catalytic cycle.
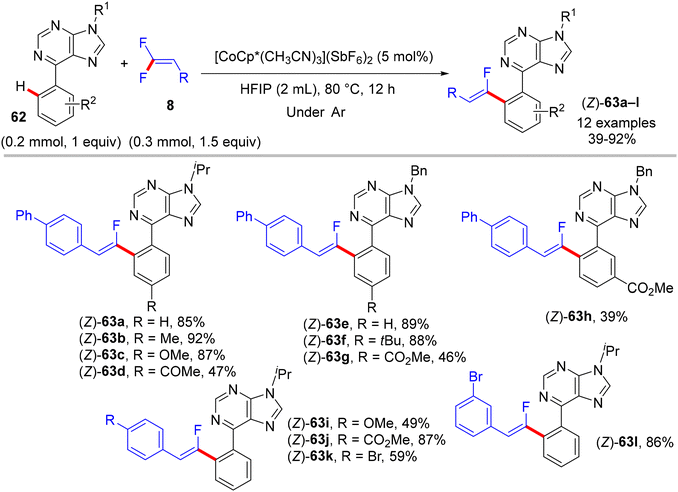 |
| | Scheme 57 Scope investigation in Co(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 62 with 8. | |
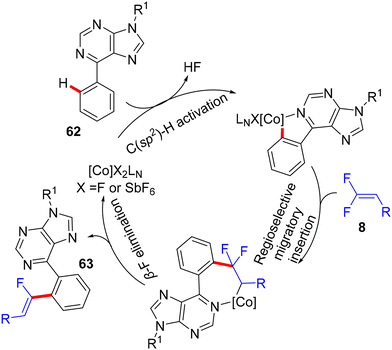 |
| | Scheme 58 Proposed catalytic cycle of Co(III)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 62. | |
3.5. C–H α-Fluoroalkenylation using Mn(0/I)
In May 2017, Ackermann et al. published a study that mimicked the previous one,41 but with a manganese(I)-based catalyst.43 Mn(I)-Catalyzed direct C(sp2)–H α-fluoroalkenylation of 20 by 8 to afford the corresponding monofluoroalkenes 21a13–a16 under these optimum conditions: [MnBr(CO)5] (10 mol%), NaOAc (20 mol%, an additive), K2CO3 (1.0 equiv.) and 1,4-dioxane solvent (0.5 mL) at 100 °C for 20 h. The desired products were installed in moderate to high yields (Scheme 59) with a Z/E ratio between 78:22 and 96:4, proving the robustness of the Co(III) protocol41 in contrast with this one.
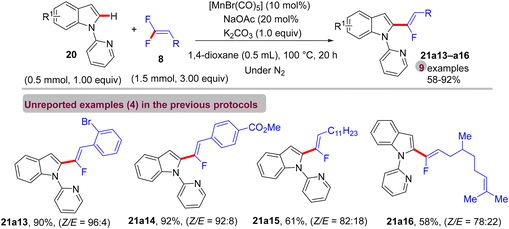 |
| | Scheme 59 Scope investigation in Mn(I)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 20 with 8. | |
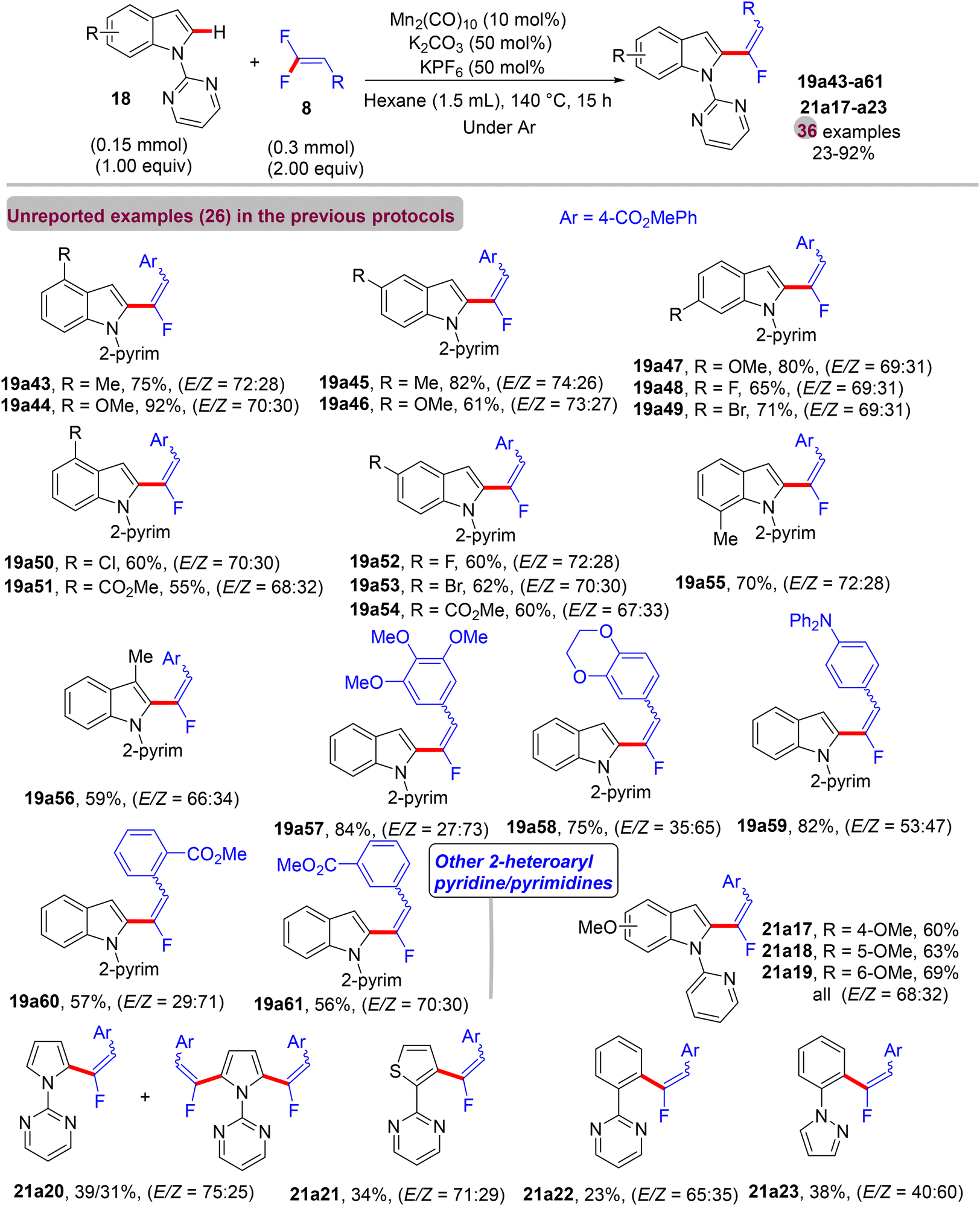
In contrast to the previous documented approaches that always showed the monofluoroalkenes in thermodynamically stable (Z)-stereoselectivity, in July 2017, a paper from Loh's lab described a redox-neutral direct C(sp2)–H α-fluoroalkenylation protocol to access mostly the E counterpart as the major products using an Mn(0)-based catalyst.44 The scope of 18/20 and 8 (Scheme 60) was investigated under the following optimal conditions: Mn2(CO)10 (10 mol%), K2CO3 base (50 mol%), and KPF6 additive (50 mol%) in hexane (1.5 mL) at 140 °C for 15 h. When this transformation was performed by the prior Ackermann's method43 with either [MnBr(CO)5] (10 mol%) or [Mn2(CO)10] (5 mol%), the (Z)-isomer was always the major product (92%) (Scheme 61a and b). Under the same conditions, albeit with hexane instead of 1,4-dioxane, the (E)-isomer percentage increased to 36–38% (Scheme 61c and d). Additionally, applying Ackermann's protocol,43 albeit with a Mn(0) catalyst and a catalytic amount of K2CO3 (50 mol%) at 140 °C for 15 h (mix between Mn protocols) in hexane provided the desired product with 43% isolated yield and an E/Z ratio of 75/25, while the reaction carried out in 1,4-dioxane gave no product (Scheme 61e and f), proving the key role of the solvent for the unusual E/Z selectivity in Loh's protocol.44
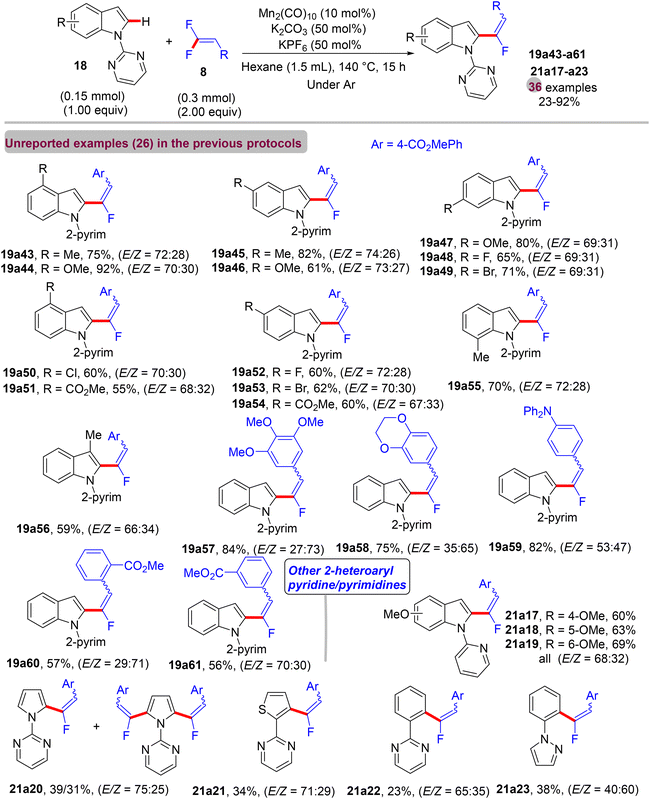 |
| | Scheme 60 Scope investigation in Mn(0)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18/20 with 8. | |
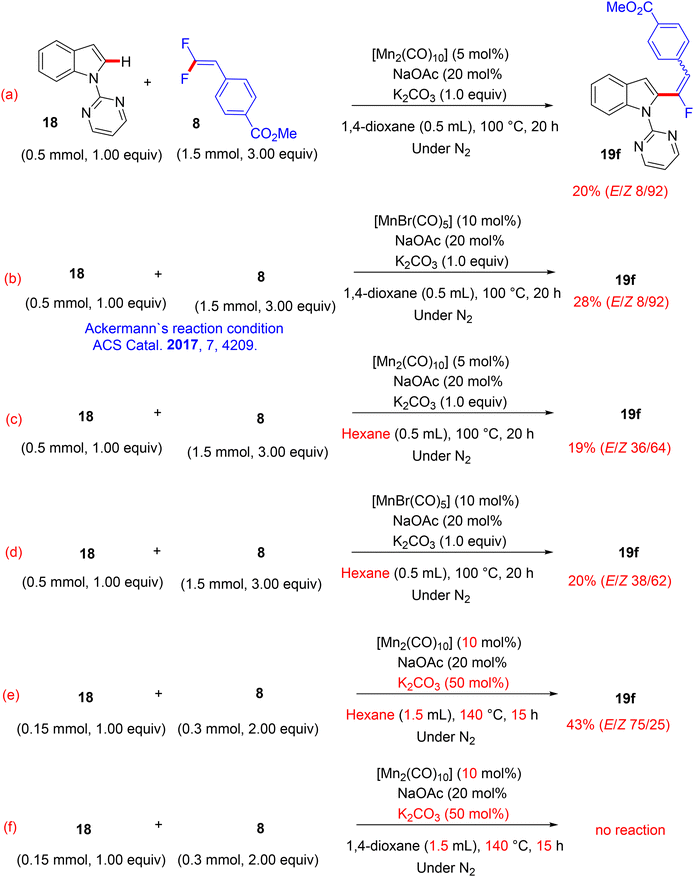 |
| | Scheme 61 Study of the E/Z selectivity of 19f in Mn(0)-catalyzed direct C(sp2)–H α-fluoroalkenylation of 18 with 8. | |
3.6. C–H α-Fluoroalkenylation using Fe(III)
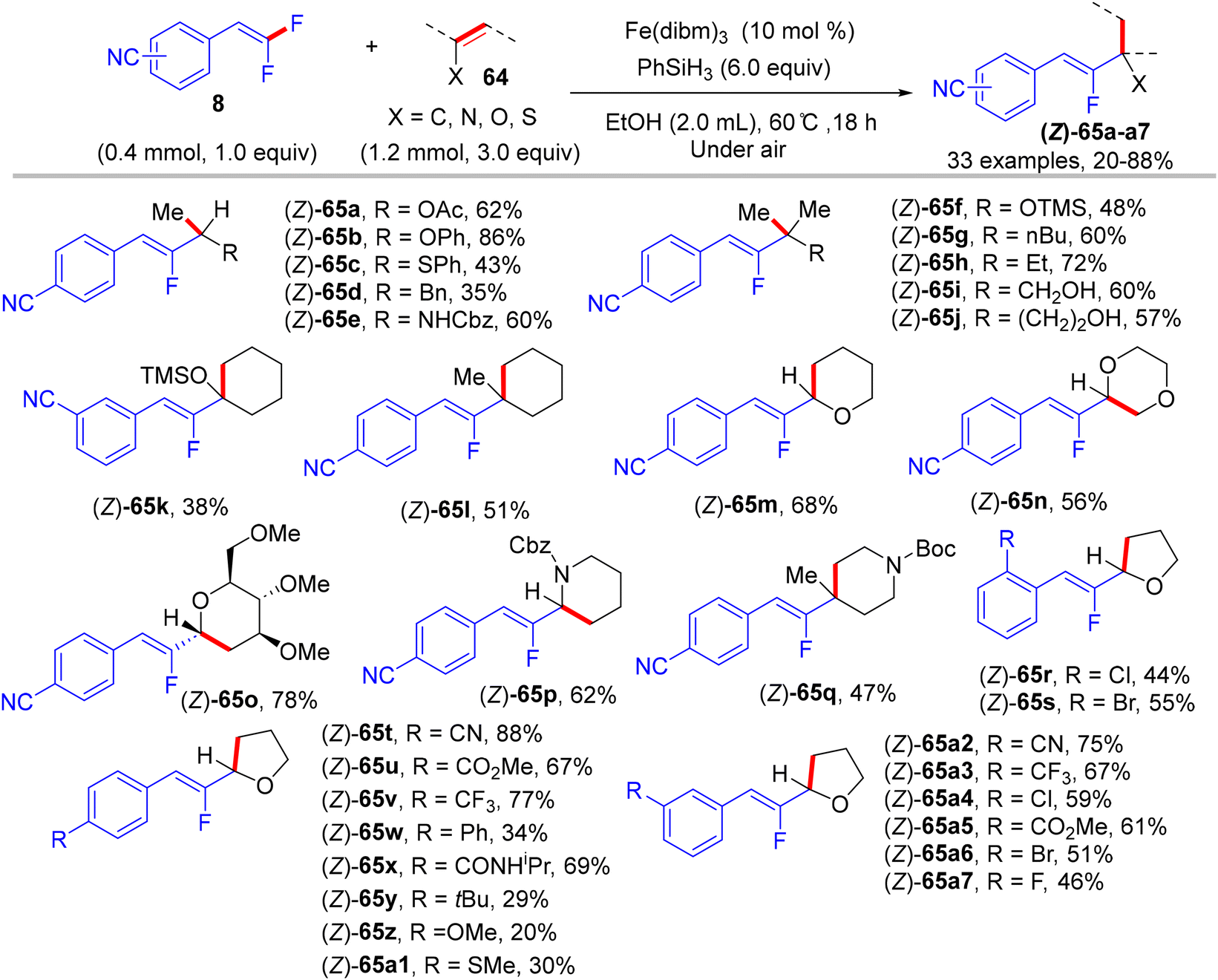
In 2018, Wang et al.'s paper documented the synthesis of alkylated monofluoroalkenes via a hydrofluoroalkenylation reaction, which was enabled by the Fe(III)/PhSiH3 catalytic system.45 This approach was a combination of the Fe(III)/PhSiH3-catalyzed hydrogen atom transfer (HAT) with the defluorinative cross-coupling strategy of various alkenes with 8. The authors hypothesized that the reaction proceeds through a free radical-based process with excellent (Z)-selectivity under air- and water-tolerant reaction conditions. Vinyl acetate and 4-cyano-gem-difluoroalkene 8 were employed for the model reaction, and the optimal conditions were obtained as follows: Fe(dibm)3 (10 mol%, dibm = diisobutyrylmethane) as a catalyst, and PhSiH3 (6.0 equiv.) as a hydrogen source in EtOH (2.0 mL) at 60 °C for 18 h under atmospheric conditions. Under the optimized conditions, a variety of alkenes, including vinyl ester, aryl vinyl ether, silyl enol ethers, endocyclic enol ethers, enecarbamates, vinyl thioether, and 1,1-disubstituted olefins, were efficiently tolerated and furnished the corresponding products (Z)-65a–a7 in 20–88% isolated yields (Scheme 62).45
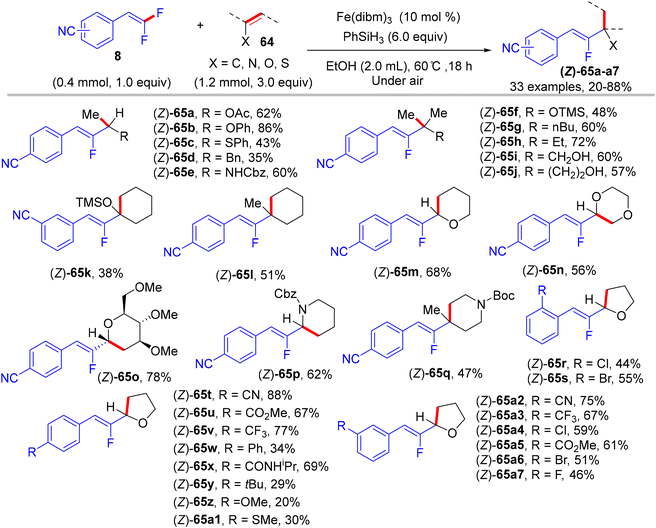 |
| | Scheme 62 Scope investigation in Fe(III)-catalyzed direct alkenyl C(sp2)–H α-hydrofluoroalkenylation of olefins (64) with 8. | |
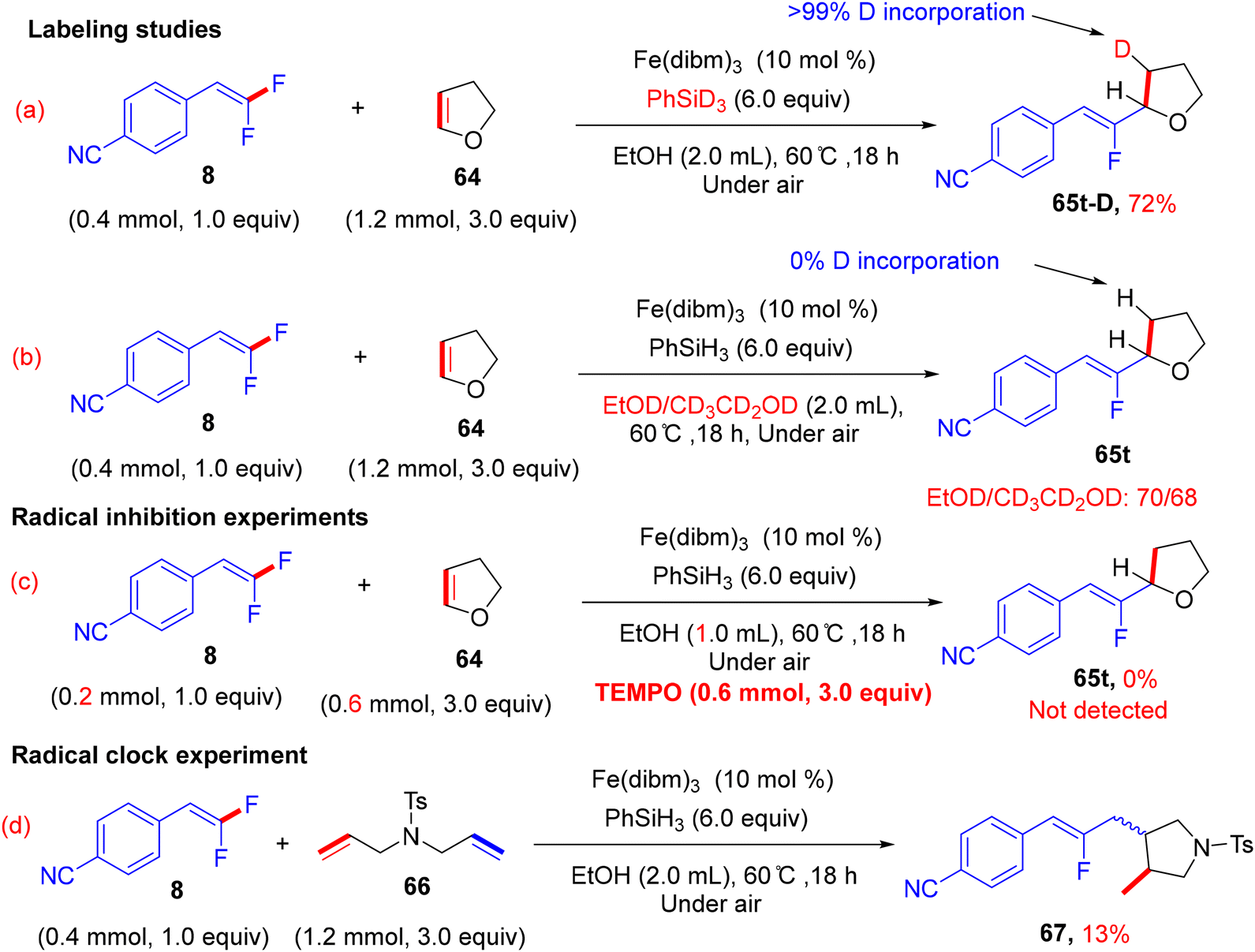
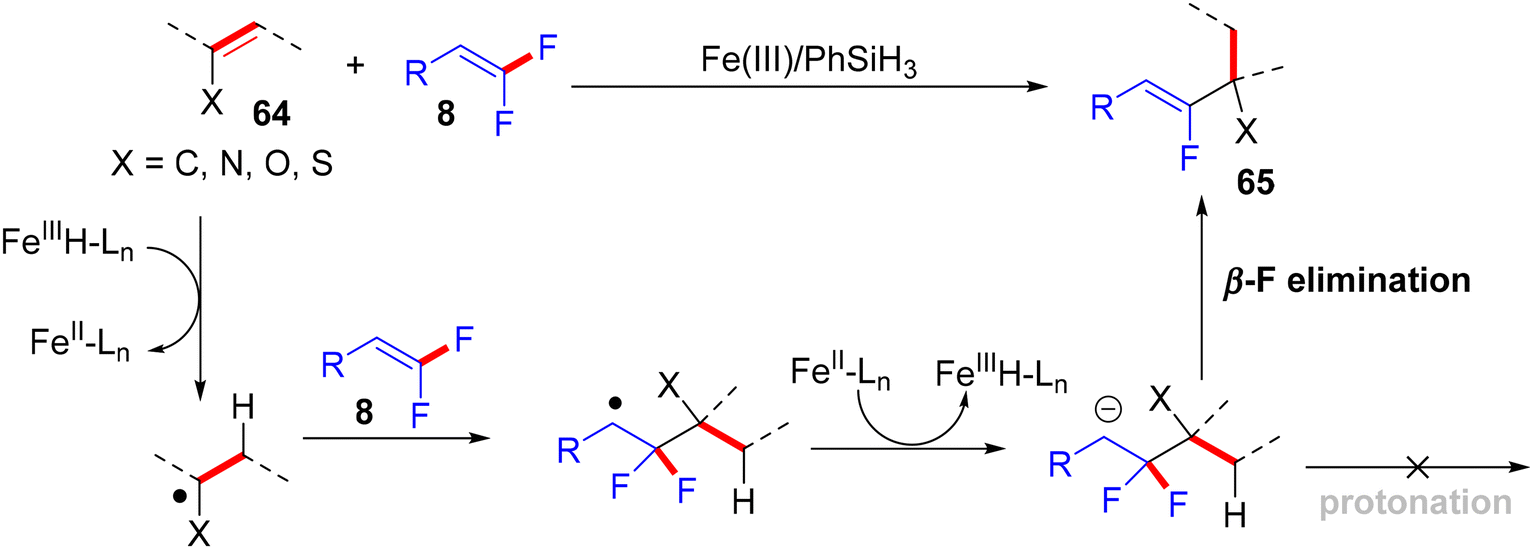
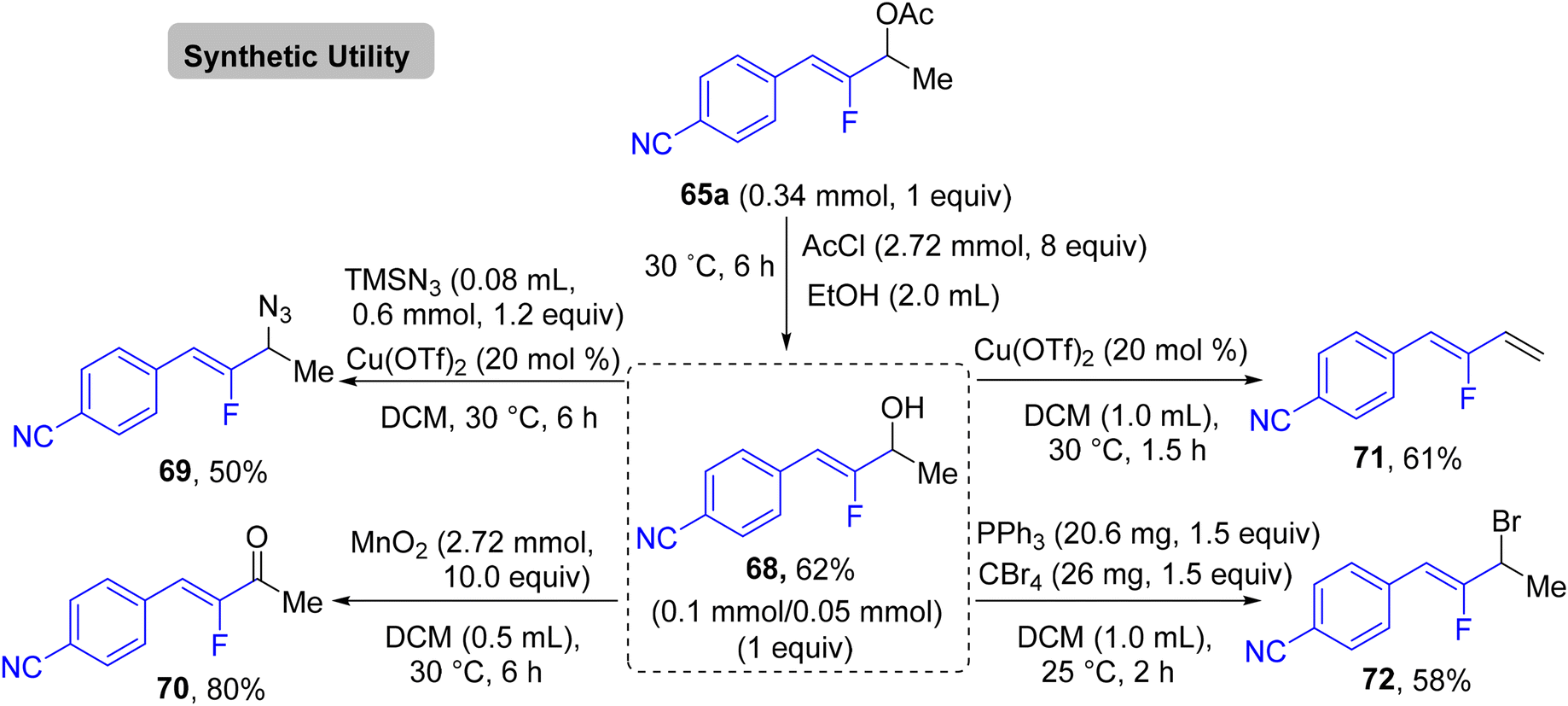
In addition, the authors performed some control experiments to gain more mechanistic insights into this transformation type, where they used PhSiD3 instead of PhSiH3, a deuterated adduct 65t-D was obtained (Scheme 63a),45 and when EtOH was changed to the deuterated analogues (EtOD or CD3CD2OD), the deuterium was not detected (Scheme 63b),45 suggesting that PhSiH3 is the sole source of hydrogen. Regarding the free radical as a hypothetical pathway, the reaction was completely inhibited when (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was utilized as a radical scavenger (Scheme 63c).45 Moreover, when N,N-diallyl-4-methylbenzenesulfonamide 66 was employed as a coupling partner, the corresponding cyclized monofluoroalkene 67 was formed (Scheme 63d).45 All these observations supported the depicted mechanism in Scheme 64,45 which illustrates two main stages, including HAT and β-F elimination rather than protonation by solvent, as reported in the literature.68–71 As a synthetic utility of this protocol, compound 65a was converted to the corresponding 2-fluoroallylic alcohol 68via the acidic hydrolysis of the acetyl group. Next, LSF of the OH group afforded some useful, structurally diverse fluorine-containing molecules (69–72) (Scheme 65).45
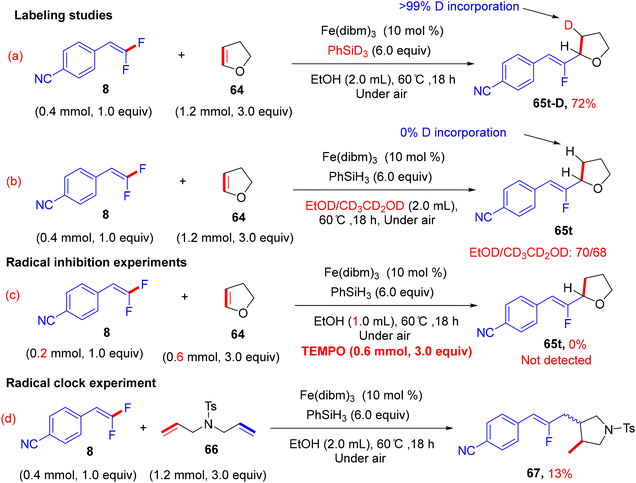 |
| | Scheme 63 Mechanistic studies of Fe(III)-catalyzed direct alkenyl C(sp2)–H α-hydrofluoroalkenylation of 64. | |
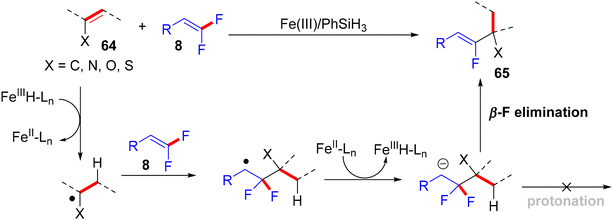 |
| | Scheme 64 Proposed mechanistic pathway of Fe(III)-catalyzed direct alkenyl C(sp2)–H α-hydrofluoroalkenylation of 64. | |
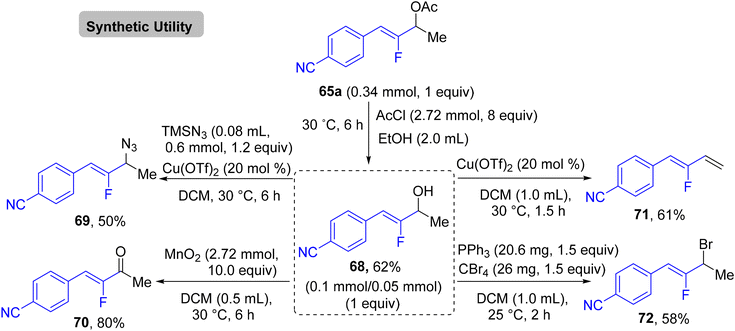 |
| | Scheme 65 Synthetic applications of α-hydrofluoroalkenylation product 65a to synthesize 68–72. | |
3.7. C–H α-Fluoroalkenylation using Cr(III)
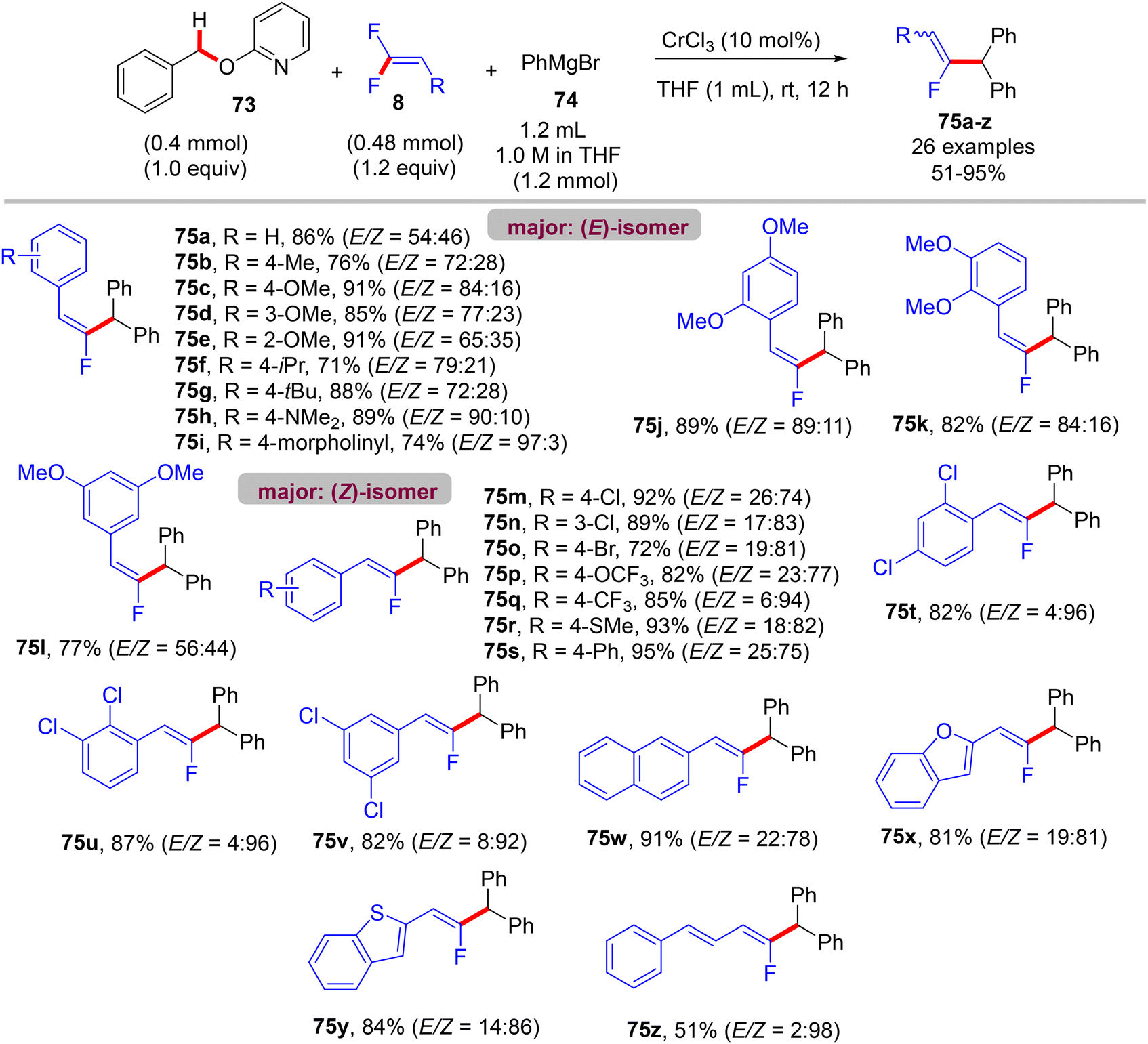
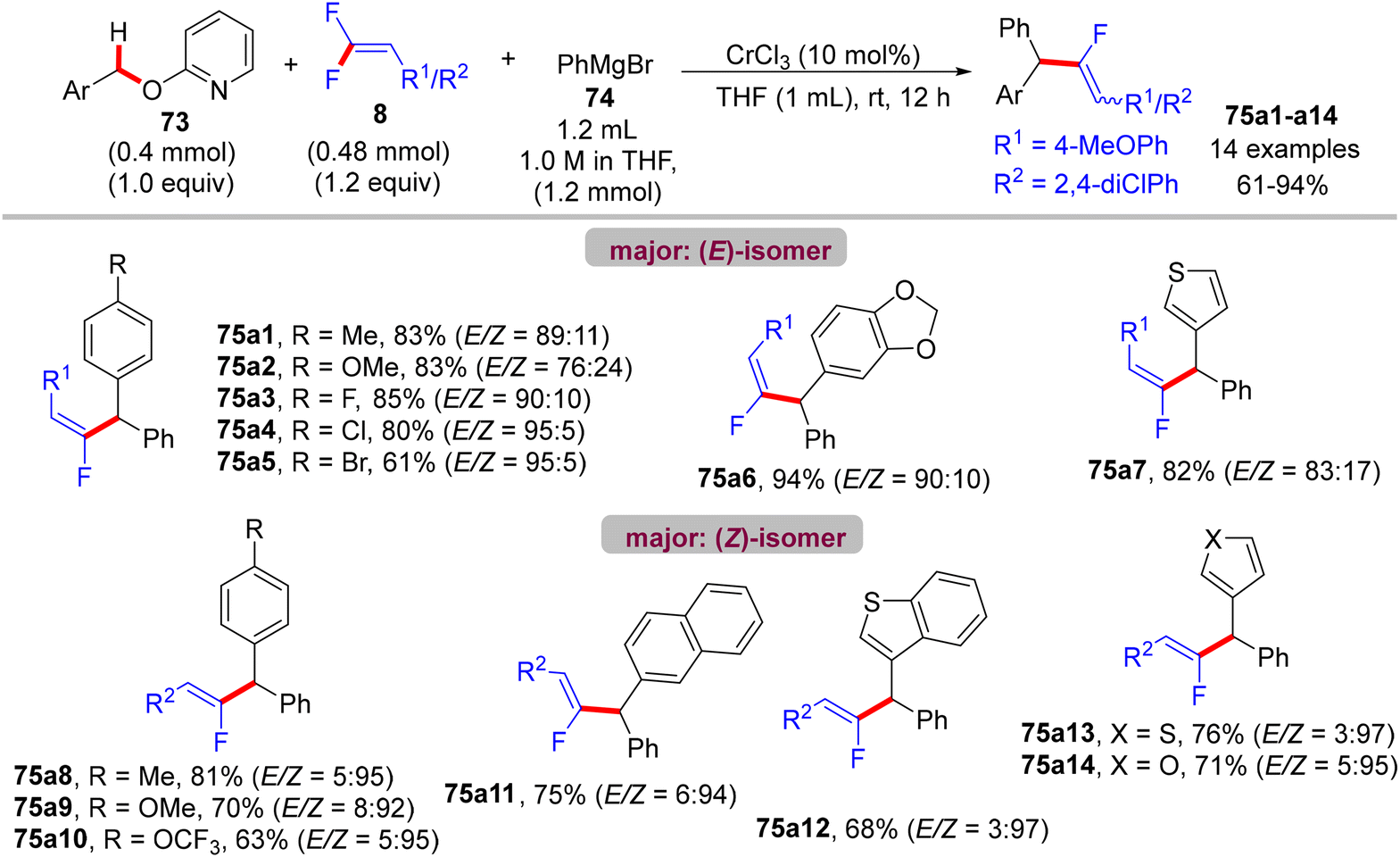
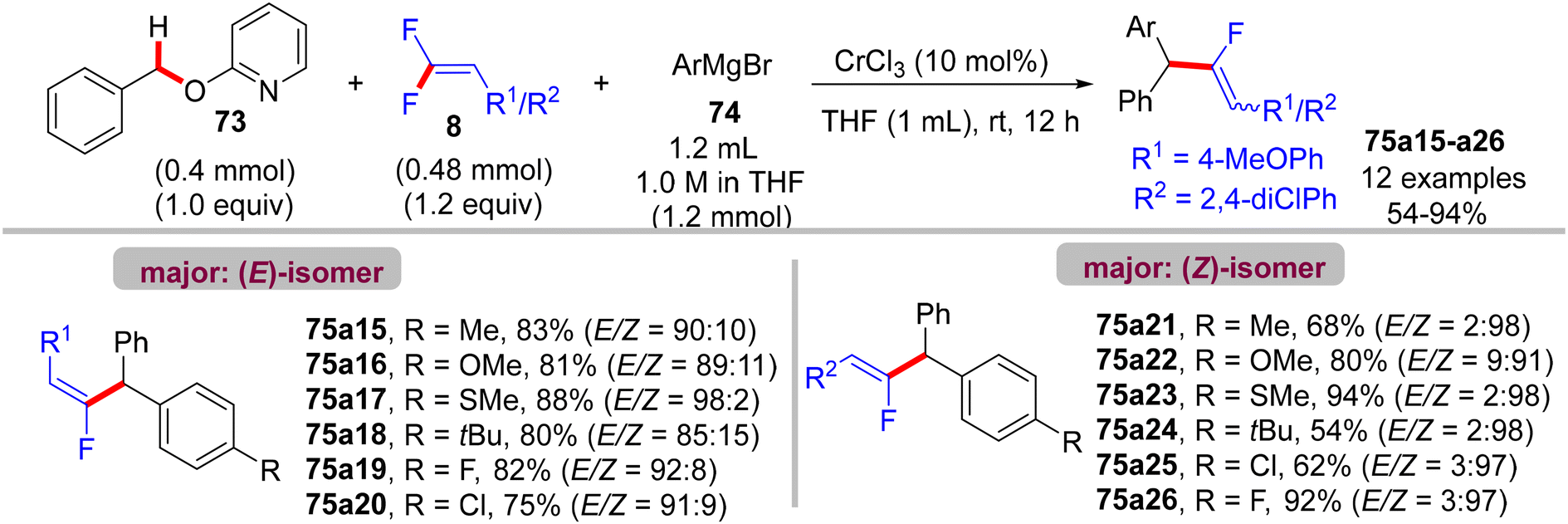
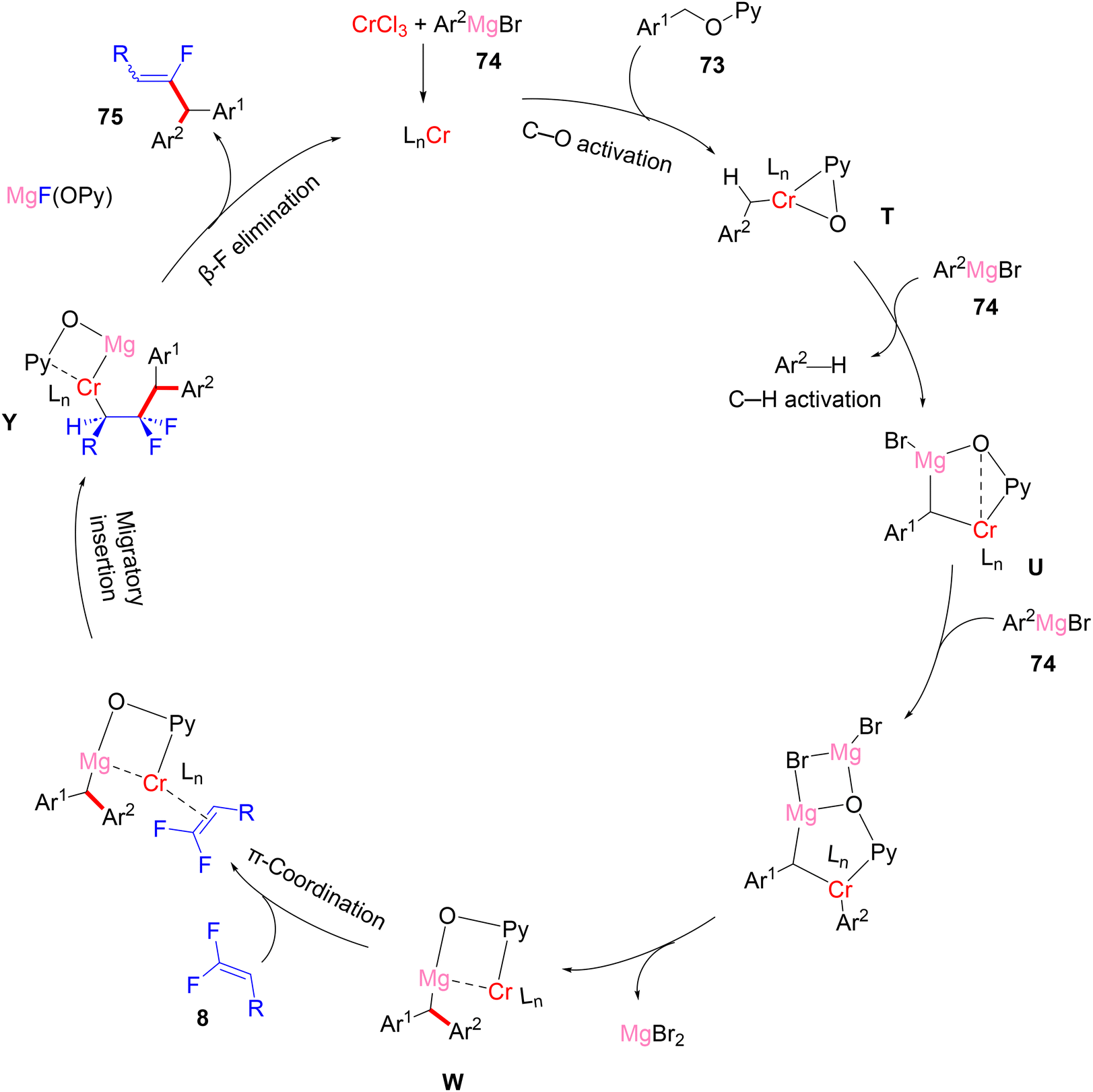
Recently (2023), Zeng et al. reported the first synthesis of diarylmethylated monofluoroalkenes 75a–ax in good yields using CrCl3 as catalyst at room temperature.46 This catalytic transformation was conducted via a step-economy three-component defluorinative cross-coupling of 8 with benzyl 2-pyridinyl ethers 73 and aryl Grignard reagents 74 in good to excellent chemo- and stereoselectivities. The reaction proceeded via the merging of C–F and C(sp3)–H/C–O activation. The authors tested several alternative metal salts (CrCl2, Cr(acac)3, CoCl2, NiCl2, and FeCl2), but CrCl3 proved the most effective, and optimal conditions were settled as follows: 8 (0.48 mmol), 73 (0.4 mmol), 74 (1.2 mmol), CrCl3 (0.04 mmol, 10 mol%), and THF (1 mL) at room temperature for 12 h. Schemes 66–68 show the substrate scope of the three-component reactants. The ratio of E/Z stereoselectivity was determined by GC analysis. Obviously, the stereoselectivity of 75a–a26 depends predominantly on the electronic profile of the substituents on the aromatic ring of the fluorinated synthons 8, where the EDGs afforded thermodynamically unfavorable (E)-isomer as a major one, and vice versa. Though the reason for (E)-isomer selectivity is not mentioned and, at present, is not clear yet.
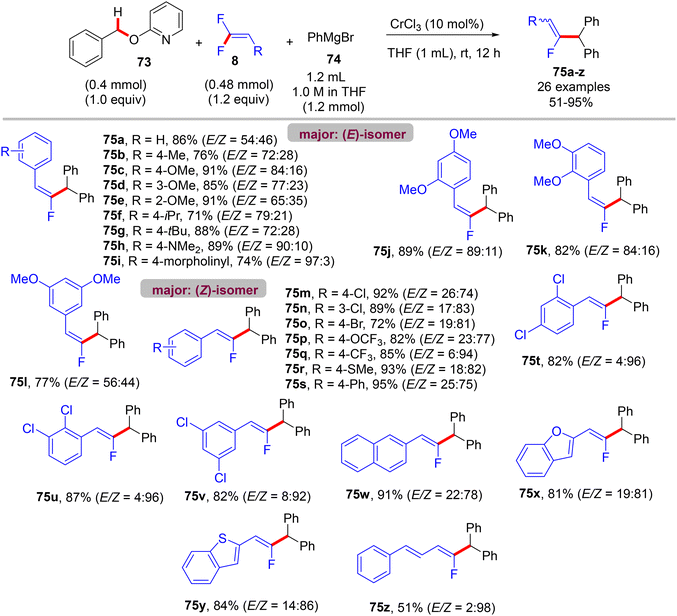 |
| | Scheme 66 Substrate scope of 8 in Cr(III)-catalyzed direct C(sp3)–H/C–O α-fluoroalkenylation reaction. | |
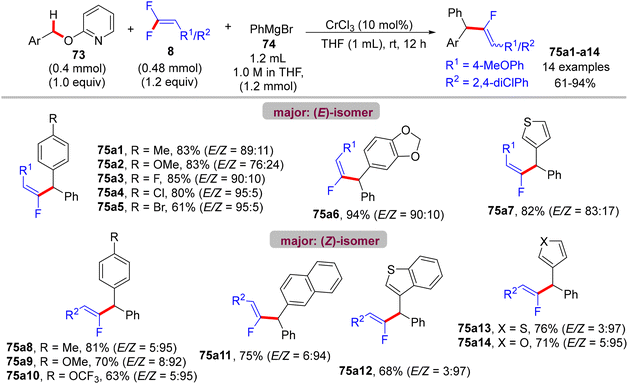 |
| | Scheme 67 Substrate scope of 73 in Cr(III)-catalyzed direct C(sp3)–H/C–O α-fluoroalkenylation reaction. | |
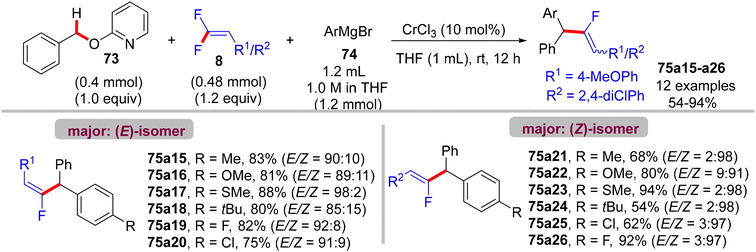 |
| | Scheme 68 Substrate scope of 74 in Cr(III)-catalyzed direct C(sp3)–H/C–O α-fluoroalkenylation reaction. | |
The gram-scale synthesis of 75c and 75t is depicted in Scheme 69.46 The authors also exhibited a plausible mechanism, which needed further experimental and theoretical studies. Initially, Grignard reagent 74 reduced the pre-catalyst CrCl3 to produce an active low-valent Cr (LnCr), probably by reductive elimination. Benzylic C–O activation produced the intermediate T. Next, deprotonation by 74 afforded the bimetalated complex U, which was followed by transmetallation with 74 to yield V. Removing MgBr2 by breaking the four-membered ring afforded the C–C coupling intermediate W. π-Coordination of olefin 8 with W yielded X, followed by migratory insertion to install Y. Finally, β-F elimination of Y exhibited the desired three-component coupling product 75 (Scheme 70).46
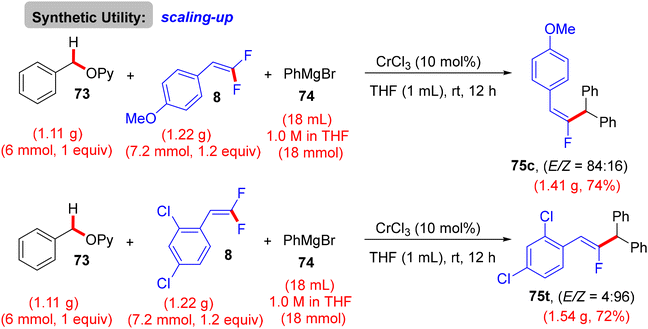 |
| | Scheme 69 Gram-scale synthesis of 75c and 75t. | |
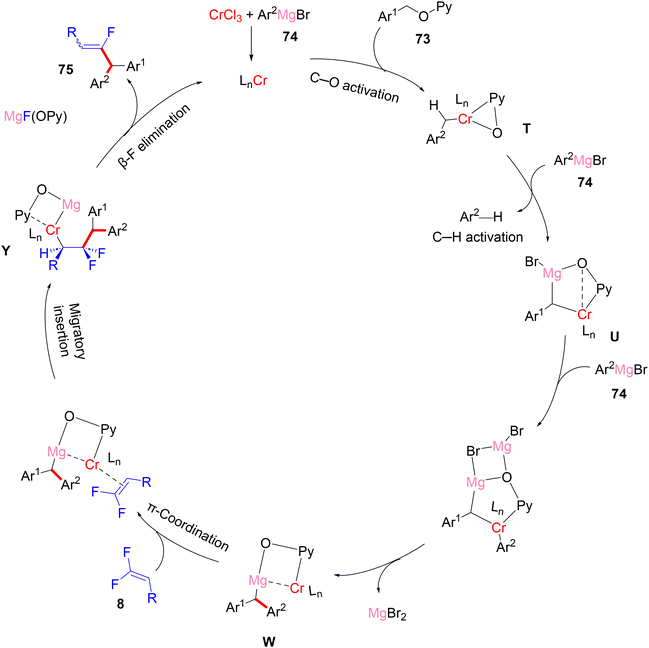 |
| | Scheme 70 Proposed catalytic cycle of Cr(III)-catalyzed direct C(sp3)–H/C–O α-fluoroalkenylation of three-component system (73, 74, and 8). | |
4. Conclusions and outlook
In the current review, we highlighted the progress of TM-catalyzed direct C–H α-fluoroolefination using easily accessible and commercially available gem-(bromo/di)fluoroolefins as fluorinated synthons. As seen throughout the reported procedures, several monofluoroalkenes are directly obtained in atom- and step-economically scalable pathways with high stereoselectivity and functional group tolerance. Importantly, some of the generated monofluoroalkenes are subjected to LSF to access distinct polyfunctionalized compounds. As a result, these approaches are suitable and applicable for the stereoselective synthesis of highly functionalized alkenes. Consequently, they can serve as a promising alternative to traditional cross-coupling transformations, the Wittig reaction or olefin metathesis. Nonetheless, numerous obstacles persist. Despite the achieved mild conditions and the use of earth-abundant TM-based protocols, such as those reported by Ackermann, Loh, and Li, the development of milder, somewhat “greener”, and low catalyst loading approaches remains imperious. As shown above, expensive Rh-based catalysts are widely used, so expanding the applicability of cost-effective first-row TM (3d) catalysts as a surrogate pathway is highly desirable. Until now, heterogenous catalysis has not been examined in the α-fluoroalkenylation reaction. The limitation of the substrate scope is another challenge that needs to be addressed by innovative procedures to expand their applicability for the synthesis of highly complex molecules. Further experimental and theoretical mechanistic investigations, such as the isolation of stable metallocyclic intermediates and the probing of the proposed individual mechanistically relevant alkene-insertion step and following eliminations, are recommended to gain more insights and achieve a better understanding of kinetics. Furthermore, only one study by Wang and Li30 reported the synthesis of cyclic monofluoroalkenes 29 and 34; thus, accessing these compounds is also desirable and needs more attention. In comparison with 8 or 25, gem-difluoroacrylate 38 as a fluorinated synthon was employed once in Loh's study.32 Therefore, it and 2 (gem-bromofluoroalkenes) need more examination in the direct α-fluoroalkenylation reaction. Worthy of note, all the reported α-fluoroalkenylation protocols are C(sp2)–H functionalization, except for the two examples of C(sp3)–H activation documented by Li34 and Zeng,46 proving the extension of this research area, is a topic of great interest. Microwave-assisted α-fluoroalkenylation is considered in none of the surveyed reports, and it is suggested that more investigation on the use of this technique might prove valuable.
Last but not least, the possible intervention of incidental soluble metal nanoparticles72 stemming from the decomposition of the metal complexes73–76 in any of the reported catalytic procedures, which are all characterized by their use of complex mixtures of reagents, was not investigated or at least mentioned, if not ruled out, to the best of our knowledge. Similarly, most of the studies lack an assessment of the role of the additives and “basic promotors”77 of the catalysis, which might be a must for further improvements away from the “trial and error” approach. This is an issue that clearly takes on a rather central relevance if one envisages the scaling to industrial levels of any of the reactions described here. The chemical speciation of the metal-containing species after the catalysis and the clear establishment of the fate of the catalyst after the catalytic event78 are particularly desirable if one seeks the responsible development of promising industrial processes that include rational waste management. The development of new catalysts under the banner of sustainability and environmental acceptability79 requires further efforts to establish the fate of the TM-catalysts after their task has been completed.
Finally, while great progress has been made, the development of the direct C–H α-fluoroolefination method seems within reach, and explorations of its chemistry might open new avenues for the synthesis of high-value organic compounds.
Abbreviations
| CMD | Concerted metalation-deprotonation |
| DCE | 1,2-Dichloroethane |
| DFT | Density functional theory |
| DG | Directing group |
| dibm | Diisobutyrylmethane |
| DMSO | Dimethyl sulfoxide |
| dppe | 1,2-Bis(diphenylphosphino)ethane |
| equiv | Equivalents |
| ERG/EDG | Electron-releasing (donating) group |
| EWG | Electron-withdrawing group |
| HAT | Hydrogen atom transfer |
| HFIP | Hexafluoroisopropanol |
| I | Inductive effect |
| KHMDS | Potassium bis(trimethylsilyl)amide |
| KIE | Kinetic isotope effect |
| LCD | Liquid crystal displays |
| LSF | Late-stage functionalization |
| MeOH | Methanol |
| NBO | Natural bond orbital |
| OTs | Tosylate group |
|
PDG | Permanent directing group |
| phen | 1,10-Phenanthroline |
|
RDG | Removal directing group |
| SNV | Vinylic nucleophilic substitution |
|
TDG | Transient directing group |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| TFE | Trifluoroethanol |
| Ts | Tosyl group |
Author contributions
The manuscript was written through the contributions of all authors. All authors have given their approval to the final version of the manuscript.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
We gratefully acknowledge the financial support from the Science, Technology, and Innovation Funding Authority (STDF) and the Embassy of France in Egypt – the French Institute of Egypt (IFE) under the Egypt-France Cooperation (STDF-IFE) – Call 9 program (project no. 46775). The authors wish to express deep thanks to the Centre National de la Recherche Scientifique, and to the University of Strasbourg for hosting and supporting this work.
References
- P. S. Steinlandt, L. Zhang and E. Meggers, Chem. Rev., 2023, 123, 4764–4794 CrossRef.
- U. Bin Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S. G. Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef PubMed.
- C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2022, 122, 3180–3218 CrossRef.
- J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef.
- J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- S. Faarasse, N. El Brahmi, G. Guillaumet and S. El Kazzouli, Molecules, 2021, 26, 5763–5800 CrossRef CAS.
- H. Azizollahi and J. A. García-López, Molecules, 2020, 25, 5900–5966 CrossRef CAS.
- R. L. Carvalho, A. S. De Miranda, M. P. Nunes, R. S. Gomes, G. A. M. Jardim and E. N. Da Silva, Beilstein J. Org. Chem., 2021, 17, 1849–1938 CrossRef CAS.
- J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1–17 Search PubMed.
- K. M. Altus and J. A. Love, Chem. Commun., 2021, 4, 1–11 CrossRef PubMed.
- R. L. Carvalho, G. G. Dias, C. L. M. Pereira, P. Ghosh, D. Maiti and E. N. Da Silva, J. Braz. Chem. Soc., 2021, 32, 917–952 CAS.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- F. L. Qing, X. Y. Liu, J. A. Ma, Q. Shen, Q. Song and P. Tang, CCS Chem., 2022, 4, 2518–2549 CrossRef CAS.
- R. Britton, V. Gouverneur, J. H. Lin, M. Meanwell, C. Ni, G. Pupo, J. C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef CAS.
- M. Z. Lu, J. Goh, M. Maraswami, Z. Jia, J. S. Tian and T. P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef PubMed.
- L. V. Hooker and J. S. Bandar, Angew. Chem., Int. Ed., 2023, 62, e202308880 CrossRef PubMed.
- D. J. Burton, Z. Y. Yang and W. Qiu, Chem. Rev., 1996, 96, 1641–1715 CrossRef.
- G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 CrossRef PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef PubMed.
- J. P. Sorrentino and R. A. Altman, Synthesis, 2021, 3935–3950 Search PubMed.
- S. Koley and R. A. Altman, Isr. J. Chem., 2020, 60, 313–339 CrossRef.
- T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- C. Schneider, D. Masi, S. Couve-Bonnaire, X. Pannecoucke and C. Hoarau, Angew. Chem., Int. Ed., 2013, 52, 3246–3249 CrossRef CAS.
- K. Rousée, C. Schneider, S. Couve-Bonnaire, X. Pannecoucke, V. Levacher and C. Hoarau, Chem. – Eur. J., 2014, 20, 1500–1504 CrossRef.
- K. Rousée, C. Schneider, J. P. Bouillon, V. Levacher, C. Hoarau, S. Couve-Bonnaire and X. Pannecoucke, Org. Biomol. Chem., 2015, 14, 353–357 RSC.
- P. Tian, C. Feng and T. P. Loh, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- J. Q. Wu, S. S. Zhang, H. Gao, Z. Qi, C. J. Zhou, W. W. Ji, Y. Liu, Y. Chen, Q. Li, X. Li and H. Wang, J. Am. Chem. Soc., 2017, 139, 3537–3545 CrossRef PubMed.
- W. W. Ji, E. Lin, Q. Li and H. Wang, Chem. Commun., 2017, 53, 5665–5668 Search PubMed.
- H. Liu, S. Song, C. Q. Wang, C. Feng and T. P. Loh, ChemSusChem, 2017, 10, 58–61 CrossRef PubMed.
- S. Song, H. Liu, L. Wang, C. Zhu, T. P. Loh and C. Feng, Chin. J. Chem., 2019, 37, 1036–1040 CrossRef.
- L. Kong, B. Liu, X. Zhou, F. Wang and X. Li, Chem. Commun., 2017, 53, 10326–10329 RSC.
- M. Tian, X. Yang, B. Zhang, B. Liu and X. Li, Org. Chem. Front., 2018, 5, 3406–3409 RSC.
- N. Wang, Q. Yang, Z. Deng, X. Mao and Y. Peng, ACS Omega, 2020, 5, 14635–14644 CrossRef.
- B. Pang, Y. Wang, L. Hao, G. Wu, Z. Ma and Y. Ji, J. Org. Chem., 2023, 88, 143–153 CrossRef.
- N. Li, J. Chang, L. Kong and X. Li, Org. Chem. Front., 2018, 5, 1978–1982 RSC.
- Y. Ji, C. Xia, X. Fu, L. Zhang, K. Deng, G. Wu, J. Yang and S. Tang, J. Org. Chem., 2020, 85, 12670–12681 CrossRef PubMed.
- L. Kong, X. Zhou and X. Li, Org. Lett., 2016, 18, 6320–6323 CrossRef CAS PubMed.
- D. Zell, V. Müller, U. Dhawa, M. Bursch, R. R. Presa, S. Grimme and L. Ackermann, Chem. – Eur. J., 2017, 23, 12145–12148 CrossRef CAS.
- N. Murakami, M. Yoshida, T. Yoshino and S. Matsunaga, Chem. Pharm. Bull., 2018, 66, 51–54 CrossRef CAS PubMed.
- D. Zell, U. Dhawa, V. Müller, M. Bursch, S. Grimme and L. Ackermann, ACS Catal., 2017, 7, 4209–4213 CrossRef CAS.
- S. H. Cai, L. Ye, D. X. Wang, Y. Q. Wang, L. J. Lai, C. Zhu, C. Feng and T. P. Loh, Chem. Commun., 2017, 53, 8731–8734 RSC.
- L. Yang, W. W. Ji, E. Lin, J. L. Li, W. X. Fan, Q. Li and H. Wang, Org. Lett., 2018, 20, 1924–1927 CrossRef CAS PubMed.
- Y. Xiong, M. Luo and X. Zeng, Org. Lett., 2023, 25, 3120–3125 CrossRef CAS.
- X. Lei, G. Dutheuil, X. Pannecoucke and J. Quirion, Org. Lett., 2004, 6, 2101–2104 CrossRef CAS.
- L. Zoute, G. Dutheuil, J. Quirion, P. Jubault and X. Pannecoucke, Synthesis, 2006, 3409–3418 CAS.
- L. Chausset-Boissarie, N. Cheval and C. Rolando, Molecules, 2020, 25, 5532–5544 CrossRef CAS PubMed.
- C. Verrier, P. Lassalas, L. Théveau, G. Quéguiner, F. Trécourt, F. Marsais and C. Hoarau, Beilstein J. Org. Chem., 2011, 7, 1584–1601 CrossRef CAS PubMed.
- P. M. Weintraub, A. K. Holland, C. A. Gates, W. R. Moore, R. J. Resvick and N. P. Peet, Bioorg. Med. Chem., 2003, 11, 427–431 CrossRef CAS PubMed.
- Y. Xiong, X. Zhang, T. Huang and S. Cao, J. Org. Chem., 2014, 79, 6395–6402 CrossRef CAS PubMed.
- K. Ichikawa, J. Wada, Y. Fujiwara and M. Kotaro Sakoda, Synthesis, 2002, 13, 1917–1936 Search PubMed.
- V. A. Online, X. Zhang, Y. Lin, J. Zhang and S. Cao, RSC Adv., 2015, 5, 7905–7908 RSC.
- K. Chen, W. Chen, F. Chen, H. Zhang and H. Xu, Org. Chem. Front., 2021, 8, 4452–4458 RSC.
- C. Zhou, L. Zhu, C. Loh and T. Feng, Chem. Commun., 2018, 54, 5618–5621 RSC.
- S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623–1631 CrossRef CAS PubMed.
- N. Wang, B. Li, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2013, 19, 358–364 CrossRef CAS PubMed.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 11363–11370 RSC.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS.
- F. Weinhold, J. Comput. Chem., 2012, 33, 2363–2379 CrossRef CAS.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- A. Pereira, C. Albornoz and O. S. Trofymchuk, Organometallics, 2022, 41, 1158–1166 CrossRef CAS.
- P. G. Chirila and C. J. Whiteoak, Dalton Trans., 2017, 46, 9721–9739 RSC.
- A. G. Chaidali and I. N. Lykakis, Eur. J. Org. Chem., 2023, 26, e202300497 CrossRef CAS.
- S. St John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC.
- L.-L. Gundersen, J. Nissen-Meyer and B. Spilsberg, J. Med. Chem., 2002, 45, 1383–1386 CrossRef CAS PubMed.
- E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428–1431 CrossRef CAS.
- H. Zhang, H. Li, H. Yang and H. Fu, Org. Lett., 2016, 18, 3362–3365 CrossRef CAS PubMed.
- S. Bordi and J. T. Starr, Org. Lett., 2017, 19, 2290–2293 CrossRef CAS.
- J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed.
- D. M. Kaphan, K. R. Brereton, R. C. Klet, R. J. Witzke, A. J. M. Miller, K. L. Mulfort, M. Delferro and D. M. Tiede, Organometallics, 2021, 40, 1482–1491 CrossRef CAS.
- K. E. Gonsalves, H. Li, R. Perez, P. Santiago and M. Jose-Yacaman, Coord. Chem. Rev., 2000, 206–207, 607–630 CrossRef CAS.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, New J. Chem., 2013, 37, 3374–3401 RSC.
- M. L. Kahn, A. Glaria, C. Pages, M. Monge, L. Saint Macary, A. Maisonnat and B. Chaudret, J. Mater. Chem., 2009, 19, 4044–4060 RSC.
- C. Antuña-Hörlein, F. Wu, C. Deraedt, C. Bouillet and J.-P. Djukic, Eur. J. Inorg. Chem., 2023, 26, e202200563 CrossRef.
- W. Yang, T. Y. Kalavalapalli, A. M. Krieger, T. A. Khvorost, I. Yu. Chernyshov, M. Weber, E. A. Uslamin, E. A. Pidko and G. A. Filonenko, J. Am. Chem. Soc., 2022, 144, 8129–8137 CrossRef CAS PubMed.
- F. Wu, C. Deraedt, Y. Cornaton, L. Ruhlmann, L. Karmazin, C. Bailly, N. Kyritsakas, N. Le Breton, S. Choua and J.-P. Djukic, Organometallics, 2021, 40, 2624–2642 CrossRef CAS.
- S. Mitchell, A. J. Martín and J. Pérez-Ramírez, Nat. Chem. Eng., 2024, 1, 13–15 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Ahmed H.
Bedair
b and
Jean-Pierre
Djukic
ab,
Ahmed H.
Bedair
b and
Jean-Pierre
Djukic