
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zn(II)-catalyzed asymmetric [3 + 2] cycloaddition of acyclic enones with azomethine ylides†
Sundaravel Vivek Kumar,
Jeremiah Olusegun and
Patrick J. Guiry *
*
Centre for Synthesis and Chemical Biology, School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: p.guiry@ucd.ie
First published on 14th June 2024
Abstract
The Zn(II)/UCD-Imphanol-catalyzed highly endo-selective [3 + 2] asymmetric cycloaddition of acyclic enones and azomethine ylides has been developed. Moderate to high yields (up to 94%) with excellent endo/exo selectivities (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and enantioselectivities up to 96.5:3.5 er were obtained.
:1) and enantioselectivities up to 96.5:3.5 er were obtained.
Introduction
The pyrrolidine ring is a pivotal core structure found in numerous natural products and bioactive compounds.1 It is considered a privileged scaffold in pharmaceutical and medicinal chemistry, serving as a foundation for the development of novel classes of bioactive and medicinally important drugs.2 Consequently various synthetic routes to assembling such types of frameworks have been developed.3 Of all of the routes developed, the transition metal-catalyzed [3 + 2] asymmetric cycloaddition reaction stands out due to its convergent and atom-economical approach for the construction of pyrrolidines with more than one chiral center.4 Various classes of dipolarophiles, such as α,β-unsaturated esters, maleimides, α,β-unsaturated nitriles, acyclic/cyclic enones, nitroalkenes, vinyl sulfones, and fullerenes, have been extensively studied. The reactivity and selectivities in these reactions are significantly influenced by the steric and electronic properties of both the dipolarophiles and dipoles, as well as the Lewis acidity of the metal–ligand complex utilized.4 More recently, dipolarophiles such as cyclobutanone,5 α-halonitroalkenes,6 and electron-rich benzofulvenes7 have also been explored. Among these, acyclic/cyclic enones have garnered significant attention due to their synthetic potential.8–14 However, achieving high endo/exo ratios and enantioselectivities with these substrates remains challenging.However, Carretero reported that the Cu(I)-Fesulphos complex catalyzed the [3 + 2] azomethine ylide cycloaddition of acyclic/cyclic enones, with the endo/exo selectivity being highly dependent on the cis or trans geometry of the enones.8 In contrast, use of Cu(I)-imFerroS or Ag-imFerroS produced endo-cycloadducts from azomethine ylide cycloaddition of cyclic/acyclic enones, respectively.9 Similarly, the Ag(I)-ThioClickFerrophos catalytic system afforded high levels of endo-selectivity in cycloadduct formation, irrespective of the cis or trans geometry of the enones.10 The application of the Ag(I)-Xingphos catalytic system demonstrated solvent-dependent selectivities.11
Nájera observed metal-dependent diastereoselectivity in the application of the Cu(I)-DM-SegPhos system in the formation of an exo-cycloadduct with 70% ee, whereas the Ag(I)-SegPhos system yielded an endo-cycloadduct with 88% ee.12 Oh reported a metal-dependent enantiodivergent [3 + 2] cycloaddition, where the Cu(I)-brucine diol system provided endo-selective products with good yields and enantioselectivities, compared to the Ag(I)-brucine diol system.13 Additionally, the Au(I)-(Sa)-Binap catalytic system was reported to deliver good yields of endo-cycloadducts with 60–80% ee, compared to the Ag(I)-(Sa)-Binap catalytic system.14
Both Cu(I) and Ag(I)-based catalysts have been extensively explored, often offering complementary results in terms of endo/exo selectivity.4 However, this trend becomes variable when acyclic enones are used as dipolarophiles.8–14 These procedures frequently encounter one or more limitations, such as modest diastereo- or enantioselectivities and/or restricted substrate scope.
On the other hand, despite its unique advantages such as cost-effectiveness, abundance (24th most abundant element), non-toxicity, and environmental friendliness, the use of zinc catalysis in azomethine ylide cycloadditions is rather limited.15 Recently, our group developed the UCD-Imphanol family of ligands, which proved highly efficient for Zn(II)-catalyzed [3 + 2] asymmetric cycloaddition of azomethine ylides with maleimides. These Zn(II)-UCD-Imphanol catalytic system produced endo-selective cycloadducts in good yields, achieving enantioselectivities of up to 99.9% ee.15c In continuation of our work, we present a highly endo-selective asymmetric [3 + 2] cycloaddition of azomethine ylides with chalcones catalyzed by Zn(II)/UCD-Imphanol (Scheme 1).
 | ||
| Scheme 1 Asymmetric [3 + 2] cycloaddition of azomethine ylides with acyclic enones. | ||
Results and discussion
We began our investigation by using (E)-chalcone 1a, ethyl (E)-2-((4-chlorobenzylidene)amino)acetate 2a as model substrates employing DABCO as base, Zn(OTf)2 as the metal catalyst to screen the series of UCD-Imphanol family of ligands including the non-planar chiral N,O-ligand 6. The use of ligand (S,S,SP)-4a gave the cycloadduct 3a in 55% yield with a 93:7 endo/exo ratio and 95:5 er (Table 1, entry 1), whereas the use of 4-methylbenzyl- or pentafluorobenzyl-substituted ligands (S,S,SP)-4b and (S,S,SP)-4c led to the cycloadduct 3a in slightly reduced yield and selectivity (Table 1, entries 2 and 3). Use of ligand 4d afforded the cycloadduct 3a in 79% yield with a 94:6 diastereoselectivity and 94.5:5.5 er (Table 1, entry 4). In contrast, the diastereomeric ligands (S,S,RP)-5a–c showed poor levels of reactivity and selectivity (Table 1, entries 5–7). The non-planar chiral N,O-ligand 6 gave a 38% yield with moderate diastereoselectivity (78:12) but poor enantioselectivity (54:46 er) (Table 1, entry 8), which indicated that the planar chiral paracyclophane unit was necessary to achieve high levels of reactivity and selectivity. The metal salts AgOAc and Cu(OTf)2 were examined; the former produced the cycloadduct 3a in 58% yield with high diastereoselectivity and a 55:45 er, while the latter yielded only a trace amount of product (for full optimization studies see the ESI†). The screening of base and solvent revealed that dichloromethane and DABCO were optimal choices of solvent and base, respectively (see ESI†).
|
|||||
|---|---|---|---|---|---|
| Entry | L | Temp. °C | Yieldb (%) | Endo/Exoc | Erd |
| a All reactions were carried out with Zn(OTf)2 (10 mol%), L (11.5 mol%), 1a (0.15 mmol), 2a (0.225 mmol), DABCO (10 mol%) in CH2Cl2 (1.5 mL) for 24 h.b Isolated yield after purification.c The endo/exo ratios were determined by analyzing the 1H NMR spectra of the crude product.d Determined by chiral HPLC analysis.e 0.3 mmol of 2a was used.f 48 h.g Zn(OTf)2 (5 mol%) and 4d (5.75 mol%) for 72 h. | |||||
| 1 | 4a | rt | 55 | 93:7 |
95:5 |
| 2 | 4b | rt | 43 | 90:10 |
90:10 |
| 3 | 4c | rt | 46 | 92:8 |
87:13 |
| 4 | 4d | rt | 79 | 94:6 |
94.5:5.5 |
| 5 | 5a | rt | 46 | 85:15 |
46:54 |
| 6 | 5b | rt | 35 | 81:19 |
49:51 |
| 7 | 5c | rt | 48 | 83:12 |
47:53 |
| 8 | 6 | rt | 38 | 78:12 |
54:46 |
| 9e | 4d | rt | 90 | 97:3 |
94.5:5.5 |
| 10e,f | 4d | 0 | 95 | 99:1 |
96.5:3.5 |
| 11g | 4d | 0 | 86 | 99:1 |
96:4 |
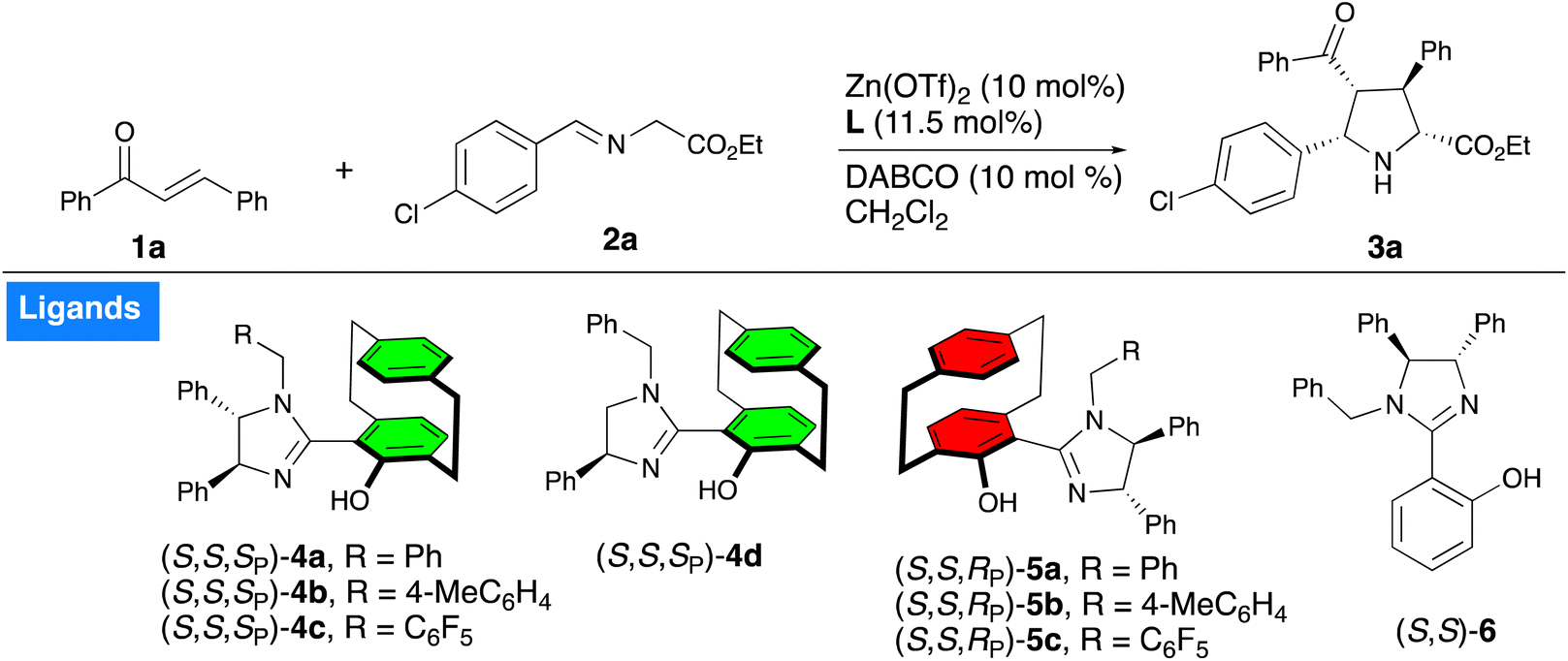
Gratifyingly, the use of an excess (2 equiv.) of 2a led to an increase in yield to 90% without affecting selectivity (Table 1, entry 9). Further, lowering the reaction temperature to 0 °C produced the cycloadduct 3a in 95% yield with an excellent endo/exo ratio and 96.5:3.5 er, our best result in this optimization study (Table 1, entry. 10). Further decreasing the catalyst loading (5 mol% of Zn(OTf)2 and 5.75 mol% of 4d) led to long reaction time with slight reduction of yield and 99:1 endo/exo ratio and 96:4 er.
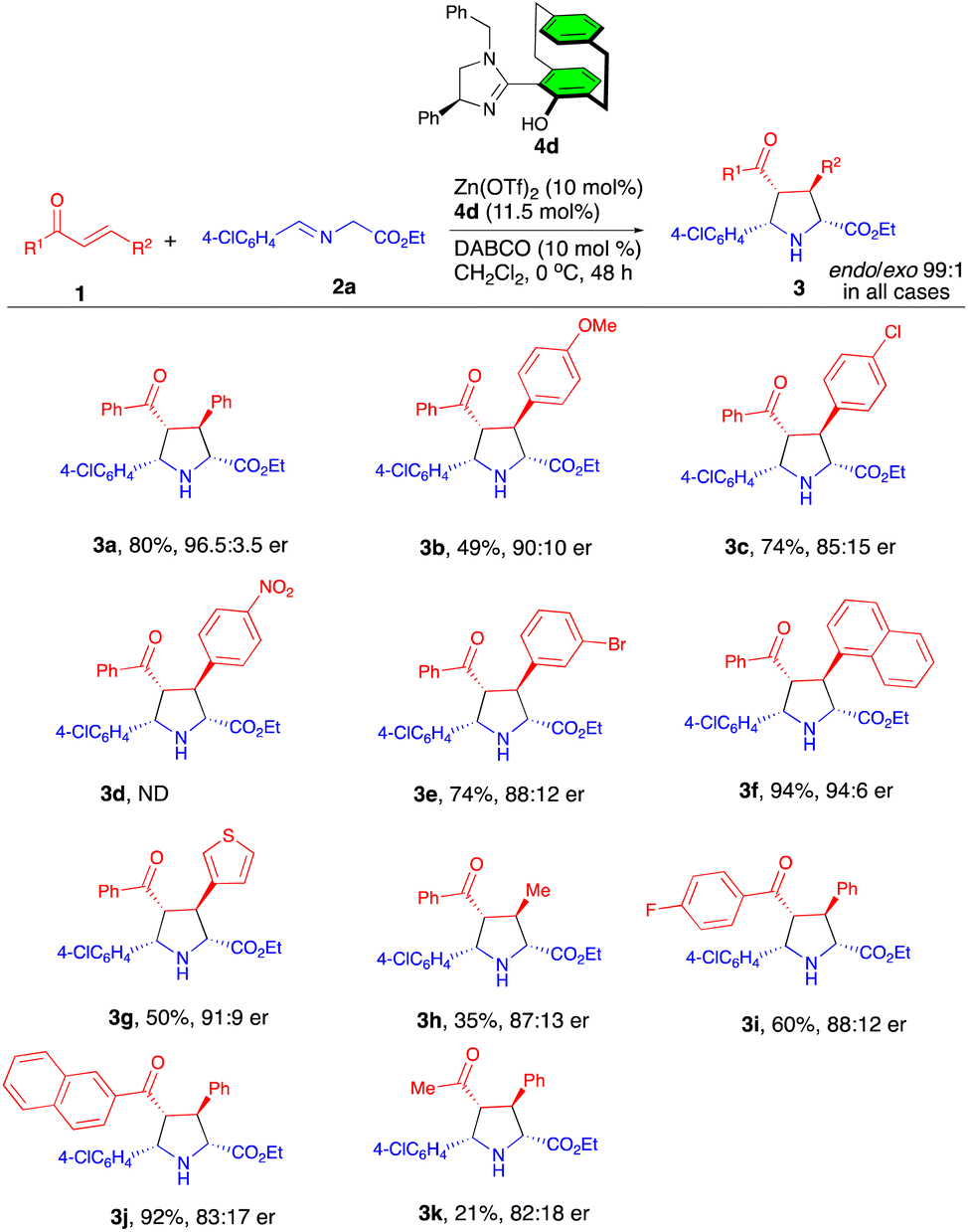
Having optimized conditions in hand, we then examined the scope of this reaction for a focused library of acyclic enones which afforded the products 3a–k with excellent levels of diastereocontrol (99:1) (Scheme 2). A series of chalcones with 4-OMe, 4-Cl, and 3-Br-substituted β-aryl groups gave the corresponding products 3b–c, 3e in moderate to good yields with enantioselectivities of 85:15–90:10 er. In contrast, the reaction of 4-NO2-substituted chalcone led to a complicated mixture; the expected product 3d was not formed, which may presumably be due to the competitive coordination of the nitro group with the Zn metal. The sterically hindered 1-naphthyl and 3-thienyl derived substrates were also tolerated under these reaction conditions to yield the corresponding cycloadducts 3f and 3g in 94% and 50% yield with 96:4 and 91:9 er, respectively. However, the reaction with β-methyl-substituted chalcone afforded the cycloadduct 3h in a lower yield (35%) with a lower level of enantioselectivity (87:13). Next, various α-benzoyl-substituted chalcones also reacted to deliver products 3i–j in moderate to good yields with 88:12 and 87:13 er's. The use of (E)-benzylidene acetone gave rise to the cycloadduct 3k in a disappointing yield of 21% with 82:18 er.
 | ||
| Scheme 2 Scope of the acyclic enones. | ||
Next, our attention turned to exploring the scope of the α-iminoesters (Scheme 3). We were pleased to find that a wide range of α-iminoesters with varying electronics and sterics were tolerated to afford the desired products 3l–w in 20–81% yields with 72:28–94:6 enantioselectivities and 99:1 endo/exo selectivities. In particular, α-iminoesters with electron-deficient and neutral substituents on the aryl ring led to the products 3m–p in 84–71% yields with enantioselectivities of 91:9–94:6 er. The α-iminoesters with sterically hindered substituents such as o-tolyl, 2-naphthyl, and ferrocenyl underwent the reaction to give the products 3q, 3s–t in 20–54% yields with 84:16–90:10 er's. Notably, the 1-naphthyl-substituted α-iminoester provided the cycloadduct 3r with the opposite stereochemistry (12:88 er) in 72% yield. Furthermore, 2-thienyl- and cyclohexyl-substituted iminoesters also showed poor reactivity (28–37% yield) for the formation of products 3u–v with enantioselectivities in the range 72:28–82:18 er's. Moreover, the α-methyl-substituted iminoester also displayed moderate reactivity, forming the product 3w in 48% yield with 75:25 er.
 | ||
| Scheme 3 Scope of the α-iminoesters. | ||
To demonstrate the synthetic potential of the present reaction, we performed the reaction on a 1 mmol scale, furnishing 3a in 86% yield and excellent diastereo- and enantioselectivity (99:1 endo/exo, 96.5:3.5 er) (Scheme 4a). Further treatment of the product with chloroacetyl chloride led to the formation of 7, which then reacted with 40% aq. MeNH2,17 affording the diketopiperazine derivative 8 in 80% yield with 96:4 er (Scheme 4b).
 | ||
| Scheme 4 Scaled-up synthesis of 3a and its derivatization. | ||
A plausible transition state was tentatively proposed based on the Zn-complex 9, which was prepared from the reaction of ligand 4a with Et2Zn, affording the homoleptic zinc(II) complex 9 (Scheme 5). The homoleptic complex was confirmed by single crystal X-ray diffraction studies (Fig. 1), and this complex would generate the heteroleptic Zn(II) complex by interaction with iminoester 2 under basic conditions that can readily react with acyclic enones.
 | ||
| Scheme 5 Preparation of complex 9. | ||
 | ||
| Fig. 1 Single crystal XRD analysis for 9. All H-atoms are omitted for clarity. | ||
The transition state contains the azomethine ylide coordinating with the Zn(II)-imidazolinyl-paracyclophanol in a tetrahedral complex (Fig. 2). The sterically crowded paracyclophane and C-4 phenyl group effectively shield the top-face approach to the Si-face of the ylide, while the Re-face is less crowded thus allowing the acyclic enone approach in an endo fashion (TSI) to deliver the products with good to moderate levels of enantioselectivity. Although it cannot be completely ruled out, the exceptional endo-selectivity that was observed may perhaps be due to the additional coordination of the α,β-unsaturated ketone carbonyl group with the metal center. Additionally, TSII may explain the noted opposite enantiomer in the case of 1-naphthyl iminoester since TSI presumably leads to a disfavored steric clash between the 1-naphthyl iminoester and the paracyclophane unit of ligand.
 | ||
| Fig. 2 Proposed stereochemical models. | ||
Conclusions
In summary, we have developed the Zn(II)-catalyzed highly endo-selective asymmetric [3 + 2] cycloaddition of acyclic enones with azomethine ylides by the use of the planar chiral UCD-Imphanol family of ligands, leading to chiral pyrrolidines in moderate to good yields with 99:1 endo/exo selectivities and enantioselectivities up to 96.5:3.5 er. A variety of acyclic enones and iminoester-derived azomethine ylides are compatible with the less expensive Zn-catalysis. Further detailed mechanistic studies with computational analysis are currently underway and will be reported in due course.
Experimental section
General procedure for the Zn(II)-catalyzed [3 + 2] azomethine ylide cycloaddition
Under nitrogen atmosphere, ligand (S,S,SP)-4d (11.5 mol%) and Zn(OTf)2 (10 mol%) was added to a flame-dried 10 mL Schlenk tube. Then dry CH2Cl2 (0.5 mL) was added and stirred for 60 min. After cooling to 0 °C, acyclic enone 1 (0.15 mmol, 1.0 equiv.) in CH2Cl2 (0.4 mL), α-iminoester 2 (0.3 mmol, 2 equiv.) in CH2Cl2, (0.4 mL) and DABCO (10 mol%) in CH2Cl2 (0.2 mL) were added sequentially. The reaction mixture was stirred at the same temperature for 48 h. After completion, the reaction mixture was directly purified by silica gel column chromatography (30% ethyl acetate in cyclohexane) to afford the desired product 3. The endo/exo ratio was determined by 1H-NMR before purification and the enantioselectivity was determined by HPLC analysis of the isolated product. The configuration of the products was assigned by comparison with the optical rotation values of prepared compounds and the reported optical rotation of the cycloadducts.12a,16 (Note: the reaction temperature (0 °C) was maintained using a cryocooler).Data availability
The data supporting this article have been included as part of the ESI.† CDC 2299375 contains the supplementary crystallographic data for Zinc complex 9.†Author contributions
PG supervised the research; S. V. K. and P. G. conceived the idea; S. V. K. and J. O. carried out all the synthetic experimental work and data acquisition. S. V. K. prepared the manuscript with contributions from J. O. and P. G. All authors approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. V. K. thanks the Irish Research Council (IRC) for the award of a Postdoctoral Scholarship (GOIPD/2019/918) and UCD School of Chemistry for the award of a Teaching Fellowship. We acknowledge the facilities provided by the Centre for Synthesis and Chemical Biology (CSCB), funded by the Higher Education Authority's Programme for Research in Third-Level Institutions. We thank Dr Yannick Ortin, Dr Patricia Fleming and Dr Guanghua Jin for NMR spectroscopy, and Dr Jimmy Muldoon for mass spectrometric analysis (supported by a Science Foundation Ireland Infrastructure Award (18/RI/5702)).References
- (a) W. J. Olivier, J. A. Smith and A. C. Bissember, Chem. Rec., 2022, 22, e202100277 CrossRef CAS PubMed; (b) M. T. Islam and M. S. Mubarak, Adv. Tradit. Med., 2020, 20, 13 CrossRef CAS; (c) A. K. Chattopadhyay and S. Hanessian, Chem. Rev., 2017, 117, 4104 CrossRef CAS PubMed; (d) C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405 RSC; (e) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC; (f) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC.
- (a) S. Poyraz, H. A. Döndaş, N. Y. Döndaş and J. M. Sansano, Front. Pharmacol., 2023, 14, 1239658 CrossRef CAS PubMed; (b) G. L. Petri, M. V. Raimondi, V. Spanò, R. Holl, P. Barraja and A. Montalbano, Top. Curr. Chem., 2021, 379, 34 CrossRef PubMed.
- Selected examples see; (a) R. Zhang, M. Sun, Q. Yan, X. Lin, X. Li, X. Fang, H. H. Y. Sung, I. D. Williams and J. Sun, Org. Lett., 2022, 24, 2359 CrossRef CAS PubMed; (b) J.- B. Qiao, Y.-Q. Zhang, Q.-W. Yao, Z.-Z. Zhao, X. Peng and X.-Z. Shu, J. Am. Chem. Soc., 2021, 143, 12961 CrossRef CAS PubMed; (c) Y.-F. Yu, C. Shu, T.-D. Tan, L. Li, S. Rafique and L.-W. Ye, Org. Lett., 2016, 18, 5178 CrossRef CAS PubMed; (d) L. Liu, M. Leutzsch, Y. Zheng, M. W. Alachraf, W. Thiel and B. List, J. Am. Chem. Soc., 2015, 137, 13268 CrossRef CAS PubMed; (e) G.-H. Hou, J.-H. Xie, P.-C. Yan and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 1366 CrossRef CAS PubMed; (f) R. L. LaLonde, B. D. Sherry, E. J. Kang and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2452 CrossRef CAS PubMed.
- Selected recent examples see; (a) S. V. Kumar and P. J. Guiry, Chem. – Eur. J., 2023, 29, e202300296 CrossRef CAS PubMed; (b) J. Adrio and J. C. Carretero, Chem. Commun., 2019, 55, 11979 RSC; (c) X. Fang and C.-J. Wang, Org. Biomol. Chem., 2018, 16, 2591 RSC; (d) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef CAS PubMed; (e) J. Adrio and J. C. Carretero, Chem. Commun., 2014, 50, 12434 RSC.
- J. Corpas, A. Ponce, J. Adrio and J. C. Carretero, Org. Lett., 2018, 20, 3179 CrossRef CAS PubMed.
- V. A. Motornov, A. A. Tabolin, Y. V. Nelyubina, V. G. Nenajdenko and S. L. Ioffe, Org. Biomol. Chem., 2021, 19, 3413 RSC.
- X. Chang, X.-T. Liu, F. Li, Y. Yang, L. W. Chung and C.-J. Wang, Chem. Sci., 2023, 14, 5460 RSC.
- J. Hernández-Toribio, R. G. Arrayás, B. Martín-Matute and J. C. Carretero, Org. Lett., 2009, 11, 393 CrossRef PubMed.
- C. Zhang, S.-B. Yu, X.-P. Hu, D.-Y. Wang and Z. Zheng, Org. Lett., 2010, 12, 5542 CrossRef CAS PubMed.
- I. Oura, K. Shimizu, K. Ogata and S. Fukuzawa, Org. Lett., 2010, 12, 1752 CrossRef CAS.
- X.-F. Bai, Z. Xu, C.-G. Xia, Z.-J. Zheng and L. W. Xu, ACS Catal., 2015, 5, 6016 CrossRef CAS.
- (a) G. S. Caleffi, O. Larrañaga, M. Martín-Rodríguez, P. R. R. Costa, C. Nájera, A. Cózar, F. P. Cossío and J. M. Sansano, J. Org. Chem., 2019, 84, 10593 CrossRef CAS PubMed; (b) A. Cayuelas, O. Larrañaga, V. Selva, C. Nájera, T. Akiyama, J. M. Sansano, A. Cózar, J. I. Miranda and F. P. Cossío, Chem. – Eur. J., 2018, 24, 8092 CrossRef CAS; (c) C. Nájera, M. G. Retamosa, M. Martín-Rodríguez, J. M. Sansano, A. Cózar and F. P. Cossío, Eur. J. Org. Chem., 2009, 5622 CrossRef.
- J.-Y. Li, H. Y. Kim and K. Oh, Adv. Synth. Catal., 2016, 358, 984 CrossRef CAS.
- M. Martín-Rodríguez, C. Nájera, J. M. Sansano, A. Cózar and F. P. Cossío, Chem. – Eur. J., 2011, 17, 14224 CrossRef.
- Zn-catalyzed iminoester-derived [3 + 2] azomethine ylide cycloaddition see; (a) A. S. Gothelf, K. V. Gothelf, R. G. Hazell and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 4236 CrossRef CAS; (b) O. Dogan, H. Koyuncu, P. Garner, A. Bulut, W. J. Youngs and M. Panzner, Org. Lett., 2006, 8, 4687 CrossRef CAS PubMed; For Zn-catalyzed [3+2] cycloadditions with other azomethine ylides see; (c) S. V. Kumar and P. J. Guiry, Angew. Chem., 2022, 134, e202205516 CrossRef; (d) Y. Yi, Y. Z. Hua, H.-J. Lu, L.-T. Liu and M.-C. Wang, Org. Lett., 2020, 22, 2527 CrossRef CAS PubMed; (e) R.-L. Wang, S.-K. Jia, Y.-J. Guo, Y. Yi, Y.-Z. Hua, H.-J. Lu and M.-C. Wang, Org. Biomol. Chem., 2021, 19, 8492 RSC; (f) P. Zhou, Y. Yi, Y. Hua, S. Jia and M. Wang, Chem. – Eur. J., 2022, 28, e202103688 CrossRef PubMed.
- Y. Zhang, J. Su, T. Lin, Z. Lin, Z. Zhou and Y. Li, J. Chem. Res., 2019, 43, 90 CrossRef CAS.
- E. García-Mingüens, V. Selva, O. Larrañaga, C. Nájera, J. M. Sansano and A. Cózar, ChemCatChem, 2020, 12, 2014 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2299375. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00854e |
| This journal is © The Royal Society of Chemistry 2024 |