
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atropisomeric 1-phenylbenzimidazoles affecting microtubule organization: influence of axial chirality†
Jana
Pospíšilová
 a,
Tomáš
Heger
b,
Ondřej
Kurka
c,
Marie
Kvasnicová
bd,
Anna
Chládková
a,
Ivan
Nemec
e,
Lucie
Rárová
*bd and
Petr
Cankař
*a
a,
Tomáš
Heger
b,
Ondřej
Kurka
c,
Marie
Kvasnicová
bd,
Anna
Chládková
a,
Ivan
Nemec
e,
Lucie
Rárová
*bd and
Petr
Cankař
*a
aDepartment of Organic Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 77900 Olomouc, Czech Republic. E-mail: petr.cankar@upol.cz
bDepartment of Experimental Biology, Faculty of Science, Palacký University Olomouc, Slechtitelu 27, 77900 Olomouc, Czech Republic. E-mail: lucie.rarova@upol.cz
cLaboratory of Growth Regulators, Institute of Experimental Botany of the Czech Academy of Sciences and Faculty of Science, Palacký University, Slechtitelu 27, Olomouc CZ-77900, Czech Republic
dLaboratory of Growth Regulators, Institute of Experimental Botany of the Czech Academy of Science, Palacký University, Slechtitelu 27, 77900 Olomouc, Czech Republic
eDepartment of Inorganic Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 77900 Olomouc, Czech Republic
First published on 8th July 2024
Abstract
Benzimidazoles are frequently used in medicinal chemistry. Their anticancer effect is among the most prominent biological activities exhibited by this scaffold. Although numerous benzimidazole derivatives have been synthesized, possible atropisomerism of ortho-substituted 1-phenylbenzimidazoles has been largely overlooked. The aim of this research was to synthesize a small library of novel atropisomeric benzimidazole derivatives and explore their biological activity in various cancer and normal human cell lines. The new unique structural motif provides an interesting 3D architecture with axial chirality, which further contributes to molecular complexity and specificity. Racemates and their separated atropisomers arrested the cell cycle, caused apoptosis, and affected microtubule organization in cancer cells in vitro at different intensities. Moreover, this phenomenon was also verified by the inhibition of endothelial cell migration. These results showed that (+)-atropisomers, especially 5n, exhibit a stronger effect and show promise as agents for cancer therapy.
Introduction
For the last few decades, research in medicinal chemistry has focused on apoptosis as a method to effectively eliminate cancer cells. Tumors are defined by deregulated cell cycles that can result in loss of cellular differentiation and uncontrolled cellular growth.1 Antiproliferative activities with different mechanisms of action including induction of apoptosis, cell cycle (G2/M) arrest, DNA alkylation, disruption of tubulin polymerization, enzyme inhibition, antiangiogenic effects, and blockage of glucose transport can be affected by inhibitors based on benzimidazoles.2,3 Nocodazole, galeterone, pracinostat, dovitinib, liarozole, abemaciclib, veliparib, glasdegib, selumetinib, bendamustine, and crenolanib are used to treat various malignancies4 (Fig. 1).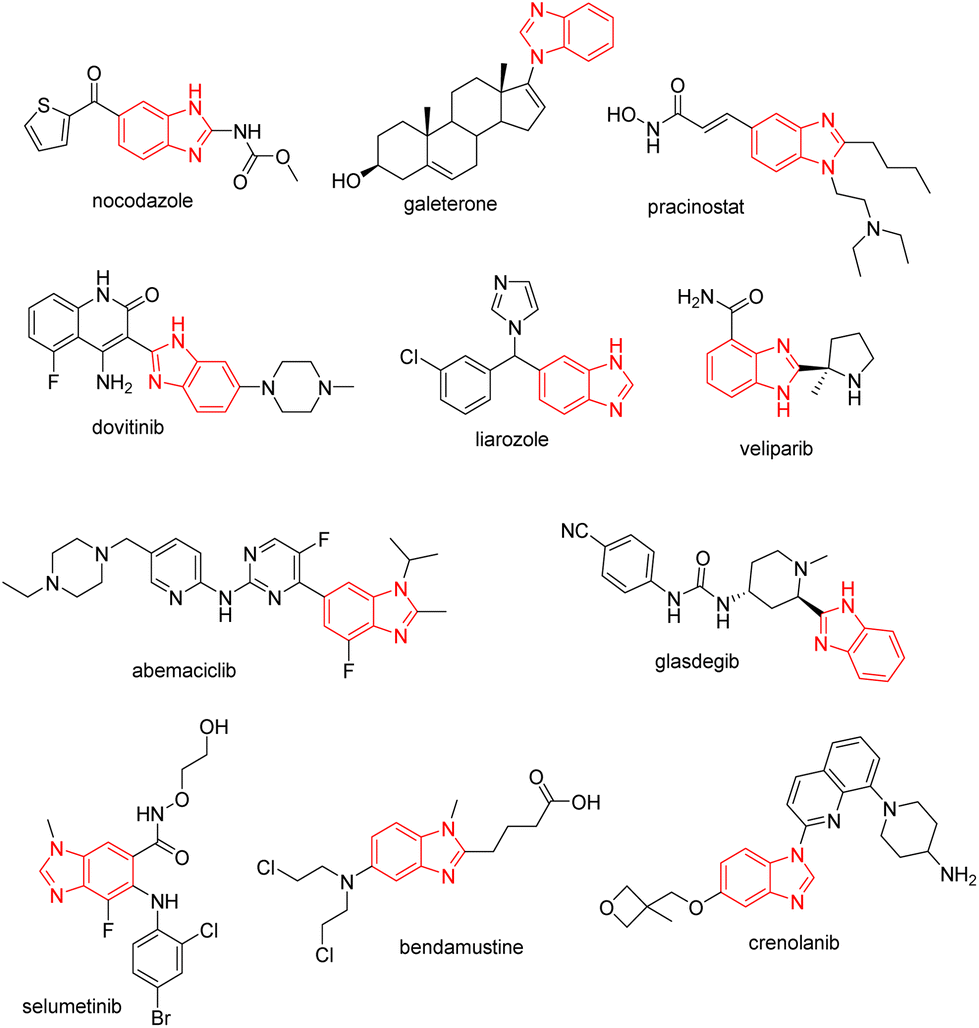 | ||
| Fig. 1 Benzimidazole scaffolds in anticancer drugs. | ||
Furthermore, aminobenzimidazoles were reported as p38α MAP kinase inhibitors5 or compounds targeting angiogenesis to impact the migration of endothelial cells.6 1-Aryl substituted benzimidazoles are also promising molecules for cancer treatment. Previous publications reported that CCL299 exhibits anticancer activity through apoptosis,7 and two older extensive SAR studies demonstrated 1-phenylbenzimidazoles as selective ATP site inhibitors.8,9 Another aryl substitution at position 2 provides also anticancer activity as a p38 kinase inhibitor,10 and moreover, other interesting activities, such as antimicrobial agents,11 COX-1 and COX-2 inhibitors,12,13 or hTRPV-1 antagonists,14 were described.
Currently, multidrug resistance in the treatment of cancer is increasing, which strengthens the need to develop novel chemotherapeutics based on benzimidazoles.1 As shown in Fig. 1, the benzimidazole scaffold can be variously substituted to obtain different structural patterns with modified anticancer activity. The structural motif, in which benzimidazole is substituted with two adjacent aryls at positions 1 and 2, was also studied.15–19 However, the substitution at these aryls was always designed to avoid atropisomerism since the stability of these atropisomers can be insufficient.20 For this reason, the aryls were usually designed to be symmetrical or not ortho-substituted. The possibility of introducing axial chirality to similar derivatives inspired us to study the impact on the cytotoxicity of benzimidazoles since this phenomenon has been poorly covered in the literature despite its importance and prevalence.
Our ongoing research is oriented towards axially chiral benzimidazoles21–24 because atropisomerism of these compounds provides an interesting three-dimensional (3D) structure, which can further enrich the chemical space in the area of benzimidazole derivatives. In this study, we focused our attention on 1-phenylbenzimidazoles, in which the ortho-substituents at the benzene ring restricts the free rotation around the single C–N bond at position 1 to generate axial chirality (Fig. 2). To obtain stable atropisomers, we chose substituents that could generate a high energy barrier and eliminate possible racemization.
 | ||
| Fig. 2 Atropisomerism of 1-phenylbenzimidazoles due to the restricted rotation around the single C–N bond. | ||
Although similar compounds have been reported as sirtuin inhibitors16,18 or anti-tubulin polymerization agents,15,17 the biological issues associated with the presence of axial chirality remained unexamined for this class of compounds. Our goal was to find novel axially chiral benzimidazole-based agents with potential antiproliferative or antiangiogenic activities. The objectives of our research were to (i) synthesize novel unique axially chiral benzimidazoles, (ii) investigate their biological activities in cancer cells in vitro (cytotoxicity and antiproliferative activity), (iii) induct cancer cell apoptosis and determine its mechanism of action, and (iv) separate the most interesting compounds to single atropisomers and explore their biological activity and stability.
Results and discussion
Chemistry
Benzimidazoles were effectively synthesized in several steps (Scheme 1). The first step was nucleophilic aromatic substitution of various fluoronitrobenzenes with o-substituted anilines using KOH in DMSO to obtain nitroanilines 3a–j in very good yields. The most convenient route to synthesize the final benzimidazoles was reducing the nitro group with catalytic hydrogenation in acetic acid (4a) or with zinc in a solution of methanol and acetic acid (4b–f), followed by cyclization with orthoesters to obtain compounds 5a–o. Moreover, we synthesized compounds 5a–f and 8a–b from intermediates after aromatic nucleophilic substitution or Suzuki–Miyaura coupling in just two steps by one-pot reduction and cyclization with trimethyl orthoformate. Compounds 3a and 3e can be modified by Suzuki–Miyaura cross-coupling with various aryl/heteroaryl boronic acids to form 6a–e or 9. Precatalyst XPhos Pd G2 was used to provide excellent yields.25,26 Reduction of the nitro group led to phenylenediamines 7a–f and 10, and an analogous cyclization protocol generated benzimidazoles 8c–l and 11a–b for the SAR study.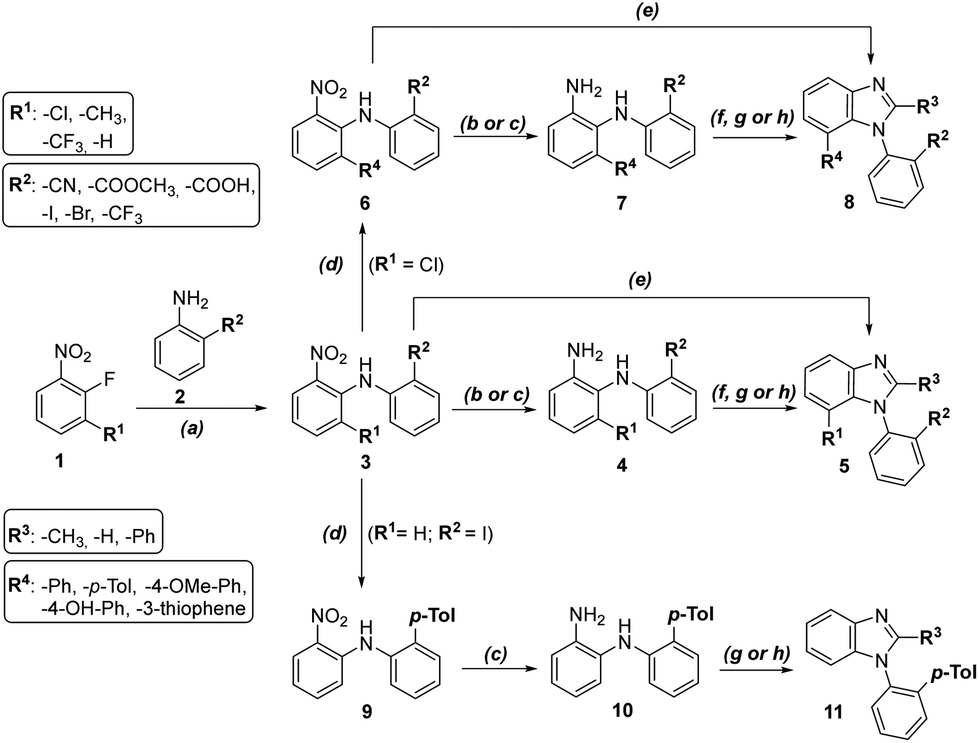 | ||
Scheme 1 General synthetic scheme. Reagents and conditions: (a) KOH, DMSO, rt, 4–12 h, 47–88%; (b) H2, Pd/C, AcOH, rt, 1.5 h, 73–88%; (c) Zn, MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH 4:1, rt, 10–60 min, 48–74%; (d) aryl/heteroarylboronic acid, K3PO4, XPhos Pd G2, dioxane:water 4:1, 100 °C, 2.5 h, 35–97%; (e) Zn, MeOH:AcOH 4:1, rt, 10–60 min, then trimethyl orthoformate, rt, overnight, 49–87%; (f) trimethyl orthoformate, pTSA, DCM, rt, overnight, 16–80%; (g) trimethyl orthoacetate, pTSA, DCM, rt, overnight, 44–81%; (h) trimethyl orthobenzoate, pTSA, DCM, rt, overnight, 40–75%. :AcOH 4:1, rt, 10–60 min, 48–74%; (d) aryl/heteroarylboronic acid, K3PO4, XPhos Pd G2, dioxane:water 4:1, 100 °C, 2.5 h, 35–97%; (e) Zn, MeOH:AcOH 4:1, rt, 10–60 min, then trimethyl orthoformate, rt, overnight, 49–87%; (f) trimethyl orthoformate, pTSA, DCM, rt, overnight, 16–80%; (g) trimethyl orthoacetate, pTSA, DCM, rt, overnight, 44–81%; (h) trimethyl orthobenzoate, pTSA, DCM, rt, overnight, 40–75%. | ||
Biology
| R1/R4 | R2 | R3 | CCRF-CEM | MV4-11 | MOLM-13 | MCF7 | HeLa | G-361 | A2780 | RPE-1 | BJ | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5a | –CH3 | –I | –H | 40.6 ± 0.0 | 27.5 ± 0.8 | 31.9 ± 4.0 | >100 | 28.0 ± 3.3 | 23.3 ± 3.4 | 25.5 ± 4.6 | >100 | — |
| 5b | –Cl | –CF3 | –H | 31.2 ± 2.4 | 14.5 ± 1.5 | 16.2 ± 0.1 | 14.2 ± 0.3 | 22.5 ± 4.2 | 59.8 ± 4.5 | 16.3 ± 2.2 | >100 | — |
| 5c | –Cl | –I | –H | 42.0 ± 1.3 | 20.5 ± 2.2 | 28.3 ± 0.1 | 26.9 ± 2.4 | 18.0 ± 0.1 | 69.9 ± 1.5 | 18.6 ± 2.5 | >100 | — |
| 5d | –CF3 | –I | –H | 60.0 ± 7.0 | 29.2 ± 8.7 | 23.8 ± 7.7 | 41.8 ± 5.4 | 22.8 ± 2.5 | 59.5 ± 0.5 | 20.8 ± 5.1 | >100 | >100 |
| 5e | –Cl | –COOCH3 | –H | >100 | >100 | >100 | >100 | >100 | >100 | 71.5 ± 5.5 | >100 | — |
| 5f | –Cl | –Br | –H | 24.1 ± 4.5 | 12.3 ± 0.2 | 15.8 ± 1.3 | 14.0 ± 0.4 | 15.7 ± 3.2 | 82.6 ± 1.1 | 13.7 ± 1.6 | >100 | — |
| 5g | –Cl | –CN | –H | >100 | 66.4 ± 14.7 | 70.9 ± 37.0 | 54.6 ± 10.4 | 42.8 ± 1.0 | >100 | 70.6 ± 1.3 | >100 | — |
| 5h | –H | –COOH | –CH3 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | — |
| 5i | –Cl | –I | –CH3 | 37.7 ± 3.0 | 19.2 ± 2.5 | 27.3 ± 0.1 | 29.3 ± 8.3 | 17.1 ± 0.6 | 58.6 ± 4.7 | 18.5 ± 2.6 | >100 | — |
| 5j | –H | –I | –CH3 | 89.4 ± 3.8 | 54.9 ± 4.2 | 88.4 ± 0.2 | >100 | >100 | >100 | 70.3 ± 7.4 | >100 | >100 |
| 5k | –Cl | –CN | –CH3 | >100 | 60.7 ± 3.8 | 81.2 ± 5.9 | 90.7 ± 7.6 | 56.8 ± 12.8 | >100 | 52.2 ± 4.2 | >100 | — |
| 5l | –Cl | –I | –Ph | 22.9 ± 3.3 | 13.3 ± 4.8 | 11.7 ± 0.8 | 33.1 ± 2.4 | 9.3 ± 2.2 | 9.8 ± 0.9 | 13.4 ± 0.1 | >100 | >100 |
| 5m | –CF3 | –I | –Ph | 17.2 ± 0.7 | 9.9 ± 0.8 | 10.0 ± 2.4 | 19.0 ± 5.8 | 11.4 ± 2.7 | 22.3 ± 6.9 | 8.6 ± 0.9 | >100 | — |
| 5n | –Cl | –CN | –Ph | 12.4 ± 2.1 | 7.8 ± 1.0 | 13.2 ± 1.1 | 15.6 ± 1.5 | 3.2 ± 0.5 | 4.4 ± 0.3 | 8.4 ± 1.8 | 23.4 ± 0.6 | >50 |
| 5o | –Cl | –H | –Ph | — | — | — | >100 | >100 | >100 | >100 | >100 | — |
| 8a | –pTol | –CN | –H | >100 | 43.7 ± 1.0 | 80.9 ± 17.6 | >100 | 71.3 ± 3.5 | >100 | 38.6 ± 10.4 | >100 | — |
| 8b | –4-OH-Ph | –CN | –H | 75.0 ± 2.2 | 39.4 ± 1.3 | 46.8 ± 11.7 | 70.9 ± 0.2 | 74.4 ± 2.3 | 96.5 ± 1.9 | 42.3 ± 7.7 | >100 | — |
| 8c | –4-OCH3-Ph | –CN | –H | >100 | >100 | >100 | >100 | >100 | >100 | 67.0 ± 6.4 | >100 | — |
| 8d | –3-thiophene | –CN | –H | >50 | — | — | — | 43.2 ± 0.8 | — | — | — | >50 |
| 8e | –Ph | –CN | –H | >100 | >100 | >100 | >100 | >100 | >100 | 87.5 ± 12.0 | >100 | — |
| 8f | –pTol | –CN | –CH3 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | — |
| 8g | –4-OCH3-Ph | –CN | –CH3 | >50 | — | — | — | >50 | — | — | — | >50 |
| 8h | –3-thiophene | –CN | –CH3 | >50 | — | — | — | >50 | — | — | — | >50 |
| 8i | –Ph | –CN | –CH3 | >100 | >100 | >100 | >100 | >100 | >100 | 74.0 ± 18.5 | >100 | >100 |
| 8j | –4-OCH3-Ph | –CN | –Ph | >50 | — | — | — | >50 | — | — | — | >50 |
| 8k | –3-thiophene | –CN | –Ph | 32.2 ± 3.3 | — | — | — | >50 | — | — | — | >50 |
| 8l | –Ph | –CN | –Ph | >50 | — | — | 85.5 | >100 | >100 | 22.1 | >100 | >50 |
| 11a | –H | –pTol | –CH3 | >100 | 93.6 ± 5.9 | 92.8 ± 3.7 | >100 | >100 | >100 | 77.0 ± 12.1 | >100 | — |
| 11b | –H | –pTol | –Ph | 44.5 ± 3.4 | 36.7 ± 0.2 | 25.3 ± 2.8 | 41.7 ± 0.3 | 13.6 ± 1.5 | 15.1 ± 4.8 | 35.4 ± 4.0 | 25.3 ± 4.5 | — |
Generally, we observed interesting trends for the SAR study. Among the compounds, those with iodine as the R2 substituent provided the best biological properties, and these compounds are not toxic toward normal cells. Benzimidazole 5n with nitrile (R2), phenyl (R3), and chlorine (R1) provided the best biological activity in cancer cells (especially HeLa = 3.2 μM, G-361 = 4.4 μM); however, while the compound was not toxic to BJ cells, it was toxic to normal RPE-1 cells (23.4 μM). Changing nitrile to iodine in derivative 5l led to very interesting biological activity against cancer cell lines (HeLa = 9.3 μM, G-361 = 9.8 μM) and no activity toward normal cells.
As the essentiality of atropisomerism for cytotoxicity was indirectly illustrated by the synthesis of non ortho-substituted benzimidazole 5o, we also decided to study separated atropisomers of the most active benzimidazoles 5l and 5n, which further enrich the 3D structure with axial chirality.
 | ||
| Fig. 3 Separation of 5l and 5n into individual atropisomers (for X-ray structures and HPLC methods see ESI†). | ||
We compared the cytotoxicity of racemates and single atropisomers in six cancer and two normal cell lines (Table 2). These cell lines were selected as the most interesting according to the IC50s values of 5l and 5n from Tables 1 and 2. Racemic 5l and 5n and separated enantiopure atropisomers were cytotoxic toward cancer cell lines (A2780, CEM, G-361, HeLa, MCF7, and MV4-11), but not toward normal skin cells (BJ) for 72 h. The most sensitive cell lines toward our racemic 5l and 5n and single enantiopure atropisomers were B-myelomonocytic leukemia MV4-11, cervical carcinoma HeLa, and malignant melanoma G-361, with activities fluctuating around similar levels. Benzimidazole 5n showed significant differences in activities between single atropisomers for all six measured cancer lines and iodo derivative 5l for HeLa, G-361, and A2780 cells. Moreover, in MV4-11 cells, the cytotoxicity of (+)-5n was 7.5 × higher than that of its (−)-atropisomer (Table 2). The IC50s values for racemates are slightly different from the results in Table 1 because other biological replicates were used, which were performed in one set with atropisomers. Experiments were repeated three times with three technical replicates.
| R1 | R2 | R3 | CCRF-CEM | MV4-11 | MCF7 | HeLa | G-361 | A2780 | RPE-1 | BJ | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5l | –Cl | –I | –Ph | 22.9 ± 3.3 | 8.4 ± 1.1 | 33.1 ± 2.4 | 7.3 ± 0.6 | 5.8 ± 2.5 | 17.1 ± 3.8 | >50 | 44.9 ± 0.4 |
| (−)-5l | –Cl | –I | –Ph | 17.8 ± 3.4 | 7.4 ± 0.7 | 16.4 ± 4.0 | 6.2 ± 0.1 | 5.0 ± 0.2 | >50 | >50 | >50 |
| (+)-5l | –Cl | –I | –Ph | 12.8 ± 4.3 | 7.2 ± 0.4 | 16.0 ± 1.7 | 7.4 ± 0.2 | 8.6 ± 1.1 | 18.3 ± 5.0 | >50 | >50 |
| 5n | –Cl | –CN | –Ph | 12.4 ± 2.1 | 8.0 ± 1.0 | 15.6 ± 1.5 | 5.1 ± 1.0 | 4.9 ± 0.6 | 15.5 ± 2.3 | 23.4 ± 0.6 | >50 |
| (−)-5n | –Cl | –CN | –Ph | 18.1 ± 1.9 | 30.7 ± 1.3 | 24.0 ± 2.1 | 3.2 ± 1.0 | 2.5 ± 0.2 | 7.0 ± 0.3 | 10.5 ± 1.1 | >50 |
| (+)-5n | –Cl | –CN | –Ph | 8.2 ± 0.5 | 4.1 ± 1.4 | 9.3 ± 1.7 | 4.9 ± 0.7 | 6.6 ± 1.1 | 11.3 ± 1.9 | 17.3 ± 0.3 | >50 |
The influence of racemates 5l and 5n along with purified atropisomers on the MV4-11 and HeLa cells after 24 h of treatment was compared in a series of experiments, including immunoblotting, cell cycle analysis, and/or immunofluorescence and caspase-3/7 activity assays. Protein extracts from cells treated with 15 μM compounds for 24 h were analyzed by SDS-PAGE followed by immunoblotting. Induction of apoptosis in MV4-11 and HeLa cells with 15 μM racemates and (+)-atropisomers 5l and 5n was detected after 24 h of treatment (Fig. 4). As a marker of apoptosis, the caspase-7 fragment was specifically found in the compound-treated cells but not in the DMSO control samples. The lowest band intensity was observed in the sample treated with atropisomer (–)-5n, whereas (+)-5n provided the densest band among benzimidazole compounds. The cleavage of caspase-7 substrate, poly (ADP-ribose) polymerase PARP, was found in the same treatments by racemates and (+)-atropisomers as caspase-7 fragment was observed. In addition, the marker of mitosis, p-histone H3 at Ser 1027 was also induced by racemates and (+)-atropisomers (Fig. 4A and B). This induction of p-histone H3 indicated G2/M arrest. The most active compounds were racemates and (+)-5l and (+)-5n.
 | ||
| Fig. 4 Induction of apoptosis detected by western blotting. HeLa cells (A) and MV4-11 (B) were treated for 24 h with 15 μM 5l and 5n and their atropisomers. β-Actin and HSP60 or GAPDH were used as loading controls. p-Histone H3 was detected by an antibody recognizing phosphorylation at Ser10. 0 + DMSO are control untreated cells with DMSO at the same level as in the treatment. The results presented here are based on a representative experiment that was repeated three times. Colchicine (colch) and paclitaxel (PTX) were used as positive controls. | ||
In MV4-11 cells stained with propidium iodide and analyzed for the DNA content by a flow cytometry, apoptotic events appeared as the sub-G1 (debris) fraction increased. In the samples treated with 15 μM compounds for 24 h, the elevated sub-G1 fraction correlated with both the caspase activity measurements and immunoblotting. The overall difference between atropisomers regarding their influence on the cell cycle was more pronounced in 5n than in 5l. The sub-G1 cell population rose rapidly along with the G2/M block observed in the treatments with higher concentrations of compounds. This effect was observed from 7.5 μM (+)-5n to higher concentrations; however, this was not observed for (–)-5n, in which the G1 phase increased with the compound concentration.
Additionally, the sub-G1 fraction was almost not elevated in (–)-5n-treated samples. Similarly, (+)-5l appeared to be the most effective, whereas (–)-5l was the least effective in increasing the sub-G1 population, which probably occured via apoptosis. The S phase cells became more abundant in higher concentrations of the racemic mixture 5l and the atropisomer (+)-5l. However, the effect diminished after the cells started to die and was not observed for (–)-5l. Additionally, 5n and its atropisomers did not show this activity (Fig. 5).
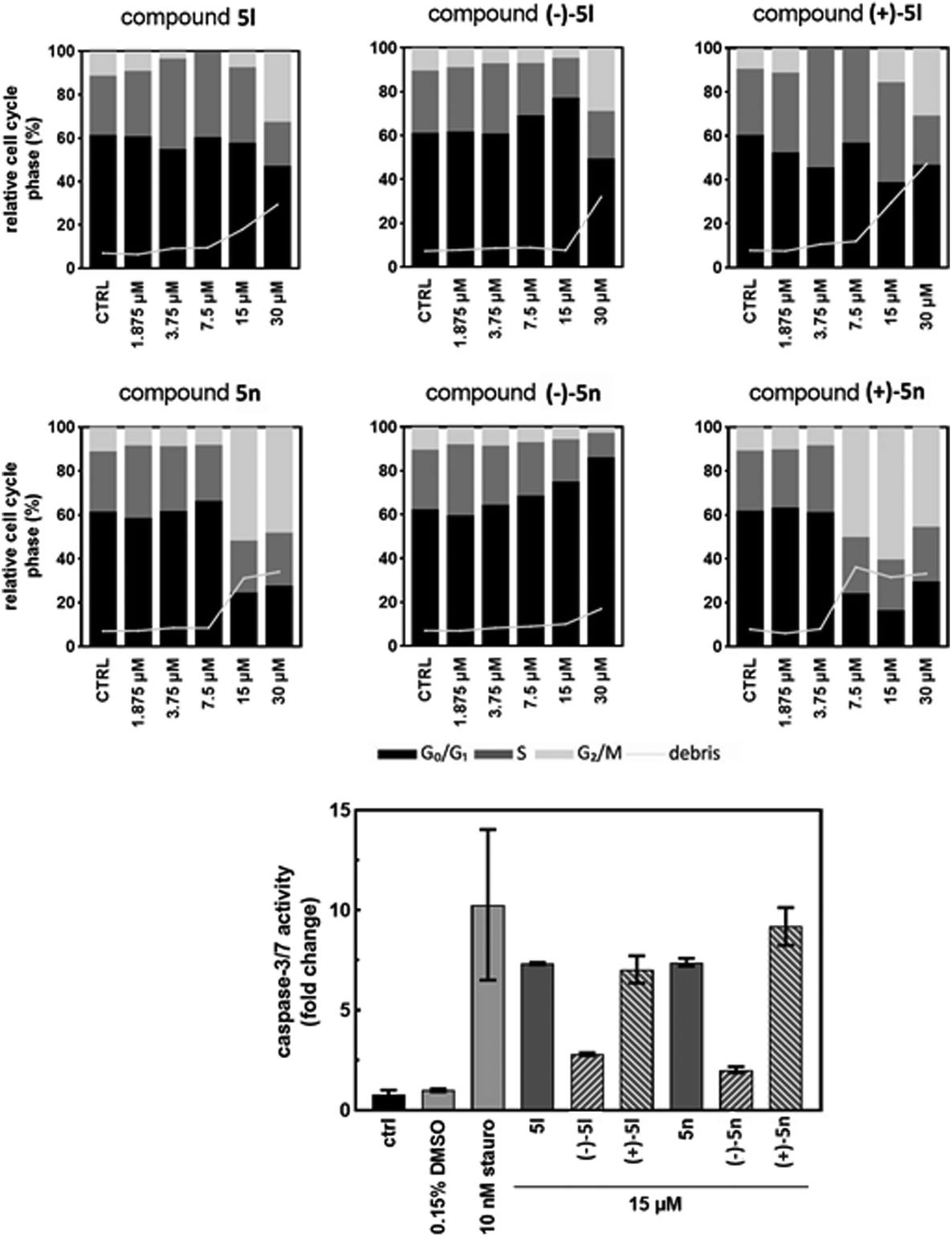 | ||
| Fig. 5 Changes in the cell cycle detected by flow cytometry and caspase activity assay. MV4-11 cells were treated for 24 h with 1.875, 3.75, 7.5, 15, and 30 μM solutions of 5l and 5n and their atropisomers. The ctrl bars are control untreated cells, and the 0.15% DMSO bars are control cells with DMSO at the same level as in the treatment. Staurosporine (stauro) was used as the positive control. The results presented here are based on a representative experiment that was repeated three times. | ||
In addition, the G2/M block caused by 5n and (+)-5n after 24 h is comparable to the cell cycle (G2/M) arrest, induction of apoptosis, and disruption of microtubule polymerization by arylpyrazole-linked benzimidazole conjugates28 and the anti-tubulin agents paclitaxel (PTX) or colchicine.29
In the caspase-3/7 assay, the proteolytic activity of both effector caspases is monitored by the release of a fluorescent product from a synthetic peptide substrate. The MV4-11 cells were treated with the 15 μM solutions of 5l, 5n, and their atropisomers for 24 h and lysed, and the lysates were mixed with the caspase-3/7 substrate. After 2 h of the substrate cleavage reaction, the fluorescence was recorded and normalized to a DMSO-treated sample to calculate the fold increase in the caspase-3/7 activity.
The activity in (–)-5l and (–)-5n-treated cells increased by 2.8- and 2.0-fold, respectively (Fig. 5). These results correlated with the cleaved caspase-7 fragment detected by immunoblotting. Opposite atropisomers, (+)-5l and (+)-5n, as well as racemic mixtures, induced a stronger response in caspase activation. The caspase-3/7 assay results were in agreement with the western blotting experiment, and both showed a clear difference in the activities of atropisomers. This demonstrates the importance of compound atropisomerism for biological activity. In the propidium iodide flow cytometry experiment, the increase in dead cells was expected, as caspase-3/7 activation is associated with the mitochondrial outer membrane permeabilization and subsequent apoptosis.30 For the most active atropisomers (+)-5l and (+)-5n, caspase-3/7 reached 7- and 9-fold activation, respectively. Whether the activation of the caspase cascade is the main mechanism of compound cytotoxicity and what triggers these events should be investigated in further research.
Based on these results, we selected only the most interesting concentrations of our compounds for additional experiments to demonstrate the cell cycle arrest and/or the influence on the microtubule organization in HeLa cells by western blotting or immunofluorescence analysis.
Moreover, we compared the effect of racemic 5l and 5n and also their enantiopure atropisomers with known microtubule disruptors (colchicine, myoseverin, nocodazole, paclitaxel, tubulysin or vincristine) on microtubules of HeLa cells after 24 h of treatment (Fig. 6).
 | ||
| Fig. 6 Changes in the cell cycle detected by flow cytometry. HeLa cells were treated for 24 h with 1.875, 3.75, 7.5, and 15 μM solutions of 5l and 5n and their atropisomers. The CTRL bars are control untreated cells, and the CTRL + DMSO bars are control cells treated with DMSO at the same level as in the treatment. Colchicine and paclitaxel were used as positive controls. | ||
We detected that both (+)-isomers 5l and 5n at 10 nM after 24 h affected microtubule organization (Fig. S4†). We also showed in Fig. 7 that 3.75 μM concentration of (+)-5n resulted in very similar clusters of microtubules, such as the depolymerization inhibitor paclitaxel at 20 nM (ref. 29) or paclitaxel analogs.34
 | ||
| Fig. 7 Effects of racemic and their enantiopure atropisomers on microtubule organization in HeLa cells treated for 24 h with 3.75 μM solutions of 5l and 5n. 0 indicates untreated cells, and 0 + DMSO indicates control cells with DMSO at the same level as in the treatment. Colchicine (Colch) and paclitaxel (PTX) were used as positive controls. | ||
Based on the results in Fig. 7, racemate 5n and its atropisomers at 3.75 and 15 μM were analyzed for the expression of proteins associated with G2/M cell cycle arrest and cytoskeleton in HeLa cells by immunoblotting (Fig. 8A). To compare the effects of all tested compounds, colchicine was used as a microtubule polymerization disruptor, and paclitaxel was used as a depolymerization inhibitor. Significant decreases in α- and β-tubulins expression were detected in cells treated with 15 μM of (+)-5n and with colchicine, the positive control. In HeLa cells treated with 5n at 15 μM and (+)-5n at both concentrations, we also detected the phosphorylation of histone H3 at Ser10, which is a specific marker of ongoing mitosis in cells. The marker is associated with the condensation and segregation of chromosomes during mitosis. Phosphorylation starts during prophase, and the highest level can be observed during metaphase; then, phosphorylation decreases.35 We clearly showed that the phosphorylation of histone H3 at Ser10 was detected after treatment with colchicine, paclitaxel, racemate (15 μM) and the (+)-atropisomer (3.75 and 15 μM), but not with the (−)-isomer of 5n (Fig. 8A). HeLa cells were treated with 15 μM of 5n, (−)-5n, (+)-5n for 24 h, then separated into soluble or polymerized fractions of tubulins.47 Using western blotting, differences between these two fractions were detected (Fig. 8B). (+)-5n decreased the level of soluble and polymerized tubulins (α and β), similarly as colchicine.
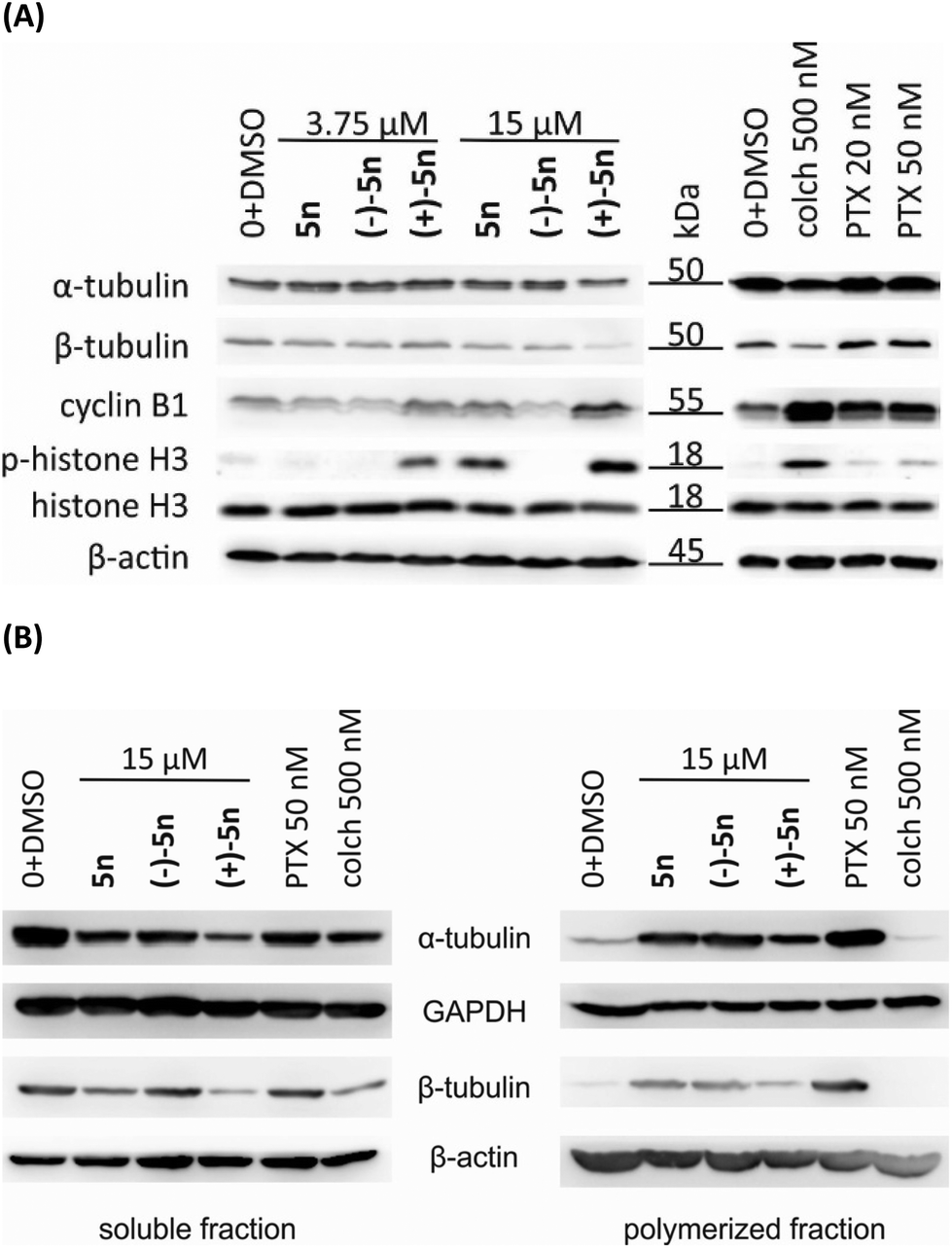 | ||
| Fig. 8 Comparison of effects of racemate 5n and its atropisomers with anti-tubulin agents by western blotting. (A) HeLa cells were treated for 24 h with the 3.75 and 15 μM solutions of 5n. (B) HeLa cells were treated for 24 h with 15 μM solutions of 5n, then separated into soluble a polymerized fractions. β-Actin or GAPDH was used as loading control. 0 + DMSO are control cells with DMSO in the same level as in the treatment. Colchicine (colch) and paclitaxel (PTX) were used as positive controls. | ||
Moreover, we determined the level of cyclin B1, which functions in complex with Cdk1 during the regulation of the G2/M phase cell cycle checkpoint; this checkpoint stops the transition to mitosis when DNA is damaged.36 This DNA defect and aberrant spindle formation can lead to mitotic catastrophe, which is a type of cell death that can occur during mitosis. Mitotic catastrophe can be characterized as a poorly defined type of apoptosis associated with abnormal activation of mitotic kinases and caspases. Mitotic catastrophe is regulated by cell cycle-specific kinases (such as the cyclin B1-dependent kinase Cdk1, polo-like kinases and Aurora kinases), cell cycle checkpoint proteins, members of the Bcl-2 family, survivin, p53, and caspases.37 The cross-talk and interdependence between Cdk1/cyclin B1-mediated phosphorylation and inactivation of antiapoptotic Bcl-2 proteins was confirmed.38 Accumulation of cyclin B1, activation of Cdc2/cyclin B1 kinase and Bcl-2 phosphorylation were tightly linked with M phase arrest but not with apoptosis.39 These effects, elevated level of cyclin B1 and phosphorylation of Bcl-2, were detected in HeLa cells treated with 3.75 and 15 μM (+)-5n, colchicine and paclitaxel (Fig. 4 and 8).
Mitotic catastrophe can be induced by DNA damage, microtubule-depolymerizing agents (such as colchicine, the Vinca alkaloids, cryptophyscins, halichondrins, and estramustine) and by microtubule-hyperpolymerizing agents (taxanes, laulimalide, docodermolide, elutherobins, epothilones, and sarcodictyins).40 Chromosomes decondense, form random clusters and accumulate in an abnormal metaphase in the presence of paclitaxel.41
In HeLa cells, paclitaxel at low concentrations (10 nM for 20 h) suppressed the dynamics of spindle microtubules and therefore arrested cells in mitosis at the metaphase/anaphase transition (by 90%). Paclitaxel did not change the mass of polymerized microtubules but blocked mitosis by kinetically stabilizing spindle microtubules.42 Colchicine blocked the assembly and polymerization of microtubules by forming tubulin-colchicine complexes. They bind to the ends of microtubules to prevent elongation of the microtubule polymer. Colchicine arrested microtubule growth at low concentrations and promoted microtubule depolymerization at higher concentrations. Colchicine could block mitotic cells in metaphase.43 Overall, we demonstrated that our compounds exhibit a similar effect to the anti-tubulin agents paclitaxel and colchicine (Fig. 4, 7, and 8). Based on these results, we would like to prove the direct effect of our compounds on tubulin polymerization using fluorescence polymerization assay. Unfortunately, the inhibition of polymerization by (+)-5n was not significant in comparison with untreated control and/or positive controls colchicine and paclitaxel (Fig. S5†). We assume that the anti-tubulin effect of (+)-5n is probably secondary arising from apoptosis after cell arresting or indirect as described in Zhou et al., 2002.44
 | ||
| Fig. 9 Inhibition of migration of HUVECs treated for 8 h with 15 and 30 μM solutions of 5l and 5n. The ctrl bars are control untreated cells, and the 0 + DMSO bars are control cells treated with DMSO at the same level. 2-Methoxyestradiol (MeO) was used as the positive control. The results presented here are based on a representative experiment that was repeated three times. | ||
Atropisomers (−)-5l, (−)-5n and (+)-5n inhibited the dose-dependent migration of HUVECs after 8 h. In comparison with the 2-aminobenzimidazole-based compound that significantly inhibited VEGF-A-induced HUVEC migration,6 our atropisomers showed a weaker effect than the reported inhibitor. Therefore, we assumed that the inhibition of migration in vitro is a side effect of their antiproliferative activity.
Conclusions
In summary, we synthesized and investigated the biological activity of new atropisomeric 1-phenylbenzimidazoles, and we revealed differences in cytotoxicity caused by atropisomerism. The association of the ortho-substituted phenyl with the presence of strong cytotoxicity in cancer cells and the preservation of inactivity on normal cells was demonstrated. The two best compounds were separated into individual atropisomers, and their good stability and different biological effects strengthen the activities of (+)-atropisomers. Racemates and single atropisomers arrested the cell cycle, caused apoptosis, and affected the microtubule organization in cancer cells in vitro with different intensities. We demonstrated that our compounds exhibit a similar effect to the anti-tubulin agents paclitaxel and colchicine. Therefore, axially chiral benzimidazole (+)-5n is promising for the future research.Experimental
Chemistry
:5:0.1. Resolution of selected axially chiral benzimidazoles and racemization stability was carried out on HPLC Agilent 1100 MWD with column Chiralpak IA-3, 3 μm, 4.6mm × 100mm; mobile phase n-heptan/ethanol 90:10, flow 0.5–1 mL min−1. For semipreparative isolation of each atropisomer (for compounds 5n and 5l) Lux Cellulose-1 column (Phenomenex) 250 × 10 mm, 5 μm particle size was used with mobile phases in isocratic mode: hexane/ethanol 9:1 (v/v), 15 min analysis time for 5l and hexane/methanol/ethanol 40:1:1 (v/v/v), 35 min analysis time for 5n.
General synthetic procedures
:hexane).
:10). The product was extracted with dichloromethane (3 × 30 mL), the organic phase was dried over MgSO4 and evaporated on a rotovap. A crude product was purified on column chromatography (EtOAc:hexane or DCM:hexane).
:hexane or DCM:MeOH).
:MeOH).
:MeOH).
:MeOH).
Biology
000g, 4 °C), the supernatant was collected and its protein concentration was determined by the Bradford reagent. Samples were mixed with the 5× Laemmli buffer, denatured for 5 min at 98 °C and stored in the −80 °C freezer. Proteins (30 μg per well) in the samples were separated by SDS-PAGE according to the Laemmli method. Polyacrylamide gels composed of 4% stacking gel and 10% or 12.5% resolving gels were used for the separation in the Mini-PROTEAN electrophoresis system (Bio-Rad, USA). Proteins were then electroblotted (2 h, 270 mA) from the gel onto a nitrocellulose membrane (0.45 μm pore size) in the Mini trans-Blot system (Bio-Rad, USA). After transfer, the membrane was stained with 0.2% Ponceau S in 1% acetic acid. Dry membrane was cut into strips defined by the target protein to be detected and a molecular weight marker. Strips were destained in TBS (20 mM Tris, 150 mM NaCl, pH 7.6) and then blocked during incubation in 5% non-fat dry milk dissolved in TBS-T (20 mM Tris, 150 mM NaCl, 0.1% TWEEN 20, pH 7.6) for 1 h at the room temperature. Membranes were incubated with the primary antibodies in the milk solution overnight at 4 °C. Specific primary antibodies were used: anti-β-actin, anti-GAPDH (0411) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-caspase-7 (zymogen, fragment), anti-PARP (46D11), anti-Mcl-1 (D35A5), anti-HSP60 (D307), anti-cyclin B1 (V152), anti-histone H3 (D1H2) (all from Cell Signaling, Danvers, MA, USA); swine anti-rabbit HRP, rabbit anti-mouse HRP, anti-p-histone H3 (Ser10), anti-α-tubulin (DM1A), anti-Bcl-2 (Bcl-2-100) (all from Merck, Darmstadt, Germany); anti-β-tubulin (abcam, Cambridge, UK). Strips were washed in TBS and TBS-T and incubated with the HRP-conjugated secondary antibody for 1 h at room temperature. After the washings, strips were covered in SuperSignal West Pico chemiluminescent reagent (Thermo Fisher Scientific, USA) and the signal was recorded by the LAS-4000 camera (Fujifilm, Japan).
400g for 10 min at 4 °C.48 Supernatants with soluble depolymerized tubulin fraction were collected. According to above-mentioned publication, pellets were lysed using modified RIPA buffers. Completely dissolved pellets were centrifuged at 14400g at 4 °C for 10 min. Supernatants with polymerized tubulin fraction were collected. The amount of proteins in both fractions was determined by the Bradford reagent. Sample preparation, electrophoresis and western blotting were performed as described in section “SDS-PAGE & western blotting”.
:1, v/v) for 10 min. The cells were subsequently labelled with primary antibody anti-α-tubulin (Merck, Darmstadt, Germany) overnight at 4 °C in the dark, washed with three changes of PBS, and incubated in appropriate fluorescently-conjugated secondary antibody (rabbit anti-mouse Alexa Fluor 488, Thermo, USA). The cells were then washed three times in PBS, in deionized water and mounted using the FluorSave (Merck, Darmstadt, Germany). Cells were visualized using a fluorescence microscope (IX51, Olympus, Japan) and compared with control untreated cells and positive controls.
Author contributions
J.P. performed almost the whole synthesis and wrote original draft together with T.H., P.C., L.R. and M.K. A.C. helped with synthesis. L.R., T.H., and M.K. – biological assay, formal analysis and visualization. I.N. – X-ray crystallography. O.K. developed chiral analytical and semi-preparative HPLC methods and separated compounds into individual atropisomers. P.C., L.R., and J.P. participated in writing – review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI† or upon request from the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Anežka Šindlerová for excellent technical assistance and Adam Přibylka and Martin Grepl for chiral HPLC analyses of atropisomers. This work was supported by the Internal Grant Agency of Palacký University (IGA_PrF_2024_028). Biological testing was supported by grant from the Czech Science Foundation Nr. 23-05474S and the European Regional Development Fund – Project ENOCH (No. CZ.02.1.01/0.0/0.0/16_019/0000868).References
- L.-S. Feng, W.-Q. Su, J.-B. Cheng, T. Xiao, H.-Z. Li, D.-A. Chen and Z.-L. Zhang, Arch. Pharm., 2022, 355, 2200051 CrossRef CAS.
- H. A. Ibrahim and H. M. Refaat, Future J. Pharm. Sci., 2020, 6, 41 CrossRef.
- S. Tahlan, S. Kumar, S. Kakkar and B. Narasimhan, BMC Chem., 2019, 13, 66 CrossRef PubMed.
- D. Hernández-Romero, S. Rosete-Luna, A. López-Monteon, A. Chávez-Piña, N. Pérez-Hernández, J. Marroquín-Flores, A. Cruz-Navarro, G. Pesado-Gómez, D. Morales-Morales and R. Colorado-Peralta, Coord. Chem. Rev., 2021, 439, 213930 CrossRef.
- A. de Dios, C. Shih, B. López de Uralde, C. Sánchez, M. del Prado, L. M. Martín Cabrejas, S. Pleite, J. Blanco-Urgoiti, M. J. Lorite, C. R. Nevill, R. Bonjouklian, J. York, M. Vieth, Y. Wang, N. Magnus, R. M. Campbell, B. D. Anderson, D. J. McCann, D. D. Giera, P. A. Lee, R. M. Schultz, L. C. Li, L. M. Johnson and J. A. Wolos, J. Med. Chem., 2005, 48, 2270–2273 CrossRef CAS.
- J.-C. Lien, C.-L. Chung, T.-F. Huang, T.-C. Chang, K.-C. Chen, G.-Y. Gao, M.-J. Hsu and S.-W. Huang, Br. J. Pharmacol., 2019, 176, 4034–4049 CrossRef CAS PubMed.
- Y. Ohno, R. Yi, A. Suganami, Y. Tamura, A. Matsumoto, S. Matsumoto, K. Saito and H. Shirasawa, Anticancer Res., 2021, 41, 699–706 CrossRef CAS PubMed.
- B. D. Palmer, J. B. Smaill, M. Boyd, D. H. Boschelli, A. M. Doherty, J. M. Hamby, S. S. Khatana, J. B. Kramer, A. J. Kraker, R. L. Panek, G. H. Lu, T. K. Dahring, R. T. Winters, H. D. H. Showalter and W. A. Denny, J. Med. Chem., 1998, 41, 5457–5465 CrossRef CAS.
- B. D. Palmer, A. J. Kraker, B. G. Hartl, A. D. Panopoulos, R. L. Panek, B. L. Batley, G. H. Lu, S. Trumpp-Kallmeyer, H. D. H. Showalter and W. A. Denny, J. Med. Chem., 1999, 42, 2373–2382 CrossRef CAS PubMed.
- R. G. Kulkarnia, S. A. Laufer, C. V. M. and A. Garlapati, Med. Chem., 2013, 9, 91–99 CrossRef.
- E. M. E. Dokla, N. S. Abutaleb, S. N. Milik, D. Li, K. El-Baz, M.-A. W. Shalaby, R. Al-Karaki, M. Nasr, C. D. Klein, K. A. M. Abouzid and M. N. Seleem, Eur. J. Med. Chem., 2020, 186, 111850 CrossRef CAS.
- D. Secci, A. Bolasco, M. D'Ascenzio, F. Della Sala, M. Yáñez and S. Carradori, J. Heterocycl. Chem., 2012, 49, 1187–1195 CrossRef CAS.
- S. N. A. Bukhari, G. Lauro, I. Jantan, C. Fei Chee, M. W. Amjad, G. Bifulco, H. Sher, I. Abdullah and N. A. Rahman, Future Med. Chem., 2016, 8, 1953–1967 CrossRef CAS.
- S. R. Fletcher, E. McIver, S. Lewis, F. Burkamp, C. Leech, G. Mason, S. Boyce, D. Morrison, G. Richards, K. Sutton and A. B. Jones, Bioorg. Med. Chem. Lett., 2016, 16, 2872–2876 CrossRef PubMed.
- Y.-L. Zhang, R. Yang, L.-Y. Xia, R.-J. Man, Y.-C. Chu, A.-Q. Jiang, Z.-C. Wang and H.-L. Zhu, Bioorg. Chem., 2019, 92, 103219 CrossRef CAS.
- Y. K. Yoon and T. S. Choon, Arch. Pharm., 2016, 349, 1–8 CrossRef CAS PubMed.
- F. Wang, X. Wang, M.-X. Zhang, Y.-H. Yang and H.-L. Zhu, RSC Adv., 2015, 5, 74425–74437 RSC.
- Y. K. Yoon, M. A. Ali, A. C. Wei, A. N. Shirazi, K. Parang and T. S. Choon, Eur. J. Med. Chem., 2014, 83, 448–454 CrossRef CAS PubMed.
- M. M. Karpińska, J. Matysiak, A. Niewiadomy, J. Wietrzyk and D. Kłopotowska, Monatsh. Chem. – Chem. Mon., 2012, 143, 269–276 CrossRef.
- S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS.
- M. Tomanová, I. Vaňková, D. Toman, A. Přibylka, I. Nemec and P. Cankař, J. Org. Chem., 2023, 88, 9265–9276 CrossRef PubMed.
- M. Kriegelstein, D. Profous, A. Lyčka, Z. Trávníček, A. Přibylka, T. Volná, S. Benická and P. Cankař, J. Org. Chem., 2019, 84, 11911–11921 CrossRef CAS PubMed.
- M. Kriegelstein, D. Profous, A. Přibylka and P. Cankař, J. Org. Chem., 2020, 85, 12912–12921 CrossRef CAS PubMed.
- K. Janíková, L. Jedinák, T. Volná and P. Cankař, Tetrahedron, 2018, 74, 606–617 CrossRef.
- L. Jedinák, R. Zátopková, H. Zemánková, A. Šustková and P. Cankař, J. Org. Chem., 2017, 82, 157–169 CrossRef.
- M. Pisár, E. Schütznerová, F. Hančík, I. Popa, Z. Trávníček and P. Cankař, Molecules, 2018, 23, 149 CrossRef.
- C. A. Belmokhtar, J. Hillion and E. Ségal-Bendirdjian, Oncogene, 2001, 20, 3354–3362 CrossRef CAS.
- A. Kamal, A. B. Shaik, S. Polepalli, G. B. Kumar, V. S. Reddy, R. Mahesh, S. Garimella and N. Jain, Bioorg. Med. Chem., 2015, 23, 1082–1095 CrossRef CAS.
- F. Naaz, M. R. Haider, S. Shafi and M. S. Yar, Eur. J. Med. Chem., 2019, 171, 310–331 CrossRef CAS PubMed.
- S. A. Lakhani, A. Masud, K. Kuida, G. A. J. Porter, C. J. Booth, W. Z. Mehal, I. Inayat and R. A. Flavell, Science, 2006, 311, 847–851 CrossRef CAS.
- S. Arora, X. I. Wang, S. M. Keenan, C. Andaya, Q. Zhang, Y. Peng and W. J. Welsh, Cancer Res., 2009, 69, 1910–1915 CrossRef CAS.
- E. Bausch, H. Kohlhof, S. Hamm, R. Krauss, R. Baumgartner and L. Sironi, PLoS One, 2013, 8, e79594 CrossRef CAS.
- N. Mundhara, A. Majumder and D. Panda, Sci. Rep., 2019, 9, 7638 CrossRef.
- A. Müller-Deku, J. C. M. Meiring, K. Loy, Y. Kraus, C. Heise, R. Bingham, K. I. Jansen, X. Qu, F. Bartolini, L. C. Kapitein, A. Akhmanova, J. Ahlfeld, D. Trauner and O. Thorn-Seshold, Nat. Commun., 2020, 11, 4640 CrossRef PubMed.
- S. J. Nowak and V. G. Corces, Trends Genet., 2004, 20, 214–220 CrossRef CAS.
- Y. W. Chan, Y. Chen and R. Y. C. Poon, Oncogene, 2009, 28, 170–183 CrossRef CAS PubMed.
- M. Castedo, J.-L. Perfettini, T. Roumier, K. Andreau, R. Medema and G. Kroemer, Oncogene, 2004, 23, 2825–2837 CrossRef CAS PubMed.
- N. Sakurikar, J. M. Eichhorn and T. C. Chambers, J. Biol. Chem., 2012, 287, 39193–39204 CrossRef CAS.
- Y. H. Ling, C. Tornos and R. Perez-Soler, J. Biol. Chem., 1998, 273, 18984–18991 CrossRef CAS.
- I. B. Roninson, E. V. Broude and B. D. Chang, Drug Resist. Update Rev., 2001, 4, 303–313 CrossRef CAS PubMed.
- M. A. Jordan, K. Wendell, S. Gardiner, W. B. Derry, H. Copp and L. Wilson, Cancer Res., 1996, 56, 816–825 CAS.
- M. A. Jordan, R. J. Toso, D. Thrower and L. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9552–9556 CrossRef CAS.
- Y. Y. Leung, L. L. Yao Hui and V. B. Kraus, Semin. Arthritis Rheum., 2015, 45, 341–350 CrossRef CAS.
- J. Zhou, D. Panda, J. W. Landen, L. Wilson and H. C. Joshi, J. Biol. Chem., 2002, 277, 17200–17208 CrossRef CAS.
- L. Rárová, J. Steigerová, M. Kvasnica, P. Bartůněk, K. Křížová, H. Chodounská, Z. Kolář, D. Sedlák, J. Oklestkova and M. Strnad, J. Steroid Biochem. Mol. Biol., 2016, 159, 154–169 CrossRef.
- V. Malínková, E. Řezníčková, R. Jorda, T. Gucký and V. Kryštof, Bioorg. Med. Chem., 2017, 25, 6523–6535 CrossRef PubMed.
- L. Rárová, D. Sedlák, J. Oklestkova, J. Steigerová, J. Liebl, S. Zahler, P. Bartůněk, Z. Kolář, L. Kohout, M. Kvasnica and M. Strnad, J. Steroid Biochem. Mol. Biol., 2018, 178, 263–271 CrossRef.
- C. Wittmann, A. S. Sivchenko, F. Bacher, K. K. H. Tong, N. Guru, T. Wilson, J. Gonzales, H. Rauch, S. Kossatz, T. Reiner, M. V. Babak and V. B. Arion, Inorg. Chem., 2022, 61, 1456–1470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2285266 and 2285267. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00863d |
| This journal is © The Royal Society of Chemistry 2024 |