
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic hydrogen bond donor/Lewis base (HBD/LB) synthesis and chiroptical properties of thiabridged [5]helicenes†
Michela
Lupi
 *a,
Mosè
Fabbri
a,
Giuseppe
Mazzeo
b,
Giovanna
Longhi
b,
Sergio
Abbate
b,
Caterina
Viglianisi
a and
Stefano
Menichetti
*a
*a,
Mosè
Fabbri
a,
Giuseppe
Mazzeo
b,
Giovanna
Longhi
b,
Sergio
Abbate
b,
Caterina
Viglianisi
a and
Stefano
Menichetti
*a
aDepartment of Chemistry “Ugo Schiff” (DICUS), University of Florence, Via della Lastruccia 13, Sesto Fiorentino (FI), 50019 Florence, Italy. E-mail: michela.lupi@unifi.it; stefano.menichetti@unifi.it
bDepartment of Molecular and Translational Medicine (DMMT), University of Brescia, V. le Europa 11, Brescia (BS), 25121 Brescia, Italy
First published on 10th July 2024
Abstract
Thiabridged [5]helicenes are obtained from thioaryl-N-phthalimido benzo[a]phenothiazines using a hydrogen bond donor/Lewis base organocatalytic approach. Resolution of [5]helicenes using either (1S)-(−)-camphanic acid as a chiral auxiliary or CSP-HPLC is reported. Thiabridged [5]helicenes show an exceptional configurational stability with racemization energy barriers higher than 40 kcal mol−1. Electronic circular dichroism and TD-DFT calculations permit the assignment of the absolute configuration, demonstrating that the sign of optical rotation is not easily related to the M or P structure. Separated enantiomers show circularly polarized luminescence with high dissymmetry ratio.
Introduction
In the last few years, helicenes have played a pivotal role in many fields, such as asymmetric synthesis,1–3 medicinal chemistry,4–6 molecular recognition7,8 and, first and foremost, material science.9–12 Indeed, several (asymmetric) syntheses of helicenes using transition metal catalysts13–18 or organocatalysts19–22 have been reported. Thiabridged [4]helicenes appear particularly attractive, due to their peculiar structure and their ability to be reversibly oxidized to the corresponding radical cations.23,24 The configurational stability, with racemization energy barriers of ≃32 kcal mol−1, allows the enantiopure forms to be handled after HPLC resolution on a chiral stationary phase.25 Thus, we previously studied the deposition of enantiopure [4]helicene radical cations on a Au(111) surface, proving that both the handedness and paramagnetism are retained after the deposition process.26 Additionally, after a chemisorbed deposition process, we demonstrated for these molecules an extremely high spin filtering capability at very low voltages.27 Additionally, the use of these systems as recyclable organophotoredox catalysts has been reported, further extending the fields of application of these thiahelicenes.28 We recently developed a new organocatalytic strategy for the synthesis of these thiahelicenes from arylthio-N-phthalimido precursors using a Lewis base/hydrogen bond donor (LB/HBD) catalytic system.29 In particular, a selection of simple selenium and sulfur-containing Lewis bases were successfully used in catalytic amounts (10% mol loading) in the presence of hexafluoro isopropanol (HFIP) as a hydrogen bond donor. This methodology allowed thiahelicenes to be obtained in good yields under very mild reaction conditions, avoiding the use, as cyclization promoters, of over-stoichiometric amounts of strong Lewis acids.The need to obtain helicenes in enantiopure form and in substantial amounts is indeed more and more urgent, possibly avoiding the drawbacks connected with HPLC resolutions.25 The preparation of enantiopure helicenes on a multi-mg scale was previously accomplished mainly for [5]-30 or [6]helicenes31 using an optical resolution approach. Nevertheless, just a few examples of optical resolution of [4]helicene derivatives are reported in the literature, due to the lower configurational stability.32
Recently, we demonstrated that chemical resolution can be achieved also for configurationally stable and properly substituted (vide infra) hydroxy thiabridged triarylamine [4]helicenes forming mixtures of diastereoisomeric esters, separable by flash column chromatography, exploiting properly selected enantiopure carboxylic acids.33 The success of the resolution depends upon a combination of the structure of the enantiopure acid used, with (1S)-(−)-camphanic acid giving the best results, and the position of the chiral auxiliary insertion. In fact, separation was effective only for the diastereomers obtained from helicene 1, bearing the hydroxyl group in the helicene bay-zone (in pink, Fig. 1, top left). Indeed, the resolution completely failed when the chiral auxiliary was inserted in the helicene 2, bearing the hydroxyl group in the cape-zone (in green, Fig. 1, top right), regardless of the structure of the chiral auxiliary used. Herein, we report the synthesis of hetero[5]helicenes 3 from arylthio-N-phthalimido benzo[a]phenothiazines 4 using Lewis base/HBD methodology, and a demonstration of how the aromatic backbone expansion improves the chiroptical properties,34 simplifies chemical resolutions, and increases configurational stability compared to the corresponding [4]helicenes.
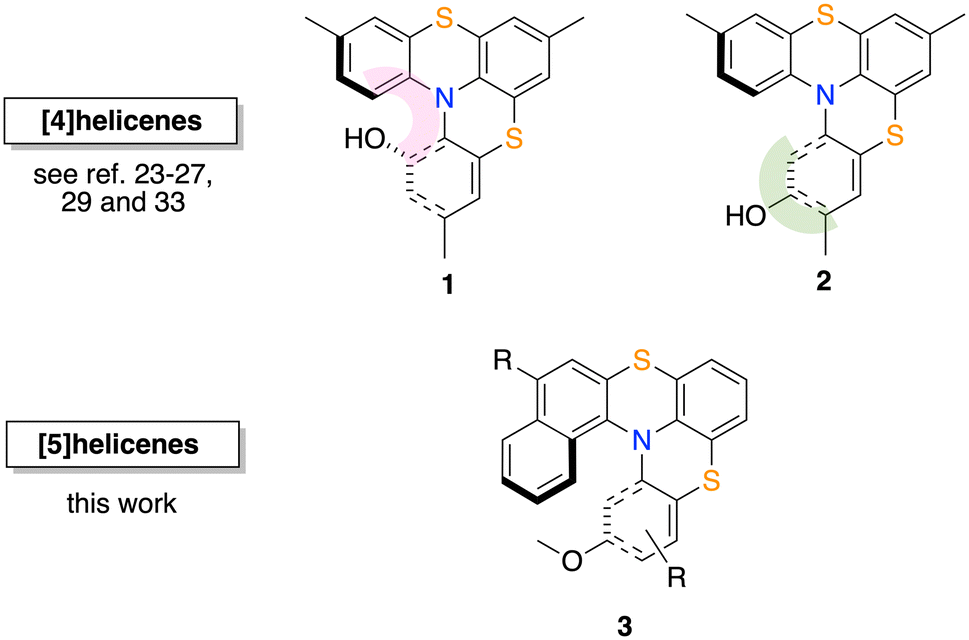 | ||
| Fig. 1 Structure of thiabridged [4]helicenes and [5]helicenes. | ||
Results and discussion
Synthesis
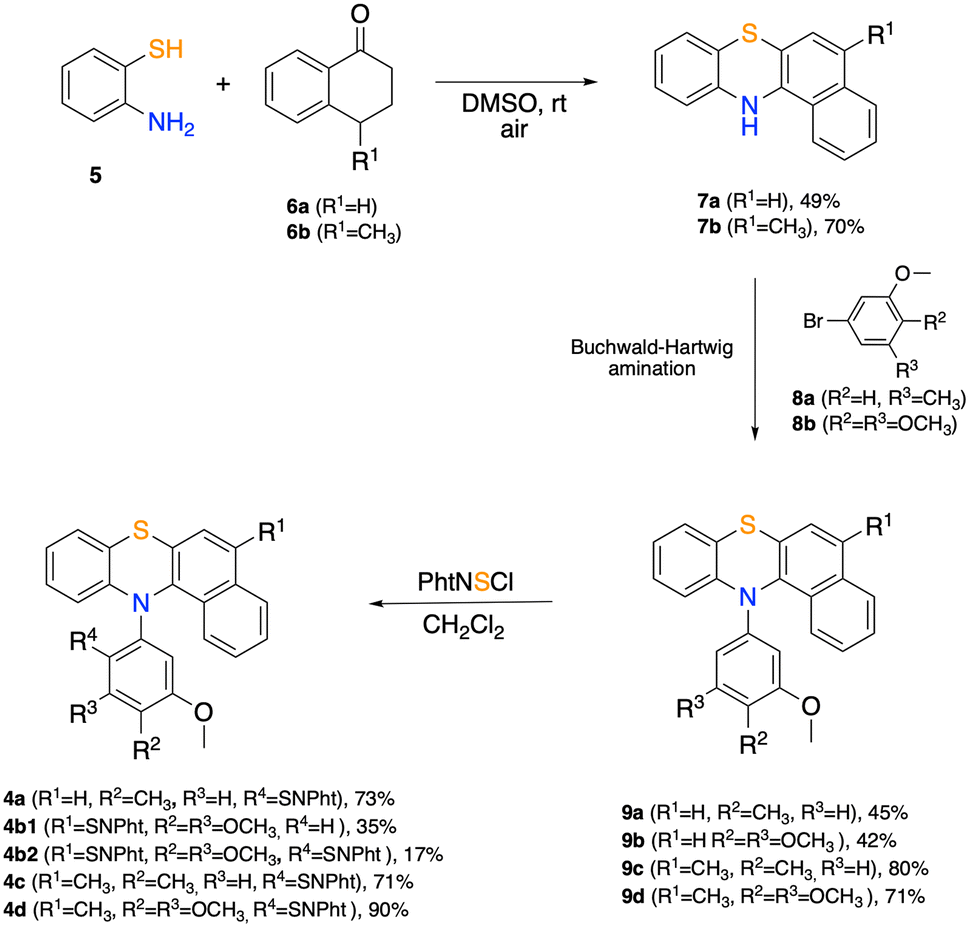 | ||
| Scheme 1 Synthetic route to arylthio-N-thiophthalimido benzo[a]phenothiazines 4. | ||
Benzo[a]phenothiazines 7 can be obtained following the metal-free procedure reported by Lin et al.35 by reacting 2-aminothiophenol 5 with tetralones366a and 6b in DMSO under an air atmosphere. Derivatives 7a and 7b undergo N-arylation by Buchwald Hartwig reaction with electron-rich aryl bromides 8a and 8b allowing access to N-aryl benzo[a]phenothiazines 9a–d in medium to good yields. Subsequent reaction of 9a–d with phthalimidesulfenyl chloride delivered sulfenylated products 4a, 4c and 4d with complete regioselectivity. It is worth mentioning that for trimethoxy-substituted derivative 4b, the naphthalene portion is particularly activated towards SEAr, causing an over-substitution process. Indeed, the reaction led to both the mono-sulfenylated product 4b1 and the bis-sulfenylated product 4b2 (Scheme 1). In fact, reacting 9d (R1 = CH3) with PhtNSCl afforded exclusively mono sulfenylated derivative 4d in 90% yield.
In contrast to the synthesis of thia[4]helicenes,23–27 the use of stoichiometric amounts of AlCl3 in CH2Cl2 or CHCl3 caused a severe decomposition of starting material 4a with very low conversion values either at 60 °C or at room temperature (Table 1, entries 1 and 2). Hence, we moved to a new synthetic procedure that exploits hexafluoro isopropanol (HFIP) as a strong HBD and a catalytic amount of a chalcogen LB.29 Therefore, a selection of Lewis bases that delivered the [4]helicenes in the highest conversions were evaluated in the presence of HFIP (see Table 1, entries 3–8). Formation of helicene 3a was initially monitored by 1H NMR, and, for satisfactory conversion values (higher than 60%), the crude mixture was purified by flash chromatography and the isolated yield of 3a was evaluated. Racemic lipoic acid (10b) and dodecyl methyl sulfide (10c) gave the best results in terms of conversion and isolated yields (Table 1, entries 5–8). However, 40% mol of lipoic acid (10b) added in four aliquots every 4 h was necessary to parallel the yield of 3a achieved using 10% mol of dodecyl methyl sulfide (10c) (entry 6 vs. entry 8).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Eq. | Solv. | T (°C) | Time | Conv.a | Yieldb |
| a Measured by 1H NMR. b Isolated yields after flash column chromatography carried out for conversions > 50%. c 0.1 M. d 40 mg of 4a in 200 μL of HFIP. e Added in four aliquots every 4 hours. | |||||||
| 1 | AlCl3 | 1.5 | CH2Cl2c | 20 | 2 | — | — |
| 2 | AlCl3 | 1.5 | CHCl3c | 60 | 2 | 11 | — |
| 3 | 10a | 0.10 | HFIPd | 20 | 48 | 5 | — |
| 4 | 10a | 0.10 | HFIPd | 50 | 48 | 6 | — |
| 5 | 10b | 0.10 | HFIPd | 20 | 24 | 17 | — |
| 6 | 10b | 0.40e | HFIPd | 50 | 24 | 80 | 55 |
| 7 | 10c | 0.10 | HFIPd | 20 | 48 | 50 | — |
| 8 | 10c | 0.10 | HFIPd | 50 | 48 | 70 | 56 |
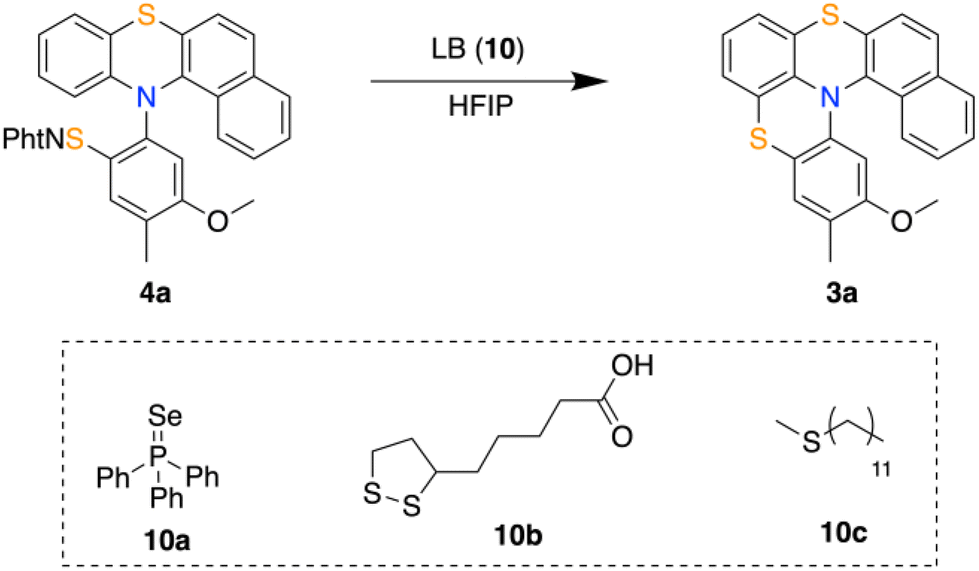
Thus, the conditions reported in entry 8 of Table 1 were chosen for the substrate scope study. When arylthio-N-phthalimido benzo[a]phenothiazines 4c and 4d were reacted under the optimized conditions, each of the desired [5]helicenes 3c and 3d, isolated respectively in 22% and 42% yield, were accompanied by a not negligible amount of a second product (Scheme 2). These unknown products were successfully isolated by flash chromatography. 1H NMR and 13C NMR analyses did not elucidate the structure of these by-products, but suggested a helical skeleton similar to that of 3c and 3d (see Experimental section and ESI†). Thus, we envisioned the formation of the pleiadene-like molecules 11c and 11d (see Scheme 2) as a result of a SEAr reaction involving the electron-rich peri position on the naphthalene ring. Eventually, together with those of helicenes 3a and 3d, suitable crystals for X-Ray analysis of the unknown products were obtained (Scheme 2). Thus, the structure of 3a and 3d was confirmed and the skeleton of the unknown derivative was disclosed as that of [5]helicene 12d. With this structure in hand, we assumed that the cyclization of 4c would afford, along with 3c, also [5]helicene 12c. A mechanism suitable to rationalize the formation of the expected helicene 3d (and 3c) together with the unexpected helicene 12d (and 12c) is highlighted in Scheme 3. Path A (pink frame): arylthio-N-phthalimido derivative 4d reacts with dodecyl methyl sulfide (10c) to form charged intermediate i1, which undergoes an intramolecular SEAr (iSEAr) to form the Wheland intermediate i2 that can evolve into helicene 3d. Path B (green frame): intermediate i2 undergoes a rearrangement to give the Wheland intermediate i3, which in the presence of catalyst 10c forms intermediate i4 (i.e. a retro-iSEAr), and eventually affords [5]helicene 12dvia intermediate i5. As a matter of fact, when helicenes 3d or 12d were placed under the HBD/LB reaction conditions, no formation of the corresponding transposed helicene was observed, indicating that, reasonably, the i2 → 3d and i5 → 12d steps are not reversible. Surprisingly, when 10c was replaced with 10b for the cyclization of 4c under the same reaction conditions as shown in Scheme 2, helicene 3c was obtained as the major product with a 1H NMR yield of 51%, while the formation of the rearranged product was observed in trace amounts (ca. 5% 1H NMR yield), indicating that the regioselectivity of the reaction can be efficiently tuned by simply changing the Lewis base catalyst.
 | ||
| Scheme 2 Cyclization reaction with the LB/HBD catalytic system. ORTEP diagrams of products 3a (CCDC 2361198), 3d (CCDC 2361200) and 12d (CCDC 2361199) are shown. Other details are reported in the ESI.† | ||
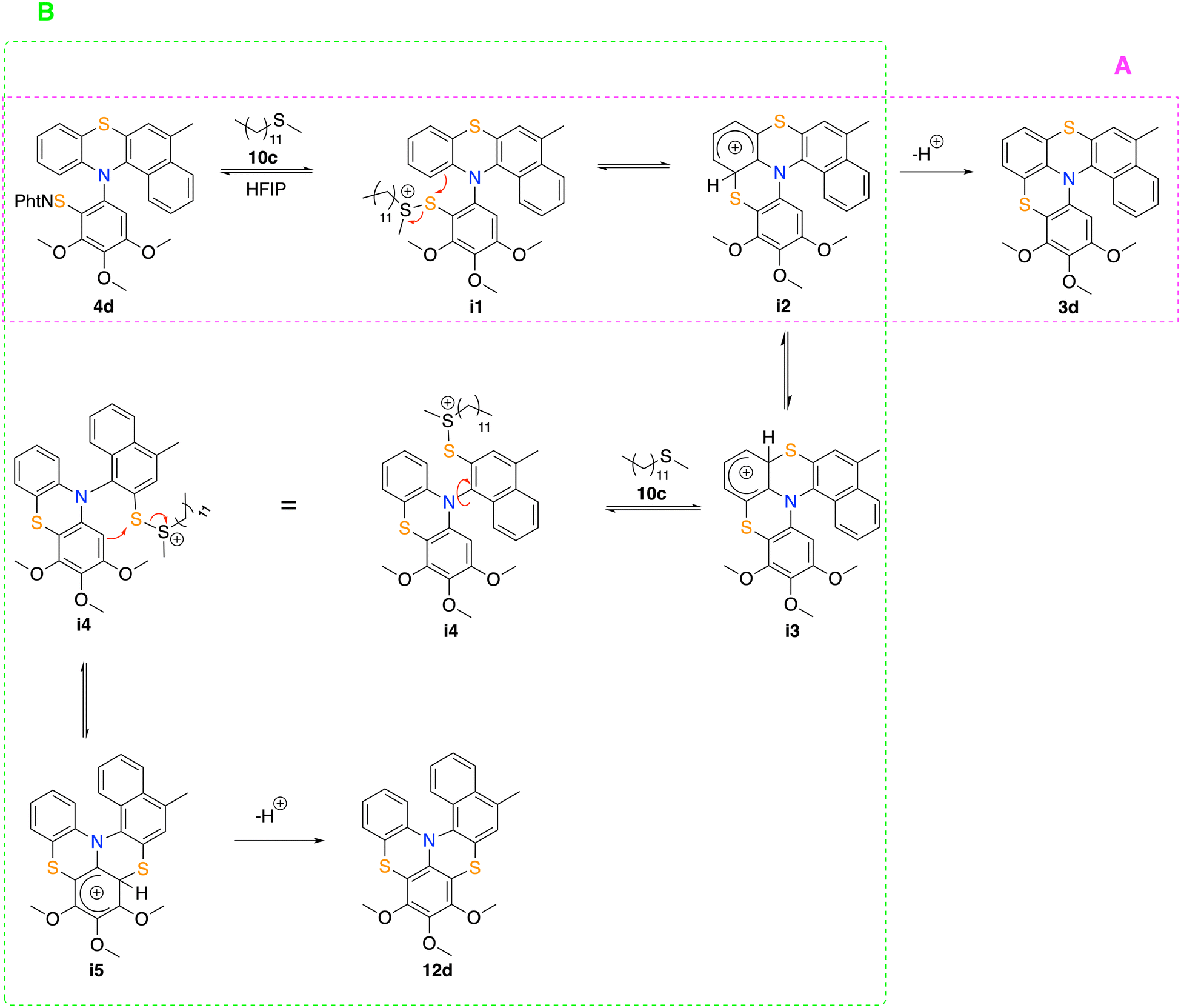 | ||
| Scheme 3 A possible rational mechanism for 3 vs. 12 thiabridged [5]helicene formation. | ||
Optical resolution
Having optimized the chemical resolution of derivatives 1,33 we envisaged that the presence of an additional aryl ring in the helical backbone could be beneficial for the resolution efficiency of compounds 3 and 12 due to steric interactions, therefore avoiding the troublesome insertion of the chiral auxiliary ortho to the nitrogen atom. Therefore, derivative 3a was demethylated with BBr3 to give phenol (rac)-3HelOH (i.e. with the –OH function meta to the nitrogen). (rac)-3HelOH was esterified with (1S)-(−)-camphanic acid (13), under the previously optimized reaction conditions,33 to give a mixture of diastereomeric esters 3D1 and 3D2 that showed a slightly different behavior on TLC with a ΔRf value of roughly 0.04. Indeed, the two esters were successfully isolated by flash chromatography on silica gel using CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :petroleum ether–2:1 as an eluent to give 3D1 (38% yield, Rf = 0.55) and 3D2 (37% yield, Rf = 0.51). Optical rotation was measured and gave [α]25D = +143 (c = 0.2, CH2Cl2) for 3D1 and [α]25D = −146 (c = 0.2, CH2Cl2) for 3D2. Basic hydrolysis of the diastereomeric esters provided helicenes (+)-3HelOH and (−)-3HelOH, respectively, in a quantitative yield. HPLC analysis with a chiral stationary phase showed that (+)-3HelOH [α]25D +104 (c = 0.2, CH2Cl2) exhibits an enantiomeric ratio of 1:99 (ee = 98%), while (−)-3HelOH [α]25D = −102 (c = 0.2, CH2Cl2) exhibits an enantiomeric ratio of 97:3 (ee = 94%). These results demonstrate that increasing the size of the helical bay by structural homologation, optical resolution can be achieved also for derivatives bearing the anchoring group not adjacent to the nitrogen. Interestingly, the 1H NMR spectra of 3D1 (see Fig. 2, inset, top) and 3D2 (see Fig. 2, inset, bottom) appear very similar, being two of the three methyl groups of the camphanic moiety, the sole functional groups that resonate (slightly) differently.
:petroleum ether–2:1 as an eluent to give 3D1 (38% yield, Rf = 0.55) and 3D2 (37% yield, Rf = 0.51). Optical rotation was measured and gave [α]25D = +143 (c = 0.2, CH2Cl2) for 3D1 and [α]25D = −146 (c = 0.2, CH2Cl2) for 3D2. Basic hydrolysis of the diastereomeric esters provided helicenes (+)-3HelOH and (−)-3HelOH, respectively, in a quantitative yield. HPLC analysis with a chiral stationary phase showed that (+)-3HelOH [α]25D +104 (c = 0.2, CH2Cl2) exhibits an enantiomeric ratio of 1:99 (ee = 98%), while (−)-3HelOH [α]25D = −102 (c = 0.2, CH2Cl2) exhibits an enantiomeric ratio of 97:3 (ee = 94%). These results demonstrate that increasing the size of the helical bay by structural homologation, optical resolution can be achieved also for derivatives bearing the anchoring group not adjacent to the nitrogen. Interestingly, the 1H NMR spectra of 3D1 (see Fig. 2, inset, top) and 3D2 (see Fig. 2, inset, bottom) appear very similar, being two of the three methyl groups of the camphanic moiety, the sole functional groups that resonate (slightly) differently.
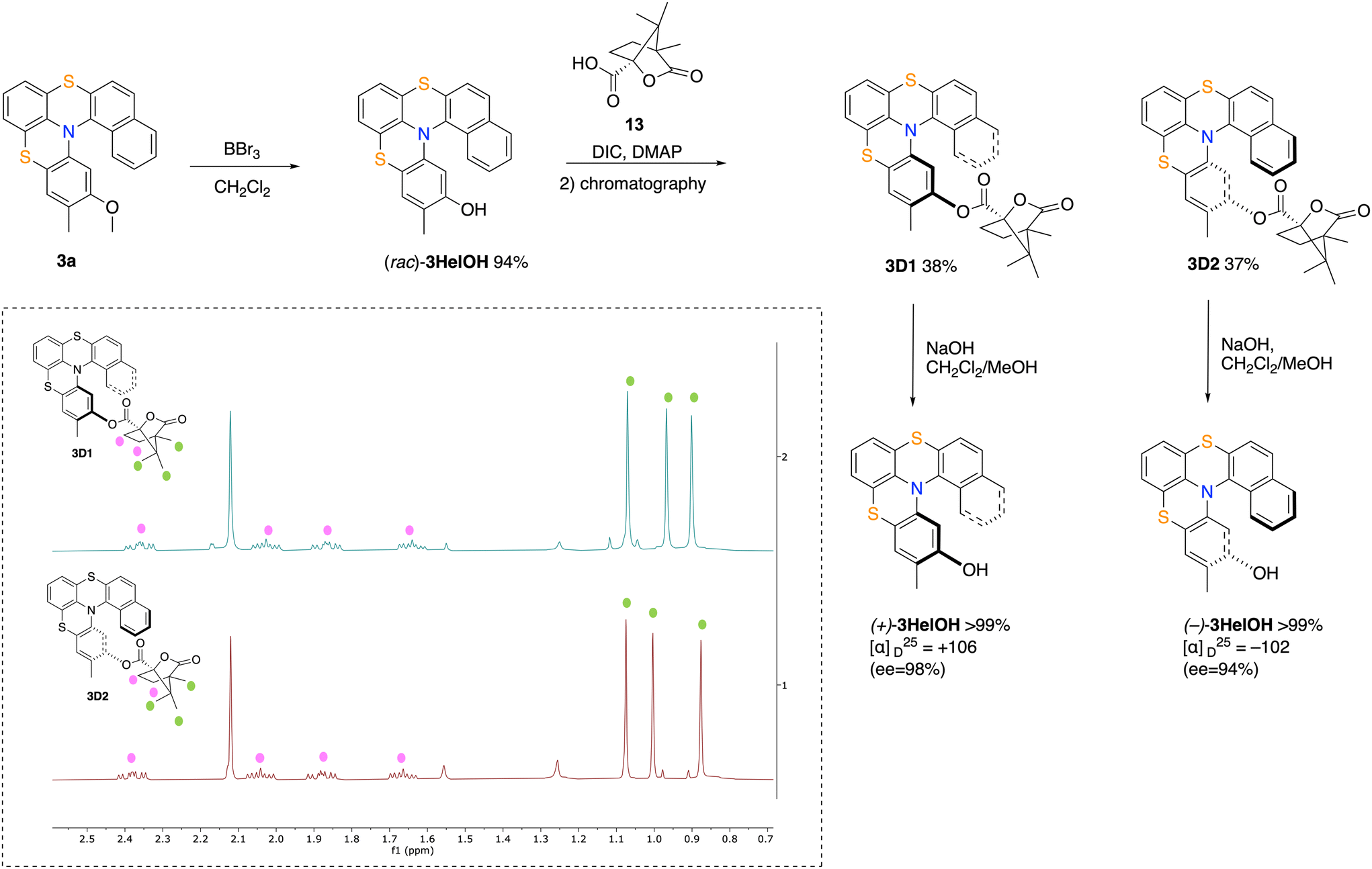 | ||
| Fig. 2 Optical resolution of helicene 3a using (1S)-camphanic acid as a chiral auxiliary. Inset: 1H NMR spectra of 3D1 (top) and 3D2 (bottom). | ||
Semipreparative CSP-HPLC resolution
Derivatives 3c and 12d were successfully resolved by semipreparative HPLC using CHIRALPAK IG as a chiral stationary phase. Optical rotation of the resolved 12d was measured and gave [α]25D = –81 (c = 0.07, CH2Cl2) for (–)-12d (first eluted, ee > 99%) and [α]25D = +76 (c = 0.07, CH2Cl2) for (+)-12d (second eluted, ee = 94%), while for 3c it gave [α]26D = +213 (c = 0.03, CH2Cl2) for (+)-3c (first eluted, ee > 99%) and [α]26D = –213 (c = 0.03, CH2Cl2) for (–)-3c (second eluted, ee > 99%). Regrettably, for derivatives 3d and 12c, the semipreparative resolution using CHIRALPAK IG as the chiral stationary phase was unsuccessful. Other CSPs are under investigation.Configurational stability of the thiabridged [5]helicenes
With the enantiopure compounds 3c and 12d in hand, we focused on the evaluation of their configurational stability. Enantiopure (+)-12d (ee > 99%) was dissolved in n-decane (1 mg mL−1) and heated at increasing temperatures, 60 °C, 80 °C, 100 °C, 120 °C, 140 °C and at reflux (174 °C) for 2 hours. The solutions were cooled at rt and CSP-HPLC analysis was carried out. To our great satisfaction, no racemization was observed, even after 2 h at reflux, indicating that thiabridged [5]helicenes are significantly more stable than the corresponding thiabridged [4]helicenes, which showed a significant racemization rate at 121 °C in n-decane.25 This allowed it to be estimated (see ESI†) that the racemization energy barrier for these [5]helicenes is higher than 40 kcal mol−1.Chiroptical properties
The chiroptical properties of [5]helicenes 3HelOH, 3c and 12d, namely ECD (electronic circular dichroism) and CPL (circularly polarized luminescence), have been investigated also with the aim to assign the absolute configuration by means of TD-DFT calculations. All spectra have been recorded in dichloromethane solutions. The ECD spectra (Fig. 3) are quite similar for the three compounds: a long wavelength broad CD band at ca. 395 nm; a band at ca. 325 nm and a band at 250 nm, both of the opposite sign to the first feature at 395 nm. In these three cases, the longest wavelength/lowest energy band presents a dissymmetry ratio gabs = ΔA/A of about 0.8–0.9 × 10−2, comparable to that of the second feature and higher than the one associated with the shorter wavelength region. Comparing the experimental and calculated ECD spectra, one can associate the positive first CD band (and the two other main negative bands) (Fig. 3, blue lines) to the M configuration (Fig. 3, black lines). In Fig. 4, we also report the experimental CPL spectra for the two eluted fractions of 3HelOH, 3c and 12d. All three compounds behave very similarly, presenting a CPL band at about 480 nm. It is worth noting that the sign of the CPL bands correlates with the sign of the longest wavelength ECD band reported in Fig. 3. The dissymmetry ratio for CPL, glum = ΔI/I = 2(IL − IR) − (IL + IR), is about 0.8 × 10−2 (similar to the gabs of the lowest energy transition) for all three compounds, which is a quite large value, since it is in the upper limit among simple organic compounds.37,38 | ||
| Fig. 3 Experimental (red and blue lines for the two enantiomers) and calculated ECD spectra for the M (black lines) configuration of 3HelOH, 3c and 12d. Calculations were performed at the TD-DFT/M06/cc-pVTZ/PCM(DCM) level of theory. | ||
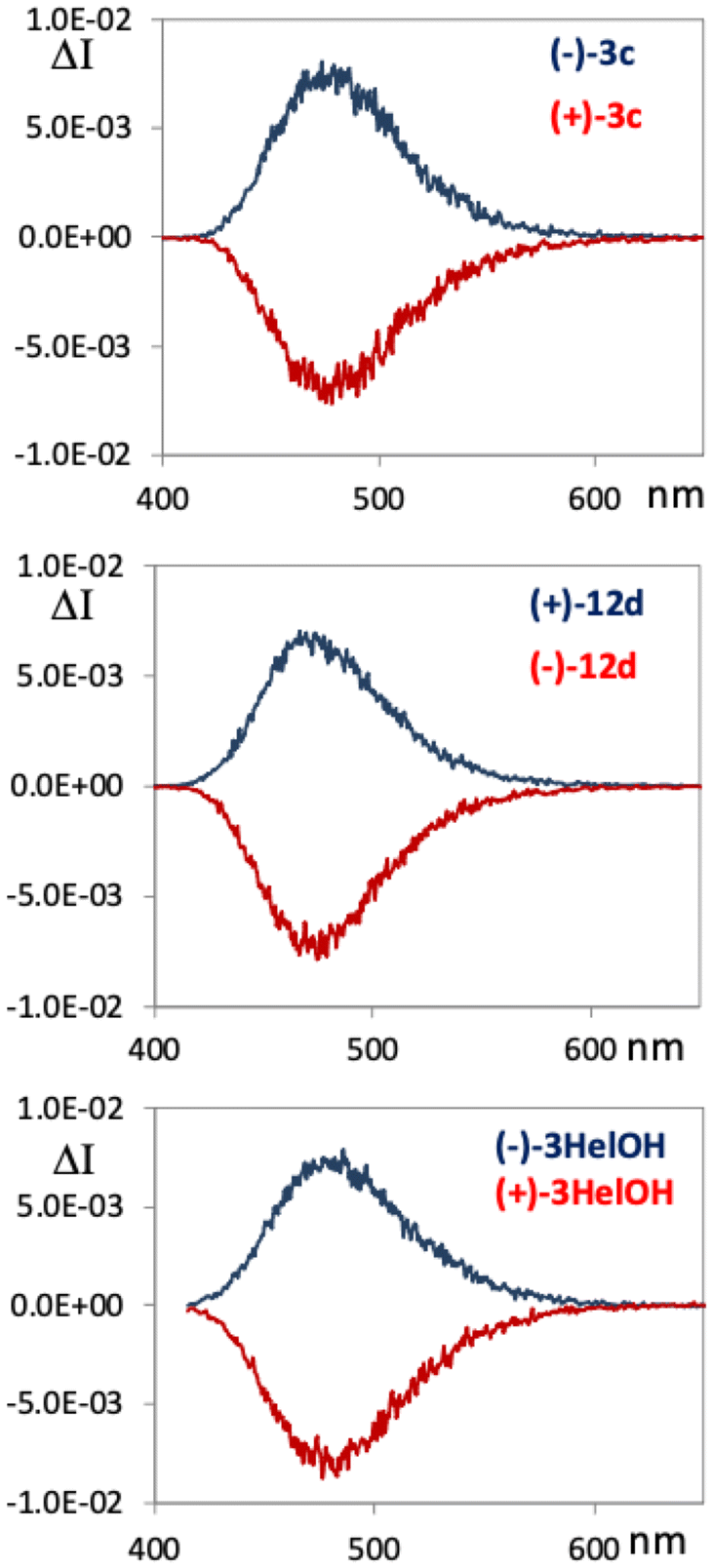 | ||
| Fig. 4 Experimental CPL spectra for the two enantiomers of 3HelOH, 3c and 12d. CPL has been plotted after normalizing the fluorescence signal recorded with the same apparatus. | ||
We recall here that the spectra of simple carbo-helicenes present helical-sense responsive and substituent-sensitive chiroptical ECD features.39 The latter, responsible also for CPL, are generally weak and dominated by vibronic contributions; on the contrary, the particular structure of the compounds under study here promotes the enhancement of the chiroptical properties: in fact the high dissymmetry ratio is in line with what is observed on an analogous thiabridged [6]helicene.40 The present work confirms that substituents do not perturb this response and that the five-membered helicenes studied here exhibit chiroptical properties comparable to the longer one. We should add some comments about optical rotation (OR): as one may notice from Fig. 3, within this set of molecules it appears difficult to correlate the OR sign with the configuration. It was previously observed that shorter analogous helicenes, i.e. thiabridged [4]helicenes,25,33 present a negative OR at 589 nm associated with the M configuration, while longer ones, in particular a thiabridged [6]helicene, present positive OR.40 In the [5]helicenes under study, we recorded negative optical rotation for 3HelOH and 3c associated with the M configuration, and positive optical rotation for 12d. As a further check, we recorded optical rotatory dispersion (ORD) for the same compounds in dichloromethane solutions (see Experimental section for details) at different wavelengths, until 436 nm. According to the Kronig–Kramers relation,41 ORD can be calculated from the whole CD spectrum, and this implies that often the optical rotation at the sodium D-line ends up with the same sign as the first CD band.42 This is not the case for 3HelOH and 3c. However, it is interesting to note that, performing OR measurements at lower wavelengths, the OR sign of 3c and 3HelOH inverts and one records positive OR at 436 nm (+4163 and +3898, respectively), in accordance with the sign of the ECD band set at 395 nm and as expected while approaching the anomalous dispersion region. Analogously, a positive value is obtained at 436 nm also for the previously mentioned [4]helicene,33 showing a negative value at 589 nm for the M configuration; that is to say, that in all examined cases approaching the first band, optical rotation takes the sign of the ECD band, which correlates with the helicene configuration. These observations suggest that a consistent set of ORD data, and not only a single OR value at the sodium D-line, is recommended to assign the absolute configuration.
Conclusions
In conclusion, thiabridged [5]helicenes can be obtained by our previously developed LB/HBD catalytic route. The resolution of these systems either chemically, by formation of diastereoisomeric (1S)-camphanic esters, or through semipreparative CSP-HPLC was, overall, simpler than the corresponding thiabridged [4]helicenes. On the other hand, [5]helicenes showed an exceptional chemical and configurational stability; in fact, after 2 hours at 174 °C in n-decane, no trace of decomposition or racemization was detected allowing a racemization ΔG≠ barrier higher than 40 kcal mol−1 to be estimated. Furthermore, the CPL activity with high dissymmetry ratio, taken together with the configurational stability, makes these compounds useful candidates for applications in material science.Experimental part
General information
1H and 13C NMR spectra were recorded with a Varian Mercury Plus 400, and a Varian Inova 400, using CDCl3 as a solvent. Residual CHCl3 at δ = 7.26 ppm and the central line of CDCl3 at δ = 77.16 ppm were used as the reference of the 1H-NMR spectra and 13C NMR spectra, respectively. FT-IR spectra were recorded with a spectrum two FT-IR spectrometer. ESI-MS spectra were recorded with a JEOL MStation JMS700. Melting points were measured with Stuart SMP50 automatic melting point apparatus. Optical rotation measurements were performed on a JASCO DIP-370 polarimeter (JASCO, Easton, MD, USA) and the specific rotation of the compounds was reported. All the reactions were monitored by TLC on commercially available precoated plates (silica gel 60 F 254) and the products were visualized with acidic vanillin solution. Silica gel 60 (230–400 mesh) was used for column chromatography. Dry solvents were obtained by The PureSolv Micro Solvent Purification System. Chloroform was washed with water several times and stored over calcium chloride. Triethylamine was freshly distilled over KOH before use. Reagents were purchased from Signa Aldrich and used as received, unless otherwise specified. Phthalimidesulfenyl chloride was prepared from the corresponding disulfide as reported elsewhere.43 Benzo[a]phenothiazine 7a was prepared according to literature procedures.35 The preparation and characterization of benzo[a]phenothiazines 7b, N-aryl benzo[a]phenothiazines 9a–9d and sulfenylated derivatives 4a–4d are reported in the ESI.†Experimental chiroptical properties
ECD/UV measurements were conducted with a Jasco 815SE instrument with 2 mm quartz cuvettes in dichloromethane. Fluorescence spectra were recorded on a Jasco FP8600 instrument and CPL spectra were recorded on a home-built apparatus44 with 10 accumulation scans using 2 mm fluorescence quartz cuvettes. ORD measurements were carried out with a JASCO P-2000 polarimeter using dichloromethane solutions at 0.07 g per 100 mL, 0.13 g per 100 mL and 0.02 g per 100 mL for 3c, 12d and 3HelOH, respectively, at four different wavelengths, 589 nm (Na lamp), 578 nm, 546 nm, and 435 nm (Hg lamp), with 10 measurements at each wavelength.Calculations
The optimized geometry and ECD spectra have been calculated through DFT and TD-DFT methods performed with the Gaussian16 suite of programs.45 The M06/cc-pVTZ level of theory, including bulk solvent effects by the conductor version of the polarizable continuum model (PCM), has been used.HPLC resolution
An analytical (250 × 4.6 mm) column packed with Chiralpak IA chiral stationary phase was purchased from Chiral Technologies Europe. A semipreparative (250 × 4.6 mm) column packed with Chiralpak IG chiral stationary phase was purchased from Chiral Technologies Europe. The HPLC resolution of the products was performed on a HPLC Waters Alliance 2695 equipped with a 200 μL loop injector and a spectrophotometer UV Waters PDA 2996. For CSP-HPLC semipreparative resolution of 3c and 12d, the mobile phase, delivered at a flow rate of 3.5 mL min−1, was hexane/CH2Cl2 80/20. For CSP-HPLC analytical resolution of [5]helicene 3c, the mobile phase, delivered at a flow rate of 0.7 mL min−1, was hexane/CH2Cl2 90/10 while for 12d the mobile phase, delivered at a flow rate of 1.0 mL min−1, was hexane/CH2Cl2 80/20.Syntheses
:petroleum ether–1:3) to obtain 3a as a light-yellow solid (90 mg, 56% yield).
m.p. 225–227 °C (dec.). IR (ATR neat) ν = 1556, 1491, 1431, 1384, 1165, 804, 775 cm−1. Anal. calcd for C24H17NOS2: C, 72.15; H, 4.29; N, 3.51; S, 16.05. Found: C, 72.34; H, 4.29; N, 3.71; S, 16.21. 1H NMR (400 MHz, CDCl3, δ): 7.78 (dd, J = 8.2, 1.2 Hz, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.58 (bd, J = 8.6 Hz, 1H), 7.39–7.26 (m, 3H), 7.15–6.98 (m, 2H), 6.11 (s, 1H), 3.35 (s, 3H), 2.17 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3, δ): 157.9, 145.0, 142.0, 135.2, 134.2, 129.3, 129.1, 128.4, 127.3, 126.9, 126.65, 126.60, 126.4, 125.9, 125.7, 125.5, 125.2, 125.0, 123.8, 123.1, 116.8, 102.8, 55.6, 15.8, ppm.
:4 to CH2Cl2:peteroleum ether–1:2) to obtain 3c (F1) as a light-yellow solid (16 mg, 22% yield) and 12c (F2) as a light-yellow solid (26 mg, 36% yield).
3c: m.p. 252–254 °C. IR (ATR neat) ν = 1598, 1555, 1483, 1430, 1231, 1163 cm−1. Anal. calcd for C25H19NOS2: C, 72.61; H, 4.63; N, 3.39; S, 15.50. Found: 72.91; H, 4.68; N, 3.52; S, 15.71. 1H NMR (400 MHz, CDCl3, δ): 7.91 (bd, J = 8.4 Hz, 1H), 7.61 (bd, J = 8.6 Hz, 1H), 7.42–7.38 (m, 1H), 7.29–7.25 (m, 1H), 7.16 (bs, 1H), 7.11 (dd, J = 7.4, 1.6 Hz, 1H), 7.08 (bs, 1H), 7.04–6.96 (m, 2H), 6.07 (s, 1H), 3.33 (s, 3H), 2.67 (s, 3H), 2.14 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3, δ): 157.9, 145.3, 142.1, 133.3, 133.2, 132.9, 129.3, 129.2, 127.1, 126.9, 126.6, 126.3, 126.0, 125.9, 125.2, 125.0, 124.9, 124.7, 124.3, 122.9, 116.6, 102.7, 55.7, 19.3, 15.8, ppm.
12c: m.p. 270 °C (dec.). IR (ATR neat) ν = 1595, 1557, 1486, 1461, 1437, 1356, 1220, 1177, 1160, 1052 cm−1. Anal. calcd for C25H19NOS2: C, 72.61; H, 4.63; N, 3.39; S, 15.50. Found: 72.82; H, 4.58; N, 3.57; S, 15.38. 1H NMR (400 MHz, CDCl3, δ): 9.28 (bd, J = 8.5 Hz, 1H), 8.44 (bd, J = 8.2 Hz, 1H), 7.87 (bd, J = 8.4 Hz, 1H), 7.62 (s, 1H), 7.40–6.08 (m, 6H), 6.97 (s, 1H), 3.70 (s, 3H), 2.65 (s, 3H), 1.89 (s, 3H), ppm. 13C NMR (50 MHz, CDCl3, δ): 157.7, 150.0, 147.1, 140.2, 134.5, 132.9, 132.7, 132.0, 131.0, 128.9, 127.9, 127.4, 127.0, 126.8, 126.7, 125.7, 125.7, 125.4, 124.9, 124.3, 112.3, 55.4, 19.3, 15.6, ppm.
:petroleum ether–7:3) to obtain 12d (F1) as a light-yellow solid (28 mg, 37% yield) and 3d (F2) as a light-yellow solid (32 mg, 42% yield).
3d: m.p. 222–223 °C. IR (ATR neat) ν = 1592, 1575, 1456, 1444, 1403, 1386, 1087, 1021 cm−1. Anal. calcd for C26H21NO3S2: C, 67.95; H, 4.61; N, 3.05; S, 13.95. Found: C, 67.86; H, 4.92; N, 2.88; S, 13.86. 1H NMR (400 MHz, CDCl3, δ): 7.90 (bd, J = 8.4 Hz, 1H), 7.59 (bd, J = 8.6 Hz, 1H), 7.42–7.38 (m, 1H), 7.30–7.26 (m, 1H), 7.16–7.14 (m, 2H), 7.04–6.96 (m, 2H), 5.93 (s, 1H), 4.03 (s, 3H), 3.83 (s, 3H), 3.36 (s, 3H), 2.66 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3, δ): 153.4, 150.1, 142.3, 142.0, 139.0, 133.2, 133.1, 133.0, 128.4, 127.1, 126.9, 126.5, 126.4, 126.1, 126.0, 125.6, 125.3, 124.9, 124.7, 124.1, 112.1, 100.0, 61.4 (2C), 56.3, 19.3, ppm.
12d: m.p. 204–207 °C. IR (ATR solid) ν = 1580, 1557, 1479, 1434, 1422, 1385, 1242, 1106, 1016 cm−1. Anal. calcd for C26H21NO3S2: C, 67.95; H, 4.61; N, 3.05; S, 13.95. Found: C, 67.66; H, 4.82; N, 2.98; S, 13.55. 1H NMR (400 MHz, CDCl3, δ): 7.91 (bd, J = 8.4 Hz, 1H), 7.52 (bd, J = 8.6 Hz, 1H), 7.41–7.37 (m, 2H), 7.26–7.22 (m, 1H), 7.19 (bs, 1H), 6.99 (td, J = 7.5, 1.4 Hz, 1H), 6.93 (td, J = 7.6, 1.6 Hz, 1H), 6.52 (dd, J = 7.9, 1.4 Hz, 1H), 3.95 (s, 3H), 3.92 (s, 3H), 3.87 (s, 3H), 2.67 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3, δ): 148.9, 148.5, 146.7, 144.0, 137.4, 133.3, 132.9, 132.6, 128.2, 127.6, 127.0, 126.8, 126.34, 126.32, 126.1, 125.6, 124.8, 124.1, 123.7, 119.1, 118.2, 115.9, 61.5, 61.4, 61.3, 19.3, ppm.
:CH2Cl2–2:3) to afford (rac)-3HelOH as a white solid (53 mg, quantitative yield). m.p. 255 °C (dec.). IR (ATR neat) ν = 3542, 1431 cm−1 Anal. calcd for C23H15NOS2: C, 71.66;%; H 3.92%; N 3.63%; found: C, 71.22;%; H 3.57%; N 3.77%.1H NMR (400 MHz, CDCl3, δ): 7.76 (d, 1H, J = 8.1 Hz), 7.64 (d, 1H, J = 8.7 Hz), 7.57 (d, 1H, J = 8.6 Hz), 7.38–7.34 (m, 1H), 7.30–7.27 (m, 2H), 7.12 (dd, 1H, J = 7.2, 1.8 Hz), 7.08 (bs, 1H), 7.04–6.97 (m, 2H), 6.06 (s, 1H), 4.44 (s, 1H), 2.16 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3, δ): 153.8, 145.3, 141.8, 134.9, 134.3, 129.8, 129.1, 128.5, 127.5, 127.0, 126.8, 126.6, 126.4, 126.0, 125.8, 125.5, 125.2, 125.1, 123.8, 120.4, 117.5, 106.8, 15.4, ppm.
3D1: IR (ATR neat) ν = 1791, 1435 cm−1 Anal. calcd for C33H27NO4S2: C, 70.07%; H 4.81%; N 2.48%; found: C, 70.49;%; H 5.14%; N 2.77%. 1H NMR (400 MHz, CDCl3, δ): 7.76 (d, 1H, J = 8.1 Hz), 7.65 (d, 1H, J = 8.6 Hz), 7.48 (d, 1H, J = 8.6 Hz) 7.39–7.35 (m, 1 H), 7.30–7.22 (m, 3H), 7.11 (dd, 1H, J = 7.2, 1.9 Hz), 7.05–6.98 (m, 2H), 6.23 (s, 1H), 2.40–2.33 (m, 1H), 2.12 (s, 3H), 2.06–1.99 (m, 1H), 1.90–1.83 (m, 1H), 1.67–1.61 (m, 1H), 1.07 (s, 3H), 0.97 (s, 3H) 0.90 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3, δ): 177.8, 165.6, 148.5, 145.1, 141.4, 134.4, 134.3, 129.9, 128.6, 128.1, 127.6, 127.0, 126.84, 126.77, 126.7, 126.02, 125.97, 125.9, 125.5, 125.41, 125.39, 125.0, 123.5, 112.8, 90.8, 54.9, 54.6, 31.2, 29.0, 16.9, 16.8, 16.3, 9.8, ppm. Opt. Rot.: [α]20D +143 (c = 0.2 in CH2Cl2) (99:1 dr).
3D2: IR (ATR neat) ν = 1791, 1435, cm−1. Anal. calcd for C33H27NO4S2: C, 70.07%; H 4.81%; N 2.48%; found: C, 70.37%; H 4.74%; N 2.78%. 1H NMR (400 MHz, CDCl3, δ): 7.77 (d, 1H, J = 8.1 Hz), 7.66 (d, 1H, J = 8.5 Hz), 7.50 (d, 1H, J = 8.5 Hz) 7.40–7.35 (m, 1 H), 7.31–7.23 (m, 3H), 7.12 (dd, 1H, J = 6.9, 2.1 Hz), 7.07–7.00 (m, 2H), 6.22 (s, 1H), 2.42–2.35 (m, 1H), 2.12 (s, 3H), 2.08–2.01 (m, 1H), 1.92–1.84 (m, 1H), 1.70–1.63 (m, 1H), 1.07 (s, 3H), 1.00 (s, 3H) 0.88 (s, 3H), ppm. 13C NMR (100 MHz, CDCl3, δ): 177.8, 165.5, 148.5, 145.1, 141.4, 134.4, 134.3, 129.9, 128.5, 128.1, 127.6, 127.0, 126.9, 126.8, 126.7, 126.00, 125.97, 125.9, 125.5, 125.44, 125.39, 125.1, 123.5, 112.8, 90.8, 54.9, 54.4, 31.1, 29.0, 16.90, 16.87, 16.2, 9.8, ppm. Opt. Rot.: [α]20D −146 (c = 0.2 in CH2Cl2) (97:3 dr).
Data availability
The data that support the findings of this study, including 1H and 13C NMR spectra, CSP-HPLC traces of all the resolved compounds, and details of the racemization energy estimation, are available in the ESI of this article. Crystallographic data for 3a, 3d, and 12d has been deposited at the Cambridge Crystallographic Data Centre under 2361198 (3a) 2361200 (3d) and 2361199 (12d) and can be obtained from https://www.ccdc.cam.ac.uk/structures/.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support provided by the MUR – Progetto Dipartimenti di Eccellenza 2023–2027 (DICUS 2.0) to the Department of Chemistry “Ugo Schiff” of the University of Florence and by the Horizon Europe Program through the ERC-Synergy project CASTLE (proj. no. 101071533) is acknowledged. The authors thank Dr Cristina Faggi for X-ray analyses. The Big & Open Data Innovation Laboratory (BODaILab) of the University of Brescia and Computing Center CINECA (Bologna, Italy) are acknowledged for providing high-performance computing facilities.References
- K. Yavari, P. Aillard, Y. Zhang, F. Nuter, P. Retailleau, A. Voituriez and A. Marinetti, Angew Chem., Int. Ed., 2014, 53, 861–865 CrossRef CAS.
- N. Takenaka, J. Chen, B. Captain, R. S. Sarangthem and A. Chandrakumar, J. Am. Chem. Soc., 2010, 132, 4536–4537 CrossRef CAS PubMed.
- D. Sakamoto, I. G. Sánchez, J. Rybáček, J. Vacek, L. Bednárová, M. Pazderková, R. Pohl, I. Císařová, I. G. Stará and I. Starý, ACS Catal., 2022, 12, 10793–10800 CrossRef CAS.
- B. Lousen, S. K. Pedersen, D. M. Răsădean, G. D. Pantoş and M. Pittelkow, Chem. – Eur. J., 2021, 27, 6064–6069 CrossRef CAS PubMed.
- P. A. Summers, A. P. Thomas, T. Kench, J. B. Vannier, M. K. Kuimova and R. Vilar, Chem. Sci., 2021, 12, 14624–14634 RSC.
- J. L. Rushworth, A. R. Thawani, E. Fajardo-Ruiz, J. C. M. Meiring, C. Heise, A. J. P. White, A. Akhmanova, J. R. Brandt, O. Thorn-Seshold and M. J. Fuchter, JACS Au, 2022, 2, 2561–2570 CrossRef CAS PubMed.
- T. R. Schulte, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2019, 58, 5562–5566 CrossRef CAS PubMed.
- A. U. Malik, F. Gan, C. Shen, N. Yu, R. Wang, J. Crassous, M. Shu and H. Qiu, J. Am. Chem. Soc., 2018, 140, 2769–2772 CAS.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. Hernández Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957–1962 CrossRef CAS PubMed.
- R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709–7719 CrossRef PubMed.
- Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
- W. Fu, V. Pelliccioli, M. von Geyso, P. Redero, C. Böhmer, M. Simon, C. Golz and M. Alcarazo, Adv. Mater., 2023, 2211279 CrossRef CAS.
- T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 5660 CrossRef CAS.
- V. Pelliccioli, T. Hartung, M. Simon, C. Golz, E. Licandro, S. Cauteruccio and M. Alcarazo, Angew. Chem., Int. Ed., 2022, 61, e202114577 CrossRef CAS PubMed.
- L. D. M. Nicholls, M. Marx, T. Hartung, E. González-Fernández, C. Golz and M. Alcarazo, ACS Catal., 2018, 8, 6079–6085 CrossRef CAS.
- P. Redero, T. Hartung, J. Zhang, L. D. M. Nicholls, G. Zichen, M. Simon, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2020, 59, 23527–23531 CrossRef CAS.
- E. González-Fernández, L. D. M. Nicholls, L. D. Schaaf, C. Farès, C. W. Lehmann and M. Alcarazo, J. Am. Chem. Soc., 2017, 139, 1428–1431 CrossRef PubMed.
- C. Li, Y.-B. Shao, X. Gao, Z. Ren, C. Guo, M. Li and X. Li, Nat. Commun., 2023, 14, 3380 CrossRef CAS PubMed.
- K. Li, S. Huang, T. Liu, S. Jia and H. Yan, J. Am. Chem. Soc., 2022, 144, 7374–7381 CrossRef CAS.
- L. Kötzner, M. J. Webber, A. Martínez, C. Defusco and B. List, Angew. Chem., Int. Ed., 2014, 53, 5202–5205 CrossRef PubMed.
- W. Liu, T. Qin, W. Xie, J. Zhou, Z. Ye and X. Yang, Angew. Chem., Int. Ed., 2023, 62, e202303430 CrossRef CAS PubMed.
- S. Menichetti, S. Cecchi, P. Procacci, M. Innocenti, L. Becucci, L. Franco and C. Viglianisi, Chem. Commun., 2015, 51, 11452–11454 RSC.
- R. Amorati, L. Valgimigli, A. Baschieri, Y. Guo, F. Mollica, S. Menichetti, M. Lupi and C. Viglianisi, ChemPhysChem, 2021, 22, 1446–1454 CrossRef CAS PubMed.
- G. Lamanna, C. Faggi, F. Gasparrini, A. Ciogli, C. Villani, P. J. Stephens, F. J. Devlin and S. Menichetti, Chem. – Eur. J., 2008, 14, 5747–5750 CrossRef CAS PubMed.
- N. Giaconi, A. L. Sorrentino, L. Poggini, M. Lupi, V. Polewczyk, G. Vinai, P. Torelli, A. Magnani, R. Sessoli, S. Menichetti, L. Sorace, C. Viglianisi and M. Mannini, Angew Chem., Int. Ed., 2021, 60, 15276–15280 CAS.
- (a) N. Giaconi, L. Poggini, M. Lupi, M. Briganti, A. Kumar, T. K. Das, A. L. Sorrentino, C. Viglianisi, S. Menichetti, R. Naaman, R. Sessoli and M. Mannini, ACS Nano, 2023, 17, 15189–15198 CrossRef CAS PubMed; (b) N. Giaconi, M. Lupi, T. K. Das, A. Kumar, L. Poggini, C. Viglianisi, L. Sorace, S. Menichetti, R. Naaman, R. Sessoli and M. Mannini, J. Mater. Chem. C, 2024, 12, 10029–10035 RSC.
- H. Ando, H. Takamura, I. Kadota and K. Tanaka, Chem. Commun., 2024, 60, 4765–4768 RSC.
- M. Lupi, O. Salmi, C. Viglianisi and S. Menichetti, Adv. Synth. Catal., 2023, 365, 1705–1712 CrossRef CAS.
- K. Usui, K. Yamamoto, T. Shimizu, M. Okazumi, B. Mei, Y. Demizu, M. Kurihara and H. Suemune, J. Org. Chem., 2015, 80, 6502–6508 CrossRef CAS.
- (a) T. Tsujihara, D. Y. Zhou, T. Suzuki, S. Tamura and T. Kawano, Org. Lett., 2017, 19, 3311–3314 CrossRef CAS; (b) T. Thongpanchang, K. Paruch, T. J. Katz, A. L. Rheingold, K. C. Lam and L. Liable-Sands, J. Org. Chem., 2000, 65, 1850–1856 CAS; (c) F. Torricelli, J. Bosson, C. Besnard, M. Chekini, T. Bürgi and J. Lacour, Angew. Chem., Int. Ed., 2013, 52, 1796–1800 CrossRef CAS; (d) E. H. Bhalodi, K. N. Patel and A. V. Bedekar, Tetrahedron, 2022, 114, 132761 CrossRef CAS.
- (a) C. Herse, D. Bas, F. C. Krebs, T. Bürgi, J. Weber, T. Wesolowski, B. W. Laursen and J. Lacour, Angew. Chem., Int. Ed., 2003, 42, 3162–3166 CrossRef CAS PubMed; (b) B. Laleu, P. Mobian, C. Herse, B. W. Laursen, G. Hopfgartner, G. Bernardinelli and J. Lacour, Angew. Chem., Int. Ed., 2005, 44, 1879–1883 CAS.
- M. Lupi, M. Onori, S. Menichetti, S. Abbate, G. Longhi and C. Viglianisi, Molecules, 2022, 27, 1160 CAS.
- G. R. Kiel, H. M. Bergman, A. E. Samkian, N. J. Schuster, R. C. Handford, A. J. Rothenberger, R. Gomez-Bombarelli, C. Nuckolls and T. Don Tilley, J. Am. Chem. Soc., 2022, 144, 23421–23427 CrossRef CAS.
- Y. Lin, G. Lu, R. Wang and W. Yi, Org. Lett., 2016, 18, 6424–6427 CrossRef CAS.
- All attempts to use methoxy-substituted tetralones as starting materials failed leading to oxidative degradation.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- G. Mazzeo, S. Ghidinelli, R. Ruzziconi, M. Grandi, S. Abbate and G. Longhi, ChemPhotoChem, 2022, 6, e202100222 CAS.
- S. Abbate, G. Longhi, F. Lebon, E. Castiglioni, S. Superchi, L. Pisani, F. Fontana, F. Torricelli, T. Caronna, C. Villani, R. Sabia, M. Tommasini, A. Lucotti, D. Mendola, A. Mele and D. A. Lightner, J. Phys. Chem. C, 2014, 118, 1682–1695 CrossRef CAS.
- G. Longhi, E. Castiglioni, C. Villani, R. Sabia, S. Menichetti, C. Viglianisi, F. Devlin and S. Abbate, J. Photochem. Photobiol., A, 2016, 331, 138–145 CrossRef CAS.
- A. Moskowitz, Optical Rotatory Dispersion, ed. C. Djerassi, McGraw-Hill Book Co, New York, 1960, vol. 151 Search PubMed.
- E. Giorgio, R. G. Viglione, R. Zanasi and C. Rosini, J. Am. Chem. Soc., 2004, 126, 12968–12976 CrossRef CAS PubMed.
- C. Viglianisi, E. Marcantoni, V. Carapacchi, S. Menichetti and L. Marsili, Eur. J. Org. Chem., 2014, 6405–6410 CrossRef CAS.
- E. Castiglioni, S. Abbate and G. Longhi, Appl. Spectrosc., 2010, 64, 1416–1419 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2361198–2361200. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00979g |
| This journal is © The Royal Society of Chemistry 2024 |