
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ammonium carboxylates in the ammonia-Ugi reaction: one-pot synthesis of α,α-disubstituted amino acid derivatives including unnatural dipeptides†
Keisuke
Tomohara
 *ab,
Satoru
Kusaba
c,
Mana
Masui
b,
Tatsuya
Uchida
acd,
Hisanori
Nambu
b and
Takeru
Nose
*ac
*ab,
Satoru
Kusaba
c,
Mana
Masui
b,
Tatsuya
Uchida
acd,
Hisanori
Nambu
b and
Takeru
Nose
*ac
aFaculty of Arts and Science, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
bFaculty and Graduate School of Pharmaceutical Science, Kyoto Pharmaceutical University, 1 Misasagishichono-cho, Yamashina-ku, Kyoto 607-8412, Japan. E-mail: tomohara47@mb.kyoto-phu.ac.jp
cGraduate School of Science, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan. E-mail: nose@artsci.kyushu-u.ac.jp
dInternational Institute for Carbon-Neutral Energy Research, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
First published on 3rd July 2024
Abstract
Despite the remarkable developments of the Ugi reaction and its variants, the use of ammonia in the Ugi reaction has long been recognized as impractical and unsuccessful. Indeed, the ammonia-Ugi reaction often requires harsh reaction conditions, such as heating and microwave irradiation, and competes with the Passerini reaction, thereby resulting in low yields. This study describes a robust and practical ammonia-Ugi reaction protocol. Using originally prepared ammonium carboxylates in trifluoroethanol, the ammonia-Ugi reaction proceeded at room temperature in high yields and showed a broad substrate scope, thus synthesizing a variety of α,α-disubstituted amino acid derivatives, including unnatural dipeptides. The reaction required no condensing agents and proceeded without racemization of the chiral stereocenter of α-amino acids. Furthermore, using this protocol, we quickly synthesized a novel dipeptide, D-Leu-Aic-NH-CH2Ph(p-F), which exhibited a potent inhibitory activity against α-chymotrypsin with a Ki value of 0.091 μM.
Introduction
Peptides have attracted global interest as new modalities of drug carriers,1 beyond Lipinski's rule of 5 (bRo5) medicines,2 and biomaterials.3 Unnatural α,α-disubstituted amino acid residues have played an important role in providing peptides with biostability and conformational stability, which in turn enhances the bioavailability, target specificity, and binding activity of peptides for in vivo applications.4 Such potential has resulted in a high demand for peptides containing unnatural α,α-disubstituted amino acid residues. Conventional peptide synthesis relies on the sequential coupling of protected amino acids on solid supports, typically using large amounts of condensing agents. During this process, introducing sterically hindered α,α-disubstituted amino acids into peptides usually requires harsh conditions, such as heating and microwave irradiation, and also uses excessive amounts of condensing reagents and N-protected α,α-disubstituted amino acids,5 the latter of which are prepared by time- and step-consuming processes including protection and deprotection steps, typically from glycine-derived Schiff bases (Fig. 1a).6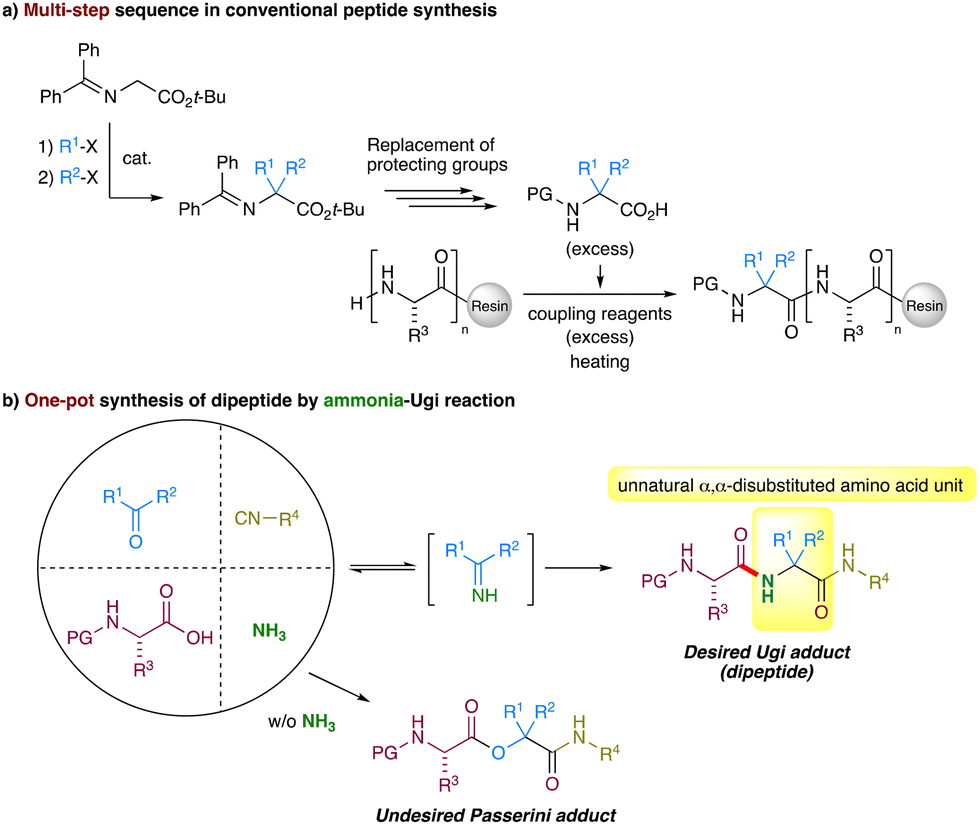 | ||
| Fig. 1 Synthetic methods of peptides containing unnatural α,α-disubstituted amino acid residues. (a) Conventional multi-step synthetic method and (b) one-pot synthesis of dipeptides by the ammonia-Ugi reaction. | ||
The Ugi reaction is a classical four-component coupling reaction involving an aldehyde or a ketone, an isocyanide, a carboxylic acid, and a primary amine to give an α-acylamino amide (α-amino acid derivative) in a single synthetic operation.7 A series of Ugi variants,8 including disrupted-Ugi9 and on-resin Ugi reactions,10 have been developed, and recently a catalytic asymmetric Ugi reaction11 and an Ugi reaction using natural product extracts as substrates12 have been introduced, and thus the Ugi reaction has now been recognized as one of the well-established methods for diversity-oriented synthesis.13 In contrast to the great expansion of the Ugi variants, the use of ammonia in the classical Ugi reaction (the ammonia-Ugi reaction) has long been recognized to be impractical and unsuccessful, although this reaction directly provides unnatural peptides containing α,α-disubstituted amino acids (Fig. 1b). Mechanistically, the ammonia-Ugi reaction begins with the reversible formation of an N-unsubstituted imine intermediate. Herein, the relatively weak nucleophilicity of ammonia, compared with primary amines, hampers the imine formation. This situation, in turn, can lead to the undesired Passerini reaction (the three-component coupling reaction of an aldehyde or ketone, an isocyanide, and a carboxylic acid).14,15 Thus, the ammonia-Ugi reaction has often required harsh conditions such as heating,16 microwave irradiation at high temperatures,17 or high dilution18 and has suffered from low yields.16a,b,18a,19 This situation becomes more serious when a ketone is used as the carbonyl component.20 Indeed, previous reports on the practical ammonia-Ugi reaction are limited where aldehydes are used as the carbonyl component.21 Overall, the applications of the ammonia-Ugi reaction toward the synthesis of unnatural α,α-disubstituted amino acid derivatives22 and peptides containing α,α-disubstituted amino acids23 have been sparingly reported.
Here, we came across a small number of successful reports on the ammonia-Ugi reaction, where the ammonia-Ugi adducts were obtained in moderate to high yields under mild reaction conditions by using commercially available ammonium formate, acetate, or benzoate as sources of both ammonia and carboxylic acids.22–24 Despite such unique potential of ammonium carboxylates, commercially unavailable ammonium carboxylates have never been applied to the ammonia-Ugi reaction and thus the effects of ammonium carboxylates remain unclear. In this context, this study systematically investigated their effects on the ammonia-Ugi reaction through establishing a method for preparing a variety of ammonium carboxylates, including the ammonium salts of α-amino acids. Using these unique reagents, a variety of α,α-disubstituted amino acid derivatives, including unnatural dipeptides, were successfully synthesized in high yields in a practical and robust manner.
Results and discussion
First, the ammonia-Ugi reaction conditions were examined using model substrates, including cyclopentanone, benzyl isocyanide, acetic acid, and ammonia, to synthesize the 1-aminocyclopentane-1-carboxylic acid (Ac5c) derivative 1a (Table 1). Previously, inorganic ammonium salts have often been used as ammonia sources;14,25 therefore, we initially tested typical inorganic ammonium salts in the ammonia-Ugi reaction at room temperature in trifluoroethanol (TFE)/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (entries 1–4). However, these reactions resulted in a low conversion of the substrates and gave both the desired ammonia-Ugi adduct Ac-Ac5c-NH-Bn (1a) and the undesired Passerini adduct (α-acyloxy amide) 2a in low yields (up to 19% yields). Among these inorganic ammonium salts, the salts of strong acids delivered 2a preferentially and gave no or trace amounts of the ammonia-Ugi adduct 1a (entries 1 and 2). In contrast, those of weak acids preferentially gave 1a over 2a (entries 3 and 4). Furthermore, the combination of NH4Cl and AcONa also resulted in the preferential production of 1a (entry 5). These results, although low yields, imply that the dissociated ammonium ions would prompt imine formation, and concomitantly the carboxylate ions would keep ketones away from the undesirable Passerini pathway, thus providing 1a preferentially. The use of aqueous NH3 greatly improved the isolated yield of 1a (55%); however, these conditions still yielded an appreciable amount of 2a (18% yield, entry 6). Finally, the use of ammonium acetate in TFE (0.5 M) at room temperature dramatically enhanced the conversion of the starting materials and concomitantly minimized the formation of the Passerini adduct 2a (6% yield), thus providing the desired Ugi adduct 1a in an excellent yield (90%, entry 7). As expected, the polar solvent methanol completely suppressed the Passerini competition; however, the nucleophilicity of methanol caused several side reactions and the reaction mixture became complicated, thereby delivering 1a in only a moderate yield (68%, entry 8). Other alcoholic solvents, ethanol, 2-propanol, and tert-butyl alcohol, also resulted in incomplete conversion of the starting materials because ammonium acetate was often insoluble in these alcohols (Table S1, entries 1–3†). Although the addition of water (up to 10%, v/v) made the reaction mixture homogeneous, it did not improve the isolated yields of 1a (Table S1, entries 4–6†). In contrast, 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) dissolved ammonium acetate well, but accelerated the Passerini reaction (Table 1, entry 9). The use of dry TFE under a N2 atmosphere had no impact on the isolated yield of 1a (entry 10), while the addition of Na2SO4 slightly increased the isolated yield of 1a (entry 11). By increasing the reaction temperature to 60 °C, the reaction time could be dramatically reduced from 30 h to 3 h with no loss of the isolated yield of 1a (entry 12). Overall, the systematic screening of a set of reagents and solvents confirmed that the use of ammonium acetate in TFE is optimal and practical for the ammonia-Ugi reaction.
:1) (entries 1–4). However, these reactions resulted in a low conversion of the substrates and gave both the desired ammonia-Ugi adduct Ac-Ac5c-NH-Bn (1a) and the undesired Passerini adduct (α-acyloxy amide) 2a in low yields (up to 19% yields). Among these inorganic ammonium salts, the salts of strong acids delivered 2a preferentially and gave no or trace amounts of the ammonia-Ugi adduct 1a (entries 1 and 2). In contrast, those of weak acids preferentially gave 1a over 2a (entries 3 and 4). Furthermore, the combination of NH4Cl and AcONa also resulted in the preferential production of 1a (entry 5). These results, although low yields, imply that the dissociated ammonium ions would prompt imine formation, and concomitantly the carboxylate ions would keep ketones away from the undesirable Passerini pathway, thus providing 1a preferentially. The use of aqueous NH3 greatly improved the isolated yield of 1a (55%); however, these conditions still yielded an appreciable amount of 2a (18% yield, entry 6). Finally, the use of ammonium acetate in TFE (0.5 M) at room temperature dramatically enhanced the conversion of the starting materials and concomitantly minimized the formation of the Passerini adduct 2a (6% yield), thus providing the desired Ugi adduct 1a in an excellent yield (90%, entry 7). As expected, the polar solvent methanol completely suppressed the Passerini competition; however, the nucleophilicity of methanol caused several side reactions and the reaction mixture became complicated, thereby delivering 1a in only a moderate yield (68%, entry 8). Other alcoholic solvents, ethanol, 2-propanol, and tert-butyl alcohol, also resulted in incomplete conversion of the starting materials because ammonium acetate was often insoluble in these alcohols (Table S1, entries 1–3†). Although the addition of water (up to 10%, v/v) made the reaction mixture homogeneous, it did not improve the isolated yields of 1a (Table S1, entries 4–6†). In contrast, 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) dissolved ammonium acetate well, but accelerated the Passerini reaction (Table 1, entry 9). The use of dry TFE under a N2 atmosphere had no impact on the isolated yield of 1a (entry 10), while the addition of Na2SO4 slightly increased the isolated yield of 1a (entry 11). By increasing the reaction temperature to 60 °C, the reaction time could be dramatically reduced from 30 h to 3 h with no loss of the isolated yield of 1a (entry 12). Overall, the systematic screening of a set of reagents and solvents confirmed that the use of ammonium acetate in TFE is optimal and practical for the ammonia-Ugi reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | NH3 source | AcOH source | Solvent | Yielda (%) | |
| 1a | 2a | ||||
| a Isolated yield. b v/v. c Starting materials remained. d Under a N2 atmosphere. e In the presence of Na2SO4 (100 mg). f At 60 °C, 3 hours. | |||||
| 1 | (NH4)2SO4 | AcOH | TFE/H2O (1:1)b |
—c | 19 |
| 2 | NH4Cl | AcOH | TFE/H2O (1:1)b |
1c | 19 |
| 3 | NH4HCO3 | AcOH | TFE/H2O (1:1)b |
17c | 7 |
| 4 | (NH4)2CO3 | AcOH | TFE/H2O (1:1)b |
19c | 8 |
| 5 | NH4Cl | AcONa | TFE/H2O (1:1)b |
22c | 9 |
| 6 | NH3 aq. | AcOH | TFE | 55 | 18 |
| 7 | AcONH4 | TFE | 90 | 6 | |
| 8 | AcONH4 | MeOH | 68 | — | |
| 9 | AcONH4 | HFIP | 53 | 25 | |
| 10 | AcONH4 | Dry TFEd | 89 | 4 | |
| 11 | AcONH4 | TFEe | 95 | 3 | |
| 12 | AcONH4 | TFEf | 92 | 4 | |

Having confirmed the potential of ammonium acetate as a key reagent for the ammonia-Ugi reaction, we then explored the scope of ketones in the synthesis of various α,α-disubstituted amino acid derivatives (Scheme 1). The six- and seven-membered ketones cyclohexanone and cycloheptanone were compatible with the reaction conditions to give the corresponding Ugi adducts 1c and 1d in excellent yields (96%), while cyclobutanone afforded the Ugi adduct 1b in a moderate yield (54%) probably because the conformationally strained four-membered ring hampered the formation of the imine intermediate. Instead, this reaction yielded a considerable amount of the Passerini adduct 2b (32%). Cyclooctanone also resulted in a low yield (53%), and the starting materials were mostly recovered. The sterically congested (−)-menthone tolerated the present reaction conditions to give 1f in 76% yield in an approximately 2:1 diastereomeric ratio (dr). The major diastereomer of 1f has a configuration of (1R), which was determined by a NOESY experiment (Fig. S1†). A series of cyclohexanone analogs containing N-Boc, O, S, and SO2 at the γ-position were well tolerated under the present reaction conditions providing the corresponding Ugi adducts 1g–1j in excellent isolated yields (86%–96%). An acetal moiety was found to be partially tolerated under the present reaction conditions to give 1k in 57% yield together with the Passerini adduct 2k (23%). Aromatic ketones resulted in low yields (1l and 1m) due to a slow conversion, while benzaldehyde exhibited a high reactivity, even at 4 °C, producing 1n in 80% yield. The ketones containing an acidic hydrogen, β-tetralone and 2-indanone, gave 1o and 1p in moderate yields (73% and 52%, respectively). The yield of 1p could be increased to 76% by using benzyl isocyanide in excess at low temperature (4 °C) in the presence of Na2SO4. The bicyclic N-Boc-nortropinone provided 1q in a moderate yield (44%), and furthermore both 2-adamantanone and 2-norbornanone were well compatible with the present reaction conditions to give 1r and 1s in high yields (86%). The hydroxy group of steroidal stanolone had no detrimental effect, affording the unusual amino acid derivative 1t in 97% yield. Acyclic ketones, i.e., acetone and 4-phenyl-2-butanone, were tolerated under these reaction conditions (1u and 1v). In contrast, the highly bulky diisopropyl ketone resulted in a low yield (1w, 16%) because of the incomplete conversion, and (+)-fenchone and hexafluoroacetone remained unreacted. The use of hexachloroacetone resulted in the isolation of the corresponding imine intermediate, which remained unreactive toward the further nucleophilic addition of isocyanide. Unless otherwise mentioned, all reactions could suppress the formation of the competing Passerini adducts to less than 5% isolated yields. Overall, these screenings clearly illustrated the generality, functional group tolerance, and utility of the developed ammonia-Ugi reaction for the one-pot synthesis of unnatural α,α-disubstituted amino acid derivatives.
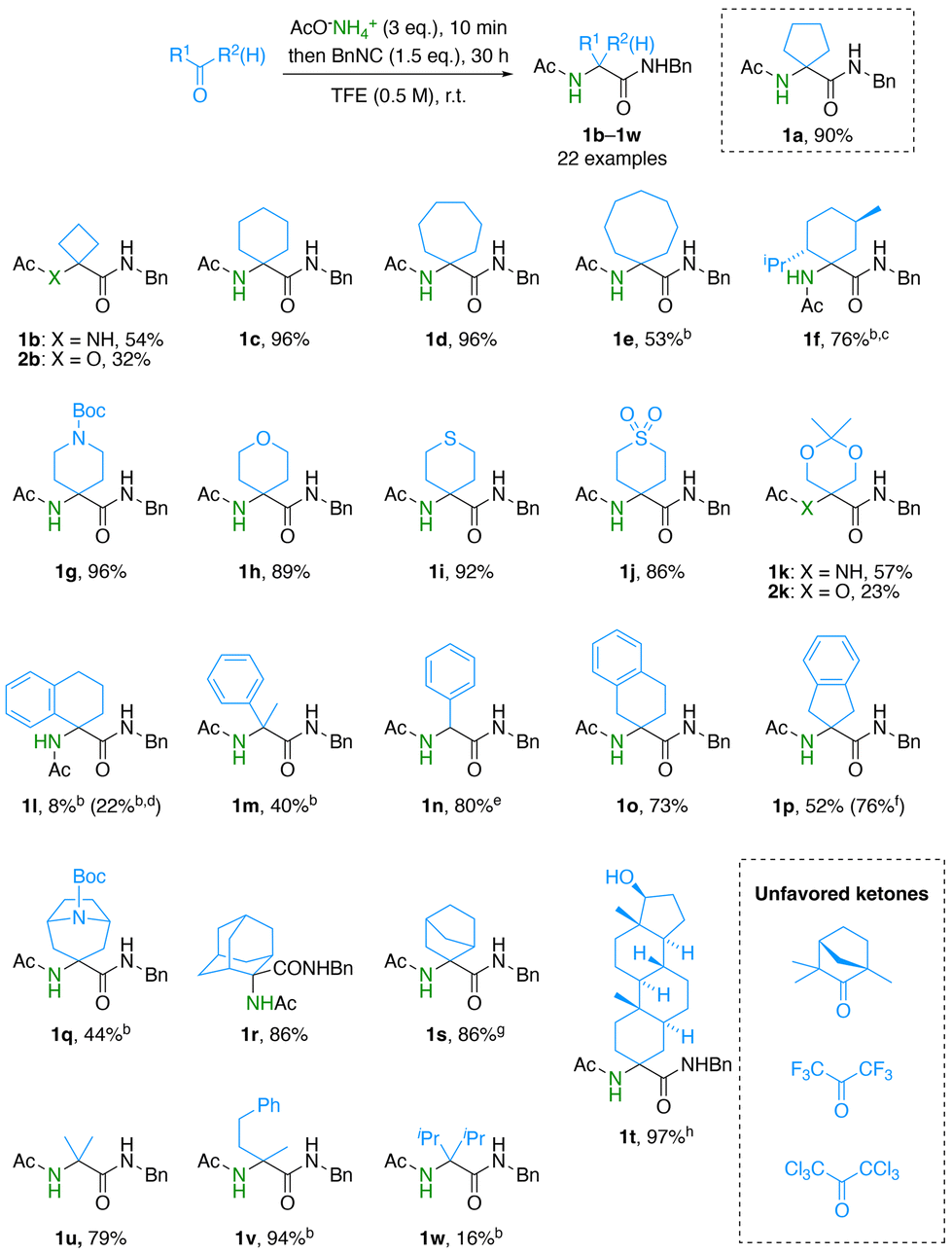 | ||
Scheme 1 Scope of ketones in the ammonia-Ugi reaction.a aIsolated yield. bStarting materials remained. cdr approx. 2:1, ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) d14 d. e3 h. fAt 4 °C in the presence of Na2SO4 (100 mg). gdr approx. 1.5:1. hdr > 20:1. d14 d. e3 h. fAt 4 °C in the presence of Na2SO4 (100 mg). gdr approx. 1.5:1. hdr > 20:1. | ||
We next investigated how the acidity of carboxylic acids affects the outcome of the ammonia-Ugi reaction, compared with the classical Ugi reaction using benzylamine (Table 2).26 Both Ugi and ammonia-Ugi reactions were performed at room temperature in TFE (0.5 M). In the Ugi reaction, upon increasing the acidity of acids, the isolated yields of the Ugi adducts 3a–3g gradually decreased from 92% to 55%, and finally, the Ugi adduct 3h derived from trifluoroacetic acid (TFA) was obtained only in a trace amount (5% yield). Thus, the Ugi reaction was found to be susceptible to acidity. In contrast, the present ammonia-Ugi reaction conditions could tolerate a variety of acids with pKa values in the range of 2.16 to 5.03 to give the ammonia-Ugi adducts 4a–4g in moderate to high yields (42%–92%). The relatively low yields of 4c and 4e (73% and 42%, respectively) were attributed to the fact that the corresponding ammonium benzoates were poorly soluble in TFE. To our delight, TFA was well compatible with the ammonia-Ugi reaction conditions to afford 4h in a moderate yield (53%). Here, ammonium acetate, benzoate, and formate were commercially available and the other ammonium carboxylates could be easily prepared as pure crystals by adding aqueous ammonia to a solution of the carboxylic acid in acetone, acetonitrile, or THF at 4 °C, followed by filtration (Table S2†). Thus, volatile acids, such as formic acid and TFA, could be handled accurately and easily as solid ammonium carboxylates in the ammonia-Ugi reaction. Taken together, in sharp contrast to the classical Ugi reaction, the present ammonia-Ugi reaction conditions allowed the use of a broad range of acids providing a variety of N-acyl α,α-disubstituted amino acid derivatives.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Acid | pKa of acid | Yielda (%) | |||
| 3 | 4 | |||||
| a Isolated yield. b Reaction protocol: a solution of cyclopentanone (0.5 mmol) and benzylamine (1.5 mmol) in TFE was stirred at room temperature for 10 min, and then carboxylic acid (1.5 mmol) and benzyl isocyanide (0.75 mmol) were successively added. c Reaction protocol: a solution of cyclopentanone (0.5 mmol) and ammonium carboxylate (1.5 mmol) was stirred at room temperature for 10 min, and then benzyl isocyanide (0.75 mmol) was added. d Quoted from Table 1, entry 6. e Starting materials remained. f Isolated yield of the corresponding Passerini adduct. | ||||||
| 1 | Pivalic acid | 5.03 | 3a | 92 | 4a | 86 |
| 2 | Acetic acid | 4.76 | 3b | 88 | 4b | 90d |
| 3 | Benzoic acid | 4.20 | 3c | 83 | 4c | 73 |
| 4 | Formic acid | 3.77 | 3d | 83 | 4d | 92 |
| 5 | p-Nitrobenzoic acid | 3.41 | 3e | 73 | 4e | 42e (12)f |
| 6 | Chloroacetic acid | 2.86 | 3f | 63 | 4f | 81 |
| 7 | o-Nitrobenzoic acid | 2.16 | 3g | 55 | 4g | 91 |
| 8 | Trifluoroacetic acid | −0.25 | 3h | 6 | 4h | 53e |

To further explore the utility of the ammonia-Ugi reaction, a variety of dipeptides containing the unnatural Ac5c residue were synthesized using a set of commercially available N-protected α-amino acids as coupling partners (Scheme 2). The ammonium salts of the N-protected α-amino acids could be carefully prepared by adding aqueous ammonia to a solution of N-protected α-amino acid in acetone or acetonitrile at 4 °C, followed by filtration (Table S2†). Typical nitrogen-protecting groups of amino acids, such as Boc, Bz, and Cbz, were well compatible with the present reaction conditions to afford the corresponding dipeptides PG-Gly-Ac5c-NH-Bn 5a–5c in high to excellent yields (72%–97%). In contrast, Ac-Phe-ONH4 was unsuitable because it was insoluble in TFE even at 0.1 M, resulting in a low yield of 5d (23%). Similar results were obtained using Fmoc-Gly-ONH4 (data not shown). Representative Boc-protected α-amino acids (Val, Phe, Pro, and Met) were successfully used to produce the corresponding dipeptides 5e–5h in high yields (78%–97%). A series of sidechain-protecting groups, such as benzyl ether, tert-butyl ester, and formyl amide, were all compatible with the present conditions to afford the dipeptides 5i–5k in high yields (73%–87%). Importantly, chiral HPLC analysis confirmed that no racemization of the chiral stereocenter of the α-amino acids had occurred under the present reaction conditions (Fig. S130–138†). Furthermore, when the reaction temperature was set at 60 °C, 5f could be obtained within 4 hours in a high yield (78%) without the loss of stereochemical integrity (>99.5:0.5 er). Notably, the sterically hindered 2-aminoisobutyric acid (Aib) was smoothly converted into the dipeptide Boc-Aib-Ac5c-NH-Bn (5l), containing two contiguous α,α-disubstituted amino acid residues, in 87% yield. Thus, the present reaction conditions enabled the one-pot synthesis of dipeptides containing unnatural α,α-disubstituted amino acid motifs.
 | ||
| Scheme 2 Scope of amino acids in the ammonia-Ugi reaction.a a Isolated yield. b Starting materials remained. c 0.1 M. | ||
To gain mechanistic insight into the role of the ammonium carboxylates, two control experiments were performed (Table 3). Compared with the optimal conditions (Table 1, entry 7), the procedure of mixing ammonia and ketone before the addition of carboxylic acid considerably reduced the isolated yield of 1a (55%), mainly because of the production of 2a (18%) as a by-product (Table 3, entry 1). In turn, the in situ formation of AcONH4 slightly improved the isolated yield of 1a (76%); however, this procedure still gave the Passerini adduct 2a (11%) probably due to the incomplete formation of AcONH4 (entry 2). Eventually, both procedures suffered from competition with the undesired Passerini pathway. These results indicated that the preparation of the ammonium carboxylates outside of the reaction vessel minimized the generation of free carboxylic acids in situ and effectively suppressed the undesired Passerini pathway (Fig. 2). Moreover, the ammonium ions are expected to promote the formation of N-unsubstituted ketiminium intermediates and guide the reaction toward the otherwise unfavored ammonia-Ugi pathway.
 | ||
| Fig. 2 Effect of ammonium carboxylates on the ammonia-Ugi reaction. | ||
| Entry | Procedure | Yielda (%) | |
|---|---|---|---|
| 1a | 2a | ||
| a Isolated yield. b Reaction procedure: a solution of cyclopentanone (0.5 mmol) and aqueous ammonia in TFE was stirred at room temperature for 10 min, and then acetic acid and benzyl isocyanide were successively added. c Reaction procedure: a solution of aqueous ammonia and acetic acid in TFE was stirred at room temperature for 10 min and then cyclopentanone was added and the mixture was stirred at room temperature for 10 min, followed by the addition of benzyl isocyanide. d Starting materials remained. | |||
| 1 | Mixing ammonia and ketone before acid additionb | 55d | 18 |
| 2 |
In situ formation of AcONH4c |
76d | 11 |
Previously, we have reported the inhibitors of the serine proteinase α-chymotrypsin.27 Among these, the dipeptidic inhibitor D-Leu-Phe-NH-CH2Ph(p-F) exhibited the most potent inhibitory activity.27b Mechanistically, its C-terminal p-fluorobenzyl group occupies the chymotrypsin S1 site where the fluorine atom forms a hydrogen bonding interaction with Ser189 and the hydrophobic core consisting of the alkyl group of D-Leu and the phenyl group of Phe binds to the S2 site.28 These unique enzyme–inhibitor interactions were essentially stabilized by the π–π stacking interaction between the phenyl group of Phe and the imidazole group of His57.29 Inspired by this inhibitory mechanism, we designed a new dipeptide analog, D-Leu-Aic-NH-R, containing the conformationally blocked residue 2-aminoindane-2-carboxylic acid (Aic), to strengthen the key π–π stacking interaction with the imidazole group of His57. Retrosynthetically, the dipeptide D-Leu-Aic-NH-R is accessible by the ammonia-Ugi reaction using Boc-D-Leu-ONH4, 2-indanone, and isocyanide as readily available building blocks (Scheme 3). Using the isocyanides 6a–6e, the ammonia-Ugi reaction followed by deprotection of the Boc group rapidly delivered a set of dipeptides 7a–7e as pure HCl salts after recrystallization in synthetically useful yields, again without the loss of the stereochemical integrity of the D-Leu residue (Fig. S134–138†). Thus, the present ammonia-Ugi reaction conditions were found to exhibit a broad scope of isocyanides. Then, the inhibitory activity of these dipeptides against α-chymotrypsin was investigated by using acetyl-tyrosine ethyl ester as the substrate and analyzed by non-linear fitting to the Michaelis–Menten equation (Table 4). Consistent with our previous results,27b the dipeptides 7a–7c, lacking the C-terminal benzyl group, exhibited no inhibitory activity even at 100 μM (entries 1–3), while dipeptide 7d, containing a C-terminal benzylamide, inhibited chymotrypsin with a Ki value of 0.78 μM, and was more effective than D-Leu-Phe-NH-CH2Ph (Ki, 3.1 μM) (entries 4 vs. 6). Fortunately, the compound 7e exhibited the most potent inhibitory activity with a Ki value of 0.091 μM, which was approximately 7-fold higher than that of the original inhibitor D-Leu-Phe-NH-CH2Ph(p-F) (Ki, 0.63 μM) (entries 5 vs. 7). These findings indicated that the conformationally rigid bicyclic system of the Aic residue30 contributed to strengthening the π–π interactions with the imidazole ring of the His57 residue, resulting in enhanced inhibitory potency against α-chymotrypsin. Thus, the ammonia-Ugi reaction enabled the rapid synthesis of potent α-chymotrypsin inhibitors from readily available building blocks. This synthetic simplicity and quickness are in sharp contrast to conventional peptide synthesis methods that begin with the preparation of the unnatural amino acid Aic.31
 | ||
| Scheme 3 Synthesis of dipeptidic inhibitors of α-chymotrypsin by the ammonia-Ugi reaction. | ||
Conclusions
In summary, we have developed a robust and practical protocol for the ammonia-Ugi reaction. Using originally prepared ammonium carboxylates in TFE, the ammonia-Ugi reaction proceeded with high yields (up to 97%) at room temperature under air, and unnatural α,α-disubstituted amino acid motifs could be constructed in situ from readily available ketones as building blocks. Furthermore, the present ammonia-Ugi reaction was used to synthesize various dipeptides containing α,α-disubstituted amino acid residues without the need for any coupling reagents and, importantly, without racemization of the chiral stereocenter of α-amino acids (99.5:0.5 er). Among them, D-Leu-Aic-NH-CH2Ph(p-F) (7e) exhibited a potent inhibitory activity against α-chymotrypsin with a Ki value of 0.091 μM. Further work will focus on the synthesis of longer peptide chains and the development of a catalytic asymmetric ammonia-Ugi reaction to construct the chiral stereocenters of α,α-disubstituted unnatural amino acids. Extensions in these directions will be reported from our laboratory in due course.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K.T. conceived this study. K.T., S.K., M.M., and T.N. performed the experiments and analyzed the data. K.T., S.K., T.U., H.N., and T.N. discussed the results. T.N. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP21K05290 and JST Adaptable and Seamless Technology transfer Program through Target-driven R&D (A-STEP) Grant Number JPMJTM22EA to KT.References
- (a) H. Takada, K. Tsuchiya and Y. Demizu, Bioconjugate Chem., 2022, 33, 1311–1318 CrossRef PubMed; (b) T. Kato, Y. Kita, K. Iwanari, A. Asano, M. Oba, M. Tanaka and M. Doi, Bioorg. Med. Chem., 2021, 38, 116111 CrossRef PubMed; (c) M. Urello, W.-H. Hsu and R. J. Christie, Acta Biomater., 2020, 117, 40–59 CrossRef PubMed; M. Oba, ChemBioChem, 2019, 20, 2041–2045 Search PubMed.
- (a) X. Wang, D. Ni, Y. Liu and S. Lu, Front. Chem., 2021, 9, 682675 CrossRef PubMed; (b) B. C. Doak, J. Zheng, D. Dobritzsch and J. Kihlberg, J. Med. Chem., 2016, 59, 2312–2327 CrossRef CAS; (c) N. Terrett, MedChemComm, 2013, 4, 474–475 RSC.
- (a) A. Boruah and A. Roy, Biomater. Sci., 2022, 10, 4694–4723 RSC; (b) T. Kato, M. Oba, K. Nishida and M. Tanaka, ACS Biomater. Sci. Eng., 2018, 4, 1368–1376 CrossRef CAS PubMed.
- (a) P. Kumar, N. G. Paterson, J. Clayden and D. N. Woolfson, Nature, 2022, 607, 387–392 CrossRef CAS PubMed; (b) M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS; (c) F. Clerici, E. Erba, M. L. Gelmi and S. Pellegrino, Tetrahedron Lett., 2016, 57, 5540–5550 CrossRef CAS.
- (a) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS; (b) T. Kato, Y. Kita, K. Iwanari, A. Asano, M. Oba, M. Tanaka and M. Doi, Bioorg. Med. Chem., 2021, 38, 116111 CrossRef PubMed.
- (a) T. Tsuji, K. Hashiguchi, M. Yoshida, T. Ikeda, Y. Koga, Y. Honda, T. Tanaka, S. Re, K. Mizuguchi, D. Takahashi, R. Yazaki and T. Oshima, Nat. Synth., 2022, 1, 304–312 CrossRef; (b) Y. Zou, J. Han, A. S. Saghyan, A. F. Mkrtchyan, H. Konno, H. Moriwaki, K. Izawa and V. A. Soloshonok, Molecules, 2020, 25, 2739 CrossRef PubMed; (c) V. A. Soloshonok and A. E. Sorochinsky, Synthesis, 2010, 2319–2344 CrossRef CAS; (d) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013–3028 CrossRef CAS PubMed.
- (a) I. Ugi, Angew. Chem., Int. Ed. Engl., 1962, 1, 8 CrossRef; (b) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef.
- Recent reviews: (a) S. E. Hooshmand and W. Zhang, Molecules, 2023, 28, 1642 CrossRef CAS; (b) N. P. Tripolitsiotis, M. Thomaidi and C. G. Neochoritis, Eur. J. Org. Chem., 2020, 6525–6554 CrossRef; (c) M. A. Fouad, H. Abdel-Hamid and M. S. Ayoup, RSC Adv., 2020, 10, 42644–42681 RSC; (d) M. Giustiniano, L. Moni, L. Sangaletti, S. Pelliccia, A. Basso, E. Novellino and G. C. Tron, Synthesis, 2018, 50, 3549–3570 CrossRef CAS.
- R. Hili, V. Rai and A. K. Yudin, J. Am. Chem. Soc., 2010, 132, 2889–2891 CrossRef CAS PubMed.
- D. G. Rivera, M. G. Ricardo, A. V. Vasco, L. A. Wessjohann and E. V. Van der Eycken, Nat. Protoc., 2021, 16, 561–578 CrossRef CAS.
- (a) B.-B. Sun, K. Liu, Q. Gao, W. Fang, S. Lu, C.-R. Wang, C.-Z. Yao, H.-Q. Cao and J. Yu, Nat. Commun., 2022, 13, 7065 CrossRef CAS PubMed; (b) J. Zhang, P. Yu, S.-Y. Li, H. Sun, S.-H. Xiang, J. Wang, K. N. Houk and B. Tan, Science, 2018, 361, eaas8707 CrossRef.
- (a) K. Tomohara, N. Ohashi, T. Uchida and T. Nose, Sci. Rep., 2022, 12, 15568 CrossRef CAS PubMed; (b) K. Tomohara, J. Maneene, N. Ohashi, T. Nose, R. Fujii, M. J. Kim, S. Sun and S. Awale, Biol. Pharm. Bull., 2023, 46, 1412–1420 CrossRef CAS.
- (a) J. Xie, N. Niu, X. Fu, X. Su, D. Wang, A. Qin, T. Han and B. Z. Tang, Chem. Sci., 2023, 14, 903–915 RSC; (b) V. A. Fernandes, R. N. Lima, Y. B. Broterson, M. Y. Kawamura, R. Echemendía, A. F. de la Torre, M. A. B. Ferreira, D. G. Rivera and M. W. Paixão, Chem. Sci., 2022, 13, 15862–15869 Search PubMed; (c) D. Yamane, S. Onitsuka, S. Re, H. Isogai, R. Hamada, T. Hiramoto, E. Kawanishi, K. Mizuguchi, N. Shindo and A. Ojida, Chem. Sci., 2022, 13, 3027–3034 RSC; (d) G. Koopmanschap, E. Ruijter and R. V. A. Orru, Beilstein J. Org. Chem., 2014, 10, 544–598 CrossRef PubMed; (e) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef; (f) J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–381 CrossRef CAS PubMed.
- M. Abbas and L. A. Wessjohann, Org. Biomol. Chem., 2012, 10, 9330–9333 RSC.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Chem. Sci., 2021, 12, 15445–15472 RSC.
- (a) Q. Wang, K. C. Mgimpatsang, X. Li and A. Dömling, J. Org. Chem., 2021, 86, 9771–9780 CrossRef CAS; (b) M. Serafini, E. Torre, S. Aprile, E. D. Grosso, A. Gesù, A. Griglio, G. Colombo, C. Travelli, S. Paiella, A. Adamo, E. Orecchini, A. Coletti, M. T. Pallotta, S. Ugel, A. Massarotti, T. Pirali and S. Fallarini, J. Med. Chem., 2020, 63, 3047–3065 CrossRef CAS PubMed.
- A. Barthelon, L. E. Kaim, M. Gizzi and L. Grimaud, Synlett, 2010, 2784–2788 CAS.
- (a) T. M. Vishwanatha, K. Kurpiewska, J. Kalinowska-Tłuścik and A. Dömling, J. Org. Chem., 2017, 82, 9585–9594 CrossRef CAS; (b) A. L. Brown, Q. L. Churches and C. A. Hutton, J. Org. Chem., 2015, 80, 9831–9837 CrossRef.
- (a) M. J. Thompson and B. Chen, J. Org. Chem., 2009, 74, 7084–7093 CrossRef; (b) R. Pick, M. Bauer, U. Kazmaier and C. Hebach, Synlett, 2005, 757–760 Search PubMed; (c) C. D. Floyd, L. A. Harnett, A. Miller, S. Patel, L. Saroglou and M. Whittaker, Synlett, 1998, 637–639 CrossRef.
- D. A.-P. de León, A. C. Sánchez-Chávez and L. A. Polindara-García, J. Org. Chem., 2019, 84, 12809–12834 CrossRef.
- (a) F. Sutanto, S. Shaabani, R. Oerlemans, D. Eris, P. Patil, M. Hadian, M. Wang, M. E. Sharpe, M. R. Groves and A. Dömling, Angew. Chem., Int. Ed., 2021, 60, 18231–18239 CrossRef; (b) S. Ackermann, H.-G. Lerchen, D. Häbich, A. Ullrich and U. Kazmaier, Beilstein J. Org. Chem., 2012, 8, 1652–1656 CrossRef PubMed.
- L. Fan, A. M. Adams, J. G. Polisar and B. Ganem, J. Org. Chem., 2008, 73, 9720–9726 CrossRef PubMed.
- (a) F. M. Mir, M. Crisma, C. Toniolo and W. D. Lubell, Chem. Sci., 2019, 10, 6908–6914 RSC; (b) S. de Castro, M.-J. Camarasa, J. Balzarini and S. Velázquez, Eur. J. Med. Chem., 2014, 83, 174–189 CrossRef PubMed; (c) R. Scheffelaar, R. A. K. Nijenhuis, M. Paravidino, M. Lutz, A. L. Spek, A. W. Ehlers, F. J. J. de Kanter, M. B. Groen, R. V. A. Orru and E. Ruijter, J. Org. Chem., 2009, 74, 660–668 CrossRef.
- (a) A. Golebiowski, D. Whitehouse, R. P. Beckett, M. V. Zandt, M. K. Ji, T. R. Ryder, E. Jagdmann, M. Andreoli, Y. Lee, R. Sheeler, B. Conway, J. Olczak, M. Mazur, W. Czestkowski, W. Piotrowska, A. Cousido-Siah, F. X. Ruiz, A. Mitschler, A. Podjarny and H. Schroeter, Bioorg. Med. Chem. Lett., 2013, 23, 4837–4841 CrossRef; (b) J. Isaacson, C. B. Gilley and Y. Kobayashi, J. Org. Chem., 2007, 72, 3913–3916 CrossRef PubMed; (c) U. Kazmaier and C. Hebach, Synlett, 2003, 1591–1594 CrossRef; (d) R. Bossio, S. Marcaccini, R. Pepino, U. Schiff, U. Firenze and V. G. Capponi, Liebigs Ann. Chem., 1990, 935–937 CrossRef.
- (a) P. Patil, K. Kurpiewska, J. Kalinowska-Tłuścik and A. Dömling, ACS Comb. Sci., 2017, 19, 343–350 CrossRef CAS; (b) P. Patil, M. de Haan, K. Kurpiewska, J. Kalinowska-Tłuścik and A. Dömling, ACS Comb. Sci., 2016, 18, 170–175 CrossRef CAS.
- S. Marcaccini and T. Torroba, Nat. Protoc., 2007, 2, 632–639 CrossRef CAS.
- (a) K. Tomohara, I. Adachi, Y. Horino, H. Kesamaru, H. Abe, K. Suyama and T. Nose, ACS Med. Chem. Lett., 2019, 10, 923–928 CrossRef CAS; (b) Y. Shimohigashi, I. Maeda, T. Nose, K. Ikesue, H. Sakamoto, T. Ogawa, Y. Ide, M. Kawahara, T. Nezu, Y. Terada, K. Kawano and M. Ohno, J. Chem. Soc., Perkin Trans. 1, 1996, 2479–2485 RSC; (c) I. Maeda, Y. Shimohigashi, K. Ikesue, T. Nose, Y. Ide, K. Kawano and M. Ohno, J. Biochem., 1996, 118, 870–877 CrossRef.
- Y. Shimohigashi, T. Nose, Y. Yamauchi and I. Maeda, Biopolymers, 1999, 51, 9–17 CrossRef CAS.
- A. Kashima, Y. Inoue, S. Sugio, I. Maeda, T. Nose and Y. Shimohigashi, Eur. J. Biochem., 1998, 255, 12–23 CrossRef CAS PubMed.
- E. Gavuzzo, G. Lucente, F. Mazza, G. P. Zecchini, M. P. Paradisi, G. Pochetti and I. Torrini, Int. J. Pept. Protein Res., 1991, 37, 268–276 CrossRef CAS.
- (a) Y. N. Belokon, N. B. Bespalova, T. D. Churkina, I. Císařová, M. G. Ezernitskaya, S. R. Harutyunyan, R. Hrdina, H. B. Kagan, P. Kočovský, K. A. Kochetko, O. V. Larionov, K. A. Lyssenko, M. North, M. Polášek, A. S. Peregudov, V. V. Prisyazhnyuk and S. Vyskočil, J. Am. Chem. Soc., 2003, 125, 12860–12871 CrossRef CAS PubMed; (b) P. W. Schiller, G. Weltrowska, T. M.-D. Nguyen, C. Lemieux, N. N. Chung, B. J. Marsden and B. C. Wilkes, J. Med. Chem., 1991, 34, 3125–3132 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental information; synthetic details; NMR spectra; HPLC charts. See DOI: https://doi.org/10.1039/d4ob00924j |
| This journal is © The Royal Society of Chemistry 2024 |