
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and properties of thienonaphtho[bc]pyridines and thienonaphtho[bc]quinolines†
Arpine
Vardanyan
,
Jonas
Polkaehn
,
Marie-Louis
Bauder
,
Alexander
Villinger
 ,
Peter
Ehlers
and
Peter
Langer
*
,
Peter
Ehlers
and
Peter
Langer
*
Universität Rostock, Institut für Chemie, A.-Einstein-Str. 3a, 18059 Rostock, Germany. E-mail: peter.langer@uni-rostock.de; Fax: +49381 498 6412; Tel: +49 381 498 6410
First published on 7th October 2024
Abstract
The incorporation of heteroatoms within polycyclic aromatic compounds has gained significant interest due to its potential to effectively alter the inherent physicochemical properties of compounds without the need for profound structural changes. We herein report the development of a modular synthesis of hitherto unknown thienonaphtho[bc]pyridines and thienonaphtho[bc]quinolines in very good yields by Brønsted acid mediated cycloisomerization, permitting selective access to two isomeric products that are isoelectronic to the parent dibenzopyrene. The photophysical and electrochemical properties of the desired compounds were extensively studied and further complemented by DFT calculations.
Over the past decades, the development of efficient optoelectronic materials based on polycyclic aromatic hydrocarbons (PAHs) has attracted wide attention and has been extensively investigated.1–8 The π-expansion of PAHs is a powerful approach to improve their photophysical and electrochemical properties. Therefore, the development of new and modular methods for the synthesis and derivatization of PAHs is of considerable current interest.9–12 In this context, the annulation and replacement of benzene rings by heterocycles is an efficient strategy to modulate the intrinsic physicochemical properties, such as band gap, redox properties, aromaticity and chemical stability of PAHs, giving rise to new and wide applications in materials science.13–17 Thus, thiophene-fused π-conjugated molecules have received significant interest as promising building blocks for the development of high-performance π-conjugated materials in diverse scientific fields, such as organic photovoltaic materials (OPVs), organic light-emitting diodes (OLEDs) and organic field-effect transistors (OFETs).18–24
Today, one of the most studied PAHs is undoubtedly pyrene, due to its unique optical and electronic properties.7,25–29 However, the incorporation of heterocycles such as electron-rich thiophene or electron-deficient pyridines into the pyrene scaffold is still in its infancy. The present synthetic methods often suffer from multi-step synthetic procedures, requirement of complex, not readily available starting materials, harsh reaction conditions, low selectivity and/or poor overall yields.30–40 For instance, the group of Otsubo obtained phenalenothiophene by flash vacuum pyrolysis. Later, Höger and coworkers used Pd-catalyzed CH-activation to obtain related benzotetracenothiophenes, which upon functionalization with certain alkynyl side chains self-assemble to ribbon- and ziggurat-like superstructures in the solid state. Diazaphenalenothiophenes were obtained by Aksenov and coworkers by the electrolysis of benzoylperimidines in the presence of H2S or its condensation with elemental sulfur at high temperature. We recently reported the synthesis of S,N-doped thienonaphthopyridines (TNP) by Brønsted acid mediated cycloisomerisation (Fig. 1).41 Incorporation of heteroatoms and alteration of the ring size within the pyrene scaffold led to substantial changes of the inherent photophysical properties.
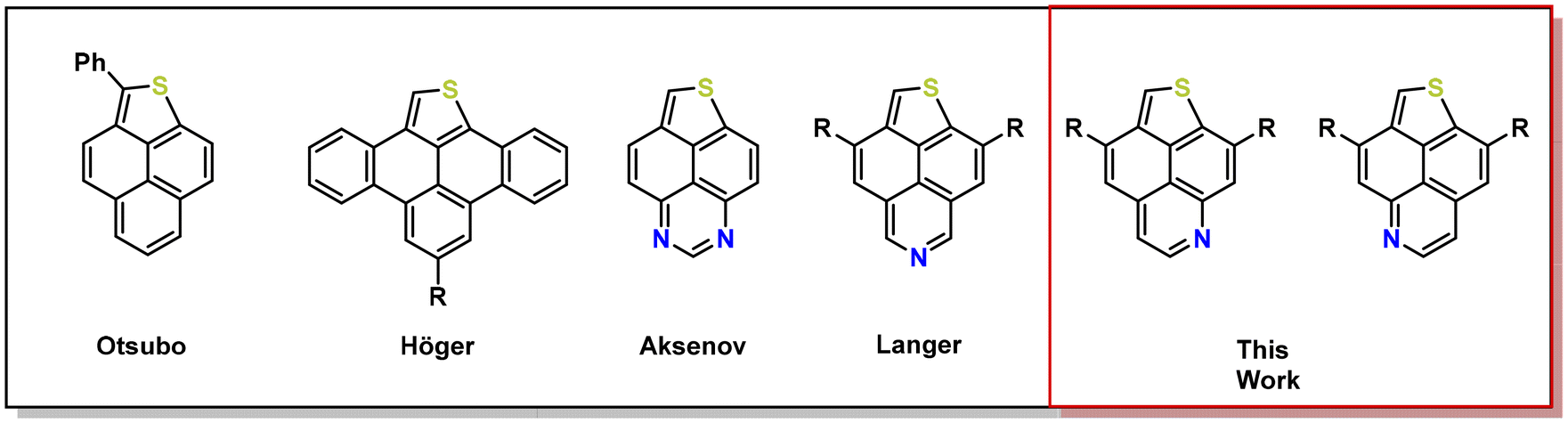 | ||
| Fig. 1 Structures of (aza)thienophenalenes. | ||
Thus, as an extension of our previous work, we were interested in the selective N-doping of the respective thienophenalenes and its impact on their photophysical and electrochemical properties. The newly developed methodology reported herein is highly modular and is based on Brønsted acid mediated cycloisomerisation, giving access to various TNP isomers including their π-expanded derivatives.
Results and discussion
Synthesis
The starting materials 1a–j and 2a–c were obtained in 41–92% isolated yields by palladium catalyzed Suzuki–Miyaura and Sonogashira reactions, respectively, adapting previously reported procedures for the respective phenyl derivatives (ESI, Scheme S1†).40Subsequently, the Brønsted acid mediated cycloisomerisation reaction was studied, utilizing 1a as a representative substrate to optimize the reaction conditions. It is important to note that direct treatment of precursor 1a with a high excess of methanesulfonic acid (MsOH) at elevated temperatures resulted in complete conversion of the starting material to give an inseparable mixture of the bis-annulated regioisomers 5a and 6a (Table 1, entries 1 and 2). Further investigations showed that employing the less acidic trifluoroacetic acid (TFA) resulted in the formation of the mono-annulated isomers 3a and 4a in moderate to very good overall yields with varying selectivities for the two products (Table 1, entries 5–9). Performing the reaction at room temperature (entry 6) resulted in an improved selectivity for the isolated isomers, but at the same time in a reduced overall yield. Hence, applying the reaction conditions at 40 °C provided the best overall results in terms of yield and selectivity of products 3a and 4a. Decreasing the amount of TFA from 120 eq. to 30 eq. proved to be ineffective as no conversion of the starting material was observed.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Acid (eq.) | Solvent | T, °C | t, h | Yielda (%) | Ratio 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a:5a:6a :4a:5a:6a |
| a Yields of isolated products. | ||||||
| 1 | MsOH (30) | — | 120 | 3 | 24:24 |
0:0:1:1 |
| 2 | MsOH (60) | — | 120 | 24 | 27:27 |
0:0:1:1 |
| 3 | MsOH (60) | — | 20 | 24 | 25:49 |
1:2:0:0 |
| 4 | MsOH (60) | — | 20 | 3 | 28:43 |
1:1.5:0:0 |
| 5 | TFA (120) | — | 70 | 24 | 81:11 |
7:1:0:0 |
| 6 | TFA (120) | — | 20 | 24 | 50:4 |
11:1:0:0 |
| 7 | TFA (120) | — | 40 | 30 |
80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :8 :8
|
10:1:0:0
|
| 8 | TFA (30) | — | 40 | 30 | — | — |
| 9 | TFA (30) | DCE | 70 | 30 | Traces | — |

In all optimization reactions, the cyclization proceeded selectively at the α-position of thiophene and no formation of the isomeric product derived from the reaction at the β-position was observed. Single crystal structures were obtained for both isomers by slow evaporation of heptane/dichloromethane solutions, which unambiguously confirmed the configuration of both isomers (ESI, Fig. S1, Table S1†). Moreover, the characteristic NMR shift of the singlet of the central benzene proton allowed for the assignment of the respective regioisomers (∼8.0 ppm for 3a and ∼7.5 ppm for 4a).
Afterwards, the preparative scope was studied and the respective mono-annulated thieno[f]quinolines 3a–j and thieno[h]isoquinolines 4a–h were obtained in moderate to very good yields (Scheme 1). Both electron-poor and electron-rich systems gave good yields regardless of the substitution pattern. Surprisingly, in the case of 1b, the formation of the bis-annulated isomer 5b instead of the expected mono-annulated product 4b was observed, albeit in only 15% yield. The reaction of 1j, containing a strongly donating dimethylamino (DMA) group, behaved differently from all other starting materials employed and provided an isomeric mixture of products derived from cyclization at the α-position and β-position of the thiophene ring. This could be explained by the formation of highly reactive charged species. The strongly acidic reaction conditions may protonate the DMA functional group and transform it into an acceptor, leading to a less stabilized vinyl carbocation upon protonation of the triple bond. In the case of 1i, an inseparable mixture of products was obtained due to the formation of phenolic by-products caused by the cleavage of the methoxy group. The use of 1f and 1g, containing electron withdrawing trifluoromethyl and cyano groups, resulted in rapid decomposition of the starting materials, most likely due to the low stability of the positively charged intermediates during the reaction. It is noteworthy that the reaction could be successfully extended to benzannulated derivatives 2a–c, resulting in the selective formation of products 7a–c as single isomers in moderate to good yields.
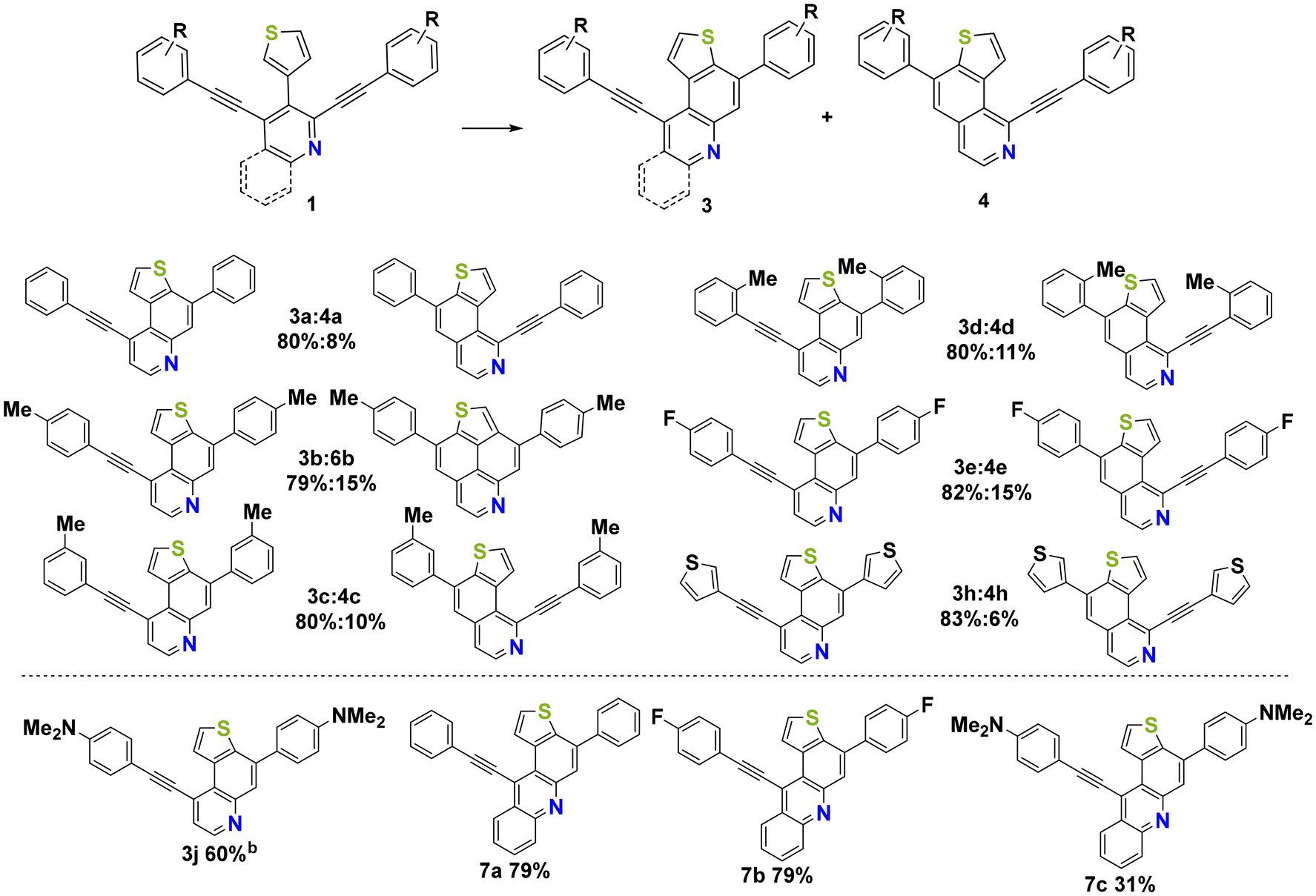 | ||
| Scheme 1 Synthesisa of 3a–j, 4a–h, 7a–c; ayields of isolated products; bmixture of isomers. | ||
With precursors 3a–j, 4a–h and 7a–c in hand, optimizations were carried out for the acid mediated cycloisomerization for the synthesis of the desired thienonaphtho[bc]pyridines and thienonaphtho[bc]quinolines (Table 2). The reaction was optimized for the transformation of the mono-cyclized precursor 3a into the bis-cyclized product 5a. The best yield of 5a (82%) was obtained using p-toluenesulfonic acid (pTsOH·H2O, 30 eq.) as the acid and xylene as the solvent at an elevated temperature (140 °C), while the use of methanesulfonic acid (MsOH) gave lower yields (Table 2, entries 1–3). Application of these optimized conditions to the mono-cyclized precursor 7a resulted in only 42% yield of the desired bis-cyclized 3,11-diphenylthieno[2′,3′,4′:4,5]naphtho[1,8-bc]quinoline 8a (Table 2, entry 4). Hence, we tested the transformation of the non-cyclized precursor 2a into 8a using MsOH at 140 °C (Table 2, entry 6). To our delight, the desired bis-annulated product 8a was formed in an improved yield (68%) directly from precursor 2a. The yield is even higher as compared to the step-wise protocol described above and avoids the tedious purification of intermediates. It is noteworthy that only one single isomer of 8a was formed.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Precursor | Acid (eq.) | Solvent | T, °C | t, h | Yield (%) |
| Yields of isolated products. | ||||||
| 1 | 3a | MsOH (30) | — | 120 | 24 | 53 |
| 2 | 3a | pTsOH·H 2 O (30) | Xylene | 140 | 24 | 82 |
| 3 | 3a | pTsOH·H2O (60) | Xylene | 140 | 24 | 76 |
| 4 | 7a | pTsOH·H2O (30) | Xylene | 140 | 24 | 42 |
| 5 | 2a | pTsOH·H2O (30) | Xylene | 140 | 24 | 30 |
| 6 | 2a | MsOH (30) | Xylene | 140 | 24 | 68 |

The optimized conditions were successfully applied to various precursors 3a–j, 4a–h and 2a–c and the desired products 5a–j, 6a–b and 8a–c were formed in moderate to very good yields, respectively (Fig. 2). The best yield (98%) was obtained for product 5e containing an electron withdrawing fluorine substituent located at the para position of the phenyl group. Using the DMA-substituted precursor 3j (used as a mixture of two isomers) afforded product 5j, albeit in only 24% yield. For comparison, an equally low yield (32%) was observed for the DMA-substituted product 8c. The reduced yield of both compounds might be explained by the protonation of the amine during the reaction and its transformation from a donor to an acceptor group. Additionally, purification by column chromatography was hampered by the low solubility of DMA-substituted products.
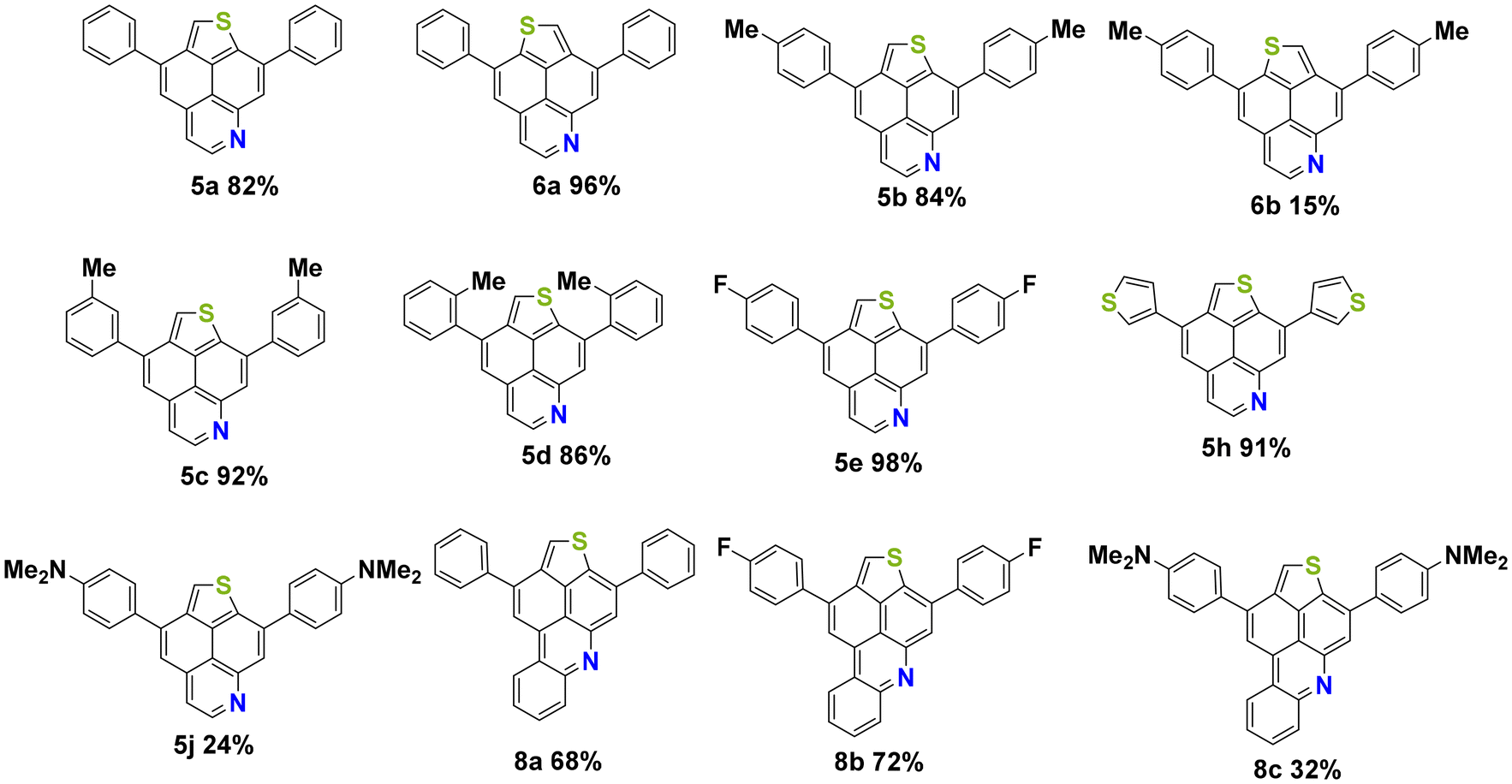 | ||
| Fig. 2 Synthesis of 5a–j, 6a–b and 8a–c; yields of isolated products. | ||
The structure of 8a was independently confirmed by X-ray crystal structure analysis. Crystals were grown from mixtures of heptane and CH2Cl2 by slow evaporation at room temperature (Fig. 3; ESI, Table S1†). Compound 8a crystallizes in a triclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The phenyl substituents are twisted out of the plane of the core structure and adopt a slipped antiparallel orientation to each other with a centroid-to-centroid distance of 3.43 Å. Furthermore, adjacent molecules form N–H hydrogen bonds with a distance of 2.70 Å.
. The phenyl substituents are twisted out of the plane of the core structure and adopt a slipped antiparallel orientation to each other with a centroid-to-centroid distance of 3.43 Å. Furthermore, adjacent molecules form N–H hydrogen bonds with a distance of 2.70 Å.
 | ||
| Fig. 3 X-ray crystal structures of 8a and intermolecular packing of 8a (determined at 123 K; thermal ellipsoids are drawn at the 50% probability level). | ||
Physical properties
Steady-state absorption and photoluminescence (PL) spectra in CH2Cl2 were recorded to obtain improved insights into the photophysical properties of the selected compounds. The key photophysical data of compounds 5a, 5j, 6a, 8a and 8c are shown in Fig. 4 and Table 3. Emission spectra were recorded at excitation wavelengths of 360 nm (5a and 6a), 400 nm (5j and 8a) and 450 nm (8c). The photophysical data are generally consistent with earlier studies on related compounds, revealing the absorption features of azapyrene and thienonaphthopyridine (TNP) with well-resolved vibrational progression bands.40,42–45 | ||
| Fig. 4 Absorption (solid lines) and emission (dashed lines) spectra of selected compounds. | ||
|
5aa |
5jb |
6aa |
8ab |
8cc |
|
|---|---|---|---|---|---|
| a λ 360em [nm]. b λ 400em [nm]. c λ 450em [nm]. d Fluorescence standard: quinine hemisulfate monohydrate in 0.05 M H2SO4 (ϕ = 0.52). e Fluorescence standard: coumarin 153 in EtOH (ϕ = 0.38). | |||||
| λ 1,abs [nm] | 373 | — | 374 | 436 | 483 |
| logελ1 |
4.30 | — | 3.85 | 4.21 | 3.83 |
| λ 2,abs [nm] | 390 | 396 | 390 | 460 | 509 |
| logελ2 |
4.25 | 4.05 | 3.86 | 4.18 | 3.81 |
| λ 3,abs [nm] | — | 501 | — | 512 | 595 |
| logελ3 |
— | 3.71 | — | 3.30 | 3.49 |
| λ 1,em [nm] | 413 | 547 | 408 | 483 | 600 |
| λ 2,em [nm] | 430 | 427 | 512 | ||
| ϕ , | 0.08 | 0.13 | 0.10 | 0.51 | 0.09 |
The lowest energy absorption band of 5a shows remarkably higher extinction coefficients (ε = 2.3 × 104 M−1 cm−1) in comparison with 6a, which also possesses more than half of the absorptivity as compared to TNP. However, the variation in the position of N-doping only has a negligible impact on the absorption edge of all three TNP derivatives. TD-DFT calculations at the B3LYP level suggest that for all three compounds, the S1 ← S0 transition originates from HOMO → LUMO with a reasonable oscillator strength and are in good agreement with experimental results (ESI, Tables S11 and S13†).46 The extension of conjugation by fusion of an additional benzene ring (8a) leads to bathochromically shifted absorption bands in comparison with 5a. Compounds 5j and 8c, containing DMA as a strong donor, show substantially altered absorption and emission spectra. In particular, 5j and 8c display a strongly red-shifted lowest energy band with loss of its absorption fine structure. The emission spectra of both compounds exhibit notable bathochromic shifts from two-band emission to a single very broad emission band, indicating the presence of intramolecular charge-transfer (ICT) features, which were also observed in previously reported amino-substituted pyrenes, 1- and 2-azapyrenes and TNP.40,42,43 Quantum yields (QYs) were determined in reference to quinine hemisulfate monohydrate in 0.05 M H2SO4 (ϕ = 0.52) and coumarin 153 in ethanol (ϕ = 0.38) as fluorescence standards (ESI, Table S2†).47 The highest fluorescence QY of 0.51 in dichloromethane was detected for derivative 8a, while derivatives 5a, 5e and 6a showed relatively low QYs of 0.08, 0.08 and 0.13, respectively. The fluorescence QY of 5j (0.13) is slightly higher than that of its unsubstituted counterpart 5a, whereas derivative 8c exhibits a decrease in QY (0.09) more than 5 times in comparison with 8a.
Hence, variation of the relative position of nitrogen and sulfur in TNP has negligible effects on the absorption edges and absorption fine structure. In comparison, benzannulation on the pyridine ring leads to strong alteration of the absorption features of the respective TNP derivatives.
To investigate the potential ICT properties, the emission spectra of derivatives 5j and 8c were measured in various solvents of different polarities (Fig. 5). The emission in acetonitrile is bathochromically shifted by 110 nm (λem,max = 605 nm 5j) and 117 nm (λem,max = 653 nm 8c) in comparison with nonpolar toluene. It is noteworthy that changing the solvent from acetonitrile to the polar and protic solvent ethanol leads to a blue shift of the emission maximum by 14 nm (λem,max = 591 nm 5j) and 18 nm (λem,max = 635 nm 8c). The emission intensities of 5j and 8c varied with changes in the solvent polarity. Nonpolar solvents cause intensity enhancements as compared to polar solvents. These results clearly suggest a certain ICT for 5j and 8c, which is characterized by stabilization of the excited state in polar environments, due to a large dipole moment in the excited state.48 Hence, the dipole moments of 5j and 8c have been calculated for their S0 and excited S1 states, confirming the experimental results. In particular, TNP 5j shows an increase in its calculated dipole moment upon excitation from 7.7 to 23.9 Debye, while for benzo-TNP 8c, the dipole moment is increased from 8.3 to 22.3 Debye.
 | ||
| Fig. 5 Emission spectra of 5j (left) and 8c (right) (c = 10−5 M) at 20 °C. | ||
Furthermore, the impact of protonation on the optical properties was studied and the changes in the absorption and emission spectra upon protonation are clearly shown in Fig. 6 for 5a and in Fig. S2 (ESI†) for 6a and 8a. All studied compounds showed red shifted absorption and emission spectra when trifluoroacetic acid (TFA) was added to a CH2Cl2 solution of 5a, 6a or 8a. For all compounds, the intensity of the low energy band is strongly enhanced, while the shape of the emission band is roughly the same. However, the intensity of the second emission band is reduced with respect to the first emission band. The bathochromic shift of both spectra might be explained by improved ICT properties from the electron-rich thiophene to the positively charged pyridinium.
 | ||
| Fig. 6 Absorption (left) and normalized emission spectra (right) of 5a in CH2Cl2 (c = 10−5 M) at 20 °C with different equivalents of TFA. | ||
The redox behavior of compounds 5a, 5j, 6a, 8a and 8c in comparison with the recently reported TNP was investigated by cyclic voltammetry experiments (Table 4; ESI, Fig. S3†). In comparison with the voltammogram of TNP (one irreversible oxidation peak at 0.92 V vs. Fc+/Fc), the voltammograms of isomers 5a and 6a display two irreversible oxidations events. However, compound 5j, bearing strong π-donating NMe2-substituents, displays a broad oxidation potential accompanied by a shoulder at a less positive potential (0.62 V vs. Fc+/Fc), which exemplifies the electron donating ability of the dimethylaniline substituents. The fusion of an additional benzene ring to the core structure results in three irreversible oxidations events (compounds 8a and 8c). Interestingly, the presence of NMe2-groups in 8c does not lead to a more facile oxidation as compared to its unsubstituted analogue 8a.
a
|
|
|
||||
|---|---|---|---|---|---|
| 5a | 5j | 6a | 8a | 8c | |
| a Measured in CH2Cl2 with 0.1 M n-Bu4NPF6 as a supporting electrolyte, glassy carbon working electrode, ANE2 as a reference electrode and Pt counter-electrode with ferrocene as a standard at a scan rate of 50 mV s−1. | |||||
| E Ox1 | 1.02 | 0.62 | 1.01 | 0.68 | 0.99 |
| E Ox2 | 1.67 | 1.62 | 1.74 | 1.47 | 1.50 |
| E Ox3 | 1.83 | 1.86 | |||


To rationalize the observed experimental results and to obtain additional insight into the intrinsic electronic characteristics of π-conjugated thienonaphtho[bc]pyridines and thienonaphtho[bc]quinolines, density functional theory (DFT) calculations were performed.46 Calculations were carried out for 5a, 5j, 6a, 8a and 8c as well as for TNP, utilizing the B3LYP functional in combination with the 6-311G(d,p) basis set and a polarizable continuum model (PCM) of dichloromethane using the integral equation formalism variant (IEFPCM) (Scheme 2, ESI, Scheme S2†).
 | ||
| Scheme 2 Calculated frontier molecular orbitals and energy levels at the B3LYP/6-311G(d,p) level of theory within IEFPCM of dichloromethane (isovalue = 0.02 a.u.). | ||
In comparison with TNP, the HOMO and LUMO energy levels of regioisomeric 5a and 6a are reduced, with 5a slightly more stabilized as compared to 6a. However, the calculated band gap remains constant. The expansion of the π-conjugated system leads to even more stabilized HOMO and LUMO levels for 8a, as corroborated by its red-shifted absorption and emission bands and consequently, its optical band gap. The DMA functional group as a strong donor leads to energetically elevated frontier molecular orbitals (5j and 8c) accompanied by reduced band gaps due to greater destabilization of the HOMO energies (ESI, Fig. S2†). Moreover, the presence of the DMA group (5c) leads to altered electron density distribution of the HOMO but does not affect the LUMO. In 5j, the electron density is mainly located on the DMA group and the edges of the TNP framework, whereas in TNP, 5a and 6a, it is mainly distributed across the polycyclic scaffold. However, for 8c, this effect is diminished, and the electron density is distributed across the entire molecule, which might explain why the DMA group has less impact on the oxidation behavior of 8c compared to 5j. In general, the calculated HOMO and LUMO energies correlate very well with experimental results.
To investigate the aromatic behavior, we performed nuclear-independent chemical shift (NICS) calculations and visualized the bond currents using the NICS2BC program developed by Gershoni-Poranne et al.49–52 The calculations were performed for the unsubstituted core structures 5′, 6′ and 8′ and the results were compared with those of TNP (Fig. 7). As shown in the depictions, all compounds exhibit a global diatropic ring current with strong variation in current density along the edges. However, the global ring currents depend on the relative position of the heteroatoms within the structure. Thus, stronger ring currents can be observed on the molecular side where the sulfur atom is located. However, it does not depend on the position of the nitrogen, as reflected by the determined NICS values of the central naphthalene unit. Each compound exhibits a semiglobal ring current of its inherent thienoquinoline or thienoacridine moiety. Local ring currents are observed for the thiophene, pyridine or quinoline rings. The central naphthalene moiety shows no semiglobal ring current and exhibits significantly different NICS values for both central benzene rings. The aryl ring involved in the semiglobal ring current of the thienoquinoline or thienoacridine moiety possesses high aromaticity, while the aromaticity of the aryl ring located outside this semiglobal ring current is strongly decreased. Thus, 6′ shows a significant difference as compared to 5′ and 8′, since the sulfur is located opposite to the nitrogen. When comparing 5′ with the π-expanded compound 8′, only slight differences in the NICS values are observed. Finally, TNP reveals almost identical NICS values as compared to 5′ and 6′.41
 | ||
| Fig. 7 NICS calculations for thieno[2′,3′,4′:4,5]naphtho[1,8-bc]pyridine (5′); thieno[4′,3′,2′:4,5]naphtho[1,8-bc]pyridine (6′); and thieno[2′,3′,4′:4,5]naphtho[1,8-bc]quinoline (8′). NICS2BC bond current graphs with NICS(1.7)ZZ values marked in black. Current was calculated from NICS(1.25)ZZ strength relative to Iref (ring current of benzene, 11.5 nA T−1).51 | ||
Conclusions
In conclusion, we have successfully developed an efficient and broadly applicable protocol for the synthesis of hitherto unknown thienonaphtho[bc]pyridines and thienonaphtho[bc]quinolines in very good yields by Brønsted acid mediated cycloisomerization. This methodology allows for selective access to regioisomeric products. The photophysical and electrochemical properties of the desired compounds were extensively studied and the experimental data were compared with theoretical results. Our studies revealed that the incorporation of the thiophene ring into the core structure plays a crucial role in the stabilization of the energy levels of the HOMO and LUMO. The orientation of the sulfur and nitrogen atoms within the structure has a negligible effect on the inherent optical and electrochemical properties of the parent TNP structure. Expansion of the π-conjugated system by the introduction of an additional benzene ring in the core structure however leads to significant effects on electronic structures, stabilizing the frontier molecular orbitals by reducing the HOMO–LUMO gap. It is noteworthy that the strong π-donating DMA unit of compounds 5j and 8c leads to strongly red-shifted, structureless absorption and emission properties and produces a large positive solvatochromism, suggestive of the strong ICT character of these compounds due to certain push–pull characteristics. Our findings are expected to have a significant impact on future investigations on the development and application of S- and N-doped PAHs.Experimental section
General information
Nuclear magnetic resonance spectra (1H/13C/19F NMR) were recorded on a Bruker AVANCE 300 III, 250 II, or 500 instrument. The analyzed chemical shifts (δ) are referenced to residual solvent signals from the deuterated solvents CDCl3 (δ = 7.26 ppm/77.16 ppm) or p-xylene-d10 (δ = 2.1 ppm/18.75 ppm). Multiplicities due to spin–spin correlation are reported as follows: s = singlet, d = doublet, dd = double doublet, and m = multiplet, and further described through their coupling constants J. Infrared spectra (IR) were measured as attenuated total reflection (ATR) experiments with a Nicolet 380 FT-IR spectrometer. The signals have been characterized through their wave numbers and their corresponding absorption as very strong (vs), strong (s), medium (m), weak (w), or very weak (vw). UV/Vis spectra were recorded on a Cary 60 UV–vis spectrophotometer and emission spectra were recorded using an Agilent Cary Eclipse fluorescence spectrophotometer. Cyclovoltammetry (CV) experiments were performed at room temperature in a three-electrode cell with CH2Cl2 as the solvent, 0.1 M nBu4NPF6 as the supporting electrolyte, a glassy carbon working electrode, ANE2 (Ag/AgNO3 0.01 M in CH3CN) as the reference electrode, and Pt as the counter electrode (0.5 mm diameter platinum wire, 6.35 mm outer diameter). Ferrocene was used as the external standard (1 mM in CH3CN), with potentials reported vs. Fc+/Fc. The potentiostat used was a PalmSense EmStat3 blue. The working electrode is a 3 mm diameter (length 80 mm, 6.35 mm outer diameter) glassy carbon disk electrode with a KeI-F coating that was polished on a polishing pad with an aqueous alumina slurry (0.03 micro alumina powder). Basic and high-resolution mass spectra (MS/HRMS) were measured on instruments that were paired with a preceding gas chromatograph (GC) or liquid chromatograph (LC). The samples were ionized through electron impact ionization (EI) on an Agilent 6890/5973 or Agilent 7890/5977 GC-MS equipped with a HP-5 capillary column using helium carrier gas or by applying electron spray ionization (ESI) on an Agilent 1200/6210 Time-of-Flight (TOF) LC–MS. X-ray single-crystal structure analysis was performed on a Bruker Apex Kappa-II CCD diffractometer. The solvents used, acetonitrile, dioxane, toluene and xylene, were obtained as dry solvents from commercial sources and employed without further purification. Solvents for extraction and column chromatography were distilled prior to use. Other reagents, catalysts, ligands, acids and bases were utilized in purchased purity. Column chromatography was performed using Merck Silica gel 60 (particle size 40–63 μm).:1), the desired product was isolated as a white solid in 51% yield (67 mg, 0.24 mmol); mp 69–71 °C. 1H NMR (250 MHz, CDCl3) δ = 8.23 (dd, 3J = 8.4 Hz, 5J = 0.6 Hz, 1H, CHAr), 8.06 (ddd, 3J = 8.4 Hz, 4J = 1.3 Hz, 5J = 0.7 Hz, 1H, CHAr), 7.79 (ddd, 3J = 8.4 Hz, 3J = 6.9 Hz, 4J = 1.5 Hz, 1H, CHAr), 7.66 (ddd, 3J = 8.3 Hz, 3J = 7.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.48 (dd, 3J = 4.9 Hz, 4J = 3.0 Hz, 1H, CHAr), 7.42 (dd, 4J = 3.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.16 (dd, 3J = 4.9 Hz, 4J = 1.3 Hz, 1H, CHAr) ppm. 13C NMR (126 MHz, CDCl3) δ = 150.7, 146.9, 143.7, 134.9 (C), 131.4, 129.0 (CHAr), 128.9 (C), 128.3, 126.4 (CHAr), 125.7 (C), 125.6, 125.0 (CHAr) ppm. MS (EI, 70 eV): m/z (%) = 283 (16), 281 (83), 280 (19), 278 ([M]+, 100), 244 (30), 209 (63), 164 (25), 45 (26). HRMS (ESI) m/z: [M + H]+ calcd for: C13H8Cl2NS 279.9754; found 279.9757. IR (ATR, cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1475 (s), 1412 (m), 1337 (m), 1296 (s), 1107 (m), 1010 (m), 841 (s), 756 (vs), 732 (s), 653 (s), 598 (s), 470 (m).
:1), the desired alkynylated products were isolated.
= 1475 (s), 1412 (m), 1337 (m), 1296 (s), 1107 (m), 1010 (m), 841 (s), 756 (vs), 732 (s), 653 (s), 598 (s), 470 (m).
:1), the desired alkynylated products were isolated.
4,4′-[(3-Chloropyridine-2,4-diyl)bis(ethyne-2,1-diyl)]dibenzonitrile was obtained as a beige solid in 38% yield (67 mg, 0.19 mmol) (heptane/EtOAc 4:1); mp 245–250 °C. 1H NMR (300 MHz, CDCl3) δ = 8.53 (d, 3J = 4.9 Hz, 1H, CHAr), 7.77–7.72 (m, 2H, CHAr), 7.71–7.69 (m, 6H, CHAr), 7.69–7.66 (m, 1H, CHAr), 7.63 (d, 3J = 3.7 Hz, 1H, CHAr), 7.44 (d, 3J = 4.9 Hz, 1H, CHAr) ppm. 13C NMR (75 MHz, CDCl3) δ = 147.7 (CHAr), 142.1 (C), 133.2, 132.9, 132.7, 132.4, 132.3 (CHAr), 131.3, 126.7, 126.4 (C), 126.3 (CHAr), 118.3, 118.2, 113.4, 113.1 (C), 97.7, 93.1, 89.3, 87.2 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 366 (8), 365 (35), 364 (28), 363 ([M]+, 100), 328 (6), 327 (9), 300 (11), 182 (10), 175 (5). HRMS (EI) m/z: [M]+ calcd for: C23H10N3Cl 363.0558; found 363.0555. Calcd for C23H10N337Cl ([M]+) 365.0528; found 365.0539. IR (ATR, cm−1): = 2225 (m), 1601 (m), 1564 (s), 1390 (m), 1271 (m), 1038 (m), 832 (vs), 550 (vs), 469 (m).
3-Chloro-2,4-bis[(thiophen-3-yl)ethynyl]pyridine was obtained as a brown solid in 90% yield (143 mg, 0.44 mmol) (Hep/EtOAc 4:1); mp 112–115 °C. 1H NMR (500 MHz, CDCl3) δ = 8.44 (s, 1H, CHAr), 7.73–7.68 (m, 2H, CHAr), 7.37–7.32 (m, 3H, CHAr), 7.32–7.29 (m, 1H, CHAr), 7.27–7.24 (m, 1H, CHAr) ppm. 13C NMR (126 MHz, CDCl3) δ = 147.3 (CHAr), 142.7, 134.4, 132.0 (C), 131.2, 131.2, 130.2, 130.0, 126.1, 125.8, 125.4 (CHAr), 121.2, 121.0 (C), 95.1, 90.4, 85.8, 83.8 (CAlkyn) ppm. MS (EI, 70°eV): m/z (%) = 328 (10), 327 (41), 326 (21), 324 ([M]+, 100), 133 (16), 69 (10), 58 (15), 45 (41). HRMS (ESI) m/z: [M + H]+ calcd for: C17H9ClNS2 325.9865; found 325.9867. IR (ATR, cm−1): = 3101 (w), 2209 (m), 1564 (s), 1445 (m), 1366 (m), 1115 (m), 826 (m), 773 (vs), 682 (m), 618 (s), 552 (s).
General procedure (A) for the synthesis of 2,4-bis(arylethynyl)-3-(thiophen-3-yl)pyridines 1a–j
An argon flushed pressure tube was loaded with 1.0 eq. of 3-chloro-2,4-bis(arylethynyl)pyridine, 2.0 eq. of K3PO4, Pd2(dba)3 (0.05 eq.), SPhos (0.1 eq.), 2.0 eq. of 3-thienylboronic acid and 2 ml of dry toluene. The pressure tube was sealed with a Teflon cap and the reaction mixture was stirred in a stainless-steel heating block for 24 h at 110 °C. After cooling to room temperature, the reaction was quenched with distilled water and extracted three times with ethyl acetate. The combined organic phases were dried over Na2SO4 and filtered, and the solvent was distilled in vacuo. After purification by column chromatography (Hep/EtOAc 4:1), the desired products 1a–j were isolated.
:1); mp 115–119 °C. 1H NMR (300 MHz, CDCl3) δ = 8.55 (d, 3J = 5.0 Hz, 1H, CHAr), 7.74 (dd, 4J = 2.9 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.52 (dd, 3J = 5.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.45 (dd, 3J = 5.0 Hz, 4J = 2.9 Hz, 1H, CHAr), 7.43 (d, 3J = 5.0 Hz, 1H, CHAr), 7.41–7.38 (m, 2H, CHAr), 7.37–7.30 (m, 8H, CHAr) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.4 (CHAr), 142.8, 136.4, 136.2 (C), 132.1, 131.9 (CHAr), 131.4 (C), 129.8, 129.4, 129.1, 128.6, 128.5, 126.5, 125.2, 124.2 (CHAr), 122.4, 122.2 (C), 97.5, 92.8, 88.7, 86.7 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 362 (20), 361 (70), 360 ([M]+, 100), 359 (37), 358 (12). HRMS (EI) m/z: [M]+ calcd for: C25H15NS 360.0841; found 360.0834. IR (ATR, cm−1): = 2921 (vs), 2851 (s), 2213 (w), 1559 (m), 1717 (m), 838 (m), 752 (s), 688 (s), 653 (s), 523 (m).
:1). 1H NMR (300 MHz, CDCl3) δ = 8.52 (d, 3J = 5.1 Hz, 1H, CHAr), 7.73 (dd, 4J = 3.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.51 (dd, 3J = 5.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.43 (dd, 3J = 5.0 Hz, 4J = 3.0 Hz, 1H, CHAr), 7.39 (d, 3J = 5.0 Hz, 1H, CHAr), 7.32–7.26 (m, 3H, CHAr), 7.25–7.23 (m, 1H, CHAr), 7.16–7.09 (m, 4H, CHAr), 2.36 (s, 3H, CH3), 2.35 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.4 (CHAr), 142.9, 139.8, 139.4, 136.5, 135.9 (C), 132.0, 131.9 (CHAr), 131.5 (C), 129.9, 129.4, 129.2, 126.5, 124.9, 124.1 (CHAr), 119.4, 119.2 (C), 97.8, 93.1, 88.3, 86.3 (CAlkyn), 21.7 (CH3) ppm. MS (EI, 70°eV): m/z (%) = 390 (30), 389 ([M]+, 100), 388 (81), 387 (16), 386 (12), 375 (11), 374 (49), 373 (28), 372 (12), 359 (12), 91 (10). HRMS (ESI) m/z: [M + H]+ calcd for: C27H20NS 390.1316; found 390.1316. IR (ATR, cm−1): = 2209 (m), 1559 (s), 1508 (s), 1417 (s), 1176 (m), 960 (m), 811 (vs), 768 (s), 677 (m), 653 (s), 525 (s).
:1). 1H NMR (300 MHz, CDCl3) δ = 8.53 (d, 3J = 5.0 Hz, 1H, CHAr), 7.74 (dd, 4J = 3.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.53 (dd, 3J = 5.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.45 (dd, 3J = 5.0 Hz, 4J = 3.0 Hz, 1H, CHAr), 7.40 (d, 3J = 5.0 Hz, 1H, CHAr), 7.24–7.11 (m, 8H, CHAr), 2.33 (s, 3H, CH3), 2.32 (s, 3H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.3 (CHAr), 142.8, 138.3, 138.1, 136.4, 136.0 (C), 132.6, 132.4 (CHAr), 131.4 (C), 130.3, 130.0, 129.9, 129.2, 129.0, 128.4, 128.3, 126.5, 125.1, 124.1 (CHAr), 122.2, 122.0 (C), 97.7, 93.0, 88.4, 86.4 (CAlkyn), 21.3, 21.3 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 390 (30), 389 ([M]+, 100), 388 (79), 387 (15), 374 (43), 373 (28), 372 (15), 359 (15), 187 (16), 140 (24), 91 (39), 89 (17), 65 (36), 45 (20), 39 (26). HRMS (EI) m/z: [M]+ calcd for: C27H19NS 389.1232; found 389.1220. IR (ATR, cm−1): = 2206 (m), 1559 (s), 1483 (m), 1417 (s), 1090 (m), 836 (m), 780 (vs), 653 (s), 544 (s), 517 (m).
:1). 1H NMR (300 MHz, CDCl3) δ = 8.57 (d, 3J = 5.1 Hz, 1H, CHAr), 7.61 (dd, 4J = 2.8 Hz, 4J = 1.5 Hz, 1H, CHAr), 7.43 (d, 3J = 5.1 Hz, 1H, CHAr), 7.42–7.40 (m, 1H, CHAr), 7.40–7.37 (m, 2H, CHAr), 7.35–7.31 (m, 1H, CHAr), 7.25–7.21 (m, 1H, CHAr), 7.21–7.18 (m, 1H, CHAr), 7.18–7.09 (m, 4H, CHAr), 2.22 (s, 6H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.6 (CHAr), 143.3, 141.0, 140.9, 136.9, 136.1 (C), 132.8, 132.5 (CHAr), 132.0 (C), 129.7, 129.6, 129.4, 129.1, 125.9, 125.8, 125.6, 125.0, 124.9 (CHAr), 122.2, 122.0 (C), 96.6, 92.2, 91.8, 90.2 (CAlkyn), 20.4, 20.4 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 390 (30), 389 ([M]+, 100), 388 (97), 387 (29), 386 (19), 374 (27), 356 (29), 355 (32), 354 (38), 353 (15), 342 (23), 341 (32), 340 (24), 298 (22), 297 (16). HRMS (ESI) m/z: [M + H]+ calcd for: C27H20NS 390.1316; found 390.1312. IR (ATR, cm−1): = 2206 (m), 1559 (s), 1485 (m), 1415 (s), 1110 (w), 836 (m), 754 (vs), 712 (s), 655 (s), 546 (m), 457 (m).
:1); mp 124–126 °C.1H NMR (250 MHz, CDCl3) δ = 8.54 (d, 3J = 5.0 Hz, 1H, CHAr), 7.70 (dd, 4J = 2.8 Hz, 4J = 1.5 Hz, 1H, CHAr), 7.47 (dd, 3J = 5.0 Hz, 4J = 2.1 Hz, 2H, CHAr), 7.41 (d, 3J = 5.0 Hz, 1H, CHAr), 7.39–7.29 (m, 4H, CHAr), 7.06–6.96 (m, 4H, CHAr) ppm. 19F NMR (282 MHz, CDCl3) δ = −108.82 (s, 1F, CF), −109.33 (s, 1F, CF) ppm. 13C NMR (63 MHz, CDCl3) δ = 165.2 (d, 1JC,F = 251.5 Hz), 163.1 (d, 1JC,F = 250.9 Hz, C), 148.5 (CHAr), 142.7, 136.4, 136.2 (C), 134.2 (d, 3JC,F = 8.5 Hz), 134.0 (d, 3JC,F = 8.5 Hz, CHAr), 131.3 (C), 129.8, 126.4, 125.0, 124.3 (CHAr), 118.5 (d, 4JC,F = 3.5 Hz), 118.3 (d, 4JC,F = 3.6 Hz, C), 116.2 (d, 2JC,F = 22.2 Hz), 115.9 (d, 2JC,F = 22.2 Hz, CHAr), 96.5, 91.8, 88.4, 86.4 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 398 (20), 397 (84), 396 ([M]+, 100), 395 (29), 75 (11). HRMS (EI) m/z: [M]+ calcd for: C25H13NSF2 396.0653; found 396.0646. IR (ATR, cm−1): = 2211 (m), 1559 (m), 1504 (s), 1228 (s), 1153 (s), 830 (vs), 764 (s), 653 (s), 647 (s), 527 (s).
:1); mp 154–156 °C. 1H NMR (300 MHz, CDCl3) δ = 8.59 (d, 3J = 5.0 Hz, 1H, CHAr), 7.70 (qt, 4J = 2.1 Hz, 1H, CHAr), 7.63–7.54 (m, 4H, CHAr), 7.51–7.41 (m, 7H, CHAr) ppm. 19F NMR (282 MHz, CDCl3) δ = −62.96 (s, 3F, CF3), −62.99 (s, 3F, CF3) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.7 (CHAr), 142.4, 136.9, 136.1 (C), 132.3, 132.2 (CHAr), 131.0 (q, 2JC,F = 32.3 Hz, C), 129.6 (CHAr), 126.6 (CHAr), 126.1 (d, 4JC,F = 1.4 Hz), 125.9 (d, 4JC,F = 1.4 Hz, C), 125.7 (C), 125.6 (q, 3JC,F = 4.2 Hz), 125.5 (q, 3JC,F = 3.9 Hz, CHAr), 124.7 (CHAr), 122.1, 122.0 (C), 95.9, 91.2, 90.5, 88.4 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 498 (34), 497 ([M]+, 100), 496 (99), 495 (18), 478 (23), 429 (21), 428 (74), 427 (47), 426 (24), 69 (47), 45 (17). HRMS (EI) m/z: [M]+ calcd for: C27H13NSF6 497.0667; found 497.0655. IR (ATR, cm−1): = 1611 (w), 1561 (m), 1318 (s), 1164 (s), 1102 (vs), 1063 (vs), 1013 (s), 836 (vs), 801 (s), 593 (s).
:1); mp 224–226 °C. 1H NMR (300 MHz, CDCl3) δ = 8.61 (d, 3J = 5.1 Hz, 1H, CHAr), 7.69 (dd, 4J = 2.9 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.66–7.58 (m, 4H, CHAr), 7.55–7.39 (m, 7H, CHAr) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.3 (CHAr), 143.8, 141.6, 137.2, 135.7 (C), 132.6, 132.5, 132.3, 132.2 (CHAr), 131.2 (C), 129.5 (CHAr), 126.9 (C), 126.8 (CHAr), 126.7 (C), 125.9, 125.0 (CHAr), 118.4, 118.3, 113.0, 112.7 (C), 96.0, 89.9 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 413 (7), 412 (20), 411 (70), 410 ([M]+, 100), 409 (18), 408 (7), 102 (6), 75 (5), 45 (7). HRMS (EI) m/z: [M]+ calcd for: C27H13N3S 410.0746; found 410.0743. IR (ATR, cm−1): = 2225 (m), 1603 (m), 1559 (m), 1502 (m), 834 (vs), 762 (s), 649 (m), 552 (vs).
:1); mp 97–100 °C. 1H NMR (250 MHz, CDCl3) δ = 8.53 (d, 3J = 5.1 Hz, 1H, CHAr), 7.73 (dd, 4J = 2.9 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.51 (dd, 3J = 5.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.49–7.46 (m, 1H, CHAr), 7.47–7.37 (m, 3H, CHAr), 7.32–7.21 (m, 2H, CHAr), 7.11–7.02 (m, 2H, CHAr) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.2 (CHAr), 142.6, 136.2, 135.8, 131.4 (C), 130.4, 129.9, 129.8, 129.7, 126.5, 125.9, 125.6, 124.9, 124.1 (CHAr), 121.5, 121.3 (C), 93.0, 88.4, 88.2, 86.4 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 375 (11), 374 (35), 373 ([M]+, 90), 372 (100), 371 (25), 340 (18), 339 (14), 328 (17), 327 (10), 69 (12), 58 (14), 45 (68). HRMS (ESI) m/z: [M + H]+ calcd for: C21H12NS3 374.0132; found 374.0132. IR (ATR, cm−1): = 2204 (m), 1561 (m), 1024 (s), 989 (s), 780 (vs), 690 (m), 622 (vs), 554 (s).
:1); mp 116–122 °C. 1H NMR (250 MHz, CDCl3) δ = 8.50 (d, 3J = 5.1 Hz, 1H, CHAr), 7.72 (dd, 4J = 2.9 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.51 (dd, 3J = 5.0 Hz, 4J = 1.3 Hz, 1H, CHAr), 7.43 (dd, 3J = 5.0 Hz, 4J = 2.9 Hz, 1H, CHAr), 7.39–7.33 (m, 2H, CHAr), 7.33–7.26 (m, 3H, CHAr), 6.89–6.79 (m, 4H, CHAr), 3.81 (s, 3H, OMe), 3.80 (s, 3H, OMe) ppm. 13C NMR (63 MHz, CDCl3) δ = 160.5, 160.3 (C), 148.3 (CHAr), 143.0, 136.6, 135.6 (C), 133.7, 133.6 (CHAr), 131.6 (C), 130.0, 126.4, 124.6, 124.0 (CHAr), 114.5, 114.3 (C), 114.3, 114.2 (CHAr), 97.9, 93.1, 87.8, 85.8 (CAlkyn), 55.5, 55.4 (OMe) ppm. MS (EI, 70 eV): m/z (%) = 422 (31), 421 ([M]+, 100), 420 (17), 406 (21), 377 (17), 335 (21), 334 (33), 333 (15). HRMS (EI) m/z: [M]+ calcd for: C27H19NSO2 421.1131; found 421.1123. IR (ATR, cm−1): = 2209 (m), 1603 (s), 1561 (s), 1506 (s), 1390 (m), 1246 (s), 1020 (m), 826 (vs), 550 (s).
:1); mp 120–125 °C. 1H NMR (250 MHz, CDCl3) δ = 8.43 (d, 3J = 5.3 Hz, 1H, CHAr), 7.75 (dd, 4J = 3.0, 4J = 1.3 Hz, 1H, CHAr), 7.54 (dd, 3J = 5.0, 4J = 1.3 Hz, 1H, CHAr), 7.43 (dd, 3J = 5.0, 4J = 3.0 Hz 1H, CHAr), 7.36–7.28 (m, 3H, CHAr), 7.25–7.19 (m, 2H, CHAr), 6.65–6.56 (m, 4H, CHAr), 3.00 (s, 6H, CH3), 2.99 (s, 6H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ = 150.8, 150.7 (C), 147.7 (CHAr), 142.9, 136.8, 134.8 (C), 133.4, 133.3 (CHAr), 132.3 (C), 130.2, 126.4, 123.8, 123.7, 111.8 (CHAr), 109.0, 108.7 (C), 99.9, 95.3, 87.4, 85.7 (CAlkyn), 40.2, 40.2 (CH3) ppm. MS (EI, 70°eV): m/z (%) = 448 (33), 447 ([M]+, 100), 446 (18), 201 (27), 69 (28), 57 (19), 44 (57), 43 (22). HRMS (EI) m/z: [M]+ calcd for: C29H25N3S 447.1763; found 447.1750. IR (ATR, cm−1): = 2206 (m), 1599 (m), 1559 (s), 1504 (s), 1226 (s), 960 (m), 828 (vs), 647 (s), 527 (s).
General procedure (B) for the synthesis of 2,4-bis(arylethynyl)-3-(thiophen-3-yl)quinolines 2a–c
An argon flushed pressure tube was loaded with 1.0 eq. of 2,4-dichloro-3-(thiophen-3-yl)quinoline (352 mg), 0.06 eq. of PdCl2(CH3CN)2, 0.04 eq. of CuI, 0.12 eq. of XPhos, 4 eq. of HNiPr2 and 2 ml of dioxane. Then 4.0 eq. of the respective alkyne was added to the solution with stirring. The pressure tube was sealed with a Teflon cap and the reaction mixture was stirred in a stainless-steel heating block for 24 h at 100 °C. After cooling to room temperature, the reaction was quenched with distilled water and extracted three times with ethyl acetate. The combined organic phases were dried over Na2SO4 and filtered, and the solvent was distilled in vacuo. After purification by column chromatography (heptane/EtOAc 5:1 (1:1 for 2c)), the desired products 2a–c were isolated.
:1); mp 167–169 °C. 1H NMR (500 MHz, CDCl3) δ = 8.40 (d, 3J = 8.2 Hz, 1H, CHAr.), 8.18 (d, 3J = 8.4 Hz, 1H, CHAr.), 7.80–7.74 (m, 2H, CHAr), 7.66 (qt, 3J = 7.6 Hz, 1H, CHAr), 7.57 (dd, 3J = 4.9 Hz, 4J = 0.7 Hz, 1H, CHAr.), 7.50 (dd, 3J = 4.9 Hz, 4J = 3.0 Hz, 1H, CHAr.), 7.48–7.41 (m, 4H, CHAr), 7.40–7.30 (m, 6H, CHAr) ppm. 13C NMR (126 MHz, CDCl3) δ = 147.1, 143.2, 137.2, 134.1 (C), 132.3, 132.0, 130.3, 130.2, 129.6, 129.5, 129.3 (CHAr.), 128.9 (C), 128.7, 128.5, 128.1 (CHAr.), 126.8 (C), 126.7, 126.4, 124.2 (CHAr.), 122.4, 122.3 (C), 102.6, 93.5, 89.3, 85.0 (CAlkyn). MS (EI, 70 eV): m/z (%) = 412 (28), 411 (80), 410 ([M]+, 100), 409 (17), 408 (18), 378 (6), 334 (10), 77 (14). HRMS (EI) m/z: [M]+ calcd for: C29H17NS 410.0998; found 410.0993. IR (ATR, cm−1): = 1492 (m), 1346 (m), 837 (m), 748 (vs), 723 (m), 684 (s), 664 (s), 610 (m), 525 (s), 495 (m).
:1); mp 190–194 °C. 1H NMR (250 MHz, CDCl3) δ = 8.34 (dd, 3J = 8.3 Hz, 4J = 1.5 Hz, 1H, CHAr.), 8.20–8.13 (m, 1H, CHAr.), 7.81–7.72 (m, 2H, CHAr), 7.65 (ddd, 3J = 8.2 Hz, 3J = 6.9 Hz, 4J = 1.3 Hz, 1H, CHAr.), 7.51 (dd, 3J = 5.0 Hz, 4J = 1.4 Hz, 1H, CHAr.), 7.49 (dd, 3J = 5.0 Hz, 4J = 2.8 Hz, 1H, CHAr.), 7.46–7.34 (m, 4H, CHAr), 7.11–6.95 (m, 4H, CHAr) ppm. 19F NMR (471 MHz, CDCl3) δ = −108.6 (s, 1F, CF), −108.9 (s, 1F, CF) ppm. 13C NMR (63 MHz, CDCl3) δ = 163.3 (d, 1JC,F = 251.7 Hz). 163.2 (d, 1JC,F = 251.3 Hz, C), 147.0, 143.0, 137.2 (C), 134.3 (d, 3JC,F = 8.6 Hz), 134.0 (d, 3JC,F = 8.5 Hz, CHAr), 130.4, 130.2, 129.6 (CHAr), 128.8 (C) 128.2 (CHAr), 126.7 (C), 126.6, 126.3, 124.3 (CHAr), 118.5 (d, 4JC,F = 3.5 Hz), 118.3 (d, 4JC,F = 3.5 Hz, C), 116.2 (d, 2JC,F = 22.2 Hz), 115.8 (d, 2JC,F = 22.2 Hz, CHAr), 101.5, 92.5 (CAlkyn), 88.9 (d, 5JC,F = 1.3 Hz), 84.8 (d, 5JC,F = 1.4 Hz, CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 449 (7), 448 (28), 447 ([M]+, 96), 446 (100), 445 (14), 444 (9), 223 (8), 222 (10), 213 (8). HRMS (ESI) m/z: [M + H]+ calcd for: C29H15F2NS 448.0971; found 448.0971. IR (ATR, cm−1): = 1504 (s), 1214 (s), 1152 (s), 830 (vs), 808 (s), 758 (vs), 528 (s), 499 (s), 453 (s).
:1); mp 112–114 °C. 1H NMR (500 MHz, CDCl3) δ = 8.39 (d, 3J = 8.1 Hz, 1H, CHAr.), 8.12 (d, 3J = 8.4 Hz, 1H, CHAr.), 7.79–7.75 (m, 1H, CHAr.), 7.71 (qt, 3J = 7.1 Hz, 1H, CHAr.), 7.62–7.57 (m, 2H, CHAr), 7.46 (dd, 3J = 5.0 Hz, 4J = 3.0 Hz, 1H, CHAr.), 7.34–7.29 (m, 4H, CHAr), 6.65–6.59 (m, 4H, CHAr), 2.99 (s, 6H, CH3), 2.98 (s, 6H, CH3) ppm. 13C NMR (126 MHz, CDCl3) δ = 150.8, 150.7, 147.1, 143.8, 137.7 (C), 133.7, 133.4 (CHAr), 132.7 (C), 130.6, 129.9 (CHAr), 129.5 (C), 129.2, 127.2, 126.6 (CHAr), 126.4 (C), 123.6, 111.8, 111.7 (CHAr), 109.0, 108.8 (C), 104.9, 95.8, 88.4, 84.1 (CAlkyn), 40.2 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 497 ([M]+, 45), 449 (52), 367 (100), 278 (91), 214 (94), 97 (66), 83 (55), 57 (53), 55 (79). HRMS (EI) m/z: [M]+ calcd for: C33H27N3S 497.1920; found 497.1923. IR (ATR, cm−1): = 2186 (s), 1601 (vs), 1519 (vs), 1341 (vs), 1183 (s), 985 (s), 944 (s), 812 (vs), 758 (vs), 674 (s), 523 (s).
General procedure (C) for the synthesis of 4-aryl-9-(arylethynyl)thieno[3,2-f]quinolines (3a–j)
:1) afforded the desired derivatives 3a–j, 4a–h and 7a–c as pure compounds.
:1); mp 149–150 °C. 1H NMR (250 MHz, CDCl3) δ = 9.26 (d, 3J = 5.6 Hz, 1H, CHAr), 8.91 (d, 3J = 4.6 Hz, 1H, CHAr), 8.07 (s, 1H, CHAr), 7.87–7.80 (m, 2H, CHAr), 7.76–7.69 (m, 4H, CHAr), 7.61–7.44 (m, 6H, CHAr) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.6 (C), 148.2 (CHAr), 140.0, 139.9, 139.3, 135.7 (C), 131.8, 129.6, 129.1, 128.9, 128.9, 128.6 (CHAr), 127.5 (C), 126.9, 126.0, 125.8, 125.7 (CHAr), 122.7, 122.6 (C), 98.8, 89.2 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 363 (5), 362 (20), 361 (100), 360 ([M]+, 69), 359 (29), 358 (12), 357 (6). HRMS (EI) m/z: [M]+ calcd for: C25H15NS 360.0842; found 360.0834. IR (ATR, cm−1): = 2197 (w), 1556 (m), 1334 (m), 875 (m), 810 (vs), 743 (s), 626 (m), 495 (s).
:1); mp 193–196 °C. 1H NMR (250 MHz, CDCl3) δ = 9.39 (d, 3J = 5.6 Hz, 1H, CHAr), 9.02 (d, 3J = 4.6 Hz, 1H, CHAr), 8.19 (s, 1H, CHAr), 7.89 (s, 1H, CHAr), 7.88–7.80 (m, 1H, CHAr), 7.74 (d, 3J = 7.8 Hz, 1H, CHAr), 7.51 (d, 3J = 8.1 Hz, 1H, CHAr), 7.41 (d, 3J = 7.5 Hz, 1H, CHAr), 2.62 (s, 3H, CH3), 2.58 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.4 (C), 147.9 (CHAr), 140.0, 139.3, 138.8, 136.9, 135.6 (C), 131.7, 129.8, 129.6, 128.5 (CHAr), 127.8 (C), 126.8, 125.7, 125.5 (CHAr), 122.6, 119.5 (C), 99.3, 88.7 (CAlkyn), 21.8, 21.5 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 391 (11), 390 (37), 389 ([M]+, 100), 387 (9), 386 (7), 374 (14), 373 (18), 372 (9), 359 (10), 186 (23). HRMS (EI) m/z: [M]+ calcd for: C27H19NS 389.1232; found 389.1224. IR (ATR, cm−1): = 2200 (w), 1555 (m), 1327 (m), 886 (m), 811 (vs), 743 (s), 694 (s), 616 (m), 498 (s).
:1); mp 135–137 °C. 1H NMR (250 MHz, CDCl3) δ = 9.25 (d, 3J = 5.6 Hz, 1H, CHAr), 8.89 (d, 3J = 4.3 Hz, 1H, CHAr), 8.06 (s, 1H, CHAr), 7.73–7.61 (m, 4H, CHAr), 7.57–7.50 (m, 2H, CHAr), 7.49–7.41 (m, 1H, CHAr), 7.40–7.28 (m, 3H, CHAr), 2.49 (s, 3H, CH3), 2.44 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.5 (C), 148.1 (CHAr), 139.9, 139.8, 139.3, 138.7, 138.6, 135.6 (C), 132.2, 130.5, 129.6, 129.3, 128.9, 128.7 (CHAr), 127.4 (C), 126.7, 125.9, 125.6 (CHAr), 122.6, 122.4 (C), 99.0, 88.8 (CAlkyn), 21.7, 21.4 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 390 (30), 389 ([M]+, 100), 388 (21), 374 (14), 373 (17), 359 (11), 186 (18). HRMS (EI) m/z: [M]+ calcd for: C27H19NS 389.1232; found 389.1225. IR (ATR, cm−1): = 1555 (m), 1481 (m), 1329 (m), 1088 (m), 873 (m), 735(s), 677 (s), 548 (m), 434 (m).
:1); mp 135–137 °C. 1H NMR (500 MHz, CDCl3) δ = 9.26 (d, 3J = 5.6 Hz, 1H, CHAr), 8.92 (d, 3J = 4.6 Hz, 1H, CHAr), 7.97 (s, 1H, CHAr), 7.77 (d, 3J = 4.6 Hz, 1H, CHAr), 7.73–7.65 (m, 2H, CHAr), 7.48–7.27 (m, 7H, CHAr), 2.65 (s, 3H, CH3), 2.22 (s, 3H, CH3) ppm. 13C NMR (126 MHz, CDCl3) δ = 147.6, 141.7, 140.8, 139.4, 139.1, 136.2, 135.0 (C), 132.3, 130.7, 130.1, 129.7, 129.5, 128.8, 127.3, 126.1, 126.1, 125.8, 125.7 (CHAr), 122.7, 122.3 (C), 92.7 (CAlkyn), 21.1, 20.0 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 390 (29), 389 ([M]+, 100), 388 (61), 387 (18), 386 (10), 374 (11), 372 (10), 298 (14), 297 (11), 91 (13). HRMS (EI) m/z: [M]+ calcd for: C27H19NS 389.1232; found 389.1222. IR (ATR, cm−1): = 2198 (w), 1555 (m), 1327 (m), 96 (m), 836 (m), 754 (vs), 729 (s), 688 (s), 560 (m), 453 (m).
:1); mp 167–169 °C. 1H NMR (300 MHz, CDCl3) δ = 9.19 (d, 3J = 5.6 Hz, 1H, CHAr), 8.90 (d, 3J = 4.6 Hz, 1H, CHAr), 8.03 (d, 3J = 2.1 Hz, 1H, CHAr), 7.84–7.75 (m, 2H, CHAr), 7.75–7.65 (m, 4H, CHAr), 7.30–7.12 (m, 4H, CHAr) ppm. 19F NMR (282 MHz, CDCl3) δ = −108.50 (s, 1F, CF), −112.73 (s, 1F, CF) ppm. 13C NMR (75 MHz, CDCl3) δ = 163.4 (d, 1JC,F = 251.8 Hz), 163.2 (d, 1JC,F = 248.3 Hz, C), 148.2 (C), 148.1 (CHAr), 140.0, 138.4 (C), 135.7 (d, 4JC,F = 3.4 Hz, C), 135.6 (C), 133.8 (d, 3JC,F = 8.6 Hz), 130.4 (d, 3JC,F = 8.2 Hz, CHAr) 127.5 (C), 127.0, 125.8, 125.6 (CHAr), 122.7 (C), 118.6 (d, 4JC,F = 3.6 Hz, C), 116.34 (d, 2JC,F = 22.2 Hz), 116.10 (d, 2JC,F = 21.6 Hz, CHAr), 98.0, 88.8 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 399 (7), 398 (27), 397 ([M]+, 100), 396 (47), 395 (26), 198 (5), 75 (4). HRMS (EI) m/z: [M]+ calcd for: C25H13NSF2 397.0731; found 397. 0725. IR (ATR, cm−1): = 2206 (w), 1599 (m), 1557 (m), 1506 (s), 1221 (s), 1156 (s), 828 (vs), 805 (s), 735 (s), 496 (s).
:1); mp 80–82 °C. 1H NMR (250 MHz, CDCl3) δ = 9.13 (d, 3J = 5.5 Hz, 1H, CHAr), 8.83 (d, 3J = 4.3 Hz, 1H, CHAr), 8.11 (s, 1H, CHAr), 7.86 (s, 1H, CHAr), 7.74–7.58 (m, 4H, CHAr), 7.53–7.30 (m, 3H, CHAr) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.2 (C), 148.1 (CHAr), 140.1, 139.1, 135.7, 133.6 (C), 130.2, 129.5, 127.8 (CHAr), 127.3 (C), 126.5, 126.4, 126.2, 125.5, 125.2, 124.0 (CHAr), 122.4, 121.6 (C), 94.2, 88.7 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 375 (13), 374 (28), 373 ([M]+, 100), 372 (42), 371 (19), 339 (7), 328 (8), 45 (16). HRMS (ESI) m/z: [M + H]+ calcd for: C21H12NS3 374.0132; found 374.0132. IR (ATR, cm−1): = 2202 (w), 1555 (s), 1314 (m), 869(m), 778 (vs), 731 (vs), 622 (s), 556 (s).
:1); mp 246–249 °C. 1H NMR (300 MHz, CDCl3) δ = 9.32 (d, 3J = 5.6 Hz, 1H, CHAr), 8.80 (d, 3J = 4.9 Hz, 1H, CHAr), 8.09 (s, 1H, CHAr), 7.81–7.74 (m, 2H, CHAr), 7.73 (d, 3J = 5.6 Hz, 1H, CHAr), 7.65 (d, 3J = 4.9 Hz, 1H, CHAr), 7.62–7.56 (m, 2H, CHAr), 6.90–6.84 (m, 2H, CHAr), 6.78–6.71 (m, 2H, CHAr), 3.06 (s, 6H, CH3), 3.06 (s, 6H, CH3) ppm. 13C NMR (75 MHz, CDCl3) δ = 151.1, 150.9, 140.3, 135.7 (C), 133.3, 129.5 (CHAr), 127.2 (C), 126.9, 125.8, 124.4 (CHAr), 122.3 (C), 112.5, 112.0 (CHAr), 108.7 (C), 88.3 (CAlkyn), 40.5, 40.3 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 450 (7), 449 (38), 448 (85), 447 ([M]+, 100), 433 (12), 432 (11), 431 (26), 430 (19), 224 (13), 223 (34). HRMS (EI) m/z: [M]+ calcd for: C29H25N3S 447.1763; found 447.1758.
:1); mp 155–157 °C. 1H NMR (300 MHz, CDCl3) δ = 9.35 (d, 3J = 5.6 Hz, 1H, CHAr), 8.67 (d, 3J = 5.4 Hz, 1H, CHAr), 7.82–7.73 (m, 6H, CHAr), 7.69 (s, 1H, CHAr), 7.61–7.50 (m, 3H, CHAr), 7.50–7.43 (m, 3H, CHAr) ppm. 13C NMR (63 MHz, CDCl3) δ = 143.3 (CHAr), 141.0, 140.5, 140.2, 139.7, 136.4, 135.2 (C), 132.1, 129.5, 129.1, 128.8, 128.6, 127.4, 125.7 (CHAr), 124.3 (C), 122.9 (CHAr), 122.6 (C), 121.7 (CHAr), 94.0, 91.2 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 363 (8), 362 (27), 361 (100), 360 ([M]+, 52), 359 (31), 358 (6), 357 (5). HRMS (ESI) m/z: [M + H]+ calcd for: C25H16NS 361.1003; found 361.1006. IR (ATR, cm−1): = 2200 (w), 1532 (m), 1256 (m), 870 (s), 681 (vs), 565 (s), 43 (m).
:1). 1H NMR (250 MHz, CDCl3) δ = 9.35 (d, 3J = 5.6 Hz, 1H, CHAr), 8.66 (d, 3J = 5.5 Hz, 1H, CHAr), 7.82–7.74 (m, 2H, CHAr), 7.69 (s, 1H, CHAr), 7.64–7.56 (m, 4H, CHAr), 7.49–7.41 (m, 1H, CHAr), 7.40–7.27 (m, 3H, CHAr), 2.49 (s, 3H, CH3), 2.43 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ = 142.9 (CHAr), 141.1, 140.6, 140.3, 139.6, 138.9, 138.5, 136.5, 135.2 (C), 132.7, 130.5, 129.9, 129.3, 129.3, 129.0, 128.7, 127.5, 125.7 (CHAr), 124.2 (C), 122.8 (CHAr), 122.3 (C), 121.7 (CHAr), 90.6 (CAlkyn), 21.7, 21.5 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 390 (30), 389 ([M]+, 100), 388 (26), 374 (15), 373 (19), 359 (11), 187 (18), 186 (13). HRMS (EI) m/z: [M]+ calcd for: C27H19NS 389.1232; found 389.1226. IR (ATR, cm−1): = 2206 (w), 1584 (m), 1522 (m), 1483 (m), 1333 (m), 1273 (m), 877 (s), 783(s), 686 (vs), 560 (s), 434 (m).
:1); mp 156–159 °C. 1H NMR (250 MHz, CDCl3) δ = 9.42 (d, 3J = 5.6 Hz, 1H, CHAr), 8.67 (d, 3J = 8.6 Hz, 1H, CHAr), 8.26 (d, 3J = 8.6 Hz, 1H, CHAr), 8.10 (s, 1H, CHAr), 7.90–7.85 (m, 2H, CHAr), 7.85–7.75 (m, 3H, CHAr), 7.71–7.47 (m, 8H, CHAr) ppm. 13C NMR (63 MHz, CDCl3) δ = 149.2, 147.2, 140.0, 139.7, 139.6, 135.5 (C), 131.8, 130.0, 129.7, 129.7, 129.0, 129.0, 128.6 (CHAr), 127.1 (C), 126.9, 126.6, 126.5, 126.2, 126.0 (CHAr), 124.4, 122.7, 122.0 (C), 105.8, 87.7 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 413 (10), 412 (32), 411 ([M]+, 100), 410 (28), 409 (21), 205 (17), 204 (12). HRMS (EI) m/z: [M]+ calcd for: C29H17NS 411.1076; found 411.1077. IR (ATR, cm−1): = 1498 (m), 1440 (m), 1341 (m), 878 (m), 746 (vs), 696 (s), 684 (s), 666 (s), 600 (m), 571 (m), 509 (m), 441 (m).
:1); mp 210–212 °C. 1H NMR (300 MHz, CDCl3) δ = 9.36 (d, 3J = 5.6 Hz, 1H, CHAr), 8.63 (ddd, 3J = 8.6 Hz, 4J = 1.4 Hz, 5J = 0.7 Hz, 1H, CHAr), 8.27 (d, 3J = 8.6 Hz, 1H, CHAr), 8.07 (s, 1H, CHAr), 7.86–7.75 (m, 5H, CHAr), 7.72–7.64 (m, 2H, CHAr), 7.29–7.18 (m, 4H, CHAr) ppm. 19F NMR (282 MHz, CDCl3) δ = −108.19 (s, 1F, CF), −112.56 (s, 1F, CF) ppm. 13C NMR (126 MHz, CDCl3) δ = 163.5 (d, 1JC,F = 252.3 Hz), 163.2 (d, 1JC,F = 248.6 Hz, C), 148.8, 146.9, 139.4, 138.9 (C), 135.6 (d, 4JC,F = 3.3 Hz, C), 135.4 (C), 133.8 (d, 3JC,F = 8.6 Hz, CHAr), 130.3 (d, 3JC,F = 8.2 Hz, CHAr), 130.1, 129.5, 127.0 (CHAr), 127.0 (C), 126.5, 126.3, 126.2, 125.8 (CHAr), 124.3, 121.8 (C), 118.7 (d, 4JC,F = 3.5 Hz, C), 116.4 (d, 2JC,F = 22.3 Hz, CHAr), 116.0 (d, 2JC,F = 21.6 Hz, CHAr), 104.7, 87.3 (CAlkyn) ppm. MS (EI, 70 eV): m/z (%) = 448 (31), 447 ([M]+, 100), 446 (29), 445 (19), 223 (13), 222 (8), 83 (9), 55 (9), 43 (11), 41 (7). HRMS (ESI) m/z: [M + H]+ calcd for: C29H16F2NS 448.0971; found 448.0966. IR (ATR, cm−1): = 1508 (vs), 1228 (s), 1156 (s), 826 (vs), 806 (s), 750 (vs), 705 (s), 670 (s), 600 (m), 523 (s), 480 (s), 457 (m).
:1); mp 124–126 °C. 1H NMR (500 MHz, CDCl3) δ = 9.55 (d, 3J = 5.5 Hz, 1H, CHAr.), 8.73 (d, 3J = 8.5 Hz, 1H, CHAr), 8.27 (d, 3J = 8.6 Hz, 1H, CHAr), 8.09 (s, 1H, CHAr), 7.85–7.78 (m, 3H, CHAr), 7.74–7.68 (m, 3H, CHAr), 7.67–7.62 (m, 1H, CHAr), 6.89 (d, 3J = 8.6 Hz, 2H, CHAr), 6.77 (d, 3J = 8.5 Hz, 2H, CHAr), 3.07 (s, 6H, CH3), 3.07 (s, 6H, CH3) ppm. 13C NMR (126 MHz, CDCl3) δ = 150.9, 150.7, 139.6, 135.6 (C), 133.2, 133.0, 129.7, 129.4 (CHAr), 127.4 (C), 126.8, 126.7 (CHAr), 126.6 (C), 126.6, 126.0, 125.7 (CHAr), 121.3, 120.2 (C), 112.3, 111.9 (CHAr), 109.0, 86.7 (CAlkyn), 40.4, 40.1 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 499 (21), 498 (43), 497 ([M]+, 100), 483 (16), 481 (17), 248 (39), 247 (15), 204 (18), 118 (32), 57 (19), 58 (18), 44 (31), 43 (21). HRMS (ESI) m/z: [M + H]+ calcd for: C33H27N3S 498.2004; found 498.2003.
General procedure (D) for the synthesis of diarylthieno[2′,3′,4′:4,5]naphtho[1,8-bc]pyridines (5a–j)
:1) afforded products 5a–j, 6a–b and 8a–c.
:1); mp 160–163 °C. 1H NMR (300 MHz, CDCl3) δ = 9.17 (d, 3J = 5.0 Hz, 1H, CHAr), 8.39 (s, 1H, CHAr), 8.21 (s, 1H, CHAr), 8.03–7.98 (m, 2H, CHAr), 7.84–7.78 (m, 2H, CHAr), 7.79–7.72 (m, 1H, CHAr), 7.68–7.51 (m, 7H, CHAr) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.9 (CHAr), 147.3, 140.7, 139.8, 139.4, 136.9, 133.1, 132.9, 132.7 (C), 129.4, 129.1, 128.9, 128.8, 128.5, 124.2, 124.2, 123.7 (CHAr), 118.3 (C), 116.2 (CHAr) ppm. MS (EI, 70 eV): m/z (%) = 363 (7), 362 (26), 361 ([M]+, 100), 360 (29), 359 (23), 358 (5), 357 (5), 180 (15). HRMS (ESI) m/z: [M + H]+ calcd for: C25H16NS 362.1003; found 362.1003. IR (ATR, cm−1): = 1578 (m), 1467 (m), 1372 (m), 1075 (m), 877 (m), 760 (s), 692 (vs), 597 (s), 465 (m).
:1); mp 221–225 °C. 1H NMR (250 MHz, CDCl3) δ = 9.15 (d, 3J = 5.0 Hz, 1H, CHAr), 8.34 (s, 1H, CHAr), 8.19 (s, 1H, CHAr), 7.90 (d, 3J = 8.1 Hz, 2H, CHAr), 7.76–7.68 (m, 3H, CHAr), 7.62 (s, 1H, CHAr), 7.47–7.34 (m, 4H, CHAr), 2.50 (s, 3H, CH3), 2.49 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.9 (CHAr), 147.5, 140.6, 139.3, 138.8, 138.8, 137.0, 136.9, 136.6, 133.1, 132.9, 132.8 (C), 130.0, 129.8, 128.7, 128.4, 124.0, 124.0, 123.4 (CHAr), 118.2 (C), 116.0 (CHAr), 21.5 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 391 (7), 390 (25), 389 ([M]+, 100), 388 (7), 373 (7), 187 (6), 186 (6). HRMS (ESI) m/z: [M + H]+ calcd for: C27H20NS 390.1316; found 390.1325. IR (ATR, cm−1): = 2901 (m), 1667 (m), 1518 (m), 1370 (s), 1162 (s), 1088(s), 842 (s), 766 (s), 712 (s), 556 (vs).
:1); mp 164–168 °C. 1H NMR (500 MHz, CDCl3) δ = 9.14 (d, 3J = 4.9 Hz, 1H, CHAr), 8.34 (s, 1H, CHAr), 8.17 (s, 1H, CHAr), 7.80 (s, 2H, CHAr), 7.71 (d, 3J = 4.9 Hz, 1H, CHAr), 7.64–7.58 (m, 3H, CHAr), 7.53–7.42 (m, 2H, CHAr), 7.33 (qt, 3J = 7.2 Hz, 2H, CHAr), 2.52 (s, 3H, CH3), 2.50 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.4 (CHAr), 146.9, 140.9, 139.6, 139.3, 139.1, 138.8, 137.0, 133.1, 132.7, 132.7 (C), 129.6, 129.6, 129.4, 129.3, 129.2, 128.9, 125.9, 125.6, 124.4, 123.7, 123.5 (CHAr), 118.1 (C), 116.0 (CHAr), 21.7, 21.7 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 391 (9), 390 (31), 389 ([M]+, 100), 388 (17), 374 (10), 373 (16), 359 (11), 187 (5), 186 (7), 179 (6), 39 (5). HRMS (ESI) m/z: [M + H]+ calcd for: C27H20NS 390.1316; found 390.1315. IR (ATR, cm−1): = 1582 (m), 1475 (m), 1397 (m), 1090 (m), 873 (m), 778 (s), 725 (s), 698 (vs), 560 (m).
:1); mp 166–171 °C. 1H NMR (250 MHz, CDCl3) δ = 9.22 (d, 3J = 5.0 Hz, 1H, CHAr), 8.20 (s, 1H, CHAr), 7.78 (d, 3J = 5.0 Hz, 1H, CHAr), 7.76 (s, 1H, CHAr), 7.57 (d, 3J = 5.9 Hz, 2H, CHAr), 7.50–7.30 (m, 7H, CHAr), 2.34 (s, 3H, CH3), 2.31 (s, 3H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ = 149.0 (CHAr), 147.1, 140.7, 139.3, 139.2, 138.6, 136.7, 136.2, 136.2, 134.7, 133.6, 132.0 (C), 130.9, 130.7, 129.8, 129.7, 128.7, 128.5, 126.2, 126.0, 125.3, 124.4, 124.2 (CHAr), 118.3 (C), 116.2 (CHAr), 20.4, 20.3 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 390 (31), 389 ([M]+, 100), 388 (61), 387 (16), 374 (15), 372 (12), 356 (11), 341 (10), 298 (14), 187 (15). HRMS (ESI) m/z: [M + H]+ calcd for: C27H20NS 390.1316; found 390.1311. IR (ATR, cm−1): = 1584 (m), 1465 (m), 1399 (m), 1046 (m), 890 (m), 758 (s), 725 (vs), 605 (s), 544 (m).
:1); mp 246–250 °C. 1H NMR (250 MHz, CDCl3) δ = 9.16 (d, 3J = 5.0 Hz, 1H, CHAr), 8.32 (s, 1H, CHAr), 8.16 (s, 1H, CHAr), 8.02–7.91 (m, 2H, CHAr), 7.83–7.73 (m, 3H, CHAr), 7.62 (s, 1H, CHAr), 7.36–7.23 (m, 4H, CHAr) ppm. 19F NMR (471 MHz, CDCl3) δ = −112.58 (s, 1F, CF) ppm. 13C NMR (63 MHz, CDCl3) δ = 165.2, 161.3 (C), 148.9 (CHAr), 147.1, 139.7, 138.4, 136.8 (C), 135.7 (d, 4JC,F = 3.4 Hz), 135.4 (d, 4JC,F = 3.4 Hz, C), 133.1, 132.8, 132.8 (C), 130.5 (d, 3JC,F = 8.1 Hz), 130.2 (d, 3JC,F = 8.2 Hz, CHAr), 124.2, 124.1, 123.8 (CHAr), 118.2 (C), 116.4 (d, 2JC,F = 21.6 Hz, CHAr), 116.3 (CHAr), 116.2 (d, 2JC,F = 21.5 Hz, CHAr) ppm. MS (EI, 70 eV): m/z (%) = 399 (12), 398 (44), 397 ([M]+, 100), 396 (48), 395 (33), 393 (5), 368 (5), 198 (9). HRMS (ESI) m/z: [M + H]+ calcd for: C25H14F2NS 398.0815; found 398.0821. IR (ATR, cm−1): = 1603 (m), 1504 (vs), 1224 (vs), 1153 (s), 1098 (s), 830 (vs), 731 (s), 531 (s), 502 (vs).
:1); mp 179–185 °C. 1H NMR (250 MHz, CDCl3) δ = 9.06 (d, 3J = 5.0 Hz, 1H, CHAr), 8.34 (s, 1H, CHAr), 8.17 (s, 1H, CHAr), 7.94 (dd, 4J = 2.9 Hz, 4J = 1.4 Hz, 1H, CHAr), 7.76–7.69 (m, 2H, CHAr), 7.59 (d, 3J = 5.1 Hz, 1H, CHAr), 7.57 (s, 1H, CHAr), 7.56–7.51 (m, 3H, CHAr) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.7 (CHAr), 146.8, 140.3, 140.1, 136.5, 134.8, 133.5, 132.7, 132.3, 132.2 (C), 127.9, 127.4, 126.9, 126.7, 124.0, 123.6, 123.5, 123.2, 122.9 (CHAr), 117.8 (C), 116.0 (CHAr) ppm. MS (EI, 70 eV): m/z (%) = 375 (15), 374 (26), 373 ([M]+, 100), 372 (22), 371 (16), 339 (6), 328 (5), 326 (5), 164 (6), 45 (12). HRMS (ESI) m/z: [M + H]+ calcd for: C21H12NS3 374.0132; found 374.0131. IR (ATR, cm−1): = 1621 (w), 1584 (m), 1465 (m), 859 (s), 774 (vs), 721 (s), 651 (vs), 533 (m).
:1); mp 185–190 °C. 1H NMR (250 MHz, CDCl3) δ = 9.08 (d, 3J = 5.1 Hz, 1H, CHAr), 8.32 (s, 1H, CHAr), 8.27 (s, 1H, CHAr), 7.99–7.91 (m, 2H, CHAr), 7.74 (d, 3J = 8.8 Hz, 2H, CHAr), 7.67 (d, 3J = 5.1 Hz, 1H, CHAr), 7.58 (s, 1H, CHAr), 6.98–6.85 (m, 4H, CHAr), 3.08 (s, 6H, CH3), 3.07 (s, 6H, CH3) ppm. 13C NMR (63 MHz, CDCl3) δ = 150.9, 133.1 (C), 129.7, 129.4 (CHAr), 127.5, 127.3, 124.4 (C), 122.0 (CHAr), 117.7 (C), 115.2, 112.8, 112.6 (CHAr), 40.6, 40.6 (CH3) ppm. MS (EI, 70 eV): m/z (%) = 448 (31), 447 ([M]+, 100), 431 (13), 223 (23), 222 (12), 82 (12). HRMS (ESI) m/z: [M + H]+ calcd for: C29H26N3S 448.1847; found 448.1841. IR (ATR, cm−1): = 1605 (vs), 1580 (s), 1510 (s), 1357 (vs), 1193 (vs), 813 (vs), 595 (s), 517 (s).
:1); mp 162–168 °C. 1H NMR (500 MHz, CDCl3) δ = 8.99 (d, 3J = 5.6 Hz, 1H, CHAr), 8.25 (s, 1H, CHAr), 8.03 (s, 1H, CHAr), 8.00–7.93 (m, 3H, CHAr), 7.91 (d, 3J = 5.6 Hz, 1H, CHAr), 7.86 (d, 3J = 7.4 Hz, 2H, CHAr), 7.65–7.50 (m, 6H, CHAr) ppm. 13C NMR (126 MHz, CDCl3) δ = 150.7 (C), 145.2 (CHAr), 140.4, 140.1, 139.6, 139.4, 134.5, 134.1, 133.6, 132.0 (C), 129.4, 129.1, 129.1, 128.9, 128.7, 128.6, 126.7, 124.5, 121.5 (CHAr), 118.6 (C), 117.3 (CHAr) ppm. MS (EI, 70 eV): m/z (%) = 363 (9), 362 (35), 361 ([M]+, 100), 360 (41), 359 (28), 358 (5), 357 (6), 180 (8). HRMS (ESI) m/z: [M + H]+ calcd for: C25H16NS 362.1003; found 362.1000. IR (ATR, cm−1): = 1578 (m), 1403 (m), 1300 (m), 1075 (m), 873 (m), 760 (s), 690 (vs), 597 (m), 562 (m).
:1); mp 182–186 °C. 1H NMR (500 MHz, CDCl3) δ = 9.35 (d, 3J = 5.6 Hz, 1H, CHAr), 8.64 (d, 3J = 5.4 Hz, 1H, CHAr), 7.80 (dd, 2J = 11.6 Hz, 3J = 5.5 Hz, 2H, CHAr), 7.75–7.67 (m, 5H, CHAr), 7.38 (d, 3J = 7.8 Hz, 2H, CHAr), 7.27 (d, 3J = 8.0 Hz, 2H, CHAr), 2.48 (s, 3H, CH3), 2.43 (s, 3H, CH3) ppm. 13C NMR (126 MHz, CDCl3) δ = 141.3, 140.2, 139.3, 136.8, 136.6, 135.3 (C), 132.2, 129.8, 129.6, 128.5, 127.8, 125.6 (CHAr), 124.0 (C), 122.6, 121.7 (CHAr), 119.2 (C) ppm. MS (EI, 70 eV): m/z (%) = 391 (9), 390 (30), 389 ([M]+, 100), 388 (23), 387 (9), 386 (7), 374 (12), 373 (15), 372 (6), 359 (9), 195 (5), 187 (12), 186 (7). HRMS (EI) m/z: [M]+ calcd for: C27H19NS 389.1232; found 389.1225. IR (ATR, cm−1): = 220 (w), 1510 (m), 1020 (m), 816 (vs), 739 (s), 692(s), 605 (m), 519 (s), 461 (s).
:1); mp 230–235 °C. 1H NMR (500 MHz, CDCl3) δ = 8.69 (d, 3J = 8.4 Hz, 1H, CHAr), 8.48–8.43 (m, 2H, CHAr), 8.36 (s, 1H, CHAr), 8.28 (s, 1H, CHAr), 8.05–8.00 (m, 2H, CHAr), 7.91–7.85 (m, 3H, CHAr), 7.71–7.67 (m, 1H, CHAr), 7.65–7.60 (m, 4H, CHAr), 7.58–7.53 (m, 2H, CHAr) ppm. 13C NMR (63 MHz, CDCl3) δ = 148.6, 148.0, 140.4, 140.3, 139.9, 139.6, 133.4, 133.1, 132.9, 131.9 (C), 130.3, 129.4, 129.3, 129.2, 129.1, 129.0, 128.8, 128.6, 125.6, 124.4, 123.5, 123.0 (CHAr), 121.7 (C), 120.0 (CHAr), 116.0 (C) ppm. MS (EI, 70 eV): m/z (%) = 413 (14), 412 (34), 411 ([M]+, 100), 410 (47), 409 (22), 407 (10), 205 (95), 204 (26). HRMS (ESI) m/z: [M + H]+ calcd for: C29H18NS 412.1160; found 412.1165. IR (ATR, cm−1): = 1453 (m), 1438 (m), 1385 (m), 1177 (m), 868 (m), 754 (s), 742 (vs), 696 (vs), 608 (s), 573 (s).
:1); mp 303–306 °C. 1H NMR (300 MHz, CDCl3) δ = 9.13 (s, 1H, CHAr), 9.01 (d, 3J = 8.3 Hz, 1H, CHAr), 8.81 (s, 1H, CHAr), 8.43–8.18 (m, 3H, CHAr), 8.06–7.95 (m, 5H, CHAr), 7.48–7.38 (m, 4H, CHAr) ppm. 19F NMR (471 MHz, CDCl3) δ = −109.4 (s, 1F, CF), −109.6 (s, 1F, CF) ppm. 13C NMR (75 MHz, CDCl3) δ = 164.7 (d, 1JC,F = 252.7 Hz, C), 164.5 (d, 1JC,F = 252.4 Hz, C), 147.9, 147.5, 141.5, 140.2, 137.7 (C), 137.4, 135.4 (CHAr), 134.0 (d, 4JC,F = 3.4 Hz, C), 133.4, 133.4, 132.8, 132.0 (C), 131.5 (d, 3JC,F = 8.6 Hz, CHAr), 131.0 (d, 3JC,F = 8.8 Hz, CHAr), 128.1, 124.7 (CHAr), 121.1 (C), 120.7 (CHAr), 120.2, 117.2 (d, 2JC,F = 22.1 Hz, CHAr), 117.1 (d, 2JC,F = 22.0 Hz, CHAr), 115.2 (C), 113.8 (CHAr) ppm. MS (EI, 70 eV): m/z (%) = 448 (29), 447 ([M]+, 71), 446 (100), 445 (17), 444 (11), 223 (40), 221 (23), 213 (17), 207 (21). HRMS (EI) m/z: [M]+ calcd for: C29H15F2NS 447.0883; found 447.0888. IR (ATR, cm−1): = 1500 (s), 1385 (m), 1335 (m), 1226 (s), 1156 (s), 1098 (m), 828 (vs), 808 (s), 746 (vs), 606 (m), 560 (s), 513 (s).
:1); mp 209–212 °C. 1H NMR (500 MHz, CDCl3) δ = 8.74 (d, 3J = 8.4 Hz, 1H, CHAr), 8.47 (d, 3J = 8.6 Hz, 1H, CHAr), 8.43 (s, 1H, CHAr), 8.37 (s, 1H, CHAr), 8.33 (s, 1H, CHAr), 7.98 (d, 3J = 8.7 Hz, 2H, CHAr), 7.87–7.81 (m, 1H, CHAr), 7.82 (d, 3J = 8.7 Hz, 2H, CHAr), 7.70–7.66 (m, 1H, CHAr), 6.95–6.90 (m, 4H, CHAr), 3.08 (s, 3H, CH3), 3.07 (s, 3H, CH3) ppm. 13C NMR (126 MHz, CDCl3) δ = 150.9, 150.9, 148.9, 148.0, 140.7, 140.5, 133.7, 133.4, 133.3, 132.1 (C), 130.1, 130.0, 129.4, 129.1 (CHAr), 127.8, 127.4 (C), 124.9, 123.6, 123.1, 122.6 (CHAr), 121.5 (C), 118.4 (CHAr), 115.5 (C), 112.7, 112.7 (CHAr), 40.6, 40.5 (CH3). MS (EI, 70 eV): m/z (%) = 512 (19), 511 (47), 498 (23), 497 ([M]+, 56), 483 (25), 248 (18), 126 (21), 56 (23), 44 (100). HRMS (ESI) m/z: [M + H]+ calcd for: C33H28N3S 498.2004; found 498.2010. IR (ATR, cm−1): = 1605 (vs), 1520 (s), 1357 (s), 1195 (s), 943 (m), 818 (s), 741 (vs), 607 (m).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- D. Zhang and L. Duan, Polycyclic Aromatic Hydrocarbon Derivatives toward Ideal Electron-Transporting Materials for Organic Light-Emitting Diodes, J. Phys. Chem. Lett., 2019, 10(10), 2528–2537, DOI:10.1021/acs.jpclett.9b00526 , Published Online: May. 6, 2019.
- C. Aumaitre and J.-F. Morin, Polycyclic Aromatic Hydrocarbons as Potential Building Blocks for Organic Solar Cells, Chem. Rec., 2019, 19(6), 1142–1154, DOI:10.1002/tcr.201900016 , Published Online: May. 20, 2019.
- Q. Li, Y. Zhang, Z. Xie, Y. Zhen, W. Hu and H. Dong, Polycyclic aromatic hydrocarbon-based organic semiconductors: ring-closing synthesis and optoelectronic properties, J. Mater. Chem. C, 2022, 10(7), 2411–2430, 10.1039/D1TC04866J.
- P. Samorí, X. Feng and D. Bonifazi, π-Conjugated Molecules: From Structure to Function, ChemPlusChem, 2019, 84(9), 1177–1178, DOI:10.1002/cplu.201900442 , Published Online: Aug. 23, 2019.
- L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J.-L. Brédas, K. N. Houk and A. L. Briseno, Unconventional, chemically stable, and soluble two-dimensional angular polycyclic aromatic hydrocarbons: from molecular design to device applications, Acc. Chem. Res., 2015, 48(3), 500–509, DOI:10.1021/ar500278w , Published Online: Dec. 2, 2014.
- X. Xu, K. Müllen and A. Narita, Syntheses and Characterizations of Functional Polycyclic Aromatic Hydrocarbons and Graphene Nanoribbons, Bull. Chem. Soc. Jpn., 2020, 93(4), 490–506, DOI:10.1246/bcsj.20190368.
- T. M. Figueira-Duarte and K. Müllen, Pyrene-based materials for organic electronics, Chem. Rev., 2011, 111(11), 7260–7314, DOI:10.1021/cr100428a . Published Online: Jul. 11, 2011.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Semiconducting π-conjugated systems in field-effect transistors: a material odyssey of organic electronics, Chem. Rev., 2012, 112(4), 2208–2267, DOI:10.1021/cr100380z , Published Online: Nov. 23, 2011.
- W. Wu, Y. Liu and D. Zhu, Pi-conjugated molecules with fused rings for organic field-effect transistors: design, synthesis and applications, Chem. Soc. Rev., 2010, 39(5), 1489–1502, 10.1039/B813123F , Published Online: Dec. 8, 2009.
- K. Ozaki, K. Kawasumi, M. Shibata, H. Ito and K. Itami, One-shot K-region-selective annulative π-extension for nanographene synthesis and functionalization, Nat. Commun., 2015, 6(1), 6251, DOI:10.1038/ncomms7251 , Published Online: Feb. 16, 2015.
- L.-J. Peng, X.-Y. Wang, Z.-A. Li and H.-Y. Gong, All Carbon Helicenes and π–Extended Helicene Derivatives, Asian J. Org. Chem., 2023, 12(12), e202300543, DOI:10.1002/ajoc.202300543.
- Y. Segawa, T. Maekawa and K. Itami, Synthesis of extended π-systems through C-H activation, Angew. Chem., Int. Ed., 2015, 54(1), 66–81, DOI:10.1002/anie.201403729 , Published Online: Sep. 26, 2014.
- H. Ito, K. P. Kawahara and K. Itami, Heteroatom-Embedding Annulative π-Extension (Hetero-APEX) Reactions: An Overview, Synthesis, 2024,(09), 1335–1354, DOI:10.1055/a-2169-4078.
- X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Heteroatom-Doped Nanographenes with Structural Precision, Acc. Chem. Res., 2019, 52(9), 2491–2505, DOI:10.1021/acs.accounts.9b00322 , Published Online: Sep. 3, 2019.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Recent Advances in Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds, Chem. Rev., 2022, 122(1), 565–788, DOI:10.1021/acs.chemrev.1c00449 , Published Online: Dec. 1, 2021.
- G. Biagiotti, I. Perini, B. Richichi and S. Cicchi, Novel Synthetic Approach to Heteroatom Doped Polycyclic Aromatic Hydrocarbons: Optimizing the Bottom-Up Approach to Atomically Precise Doped Nanographenes, Molecules, 2021, 26(20), 6306, DOI:10.3390/molecules26206306 , Published Online: Oct. 19, 2021.
- Q. Li, Y. Wu, X. Wang, Q. Yang, J. Hu, R. Zhong, S. Shao and L. Wang, Boron-, Sulfur- and Nitrogen-Doped Polycyclic Aromatic Hydrocarbon Multiple Resonance Emitters for Narrow-Band Blue Emission, Chem. – Eur. J., 2022, 28(12), e202104214, DOI:10.1002/chem.202104214 , Published Online: Feb. 3, 2022.
- W. Huang, M. Qiu, F. Yu, J. Chen, X. Li, Q. Wei, G. Xing, B. Wu, W. Chen and Q. Zhang, Synthesis, Characterization and Singlet Fission Behaviors of Heteroatom-Doped Polycyclic Aromatic Hydrocarbons with (β, β) Connected Furan/Thiophene Ring, Chem. – Eur. J., 2023, 29(29), e202203947, DOI:10.1002/chem.202203947 , Published Online: Apr. 5, 2023.
- D. J. Schipper and K. Fagnou, Direct Arylation as a Synthetic Tool for the Synthesis of Thiophene-Based Organic Electronic Materials, Chem. Mater., 2011, 23(6), 1594–1600, DOI:10.1021/cm103483q.
- C. Zhang and X. Zhu, Thieno3,4-bthiophene-Based Novel Small-Molecule Optoelectronic Materials, Acc. Chem. Res., 2017, 50(6), 1342–1350, DOI:10.1021/acs.accounts.7b00050 , Published Online: Apr. 4, 2017.
- Y. Wang, B. Liu, C. W. Koh, X. Zhou, H. Sun, J. Yu, K. Yang, H. Wang, Q. Liao, H. Y. Woo and X. Guo, Facile Synthesis of Polycyclic Aromatic Hydrocarbon (PAH)–Based Acceptors with Fine–Tuned Optoelectronic Properties: Toward Efficient Additive–Free Nonfullerene Organic Solar Cells, Adv. Energy Mater., 2019, 9(24), 1803976, DOI:10.1002/aenm.201803976.
- J. Wang, G. Ouyang, D. Wang, J. Li, J. Yao, W.-S. Li and H. Li, Enhanced Photocatalytic Performance of Donor–Acceptor-Type Polymers Based on a Thiophene-Contained Polycyclic Aromatic Unit, Macromolecules, 2021, 54(6), 2661–2666, DOI:10.1021/acs.macromol.1c00316.
- X. Yang, F. Rominger and M. Mastalerz, Contorted Heteroannulated Tetraarenoa,d,j,mcoronenes, Chem. – Eur. J., 2021, 27(57), 14345–14352, DOI:10.1002/chem.202102112 , Published Online: Sep. 9, 2021.
- Y. Chen, W. Chen, Y. Qiao and G. Zhou, B2 N2 -Embedded Polycyclic Aromatic Hydrocarbons with Furan and Thiophene Derivatives Functionalized in Crossed Directions, Chem. – Eur. J., 2019, 25(39), 9326–9338, DOI:10.1002/chem.201901782 , Published Online: Jun. 24, 2019.
- M. M. Islam, Z. Hu, Q. Wang, C. Redshaw and X. Feng, Pyrene-based aggregation-induced emission luminogens and their applications, Mater. Chem. Front., 2019, 3(5), 762–781, 10.1039/C9QM00090A.
- F. P. Kinik, A. Ortega-Guerrero, D. Ongari, C. P. Ireland and B. Smit, Pyrene-based metal organic frameworks: from synthesis to applications, Chem. Soc. Rev., 2021, 50(5), 3143–3177, 10.1039/D0CS00424C.
- J. Li, S. Chen, Z. Wang and Q. Zhang, Pyrene-fused Acenes and Azaacenes: Synthesis and Applications, Chem. Rec., 2016, 16(3), 1518–1530, DOI:10.1002/tcr.201600015 , Published Online: May. 24, 2016.
- W. Yang, J. H. S. K. Monteiro, A. de Bettencourt-Dias, V. J. Catalano and W. A. Chalifoux, Pyrenes, Peropyrenes, and Teropyrenes: Synthesis, Structures, and Photophysical Properties, Angew. Chem., 2016, 128(35), 10583–10586, DOI:10.1002/ange.201604741.
- X. Feng, X. Wang, C. Redshaw and B. Z. Tang, Aggregation behaviour of pyrene-based luminescent materials, from molecular design and optical properties to application, Chem. Soc. Rev., 2023, 52(19), 6715–6753, 10.1039/D3CS00251A , Published Online: Oct. 2, 2023.
- A. S. Lyakhovnenko, A. V. Aksenov, A. V. Andrienko and I. V. Aksenova, Synthesis of 1-Thia-5,7-Diazacyclopenta-[c,d]Phenalenes, A New Heterocyclic System, Chem. Heterocycl. Compd., 2010, 46(8), 1029–1030 CrossRef CAS.
- A. V. Aksenov, S. V. Shcherbakov, D. A. Lobach, N. N. Letichevskaya and E. A. Vasiléva, Synthesis of 1-Thia-5,7-Diazacyclopenta[c,d]-Phenalenes from 6(7)-Derivatives of Perimidine, Chem. Heterocycl. Compd., 2014, 50(5), 677–684 CrossRef CAS.
- T. Otsuo, Novel Heteroarenes Forming Conducting Molecular Complexes, Synlett, 1997, 544–550 CrossRef.
- X. H. Cheng, S.-S. Jester and S. Höger, Synthesis and Aggregates of Phenylene–Ethynylene Substituted Polycyclic Aromatic Compounds, Macromolecules, 2004, 37(19), 7065–7068, DOI:10.1021/ma048728d.
- O. S. Taniya, A. F. Khasanov, M. V. Varaksin, E. S. Starnovskaya, A. P. Krinochkin, M. I. Savchuk, D. S. Kopchuk, I. S. Kovalev, G. A. Kim, E. V. Nosova, G. V. Zyryanov and O. N. Chupakhin, Azapyrene-based fluorophores: synthesis and photophysical properties, New J. Chem., 2021, 45(45), 20955–20971, 10.1039/D1NJ03531B.
- S. Geib, S. C. Martens, U. Zschieschang, F. Lombeck, H. Wadepohl, H. Klauk and L. H. Gade, 1,3,6,8-Tetraazapyrenes: synthesis, solid-state structures, and properties as redox-active materials, J. Org. Chem., 2012, 77(14), 6107–6116, DOI:10.1021/jo300894p , Published Online: Jul. 5, 2012.
- Y. Omura, Y. Tachi, K. Okada and M. Kozaki, Synthesis and Properties of Nitrogen-Containing Pyrenes, J. Org. Chem., 2019, 84(4), 2032–2038, DOI:10.1021/acs.joc.8b02962 , Published Online: Jan. 31, 2019.
- M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, 10a-aza-10b-borapyrenes: heterocyclic analogues of pyrene with internalized BN moieties, Angew. Chem., Int. Ed., 2007, 46(26), 4940–4943, DOI:10.1002/anie.200700591.
- M. Li, Y. Yuan and Y. Chen, Bischler–Napieralski Cyclization: A Versatile Reaction towards Functional Aza–PAHs and Their Conjugated Polymers †, Chin. J. Chem., 2021, 39(11), 3101–3115, DOI:10.1002/cjoc.202100419.
- A. Vardanyan, M. A. Argüello Cordero, S. Lochbrunner, A. Villinger, P. Ehlers and P. Langer, Synthesis and Properties of 4- and 10-Benzoyl-1-azapyrenes, J. Org. Chem., 2024, 89(4), 2155–2168, DOI:10.1021/acs.joc.3c01752 , Published Online: Jan. 31, 2024.
- A. Vardanyan, S. Boldt, A. Villinger, P. Ehlers and P. Langer, Synthesis and Properties of 1-Azapyrenes, J. Org. Chem., 2022, 87(17), 11296–11308, DOI:10.1021/acs.joc.1c02394 , Published Online: Mar. 16, 2022.
- R. Molenda, J. Polkaehn, M. A. Argüello Cordero, A. Villinger, P. Ehlers, S. Lochbrunner and P. Langer, Synthesis and Properties of Thieno2′,3′,4′:4,5naphtho1,8-cdpyridines, J. Org. Chem., 2023, 88(13), 8802–8824, DOI:10.1021/acs.joc.3c00632 , Published Online: Jun. 6, 2023.
- R. Molenda, J. Polkaehn, M. A. Argüello Cordero, A. Villinger, P. Ehlers, S. Lochbrunner and P. Langer, Synthesis and Properties of Thieno2′,3′,4′:4,5naphtho1,8-cdpyridines, J. Org. Chem., 2023, 88(13), 8802–8824, DOI:10.1021/acs.joc.3c00632 , Published Online: Jun. 6, 2023.
- R. Molenda, S. Boldt, A. Villinger, P. Ehlers and P. Langer, Synthesis of 2-Azapyrenes and Their Photophysical and Electrochemical Properties, J. Org. Chem., 2020, 85(20), 12823–12842, DOI:10.1021/acs.joc.0c01622 , Published Online: Sep. 9, 2020.
- E. W. Thulstrup, J. W. Downing and J. Michl, Excited singlet states of pyrene. Polarization directions and magnetic circular dichroism of azapyrenes, Chem. Phys., 1977, 23(2), 307–319, DOI:10.1016/0301-0104(77)89013-7.
- J. Merz, J. Fink, A. Friedrich, I. Krummenacher, H. H. Al Mamari, S. Lorenzen, M. Haehnel, A. Eichhorn, M. Moos, M. Holzapfel, H. Braunschweig, C. Lambert, A. Steffen, L. Ji and T. B. Marder, Pyrene Molecular Orbital Shuffle-Controlling Excited State and Redox Properties by Changing the Nature of the Frontier Orbitals, Chem. – Eur. J., 2017, 23(53), 13164–13180, DOI:10.1002/chem.201702594 , Published Online: Aug. 21, 2017.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel and G. E. Scuseria, Gaussian 09, Gaussian, Inc., 2013 Search PubMed.
- A. M. Brouwer, Standards for photoluminescence quantum yield measruements in soltion, Pure Appl. Chem., 2011, 83(12), 2213–2228 CrossRef CAS.
- C. Reichardt, Solvatochromic Dyes as Solvent Polarity Indicators, Chem. Rev., 1994, 94(8), 2319–2358, DOI:10.1021/cr00032a005.
- A. Stanger, NICS – Past and Present, Eur. J. Org. Chem., 2020,(21), 3120–3127, DOI:10.1002/ejoc.201901829.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Nucleus-independent chemical shifts (NICS) as an aromaticity criterion, Chem. Rev., 2005, 105(10), 3842–3888, DOI:10.1021/cr030088.
- E. Paenurk and R. Gershoni-Poranne, Simple and efficient visualization of aromaticity: bond currents calculated from NICS values, Phys. Chem. Chem. Phys., 2022, 24(15), 8631–8644, 10.1039/D1CP05757J , Published Online: Apr. 13, 2022.
- R. Gershoni-Poranne and A. Stanger, Magnetic criteria of aromaticity, Chem. Soc. Rev., 2015, 44(18), 6597–6615, 10.1039/C5CS00114E , Published Online: Jun. 2, 2015.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of starting materials, experimental procedures and analytical data, photophysical and electrochemical data, single crystal X-ray data, 1H-, 19F-, and 13C-NMR spectra of isolated compounds, and computational details. CCDC 2363583–2363585. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01023j |
| This journal is © The Royal Society of Chemistry 2024 |