
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLeveraging in situ N-tosylhydrazones as diazo surrogates for efficient access to pyrazolo-[1,5-c]quinazolinone derivatives†
Jun
Yan
a,
Pascal
Retailleau
b,
Christine
Tran
*a and
Abdallah
Hamze
 *a
*a
aDepartment of Chemistry and Medicinal Chemistry, Université Paris-Saclay, CNRS, BioCIS, 91400 Orsay, France. E-mail: abdallah.hamze@universite-paris-saclay.fr
bDepartment of Chemistry and Natural Products, ICSN, Université Paris-Saclay, UPR 2301, 91198, Gif-sur-Yvette, France
First published on 21st June 2024
Abstract
We developed a transition metal-free methodology for the construction of pyrazoloquinazolinone derivatives. The strategy involves a one-pot reaction wherein the N-tosylhydrazone and its corresponding diazo derivative are generated in situ, followed by an intramolecular 1,3-dipolar cycloaddition–ring expansion to provide the pyrazolo-[1,5-c]quinazolinone motif. This approach enables straightforward access to a diverse range of highly functionalized N-heterocyclic compounds in good yields (up to 92%).
Introduction
N-Heterocycles have been a focal point for researchers for several decades, particularly due to their diverse pharmaceutical and agrochemical applications.1,2 The synthesis of these scaffolds has garnered significant interest among organic chemists, leading to the development of novel synthetic methodologies.2–5 Among these structures, pyrazolo-[1,5-c]quinazolines, with a N-heterocyclic nucleus fusing quinazolines and pyrazoles, have recently emerged as pivotal compounds within the biology and chemistry communities.These compounds have a range of biological properties, as illustrated in Fig. 1.1 Specifically, molecules I act as antagonists for glycine/NMDA (N-methyl-D-aspartic acid) receptors,6 as well as for AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and kainate7 receptors, demonstrating high affinities and selectivities toward the corresponding amino acid receptors.8,9 Additionally, pyrazolo-[1,5-c]quinazolinone derivatives II also exhibited comparable activities as antagonists for adenosine receptors.10
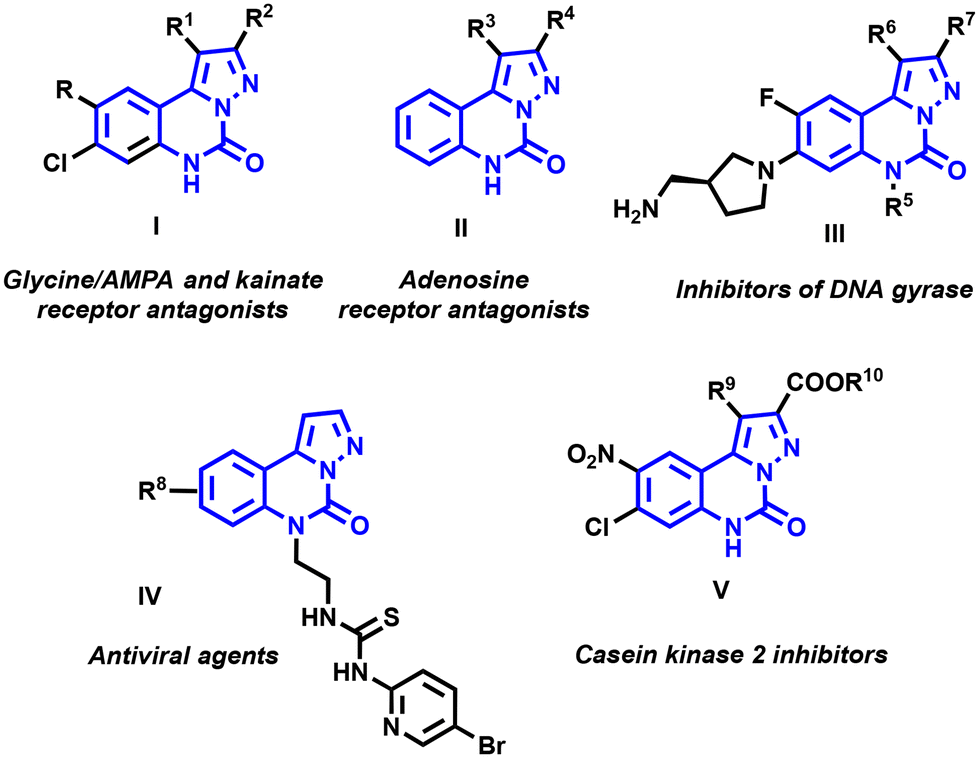 | ||
| Fig. 1 Biologically active compounds displaying the pyrazolo-[1,5-c]quinazolinone scaffold. | ||
Pyrazolo-[1,5-c]quinazolinones III emerge as promising antibacterial agents due to their function as DNA gyrase inhibitors.11 Campiani et al. reported molecules IV as potent reverse transcriptase inhibitor-type antiviral agents.12 These aza-heterocycles also acted as efficient ligands with high binding affinities towards benzodiazepine and GABAA receptors.13,14 Through virtual screening, Moro and coworkers identified pyrazolo-[1,5-c]quinazolinones V as novel casein kinase 2 inhibitors.15
The Xu group described similar structures with significant antitumor properties and inhibitory activity against cyclin-dependent kinases CDK9 and CDK2.16 More recently, the photophysical properties of pyrazolo-[1,5-c]quinazolines have been scrutinized by Sutherland et al.17 These platforms appeared to be interesting chromophores with high fluorescence quantum yields, paving the way for possible bioimaging applications.
Alongside the widely diverse properties of pyrazolo-[1,5-c]quinazolinones depicted above, numerous synthetic strategies have been explored for constructing this N-heterocyclic ring (Scheme 1). One of the earliest methods involves a multi-step synthesis, wherein 4,5-dihydro-3,5-diarylpyrazoles are formed by reacting hydrazine hydrate and 1,3-diaryl-2-propenones, and the reaction is completed with a condensation step using triphosgene (Scheme 1a).8,10,14,17 Tang and Cao combined 3-diazoindolinones with methyl β-fluoroalkylpropionates to obtain a mixture of two regioisomers of pyrazolo-[1,5-c]quinazolinone (Scheme 1b).18,19 Also, a mixture of three compounds was observed when the reaction was performed between diazooxindole and enaminones (Scheme 1c).20 Cheng and Zhai developed a [3 + 2] dipolar cycloaddition of arynes with 3-diazoindolin-2-ones in the presence of TBAT (tetrabutylammonium triphenyldifluorosilicate), leading to spiro[indazole-3,3′-indolin]-2′-ones. Their thermal isomerization obtained at 120 °C readily yields indazolo[2,3-c]quinazolin-6(5H)-ones (Scheme 1d).21,22 Mohanan et al. used the diazo derivative with the Bestmann–Ohira reagent, and the reaction with an isatin derivative afforded the spiropyrazoline derivative through a 1,3-dipolar cycloaddition followed by a 1,3-H-shift, and then a spontaneous air-oxidation in the presence of methanol delivered the phosphonated pyrazolo-[1,5-c]quinazolinones (Scheme 1e).23 Nagendra Babu et al. used a domino reaction with 3-ylideneoxindoles and diazo partners, leading to pyrazoloquinazolinones (Scheme 1f).24
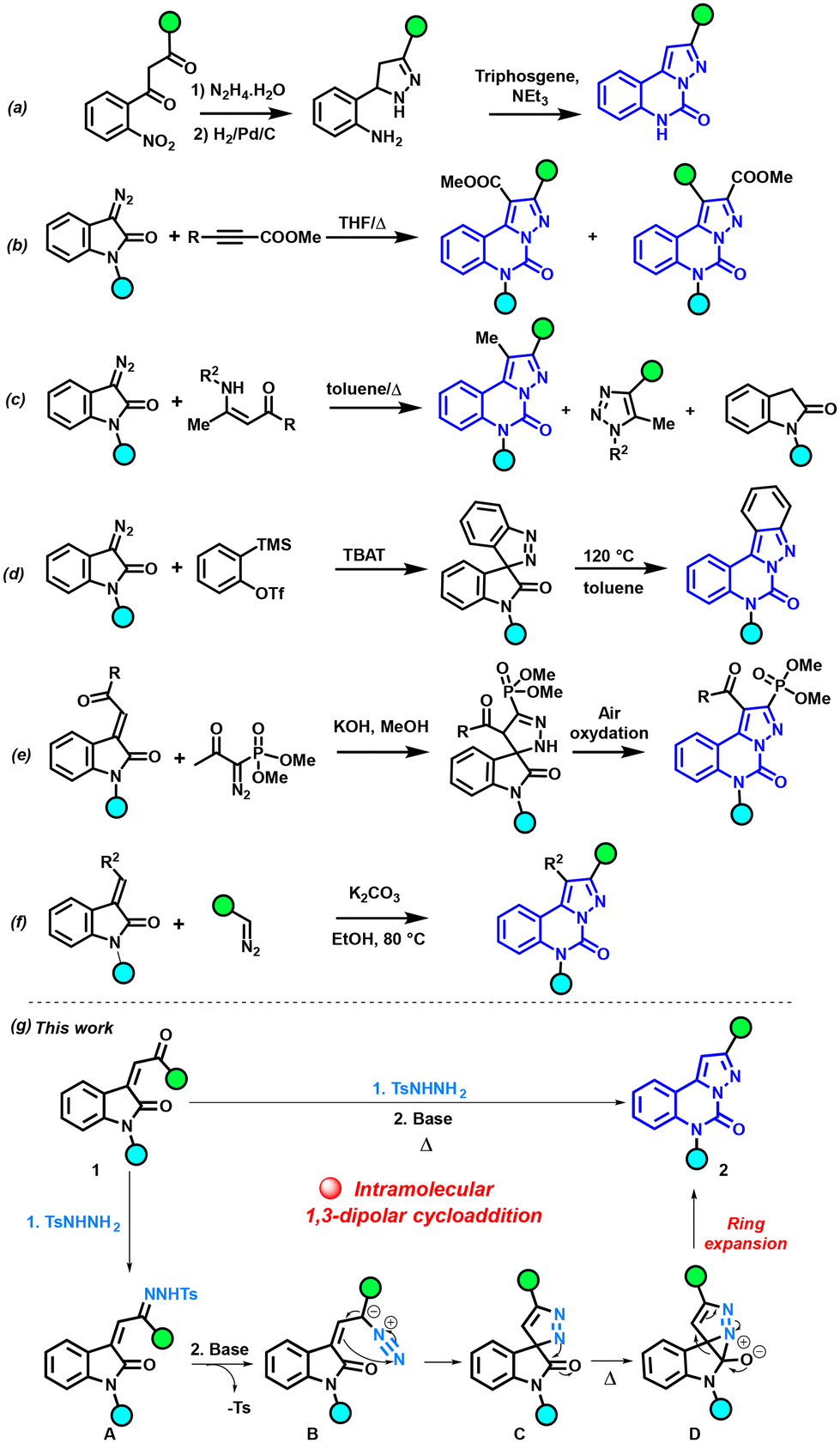 | ||
| Scheme 1 Reported approaches for synthesizing pyrazolo-[1,5-c]quinazolinones, and our approach involving intramolecular 1,3-dipolar cycloaddition for their construction. | ||
Given our sustained interest in studying the reactivity of N-tosylhydrazones (NTHs),25–32 we formulated plans to investigate reactions involving these reactive species for the construction of N-heterocyclic moieties, specifically the pyrazolo-[1,5-c]quinazolinone scaffold. Within this framework, we conceived an original strategy for pyrazolo-[1,5-c]quinazolinone synthesis through intramolecular cycloaddition (Scheme 1g). Our protocol was conducted under basic conditions and relied on the transition metal-free one-pot reaction between enone 1 and p-toluenesulfonyl hydrazide. The initial condensation of 1 with p-toluenesulfonyl hydrazide led to the NTH intermediate A. Subsequently, under the influence of a base, A was converted into the diazo species B. B then underwent an intramolecular 1,3-dipolar cycloaddition to generate C. Through thermal heating, a nucleophilic attack of the azo on the carbonyl group forms the 5/3 fused heterocyclic scaffold D.22 The final step involved the ring expansion of D, resulting in the formation of the pyrazolo-[1,5-c]quinazolinone 2.
Our approach uniquely relies on p-toluenesulfonyl hydrazide for the intramolecular 1,3-dipolar cycloaddition, without the need for any co-substrates. In comparison with prior reports, which are often limited to electron-withdrawing group (EWG)-stabilized diazo compounds, this methodology should be applicable for NTHs with electron-donating groups (EDGs). Additionally, this methodology produces only one regioisomer and eliminates the need for toxic reagents such as triphosgene, providing significant advantages for this reaction.
Results and discussion
We initiated the optimization of the reaction using enone 1a as the substrate (Table 1). The utilization of a strong base, such as tBuLi, resulted in the formation of the desired product 2a, albeit in a low yield of 18% (Table 1, entry 1). It is worth noting that the structure of 2a was fully confirmed through X-ray crystal analysis (see the ESI† for further details).33|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | 1HNMR yield (%) | Yieldb (%) |
| a Enone 1a (0.20 mmol, 1.0 equiv.) and p-toluenesulfonyl hydrazide (0.24 mmol, 1.2 equiv.) were dissolved in dioxane (2 mL). The reaction was heated in an oil bath at 90 °C and stirred for 2 h. Then, base (0.40 mmol, 2.0 equiv.) was added, and the reaction mixture was stirred at 110 °C for 3 h. b Isolated yield after column chromatography. c n.d. = not determined. d The second step was conducted at 90 °C. | ||||
| 1 | tBuLi | Dioxane | 18 | n.d.c |
| 2 | LiOtBu | Dioxane | 20 | n.d.c |
| 3 | Cs2CO3 | Dioxane | 80 | 72 |
| 4 | K3PO4 | Dioxane | 98 | 92 |
| 5 | K3PO4 | Toluene | 90 | 85 |
| 6 | K3PO4 | 2-Propanol | 88 | 80 |
| 7 | K2CO3 | Dioxane | 92 | 87 |
| 8 | K3PO4 | Dioxane | 91d | 86 |
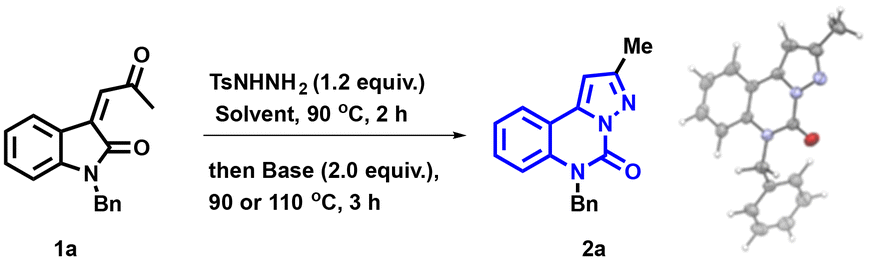
Employing a relatively weaker base, LiOtBu, did not result in a significant enhancement in reaction efficiency (entry 2). Subsequently, the exploration continued with an inorganic base such as Cs2CO3 (entry 3), which delivered a noteworthy yield of 72%. A further substantial improvement in yield was observed when dioxane was employed, coupled with a switch to K3PO4 (entry 4). Compound 2a was obtained in an excellent NMR yield of 98% and an isolated yield of 92%. Varying the solvent demonstrated the versatility of this transformation in both polar and non-polar solvents (entries 4–6). With a view to improve the sustainability of our system, the green solvent 2-propanol was employed as the reaction medium,34 resulting in the corresponding pyrazolo-[1,5-c]quinazolinones 2a in a yield of 80%. Transitioning from K3PO4 to K2CO3 as the base resulted in a slight decrease in the yield (entry 7). Furthermore, lowering the temperature to 90 °C during the cyclization step led to a marginal decrease in yield to 91% (entry 8).
Consequently, we established the optimal conditions for this transformation, utilizing K3PO4 as the base and dioxane as the solvent.
We then began to explore the scope of the reaction. First, we investigated the modification of the amino-protecting groups of enones 1. As illustrated in Scheme 2, the transformation proved to be well suited for N-ethyl, -allyl, -propynyl and -phenyl substrates and afforded the corresponding pyrazolo-[1,5-c]quinazolinones 2b–i in good yields. To our satisfaction, the unprotected enone 1e, under the optimal reaction conditions, provided the desired product 2e in 72% yield. In contrast, the expected heterocycles 2f and 2g with electron withdrawing protecting groups were not detected. Instead, the unprotected compound 2e was isolated in satisfactory yields (77% and 67% for acetyl and Boc, respectively), possibly due to the basic conditions of the cyclization step.35
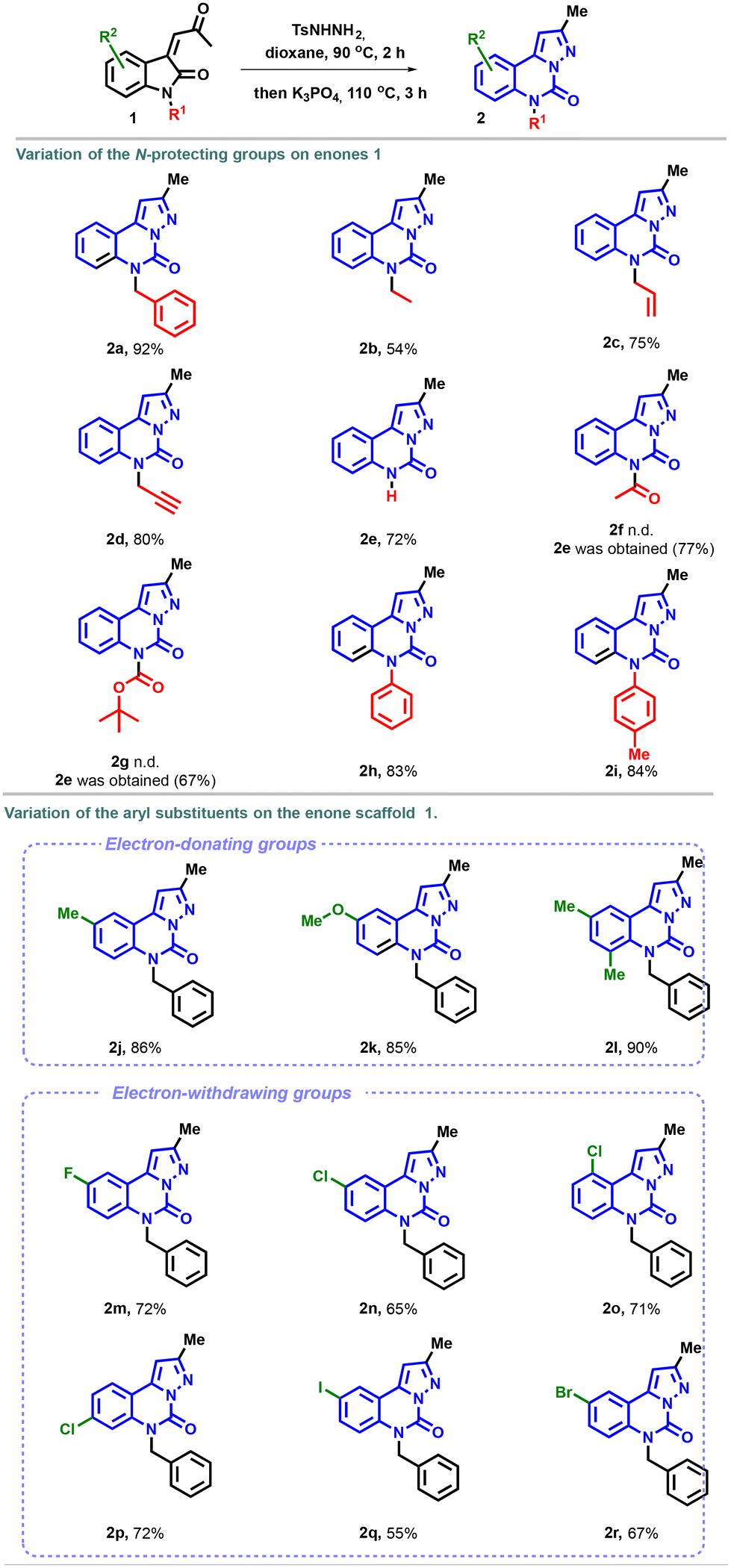 | ||
Scheme 2 Substrate scope. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: enone 1 (0.20 mmol, 1.0 equiv.) and p-toluenesulfonyl hydrazide (0.24 mmol, 1.2 equiv.) were dissolved in dioxane (2 mL). The reaction was heated in an oil bath at 90 °C, and stirred for 2 h. K3PO4 (0.40 mmol, 2.0 equiv.) was then added to the reaction mixture, which was stirred at 110 °C for 3 h. Isolated yield after chromatographic purification. Reaction conditions: enone 1 (0.20 mmol, 1.0 equiv.) and p-toluenesulfonyl hydrazide (0.24 mmol, 1.2 equiv.) were dissolved in dioxane (2 mL). The reaction was heated in an oil bath at 90 °C, and stirred for 2 h. K3PO4 (0.40 mmol, 2.0 equiv.) was then added to the reaction mixture, which was stirred at 110 °C for 3 h. Isolated yield after chromatographic purification. | ||
Next, the variation of the aryl substituents on the enone scaffold was examined (R2 group) (Scheme 2). A good tolerance was observed with methyl and methoxy groups, with yields of 86 and 85%, respectively, for molecules 2j and 2k. In particular, the presence of electron-donating substituents promoted the formation of the pyrazolo-[1,5-c]quinazolinones 2l. Similarly, the reaction exhibited good compatibility with diverse electron-poor groups, with yields up to 72%. Higher yields were obtained with fluoro (2m), chloro (2n), and bromo (2r) substituents compared to the results observed with the iodo substituent 2q. The reaction displayed good efficiency with meta, ortho, and para-substituted chloroenones, and compounds 2n, 2o, and 2p were obtained in a good yield.
Next, we expanded the scope of our studies by varying the ketone substituents of enones 1 (Scheme 3).
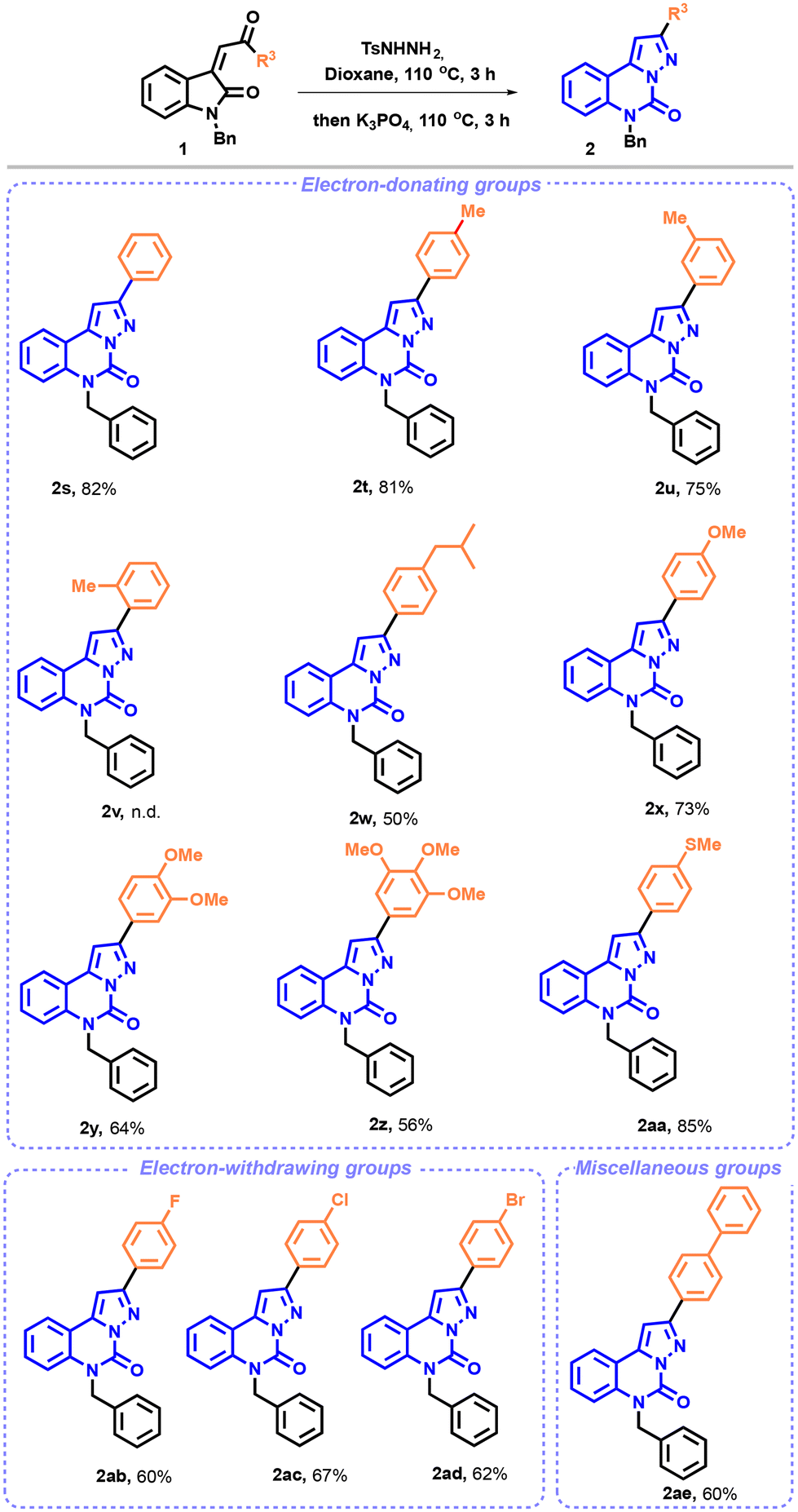 | ||
| Scheme 3 Substrate scope: variation of the ketone substituents on enones 1. aReaction conditions: enone 1 (0.20 mmol, 1.0 equiv.) and p-toluenesulfonyl hydrazide (0.24 mmol, 1.2 equiv.) were dissolved in dioxane (2 mL). The reaction was heated in an oil bath at 110 °C and stirred for 2 h. K3PO4 (0.40 mmol, 2.0 equiv.) was then added to the reaction mixture, which was stirred at 110 °C for 3 h. Isolated yield after chromatographic purification. | ||
In order to facilitate the formation of the pyrazolo-[1,5-c]quinazoline, we slightly modified the reaction conditions. The NTH synthesis was thus carried out at 110 °C for 3 h, instead of 90 °C for 2 h. Upon first examination, as shown in Scheme 3, the reaction proceeded smoothly with both electron-rich and -poor groups. The phenyl group afforded the desired compound 2s in 82% yield.
Surprisingly, the effectiveness of the reaction decreased upon the addition of a methyl or an isopropyl substituent to the phenyl moiety. While in the presence of the para and meta-methylated pyrazolo-[1,5-c]quinazolinones the desired compounds (2t and 2u) were obtained in good yields, the ortho-substituted pyrazolo-[1,5-c]quinazolinone 2v was not isolated under our reaction conditions, probably due to steric hindrance. Only degradation products could be seen on TLC and crude 1H NMR. Moderate to good yields were achieved with a methoxy or a methylthio group (2x and 2aa) or even in the presence of several electron-donating groups on the phenyl scaffold (2y and 2z). Consistent with the observations made regarding the aryl substituents on substrate 1, a similar trend was noted for electron-withdrawing substituents, resulting in the formation of compounds 2ab–2ad in yields ranging from 60 to 67%. Furthermore, we explored a biphenyl substrate, 1ae, which delivered the expected compound 2ae in 60% yield.
To confirm the viability of our methodology, we successfully performed a gram-scale reaction with substrate 1r (5 mmol), delivering the desired cyclized product 2r in a yield of 68% (1.2 g) (Scheme 4, eqn (1)). Compound 2r was then subjected to various post-functionalization reactions. A Suzuki–Miyaura cross-coupling was performed with (3-methoxyphenyl)boronic acid, K3PO4 as the base and PdCl2(PPh3)2 as the catalyst in DME/H2O/EtOH31 to afford 3 in 83% yield (Scheme 4, eqn (2)).
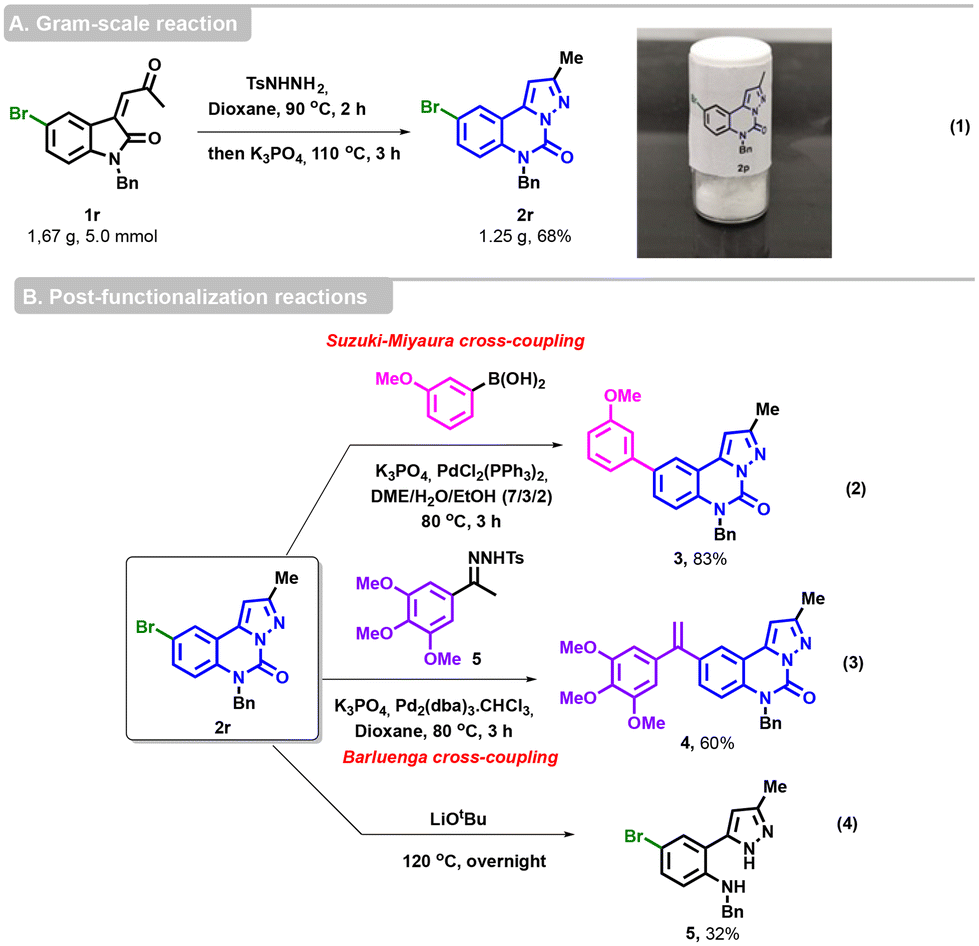 | ||
| Scheme 4 Gram-scale reaction and post-functionalization reactions. | ||
The Barluenga–Valdés coupling reaction was carried out under the standard conditions between the bromo-derived pyrazoloquinazolinone 2r and 3,4,5-trimethoxyphenyl NTH 5, allowing access to the alkene derivative 4 in a satisfactory yield (Scheme 4, eqn (3)). Finally, the hydrolysis of the urea group was also achieved in the presence of LiOtBu at 120 °C, furnishing the expected pyrazole 5 (Scheme 4, eqn (4)).
Conclusions
In summary, the synthesis of pyrazolo-[1,5-c]quinazolinone derivatives has been achieved using a convenient methodology. The process relied on the in situ formation of NTH, followed by the generation of the diazo derivative in a basic medium. This intermediate underwent an intramolecular 1,3-dipolar cycloaddition, leading to the expected pyrazolo-[1,5-c]quinazolinones. The wide functional group compatibility (29 diversely functionalized products synthesized in moderate to excellent yields), the transition metal-free process, the bench-stability of substrates, and the step economy of the intramolecular process are significant advantages of this reaction. This one-pot transformation was also validated with the gram-scale synthesis of 2r in 68% yield. Additionally, various post-functionalization reactions, including pallado-catalyzed cross-couplings, such as the Suzuki–Miyaura and Barluenga–Valdés reactions, were successfully performed with compound 2r. Further studies are underway to investigate the biological activities of the prepared pyrazolo-[1,5-c] quinazolinones.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the support provided to this project by CNRS, Paris-Saclay University and La Fondation ARC pour la recherche sur le cancer ARCPJA2022060005209. The authors also thank the China Scholarship Council for a fellowship (CSC) to J. Y.References
- M. Garg, M. Chauhan, P. K. Singh, J. M. Alex and R. Kumar, Pyrazoloquinazolines: Synthetic strategies and bioactivities, Eur. J. Med. Chem., 2015, 97, 444–461 CrossRef CAS PubMed.
- V. K. Singh, A. K. Tiwari and M. Faheem, in N-Heterocycles: Synthesis and Biological Evaluation, ed. K. L. Ameta, R. Kant, A. Penoni, A. Maspero and L. Scapinello, Springer Nature Singapore, Singapore, 2022, pp. 313–329, DOI:10.1007/978-981-19-0832-3_8.
- Y. Lv, J. Meng, C. Li, X. Wang, Y. Ye and K. Sun, Update on the Synthesis of N-Heterocycles via Cyclization of Hydrazones (2017–2021), Adv. Synth. Catal., 2021, 363, 5235–5265 CrossRef CAS.
- C.-V. T. Vo and J. W. Bode, Synthesis of Saturated N-Heterocycles, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed.
- W. Guo, M. Zhao, W. Tan, L. Zheng, K. Tao and X. Fan, Developments towards synthesis of N-heterocycles from amidines via C–N/C–C bond formation, Org. Chem. Front., 2019, 6, 2120–2141 RSC.
- F. Varano, D. Catarzi, V. Colotta, F. R. Calabri, O. Lenzi, G. Filacchioni, A. Galli, C. Costagli, F. Deflorian and S. Moro, 1-Substituted pyrazolo[1,5-c]quinazolines as novel Gly/NMDA receptor antagonists: Synthesis, biological evaluation, and molecular modeling study, Bioorg. Med. Chem., 2005, 13, 5536–5549 CrossRef CAS PubMed.
- F. Varano, D. Catarzi, V. Colotta, O. Lenzi, G. Filacchioni, A. Galli and C. Costagli, Novel AMPA and kainate receptor antagonists containing the pyrazolo[1,5-c]quinazoline ring system: Synthesis and structure–activity relationships, Bioorg. Med. Chem., 2008, 16, 2617–2626 CrossRef CAS PubMed.
- F. Varano, D. Catarzi, V. Colotta, G. Filacchioni, A. Galli, C. Costagli and V. Carlà, Synthesis and Biological Evaluation of a New Set of Pyrazolo[1,5-c]quinazoline-2-carboxylates as Novel Excitatory Amino Acid Antagonists, J. Med. Chem., 2002, 45, 1035–1044 CrossRef CAS PubMed.
- F. Varano, D. Catarzi, V. Colotta, D. Poli, G. Filacchioni, A. Galli and C. Costagli, Synthesis and Biological Evaluation of a New Set of Pyrazolo[1,5-c]quinazolines as Glycine/N-Methyl-D-aspartic Acid Receptor Antagonists, Chem. Pharm. Bull., 2009, 57, 826–829 CrossRef CAS PubMed.
- D. Catarzi, V. Colotta, F. Varano, D. Poli, L. Squarcialupi, G. Filacchioni, K. Varani, F. Vincenzi, P. A. Borea, D. Dal Ben, C. Lambertucci and G. Cristalli, Pyrazolo[1,5-c]quinazoline derivatives and their simplified analogues as adenosine receptor antagonists: Synthesis, structure–affinity relationships and molecular modeling studies, Bioorg. Med. Chem., 2013, 21, 283–294 CrossRef CAS PubMed.
- A. L. Aguirre, P. R. Chheda, S. R. C. Lentz, H. A. Held, N. P. Groves, H. Hiasa and R. J. Kerns, Identification of an ethyl 5,6-dihydropyrazolo[1,5-c]quinazoline-1-carboxylate as a catalytic inhibitor of DNA gyrase, Bioorg. Med. Chem., 2020, 28, 115439 CrossRef CAS PubMed.
- G. Campiani, F. Aiello, M. Fabbrini, E. Morelli, A. Ramunno, S. Armaroli, V. Nacci, A. Garofalo, G. Greco, E. Novellino, G. Maga, S. Spadari, A. Bergamini, L. Ventura, B. Bongiovanni, M. Capozzi, F. Bolacchi, S. Marini, M. Coletta, G. Guiso and S. Caccia, Quinoxalinylethylpyridylthioureas (QXPTs) as Potent Non-Nucleoside HIV-1 Reverse Transcriptase (RT) Inhibitors. Further SAR Studies and Identification of a Novel Orally Bioavailable Hydrazine-Based Antiviral Agent, J. Med. Chem., 2001, 44, 305–315 CrossRef CAS PubMed.
- G. Guerrini, G. Ciciani, S. Ciattini, L. Crocetti, S. Daniele, C. Martini, F. Melani, C. Vergelli and M. P. Giovannoni, Pyrazolo[1,5-a]quinazoline scaffold as 5-deaza analogue of pyrazolo[5,1-c][1,2,4]benzotriazine system: synthesis of new derivatives, biological activity on GABAA receptor subtype and molecular dynamic study, J. Enzyme Inhib. Med. Chem., 2016, 31, 195–204 CrossRef CAS PubMed.
- V. Colotta, D. Catarzi, F. Varano, G. Filacchioni, L. Cecchi, A. Galli and C. Costagli, Synthesis and Binding Activity of Some Pyrazolo[1,5-c]quinazolines as Tools To Verify an Optional Binding Site of a Benzodiazepine Receptor Ligand, J. Med. Chem., 1996, 39, 2915–2921 CrossRef CAS PubMed.
- S. Moro, F. Varano, G. Cozza, M. A. Pagano, G. Zagotto, A. Chilin, A. Guiotto, D. Catarzi, V. Calotta and L. A. Pinna, Pyrazoloquinazoline Tricyclic System as Novel Scaffold to Design New Kinase CK2 Inhibitors, Lett. Drug Des. Discovery, 2006, 3, 281–284 CrossRef CAS.
- D. Zheng, C. Yang, X. Li, D. Liu, Y. Wang, X. Wang, X. Zhang, Y. Tan, Y. Zhang, Y. Li and J. Xu, Design, Synthesis, Antitumour Evaluation, and In Silico Studies of Pyrazolo-[1,5-c]quinazolinone Derivatives Targeting Potential Cyclin-Dependent Kinases, Molecules, 2023, 28, 6606 CrossRef CAS PubMed.
- J. D. Bell, A. H. Harkiss, D. Nobis, E. Malcolm, A. Knuhtsen, C. R. Wellaway, A. G. Jamieson, S. W. Magennis and A. Sutherland, Conformationally rigid pyrazoloquinazoline α-amino acids: one- and two-photon induced fluorescence, Chem. Commun., 2020, 56, 1887–1890 RSC.
- L.-Y. Qiu, N. Ren, Z. Deng, J. Chen, H. Deng, H. Zhang, W. Cao and X.-J. Tang, The Practical Access to Fluoroalkylated Pyrazolo[1,5-c]quinazolines by Fluoroalkyl-Promoted [3 + 2] Cycloaddition Reaction, J. Org. Chem., 2023, 88, 10180–10189 CrossRef CAS PubMed.
- Q. Chen, K. Li, T. Lu and Q. Zhou, Phosphine-catalyzed domino reactions of alkynyl ketones with sulfonylhydrazones: construction of diverse pyrazoloquinazoline derivatives, RSC Adv., 2016, 6, 24792–24796 RSC.
- R. Augusti and C. Kascheres, Reactions of 3-diazo-1,3-dihydro-2H-indol-2-one derivatives with enaminones. A novel synthesis of 1,2,3-triazoles, J. Org. Chem., 1993, 58, 7079–7083 CrossRef CAS.
- B. Cheng, B. Zu, B. Bao, Y. Li, R. Wang and H. Zhai, Synthesis of Spiro[indazole-3,3′-indolin]-2′-ones via [3 + 2] Dipolar Cycloaddition of Arynes with 3-Diazoindolin-2-ones and Indazolo[2,3-c]quinazolin-6(5H)-ones by Subsequent Thermal Isomerization, J. Org. Chem., 2017, 82, 8228–8233 CrossRef CAS PubMed.
- B. Cheng, Y. Li, B. Zu, T. Wang, R. Wang, Y. Li and H. Zhai, Syntheses of spiro[indazole-3,3′-indolin]-2′-ones and spiro[indazole-3,3′-indolin]-2′-imines via 1,3-dipolar cycloadditions of arynes and studies on their isomerization reactions, Tetrahedron, 2019, 75, 130775 CrossRef.
- A. K. Gupta, S. Ahamad, E. Gupta, R. Kant and K. Mohanan, Substrate-controlled product-selectivity in the reaction of the Bestmann–Ohira reagent with N-unprotected isatin-derived olefins, Org. Biomol. Chem., 2015, 13, 9783–9788 RSC.
- G. Ramu, N. Hari Krishna, G. Pawar, K. N. Visweswara Sastry, J. B. Nanubolu and B. Nagendra Babu, Solvent-Controlled, Tunable Domino Reaction of 3-Ylideneoxindoles with in Situ-Generated α-Aryldiazomethanes: A Facile Access to 3-Spirocyclopropyl-2-oxindole and Pyrazoloquinazolinone Scaffolds, ACS Omega, 2018, 3, 12349–12360 CrossRef CAS.
- A. Hamze, B. Tréguier, J.-D. Brion and M. Alami, Copper-catalyzed reductive coupling of tosylhydrazones with amines: A convenient route to α-branched amines, Org. Biomol. Chem., 2011, 9, 6200–6204 RSC.
- J. Aziz, J.-D. Brion, A. Hamze and M. Alami, Copper Acetoacetonate [Cu(acac)2]/BINAP-Promoted Csp3
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N Bond Formation via Reductive Coupling of N-Tosylhydrazones with Anilines, Adv. Synth. Catal., 2013, 355, 2417–2429 CrossRef CAS.
N Bond Formation via Reductive Coupling of N-Tosylhydrazones with Anilines, Adv. Synth. Catal., 2013, 355, 2417–2429 CrossRef CAS. - M. Roche, J. Bignon, J.-D. Brion, A. Hamze and M. Alami, Tandem One-Pot Palladium-Catalyzed Coupling of Hydrazones, Haloindoles, and Amines: Synthesis of Amino-N-vinylindoles and Their Effect on Human Colon Carcinoma Cells, J. Org. Chem., 2014, 79, 7583–7592 CrossRef CAS PubMed.
- K. Zhang, O. Provot, C. Tran, M. Alami and A. Hamze, Copper-catalyzed sulfonylation of N-tosylhydrazones followed by a one-pot C–N bond formation, Org. Biomol. Chem., 2021, 19, 5358–5367 RSC.
- K. Zhang, O. Provot, M. Alami, C. Tran and A. Hamze, Pd-Catalyzed Coupling of N-Tosylhydrazones with Benzylic Phosphates: Toward the Synthesis of Di- or Tri-Substituted Alkenes, J. Org. Chem., 2022, 87, 1249–1261 CrossRef CAS PubMed.
- J. Yan, C. Tran, J. Bignon, O. Provot and A. Hamze, Synthesis of Dihydro-5H-Benzo[c]-Fluorenes, Dihydroindeno[c]-Chromenes and Thiochromenes via Intramolecular Cyclization and their Effect on Human Leukemia Cells, Adv. Synth. Catal., 2022, 364, 1613–1619 CrossRef CAS.
- J. Yan, C. Tran, P. Retailleau, M. Alami and A. Hamze, Catalyst-Free Synthesis of Functionalized 4-Substituted-4H-Benzo[d][1,3]oxazines via Intramolecular Cyclization of ortho-Amide-N-tosylhydrazones, J. Org. Chem., 2023, 88, 8636–8642 CrossRef CAS PubMed.
- X. Liu, O. Provot, R. Franco, P. Retailleau, M. Alami, V. Gandon, C. Tran and A. Hamze, Synthesis of Aza-Heterocyclic Compounds with N-Tosylhydrazones: Formation of Bi-Indoles via Reductive Molybdenum Catalysis, Adv. Synth. Catal., 2023, 365, 3155–3161 CrossRef CAS.
- CCDC 2341570 (2a)† contains the supporting crystallographic data for this paper.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green chemistry tools to influence a medicinal chemistry and research chemistry based organisation, Green Chem., 2008, 10, 31–36 RSC.
- S. R. Dandepally and A. L. Williams, Microwave-assisted N-Boc deprotection under mild basic conditions using K3PO4·H2O in MeOH, Tetrahedron Lett., 2009, 50, 1071–1074 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for new compounds, and crystallographic data. CCDC 2341570 (compound 2a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00950a |
| This journal is © The Royal Society of Chemistry 2024 |