Highly regio- and stereoselective (3 + 2) annulation reaction of allenoates with 3-methyleneindolin-2-ones catalyzed by a planar chiral [2.2]paracyclophane-based bifunctional phosphine–phenol catalyst†
Received
20th May 2024
, Accepted 4th July 2024
First published on 15th July 2024
Abstract
A planar chiral [2.2]paracyclophane-based phosphine–phenol catalyst catalyzed the (3 + 2) annulation reaction of ethyl 2,3-butadienoate with 3-methyleneindolin-2-ones to produce 2,5-disubstituted cyclopentene-fused C3-spirooxindoles in high yields with high regio-, diastereo-, and enantioselectivities. This catalyst was suitable for reactions of not only benzylideneindolinones but also alkylideneindolinones, the chiral phosphine-catalyzed reactions of which have not yet been reported. Density functional theory calculations suggested that the formation of hydrogen bonds between the phenolic OH group of the catalyst and the allenoate carbonyl group, rather than between the OH group and the carbonyl group of indolinone, contributed to the formation of an efficient reaction space at the enantiodetermining step.
Introduction
Phosphine catalysts perform nucleophilic attacks on activated alkynes, alkenes, and allenes, and thus these catalysts have been used for a variety of transformations.1 Lu and colleagues reported the first phosphine-catalyzed (3 + 2) annulation reactions between allenoates or alkynoates2a or Morita–Baylis–Hillman (MBH) adduct analogues2b as three-carbon synthons and electron-deficient alkenes as two-carbon synthons. Since then, this type of reaction has been one of the most reliable methods for constructing highly substituted cyclopentane derivatives, and the asymmetric variants of this reaction have been thoroughly studied. Substituents on the phosphorus atom enable efficient control of the reactivity and chemo-, regio-, and stereoselectivities in the phosphine-catalyzed reactions. Chiral phosphine molecules produce enantiomerically enriched compounds by forming a proper asymmetric environment based on a chiral phosphorus center or a chiral backbone.3 Hybridized phosphine catalysts with an acid functionality have expanded the reaction to produce the desired regio- and stereoisomer exclusively through the multiple recognition of the substrates and reactants. Many chiral phosphines, such as axially chiral binepine,4 spirocyclic SITCP,5 planar chiral FerroPHANE,6 and amino acid-derived multifunctional phosphines (Fig. 1),7 have been reported as excellent catalysts for various useful reactions. However, the development of new types of phosphine catalyst is still highly desirable because of the diversity of phosphine catalysis.
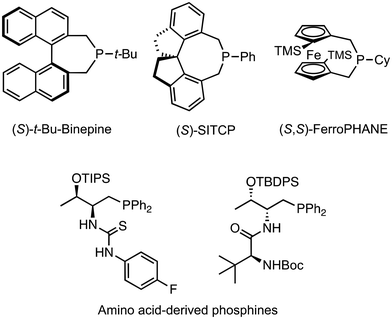 |
| | Fig. 1 Chiral phosphine catalysts. | |
Spirocycles are produced by annulation with three-carbon synthons, using exo-methylenecycles as the two-carbon synthons. Chiral cyclopentane-fused C3-spirooxindoles with multiple stereocenters are found in many natural products and biologically active compounds (Fig. 2).8 Several organocatalytic and metal complex-catalyzed methods for the simultaneous creation of spiro centers and other chiral centers on this privileged structure have been developed.9 The phosphine-catalyzed (3 + 2) annulation reaction of three-carbon synthons and 3-methyleneindolin-2-ones is one of the most efficient synthetic methods for accessing cyclopentane-spirooxindoles.10 Marinetti and colleagues11a,b,d first reported the highly enantioselective (3 + 2) annulation reaction of allenoates with 3-methyleneindolinones catalyzed by chiral phosphines (Scheme 1a).11 A similar reaction was developed independently using MBH carbonates12 (Scheme 1b). Reactions using alkynoates (Scheme 1a)13 as three-carbon synthons and a Rauhut–Currier reaction with diene derivatives14 have also been reported. However, in the syntheses of cyclopentene-spirooxindoles, the substituent on the methylene carbon in the substrates is limited to an aryl group (Type A in Scheme 1) or electron-withdrawing group (Type B in Scheme 1). This limitation is probably caused by the low reactivity of alkyl-substituted methyleneindolinones15 for the Michael addition reaction of the phosphonium enolates or phosphonium ylides, which is the first step of (3 + 2) annulation. For allenoates, Marinetti and colleagues’ (3 + 2) annulation method using t-Bu-binepine or FerroPHANE is suitable for reactions of ethyl 2,3-butadienoate and benzylideneindolinones containing various types of aromatic groups on the methylene carbon. The reaction efficiently gives 5-aryl-substituted cyclopentene-spirooxindoles (γ-adduct), which are obtained via γ-addition of the phosphonium enolate to the β-position of methyleneindolinone, in high yields with high γ/α (regio)- and stereoselectivities (Scheme 1a).11a However, product yields and regioselectivities are moderate in some cases, especially the reaction of furyl- and alkynyl-substituted methyleneindolinones. Reactions of alkylideneindolinones are not found in the literature. Wang et al. also reported SITCP-catalyzed (3 + 2) cyclization of allenoates with oxindoles, but the substrates used were methyleneindolinones with a phenyl or naphthyl group.11c To the best of our knowledge, only one example of a reaction of hexylideneindolinone with 4-phenyl-2,3-butadienoate catalyzed by Ph3P has been reported.11b,16 In searching for attractive bioactive compounds, there is a need to develop new catalysts that activate both allenes and alkenes, allowing access to greater structural diversity.
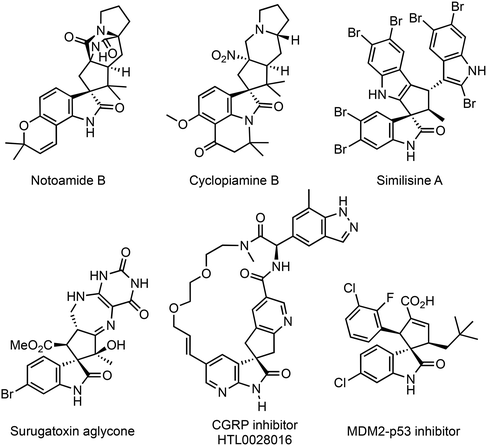 |
| | Fig. 2 Examples of natural products and biologically active compounds containing the cyclopentane-spirooxindole scaffold. | |
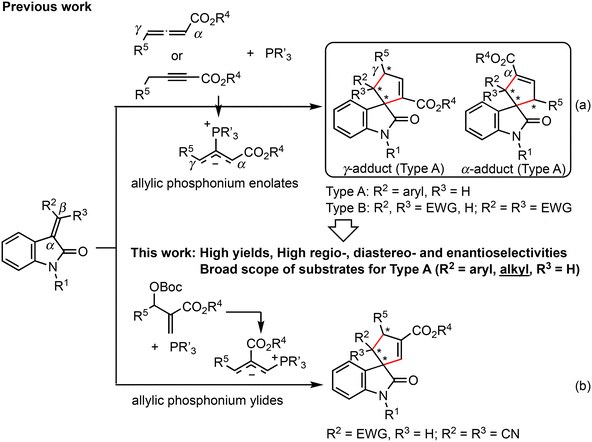 |
| | Scheme 1 Typical organocatalytic (3 + 2) annulations of 3-methyleneindolin-2-ones. Type A: Annulation reaction proceeds via Michael addition of phosphonium enolates at the β-position of methyleneindolinones. Type B: Michael reaction occurs at the α-position of methyleneindolinones. | |
Planar chiral ligands with a [2.2]paracyclophane (pCp) backbone are effective in many asymmetric catalysis reactions.17 However, the potential of pCp as a planar chiral organocatalyst backbone has yet to be investigated sufficiently.18 There are few examples of highly enantioselective reactions catalyzed by pCp-based chiral molecules.19 Therefore, we developed pCp-based bifunctional phosphine–phenol catalysts, in which one of the aryl groups on the phosphorus atom of the triarylphosphine has a pseudo-ortho-hydroxy-substituted pCp group at the meta position (Fig. 3). In other words, one of the aryl groups in the triarylphosphine serves as a spacer, which offers conformational flexibility and makes the distance between the two functional groups suitable for performing dual activation of the substance and reactant. Catalyst (Sp)-1a (R1 = H, R2 = Me) showed exceptionally high reactivity and good enantioselectivity in the aza-MBH reaction of N-tosylaldimines with various vinyl ketones.20 The importance of the phenolic OH group at the pseudo-ortho position for the reactivity and enantioselectivity of the reaction has been demonstrated by comparison with the results using methylated (Sp)-1b (R1 = R2 = Me).20b Catalyst 1a also worked well in the (3 + 2) annulation reaction of allenoates with N-tosylaldimines21 and of MBH carbonates with dicyanomethyleneindolinones.12e
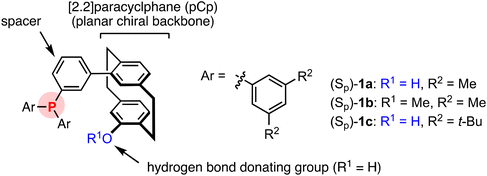 |
| | Fig. 3 [2.2]Paracyclophane-based planar chiral triarylphosphine catalysts. | |
Here, we report that pCp-phosphine–phenol (Sp)-1c (R1 = H, R2 = t-Bu)21 efficiently catalyzes the (3 + 2) annulation of allenoates with not only benzylideneoxindoles but also alkylideneoxindoles to produce 2,5-disubstituted cyclopentene-fused C3-spirooxindoles in a highly regio-, diastereo-, and enantioselective manner (Scheme 1a). Density functional theory (DFT) studies elucidate the origin of the enantioselectivity and the role of the phenolic OH group on the chiral pCp backbone in the (3 + 2) annulation.
Results and discussion
We started by evaluating pCp-phosphine–phenol catalyst (Sp)-1a with two xylyl groups in the reaction of (E)-enriched N-acetyl-3-benzylideneindolinone (E/Z = >20/1) (2a) and ethyl 2,3-butadienoate (3) (Table 1). A reaction with 5 mol% (Sp)-1a in dichloromethane at room temperature was complete within 30 min to provide a 8.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of γ- and α-adducts trans-4a in quantitative yield with 51% and 3% enantiomeric excess (ee), respectively (entry 1). Their diastereomers were not detected in the reaction mixture. Using a more polar solvent, such as THF or acetonitrile, slowed the reaction rate and decreased the enantioselectivity (entries 2–4). Among the solvents examined, toluene gave the highest reactivity and enantioselectivity (entry 6). Masked hydroxy group catalyst (Sp)-1b resulted in markedly lower reactivity and enantioselectivity, which suggested that the hydroxy group is an important feature of catalyst (Sp)-1a (entry 7). Catalyst (Sp)-1c with two bulky tert-butyl groups21 improved the γ-selectivity and the ee of the major product. Lowering the reaction temperature increased the enantioselectivity (up to 93% ee), although a catalyst loading of 10 mol% was needed for a smooth reaction (entries 10 and 11).
:1 mixture of γ- and α-adducts trans-4a in quantitative yield with 51% and 3% enantiomeric excess (ee), respectively (entry 1). Their diastereomers were not detected in the reaction mixture. Using a more polar solvent, such as THF or acetonitrile, slowed the reaction rate and decreased the enantioselectivity (entries 2–4). Among the solvents examined, toluene gave the highest reactivity and enantioselectivity (entry 6). Masked hydroxy group catalyst (Sp)-1b resulted in markedly lower reactivity and enantioselectivity, which suggested that the hydroxy group is an important feature of catalyst (Sp)-1a (entry 7). Catalyst (Sp)-1c with two bulky tert-butyl groups21 improved the γ-selectivity and the ee of the major product. Lowering the reaction temperature increased the enantioselectivity (up to 93% ee), although a catalyst loading of 10 mol% was needed for a smooth reaction (entries 10 and 11).
Table 1 Optimization of reaction conditionsa
To increase the enantioselectivity, we performed simple protecting group manipulations of the benzylideneindolinone and allenoate using 5 mol% (Sp)-1a in toluene at room temperature (Table S1, see ESI†). However, replacing the acetyl group in 2a with a Boc or alkyl group decreased the regio- and enantioselectivities, whereas a Ts group gave a similar yield and enantioselectivity but much lower regioselectivity. Increased steric bulk of alkoxy group or installation of γ-substituents on the allenoates gave poor results.
Having ascertained the effectiveness of the combination of N-acetylindolinone 2a and ethyl 2,3-butadienoate (3), we investigated the scope of the reaction with respect to the substituent on the benzene ring of the benzylidene group of N-acetyl-3-benzylideneindolinones 2 (E/Z = 7.3/1 to >20/1) with the optimal reaction conditions and catalyst (Sp)-1c (Scheme 2). Both electron-withdrawing and -donating substituents at the para position on the benzene ring were compatible with the reaction conditions (Scheme 2, trans-4b–4f). The position of the substituent did not affect the product yield and selectivities (Scheme 2, trans-4g, 4h). The naphthyl and heteroaryl-substituted methyleneindolinones also reacted smoothly to give the desired products in excellent yields and high regio- and stereoselectivities (Scheme 2, trans-4i–4k).
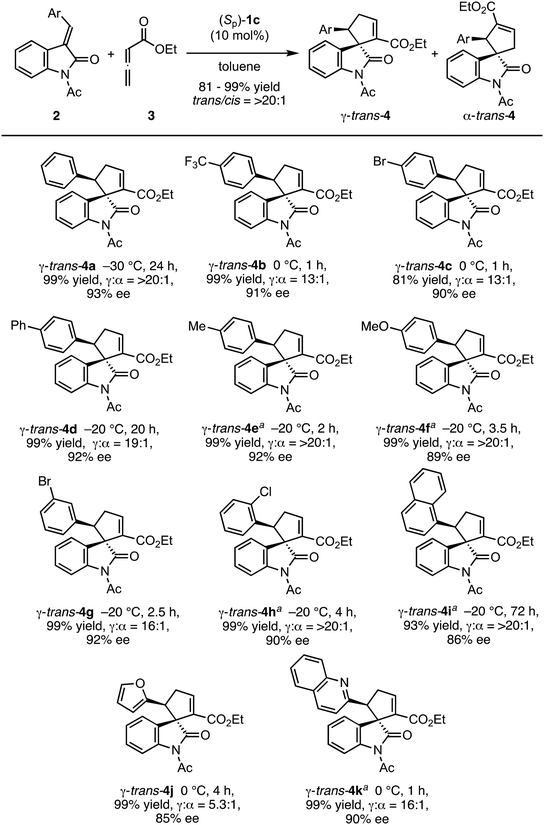 |
| | Scheme 2 Substrate scope (benzylideneindolinones). Reaction conditions: 0.05 mmol 2, 0.1 mmol 3, and 10 mol% catalyst (Sp)-1c in toluene (0.05 M) at the suggested temperature and for the specified time. Yields were of a γ/α mixture after column chromatography. The diastereomer ratio was determined by 1H NMR of the crude product. Enantiomeric excess was determined by chiral HPLC analysis. aPerformed at a concentration of 0.03 M. | |
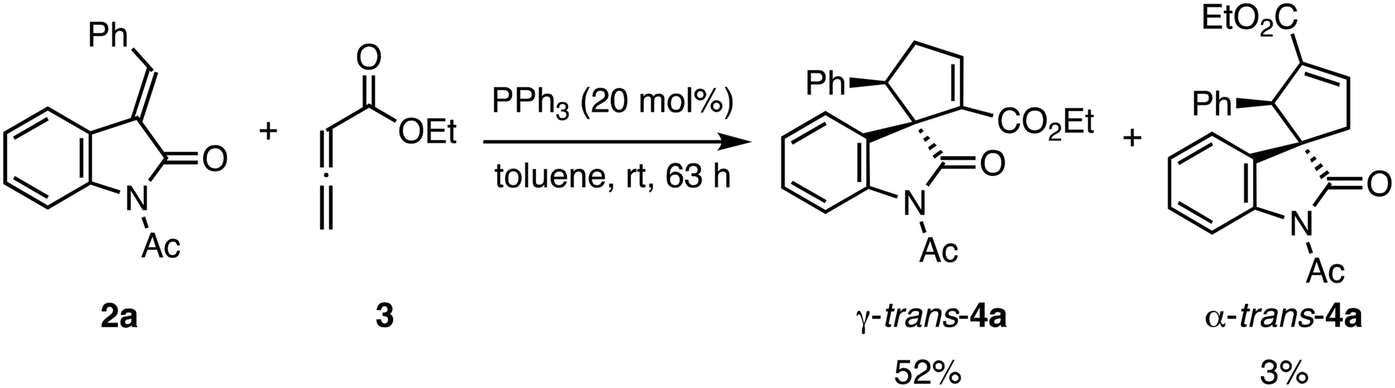
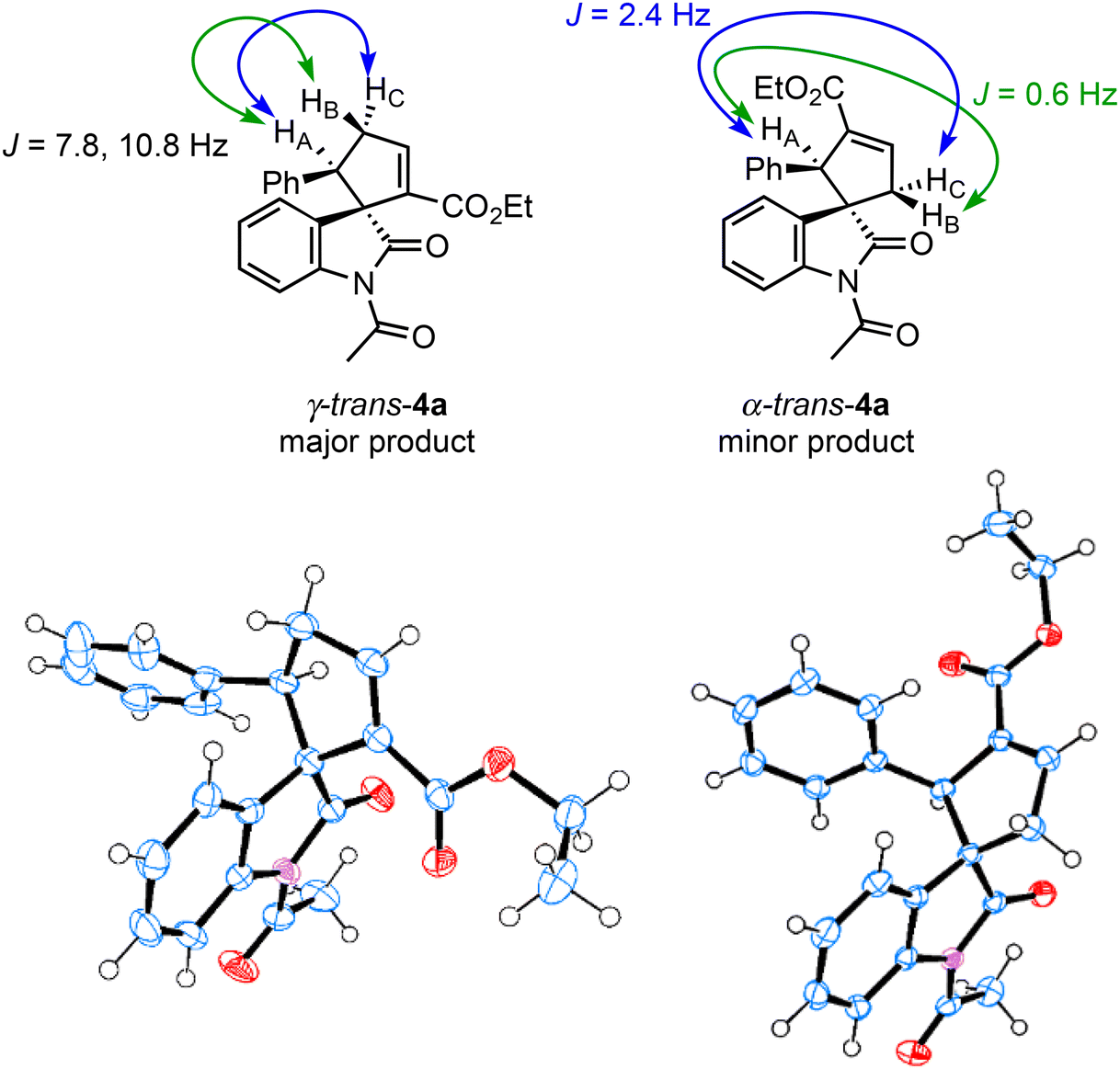
The structural determination of the minor product of this reaction using γ-unsubstituted allenoate has not yet been reported. Therefore, we conducted the triphenylphosphine-catalyzed (3 + 2) cyclization reaction of 2a and 311a on a large scale and purified the crude products using column chromatography and preparative thin-layer chromatography to separate the two cyclized products (Scheme 3). The structures of the two isolated products were determined by NMR analysis, including 1H–1H COSY, and substantiated further by X-ray structural analysis of both compounds. The major product was γ-trans adduct 4a, as reported in the literature,11a,c and the minor product, not specified in the literature, was α-trans adduct 4a (Fig. 4).22 In addition, the absolute configuration of γ-trans-4a was (1S,5R) based on comparing the optical rotation with literature data.11a We assigned the other products by analogy.
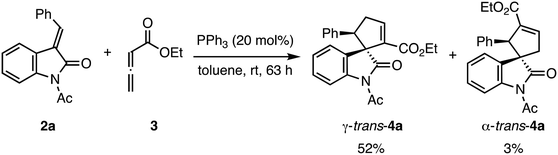 |
| | Scheme 3 Preparation of racemic products 4a for X-ray crystallographic analysis. | |
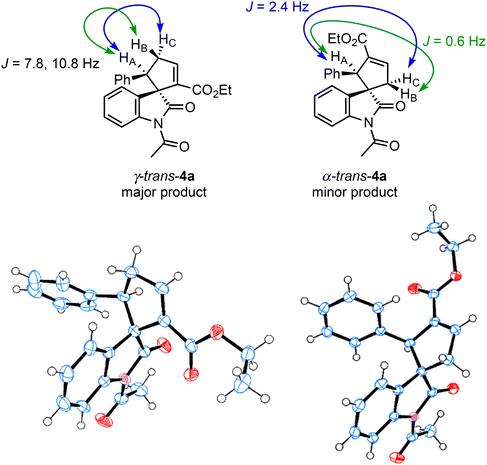 |
| | Fig. 4 X-ray crystal structures of γ-trans-4a and α-trans-4a. | |
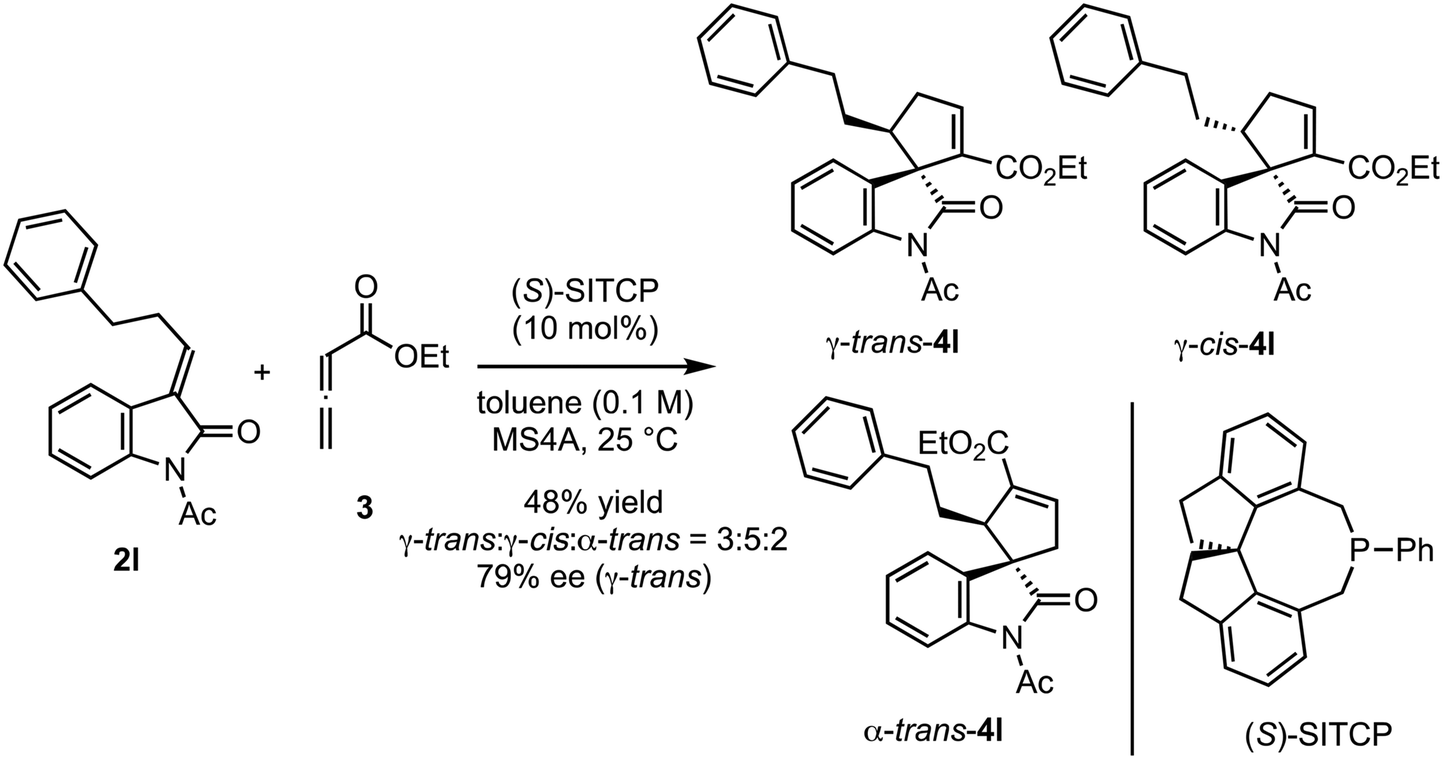
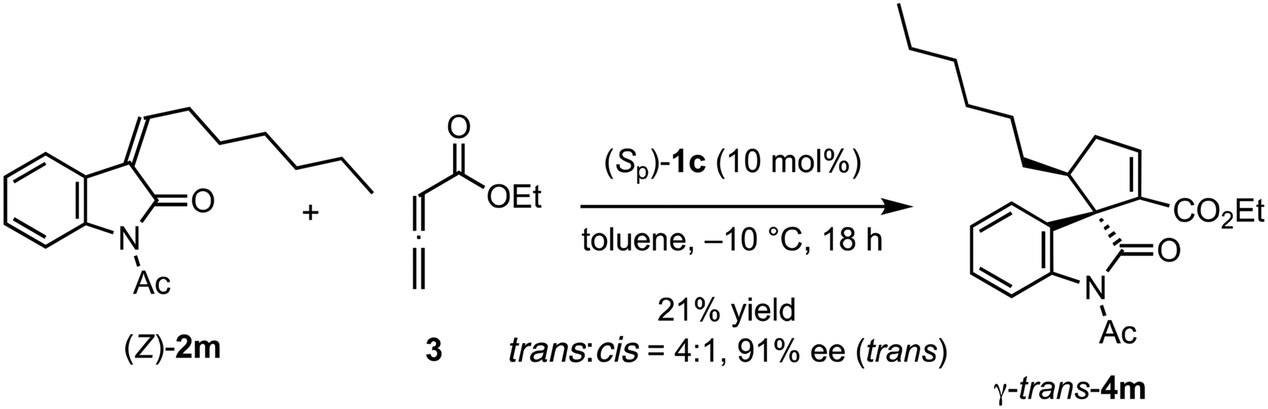
The reactions of various methyleneindolinones 2 bearing carbon substituents other than aryl groups on the methylene carbon (E/Z = >20/1) with allenoate 3 and 10 mol% (Sp)-1c were examined (Scheme 4). Phenethyl-substituted methyleneindolinone 2l reacted with 3 at room temperature to produce γ-trans-adduct 4l as a 10:1 mixture of trans/cis diastereomers in 65% yield with 91% ee for the trans-isomer. The structure of minor product 4l was determined to be the γ-cis-adduct by 1H NMR analysis. The corresponding α-isomer was not detected in the crude reaction product. Using (S)-SITCP in the reaction of 2l gave 4l in low yield, low regio- and diastereoselectivity, and moderate enantioselectivity under the reported11c conditions (Scheme 5). The effects of the geometry of alkylidene group were investigated using hexyl-substituted substrate 2m because the E/Z-isomers of 2m were separated easily by column chromatography. Reaction of (E)-2m with 3 in toluene at −10 °C was complete after 4 h and yielded γ-4m quantitatively as a 9:1 mixture of trans/cis isomers with 92% ee for the trans-isomer (Scheme 4). In contrast, the reaction of (Z)-2m under the same conditions gave γ-adduct 4m in only 21% yield, even after 18 h, and starting material 2m was recovered as an E/Z mixture (Scheme 6). Thus, the γ-adduct was obtained as a 4:1 mixture of trans/cis isomers and the ee of the major trans-isomer was 91%, which was similar to that of the reaction product of (E)-2m. This result suggested that in the (Sp)-1c-catalyzed reaction of methyleneindolinones 2 with 3, the Z-isomer of 2 was less reactive than the E-isomer, and the E-isomer, which was produced by the isomerization of the Z-isomer under the conditions,11b mainly reacted with 3 to give γ-trans-adduct 4. In this reaction, alkene, nitro, and ester groups were tolerated (Scheme 4, 4o–4r). Sterically demanding cyclohexyl and tert-butyl group-substituted substrates 2s and 2t, respectively, also exhibited high reactivity for the (3 + 2) annulation reaction to give corresponding γ-trans-adducts 4s and 4t in excellent yields with high regio- and stereoselectivity.
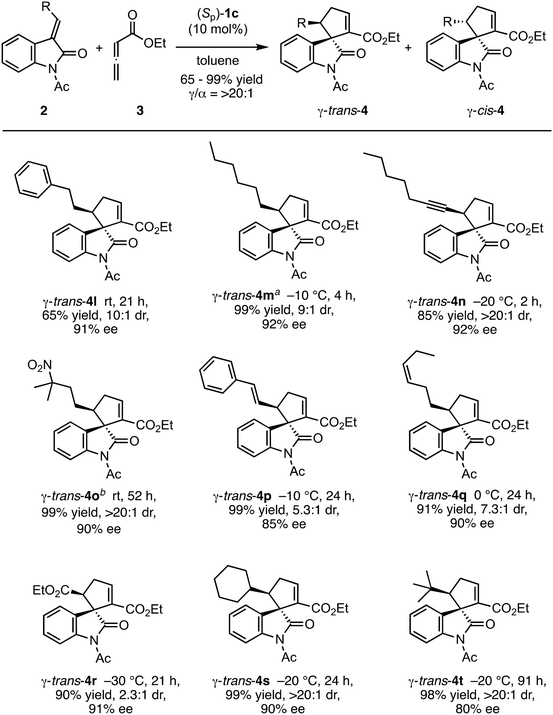 |
| | Scheme 4 Substrate scope (alkylideneindolinones). Reaction conditions: 0.05 mmol 2, 0.1 mmol 3, and 10 mol% catalyst (Sp)-1c in toluene (0.05 M) at the suggested temperature and for the specified time. Yields were of a trans/cis mixture after column chromatography. The diastereomer ratio was determined by 1H NMR of the crude product. Enantiomeric excess was determined by chiral HPLC analysis. aPerformed at a concentration of 0.03 M. bFurther catalyst (10 mol%) was added after 24 h. | |
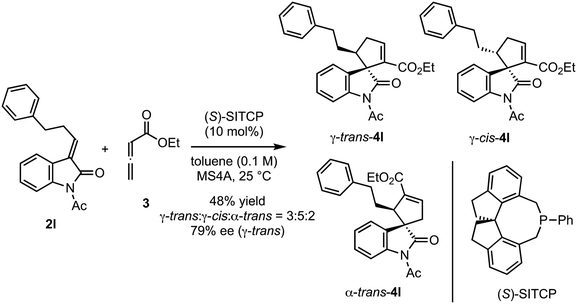 |
| | Scheme 5 Reaction of 2l with 3 in the presence of (S)-SITCP. | |
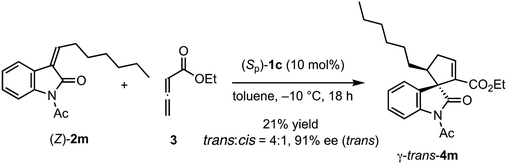 |
| | Scheme 6 Reaction of (Z)-2m with 3 in the presence of (Sp)-1c. | |
Scheme 7 shows the five-step catalytic cycle for the (3 + 2) annulation of allenoates and electron-deficient alkenes proposed in earlier reports.11,6b,23 The catalyst–allenoate complex is first formed by nucleophilic attack of the catalyst phosphorus on the sp-hybridized allenoate carbon (step 1). The cycloaddition proceeds stepwise to afford the ylide intermediate (steps 2 and 3), and the following proton transfer and elimination of the catalyst gives the final product (steps 4 and 5). The 1,4-addition of the γ-carbon of the anionic complex during step 2 gives the γ-adduct and the reaction of the α-anion produces the α-adduct. To establish the origin of the stereoselectivity, the DFT calculations were performed on the reaction pathway of steps 2 and 3 in the reaction of 2a (Scheme 7, R = Ph)24 because previous studies by other groups showed that the enantiodetermining step was step 2.23i,l Because we had obtained crystals of catalyst 1a suitable for X-ray diffraction (Fig. 5),25 the X-ray data of 1a were used as a reference for the initial structure for the catalyst conformation.
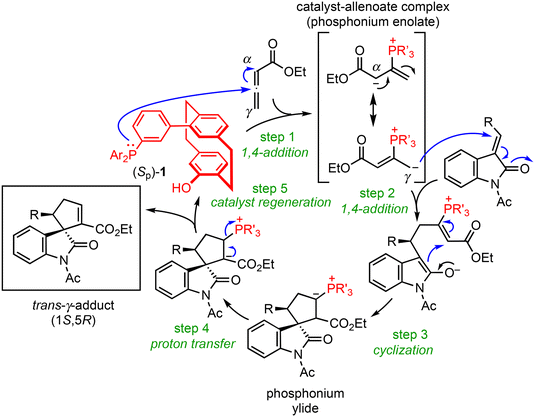 |
| | Scheme 7 Plausible mechanism for the (3 + 2) cyclization. | |
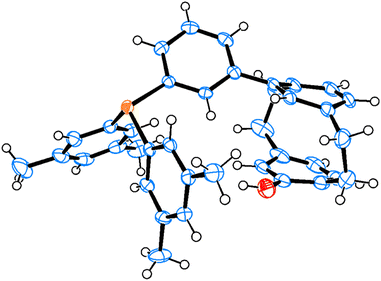 |
| | Fig. 5 X-ray crystal structure of 1a. | |
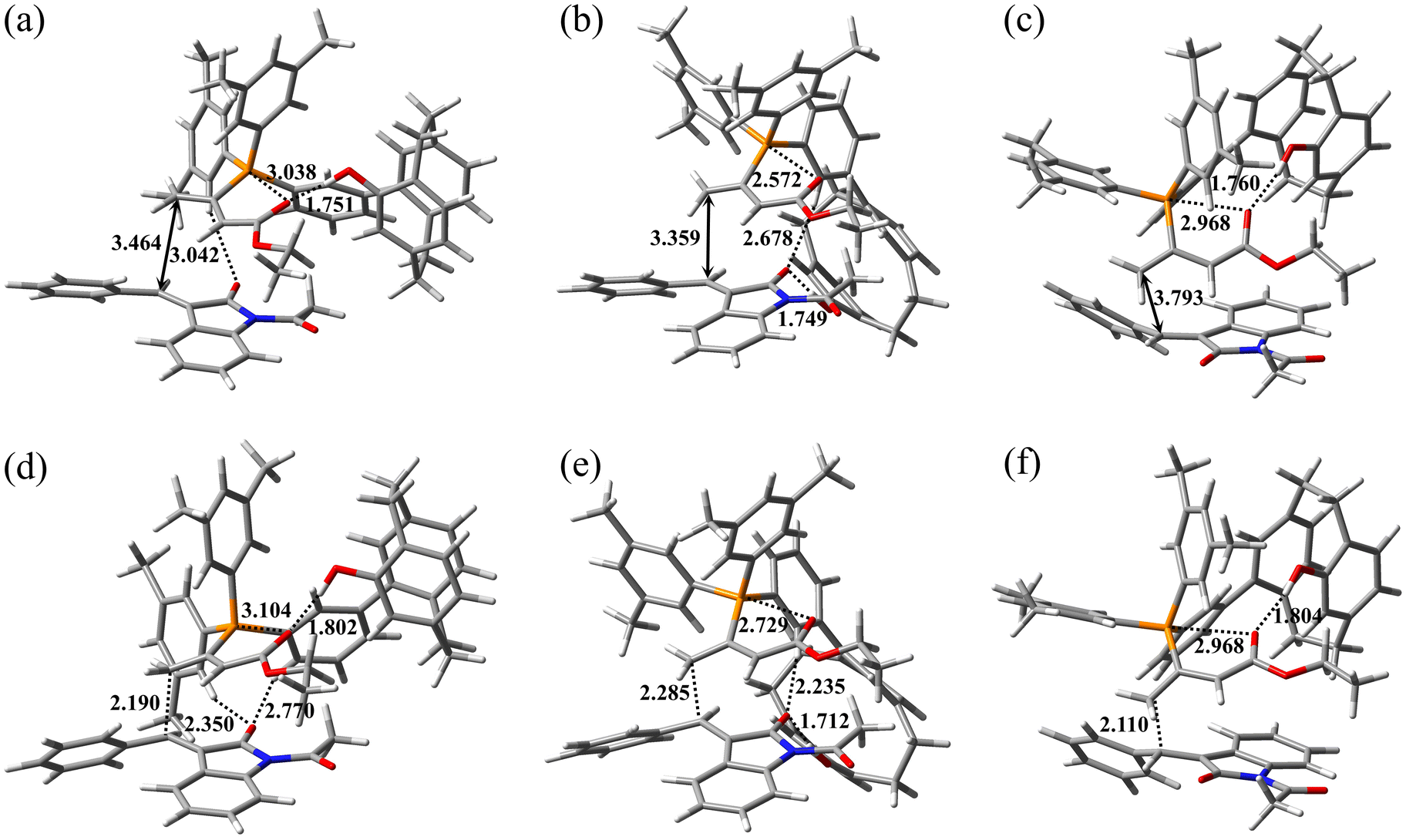
The optimized geometries of three assumed reactant complexes, RC-A, RC-B, and RC-C, stabilized by an intramolecular or intermolecular hydrogen bond are presented in Fig. 6a, b, and c, respectively. RC-A and RC-B are reactant complexes on the major enantiomer formation pathway, and RC-C is the reactant complex on the minor enantiomer formation pathway. The catalyst–allenoate complex was arranged on the re-face side of 2a in RC-A and RC-B, and on the si-face side of 2a in RC-C, leading to different enantiomer formation. The reaction pathways initiated by RC-A, RC-B, and RC-C were defined as pathways A, B, and C, respectively. The phenolic OH group intramolecularly formed hydrogen bonds with the carbonyl groups of the allenoate moiety (1.751 and 1.760 Å, respectively) in RC-A and RC-C, and intermolecularly formed hydrogen bonds with the carbonyl groups of 2a (1.749 Å) in RC-B. In these conformations, the phosphonium is near to the carbonyl oxygen of the allenoate moiety (3.038, 2.572, and 2.968 Å in RC-A, RC-B, and RC-C, respectively), allowing the formation of an electrostatic interaction between the two, which helps to stabilize the conformation of the catalyst–allenoate complex.23 In RC-A, RC-B, and RC-C, the distances between the γ carbon of the allenoate moiety in the catalyst–allenoate complex and the β carbon in 2a were 3.464, 3.359, and 3.793 Å, respectively. In RC-A, a weak CH⋯O hydrogen bond was observed between an acidic hydrogen of a xylyl group in the catalyst–allenoate complex and the carbonyl oxygen of 2a (3.042 Å). In RC-B, the carbonyl oxygen of 2a formed the CH⋯O hydrogen bonds with the spacer aryl group CH of the catalyst–allenoate complex in addition to the phenolic OH group. In contrast, the carbonyl oxygen of 2a did not form hydrogen bonds with the catalyst–allenoate complex in RC-C.
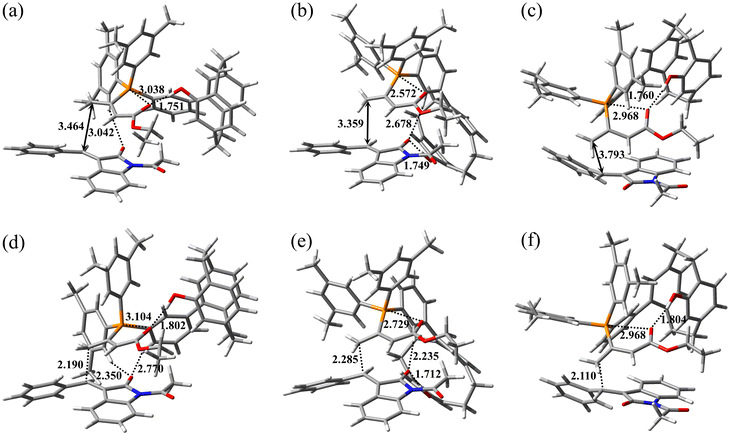 |
| | Fig. 6 Optimized geometries of (a) RC-A, (b) RC-B, (c) RC-C, (d) TS1-A, (e) TS1-B, and (f) TS1-C. Selected interatomic distances are shown in angstroms. The carbon, hydrogen, nitrogen, oxygen, and phosphorus atoms are shown in gray, white, blue, red, and orange, respectively. The cartesian coordinates of the atoms in these geometries are provided in separated xyz format file. | |
The calculated energy profile is presented in Fig. 7. The energy of RC-B was 6.09 kcal mol−1 higher than that of RC-A, and that of RC-C was 3.82 kcal mol−1 lower than that of RC-A. The energies of the transition states (TSs) of step 2 (TS1) were higher than those of the TSs of step 3 (TS2) for all pathways. The relative energies of TSs of pathways A-C (TS1-A, TS1-B, and TS1-C) with respect to RC-A were 5.99, 12.9, and 8.68 kcal mol−1, respectively.
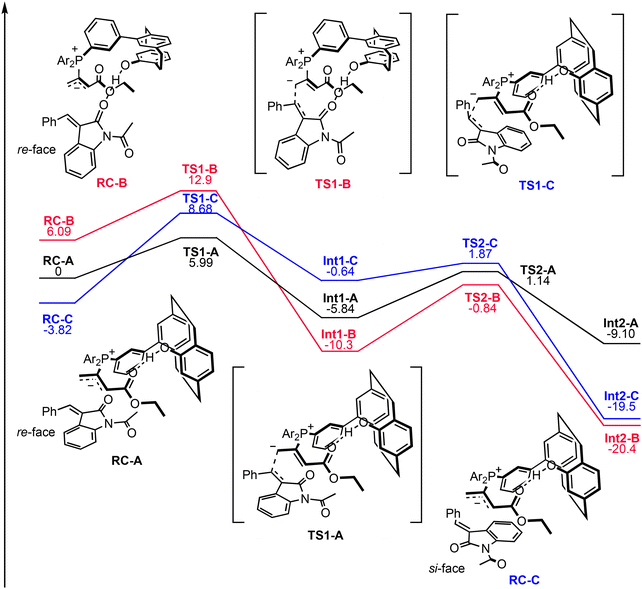 |
| | Fig. 7 Calculated energy profiles for pathways A–C. Pathways A, B, and C are shown in black, red, and blue, respectively. Relative energies are shown in kilocalories per mole (M06-2X/6-311+G(d)//B3LYP/6-31G(d)). | |
Optimized geometries of TS1-A, TS1-B, and TS1-C are presented in Fig. 6d, e, and f, respectively. In TS1-A, TS1-B, and TS1-C, the distances between the γ carbon of the allenoate moiety in the catalyst–allenoate complex and the β carbon in 2a were 2.190, 2.285, and 2.110 Å, respectively. During the formation of TS1-A and TS1-B from RC-A and RC-B, respectively, hydrogen bonds between the catalyst–allenoate complex and 2a were strengthened and formed. In general, the formation and strengthening of hydrogen bonds stabilizes the TS. In TS1-A, the CH⋯O hydrogen bond between the xylyl acidic hydrogen of catalyst–allenoate complex and carbonyl oxygen of 2a was strengthened, and the hydrogen bond distance was shortened to 2.350 Å. Additionally, a weak CH⋯O hydrogen bond was newly formed between the spacer aryl moiety of the catalyst–allenoate complex and carbonyl group of 2a (2.770 Å). In TS1-B, the two hydrogen bonds between the phenolic OH group of the catalyst–allenoate complex and the carbonyl group of 2a and between the spacer aryl moiety and carbonyl group of 2a were strengthened, with hydrogen bond distances of 1.712 and 2.235 Å, respectively. In contrast, during the formation of TS1-C from RC-C, no new hydrogen-bond formation occurred and the indolinone approached the pCp moiety, suggesting that steric repulsion between the two destabilizes TS1-C.
The energy of TS1-A was substantially lower than that of TS1-C (2.69 kcal mol−1), which could explain the experimentally observed enantioselectivity (82% ee at 0 °C, data not shown). Two strengthened CH⋯O hydrogen bonds were observed between the catalyst–allenoate complex and 2a in TS1-A; however no strengthened hydrogen bonds were observed in TS1-C. The importance of the strengthened hydrogen bonds between the catalyst–allenoate complex and substrate in controlling the enantioselectivities is noted in previous studies by other groups.23i,l However, the energy of TS1-B was substantially larger than those of TS1-A and TS1-C despite the strengthening of the two hydrogen bonds (one of which was a CH⋯O hydrogen bond). This computational result suggests that the formation of hydrogen bonds between the phenolic OH group and carbonyl group of 2a did not contribute greatly to the TS stabilization in our reaction system. The catalyst–allenoate complex was paced on the re-face side of 2a in TS1-A and TS1-B, and the carbonyl oxygen atom of the N-acetyl group in 2a faces in the opposite direction of the phenolic OH group of catalyst–allenoate complex. Conformational isomers with the carbonyl oxygen atom facing the phenolic OH group are also possible, and the formation of an additional hydrogen bond between the two was expected to contribute to stabilizing the TS. However, contrary to our intention, the energy of this TS was greater than that of TS1-A, and the additional hydrogen-bond formation did not dramatically stabilize the TS. Therefore, the main role of the phenolic OH group was probably to create the proper structure for substrate binding by forming an intramolecular hydrogen bond with the allenoate moiety23l to anchor the pCp in the appropriate position. As shown in Fig. 6, 2a was placed on the lower side of the catalyst–allenoate complex. The xylyl groups of catalyst–allenoate complex were on the upper side, and the steric hindrance of the xylyl groups prevented 2a from being placed on the upper side. That is, the xylyl groups could be involved in facilitating proper substrate binding to the catalyst–allenoate complex.
Conclusions
pCp-phosphine–phenol (Sp)-1c (R1 = H, R2 = t-Bu) catalyzed the (3 + 2) annulation of ethyl 2,3-butadienoate with various 3-methyleneindolinones to produce 2,5-disubstituted cyclopentene-fused C3-spirooxindoles in a highly regio-, diastereo-, and enantioselective manner. This catalyst could be applied to the reaction of not only benzylideneoxindoles but also alkylideneoxindoles, the enantioselective reaction of which has not been reported previously. DFT studies suggested that a phenolic OH group on the chiral pCp backbone interacted intramolecularly with the anionic oxygen atom of the phosphonium enolate by hydrogen bonding, creating a reaction space that resulted in high selectivities.
Experimental section
General information
Infrared spectra (IR) were measured in CHCl3 using a JASCO FT/IR-4100 spectrometer; absorptions are reported in reciprocal centimeters (cm−1). 1H NMR and 13C NMR spectra were recorded with a Bruker Avance III 600 spectrometer operating at 600 MHz (150 MHz for 13C NMR) at 25 °C with tetramethylsilane (δ = 0.0 ppm) as an internal standard. The data are reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet), integration, and coupling constant (Hz). 31P NMR spectra were recorded with 85% H3PO4 (δ = 0.0 ppm) as an external standard. High-resolution mass spectra were measured with a Thermo Scientific Exactive Plus Orbitrap. Analytical thin-layer chromatography was performed on MERCK silica gel, grade 60 F254. The spots and bands were detected under UV light (254 nm) and/or by staining with 5% phosphomolybdic acid followed by heating. Products were isolated by column chromatography using KANTO Sillica Gel 60 (230–400 mesh). HPLC analyses were performed using JASCO UV-2075 Plus and UV-4075 Intelligent UV/VIS detectors. The chiral columns used included Chiralpak IA-3, IB-3, and IC-3 (Daicel Chemical Industries, Ltd, 0.46 Φ × 25 cm). Optical rotations were measured on a JASCO P-2200 polarimeter.
Experimental details
General procedure for (3 + 2) annulation (Table 1: entry 11)
The following reaction was carried out under N2. Compound 3 (13.4 mg, 13.8 μL, 0.12 mmol) was added to a cooled (−30 °C) stirred solution of 2a (15.8 mg, 0.0601 mmol) and (Sp)-1c (4.3 mg, 6.0 × 10−3 mmol) in toluene (1.2 mL). After stirring at −30 °C for 24 h, the mixture was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (EtOAc/hexane, 1:10) to provide 4a (22.4 mg, 99%, γ/α = >20:1) as slightly yellow solid.
Ethyl (1S,5R)-1′-acetyl-2′-oxo-5-phenylspiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4a)11a
[α]25D = –95.6 (c = 0.425 in CHCl3) (lit.: [α]25D = −113 (c = 2.0 in CHCl3) for (1S,5R)-4a, >99% ee, γ/α = >95:5). IR 3031, 2940, 1753, 1708, 1635, 1606 cm−1. 1H NMR (600 MHz, C6D6): δ 0.68 (t, 3H, J = 7.2 Hz), 2.34 (ddd, 1H, J = 3.0, 7.8, 18.0 Hz), 2.67 (s, 3H), 2.69 (ddd, 1H, J = 1.8, 10.8, 18.0 Hz), 3.61–3.71 (m, 2H), 4.38 (dd, 1H, J = 7.8, 10.8 Hz), 6.66 (dd, 1H, J = 1.2, 7.8 Hz), 6.72 (dt, 1H, J = 1.2, 7.8 Hz), 6.77 (m, 1H), 6.82–6.87 (m, 5H), 6.97 (t, 1H, J = 2.4 Hz), 8.36 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.9, 26.8, 36.4, 56.3, 60.9, 65.2, 116.3, 123.9, 124.5, 127.4, 127.7 (2C), 127.9, 128.2 (2C), 128.7, 136.4, 138.4, 139.6, 146.6, 162.5, 170.8, 180.1. HRMS (DART) calcd for C23H22NO4 [M + H]+, 376.1543; found, 376.1542. HPLC analysis (column, Daicel Chiralpak IC-3, 2-propanol/hexane = 10:90, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 13.6 (major: γ-trans-4a), 42.0 (minor: ent-γ-trans-4a). Compound γ-trans-4a was determined to have 93% ee.
Ethyl (1S,5R)-1′-acetyl-2′-oxo-5-(4-(trifluoromethyl)phenyl)spiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4b)11a
Yellowish oil (99%, γ/α = 13:1): [α]25D = −72.9 (c = 0.995 in CHCl3) (lit.: [α]26D = −76 (c = 0.9 in CHCl3) for (1S,5R)-4b, 99% ee, γ/α = 85:15). IR 3032, 2991, 1751, 1715, 1647, 1620 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.07 (t, 3H, J = 7.0 Hz), 2.72 (s, 3H), 3.08 (ddd, 1H, J = 3.0, 7.8, 18.0 Hz), 3.26 (ddd, 1H, J = 1.8, 10.8, 18.0 Hz), 4.00 (q, 2H, J = 7.2 Hz), 4.36 (dd, 1H, J = 7.8, 10.8 Hz), 6.95 (dd, 1H, J = 1.2, 7.8 Hz), 7.01 (t, 1H, J = 7.2 Hz), 7.08 (d, 2H, J = 7.8 Hz), 7.16 (dt, 1H, J = 1.8, 7.2 Hz), 7.31–7.33 (m, 3H), 8.01 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.7, 36.4, 55.5, 60.9, 64.9, 116.5, 123.6, 123.8 (J = 271 Hz), 124.6, 125.1 (2C, J = 4 Hz), 127.3, 128.1 (2C), 129.1, 129.6 (J = 32 Hz), 138.5, 139.5, 140.6, 145.9, 162.2, 170.6, 179.7. HRMS (DART) calcd for C24H21F3NO4 [M + H]+, 444.1417; found, 444.1418; HPLC analysis (column, Daicel Chiralpak IB-3, 2-propanol/hexane = 5:95, flow rate = 0.5 mL min−1): λ = 220 nm, tR (min) = 17.9 (major), 22.9 (minor). Compound γ-trans-4b was determined to have 91% ee.
Ethyl (1S,5R)-1′-acetyl-5-(4-bromophenyl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4c)11a
Yellowish oil (81%, γ/α = 13:1): [α]25D = −67.4 (c = 0.93 in CHCl3) (lit.: [α]26D = −60 (c = 1.1 in CHCl3) for (1S,5R)-4c, >99% ee, γ/α = 90:10). IR 3033, 2992, 1750, 1715, 1635 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.06 (t, 3H, J = 7.2 Hz), 2.71 (s, 3H), 3.03 (ddd, 1H, J = 3.0, 7.8, 18.0 Hz), 3.19 (ddd, 1H, J = 2.4, 10.8, 18.0 Hz), 4.00 (q, 2H, J = 7.2 Hz), 4.26 (dd, 1H, J = 7.8, 10.8 Hz), 6.83 (d, 2H, J = 8.4 Hz), 6.94 (dd, 1H, J = 1.2, 7.8 Hz), 7.03 (dt, 1H, J = 1.2, 7.8 Hz), 7.14–7.18 (m, 3H), 7.31 (dd, 1H, J = 1.8, 3.0 Hz), 8.01 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.7, 36.5, 55.5, 60.9, 64.9, 116.5, 121.3, 123.7, 124.6, 127.5, 129.0, 129.4 (2C), 131.3 (2C), 135.5, 138.5, 139.5, 146.1, 162.3, 170.6, 179.8. HRMS (DART) calcd for C23H21BrNO4 [M + H]+, 454.0648; found, 454.0649; HPLC analysis (column, Daicel Chiralpak IC-3, 2-propanol/hexane = 2:98, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 28.5 (major), 80.6 (minor). Compound γ-trans-4c was determined to have 90% ee.
Ethyl (1S,5R)-1′-acetyl-5-(biphenyl-4-yl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4d)11a
Yellow solid (99%, γ/α = 19:1): [α]25D = −50.4 (c = 0.128 in CHCl3) (lit.: [α]22D = −159 (c = 0.5 in CHCl3) for (1S,5R)-4d, 99% ee, γ/α = 90:10). IR 3028, 2995, 2948, 1750, 1715, 1603 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.08 (t, 3H, J = 7.2 Hz), 2.75 (s, 3H), 3.07 (ddd, 1H, J = 3.0, 7.8, 18.0 Hz), 3.29 (ddd, 1H, J = 1.8, 10.2, 18.0 Hz), 4.01 (q, 2H, J = 7.2 Hz), 4.37 (dd, 1H, J = 7.8, 10.2 Hz), 6.98–7.02 (m, 2H), 7.03 (d, 2H, J = 8.4 Hz), 7.12 (dt, 1H, J = 1.8, 8.4 Hz), 7.27–7.37 (m, 6H), 7.45 (d, 2H, J = 8.4 Hz), 8.01 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.7, 36.6, 55.9, 60.8, 65.0, 116.3, 123.8, 124.4, 126.7, 126.8, 127.3, 127.8, 128.1, 128.6, 128.7, 135.4, 138.4, 139.6, 140.0, 140.3, 146.4, 162.3, 170.7, 180.0. HRMS (DART) calcd for C29H26NO4 [M + H]+, 452.1856; found, 452.1859; HPLC analysis (column, Daicel Chiralpak IA-3, 2-propanol/hexane = 5:95, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 16.9 (minor), 18.0 (major). Compound γ-trans-4d was determined to have 92% ee.
Ethyl (1S,5R)-1′-acetyl-2′-oxo-5-(p-tolyl)spiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4e)11a
Yellowish oil (99%, γ/α = >20:1): [α]25D = −144 (c = 0.152 in CHCl3) (lit.: [α]22D = −102 (c = 2.0 in CHCl3) for (1S,5R)-4e, 97% ee, γ/α = 90:10). IR 3030, 2946, 1750, 1707, 1603 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.06 (t, 3H, J = 7.2 Hz), 2.15 (s, 3H), 2.71 (s, 3H), 3.00 (ddd, 1H, J = 3.0, 7.8, 18.0 Hz), 3.22 (ddd, 1H, J = 2.4, 10.2, 18.0 Hz), 3.99 (q, 2H, J = 7.2 Hz), 4.29 (dd, 1H, J = 7.8, 10.2 Hz), 6.82–6.85 (m, 4H), 6.96 (dd, 1H, J = 1.2, 7.8 Hz), 7.00 (dt, 1H, J = 1.2, 7.8 Hz), 7.12 (dt, 1H, J = 1.2, 7.8 Hz), 7.32 (dd, 1H, J = 2.4, 3.0 Hz), 7.99 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 20.9, 26.7, 36.6, 56.0, 60.7, 65.0, 116.2, 123.8, 124.3, 127.6 (2C), 127.9, 128.6, 128.8 (2C), 133.2, 136.9, 138.4, 139.5, 146.5, 162.4, 170.7, 180.1. HRMS (DART) calcd for C24H24NO4 [M + H]+, 390.1700; found, 390.1698; HPLC analysis (column, Daicel Chiralpak IB-3, 2-propanol/hexane = 2:98, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 9.6 (major), 11.9 (minor). Compound γ-trans-4e was determined to have 92% ee.
Ethyl (1S,5R)-1′-acetyl-2′-oxo-5-(p-anisyl)spiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4f)
Yellowish oil (99%, γ/α = >20:1): [α]25D = −87.8 (c = 0.934 in CHCl3). IR 3033, 2939, 1750, 1706, 1653, 1635 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.06 (t, 3H, J = 7.2 Hz), 2.71 (s, 3H), 3.00 (ddd, 1H, J = 3.0, 8.4, 18.6 Hz), 3.18 (ddd, 1H, J = 2.4, 10.8, 18.6 Hz), 3.65 (s, 3H), 3.99 (q, 2H, J = 7.2 Hz), 4.26 (dd, 1H, J = 8.4, 10.8 Hz), 6.57 (d, 2H, J = 8.4 Hz), 6.87 (d, 1H, J = 8.4 Hz), 6.95 (dd, 2H, J = 1.2, 7.8 Hz), 7.01 (dt, 1H, J = 0.6, 7.8 Hz), 7.14 (dt, 1H, J = 1.2, 8.4 Hz), 7.32 (dd, 1H, J = 2.4, 3.0 Hz), 7.99 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.7, 36.7, 55.0, 55.7, 60.7, 65.0, 113.4 (2C), 116.2, 123.7, 124.4, 127.9, 128.3, 128.6, 128.7 (2C), 138.3, 139.5, 146.5, 158.6, 162.4, 170.7, 180.1. HRMS (DART) calcd for C24H24NO5 [M + H]+, 406.1649; found, 406.1649; HPLC analysis (column, Daicel Chiralpak IB-3, 2-propanol/hexane = 2:98, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 15.5 (major), 18.2 (minor). Compound γ-trans-4f was determined to have 89% ee.
Ethyl (1S,5R)-1′-acetyl-5-(3-bromophenyl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4g)11a
Yellowish oil (99%, γ/α = 16:1): [α]25D = −62.6 (c = 1.20 in CHCl3) (lit.: [α]25D = −56 (c = 1.2 in CHCl3) for (1S,5R)-4g, >99% ee, γ/α = 85:15). IR 3034, 2941, 1750, 1715, 1653, 1635 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.07 (t, 3H, J = 7.2 Hz), 2.72 (s, 3H), 3.03 (ddd, 1H, J = 3.0, 7.8, 18.0 Hz), 3.20 (ddd, 1H, J = 2.4, 10.2, 18.0 Hz), 4.00 (q, 2H, J = 7.2 Hz), 4.26 (dd, 1H, J = 7.8, 10.2 Hz), 6.84 (d, 1H, J = 7.8 Hz), 6.91 (t, 1H, J = 7.8 Hz), 6.94 (dd, 1H, J = 1.2, 7.8, Hz), 7.05 (dt, 1H, J = 1.0, 7.5 Hz), 7.12–7.14 (m, 1H), 7.16 (dt, 2H, J = 1.0, 7.8 Hz), 7.31 (t, 1H, J = 2.4 Hz), 8.01 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.7, 36.3, 55.6, 60.9, 65.0, 116.4, 122.2, 123.7, 124.6, 126.4, 127.3, 129.0, 129.7, 130.5, 130.9, 138.3, 138.8, 139.5, 146.1, 162.2, 170.6, 179.7. HRMS (DART) calcd for C23H21BrNO4 [M + H]+, 454.0648; found, 454.0645; HPLC analysis (column, Daicel Chiralpak IA-3, 2-propanol/hexane = 5:95, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 12.4 (major), 16.6 (minor). Compound γ-trans-4g was determined to have 92% ee.
Ethyl (1S,5S)-1′-acetyl-5-(2-chlorophenyl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4h)
Yellowish oil (99%, γ/α = >20:1): [α]25D = −60.8 (c = 0.178 in CHCl3). IR 3025, 2948, 1760, 1714, 1603 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.00 (t, 3H, J = 7.2 Hz), 2.70 (s, 3H), 3.17 (ddd, 1H, J = 2.4, 10.2, 19.2 Hz), 3.22 (ddd, 1H, J = 3.0, 9.0, 19.2 Hz), 3.94–3.99 (m, 2H), 4.81 (t, 1H, J = 8.4 Hz), 6.68 (dd, 1H, J = 1.2, 7.8 Hz), 6.88 (dt, 1H, J = 1.2, 7.8 Hz), 6.99 (dt, 1H, J = 1.8, 7.8 Hz), 7.02 (dt, 1H, J = 1.8, 7.8 Hz), 7.10–7.13 (m, 2H), 7.19 (dd, 1H, J = 1.2, 7.8 Hz), 7.36 (dd, 1H, J = 2.4, 3.0 Hz), 8.03 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 12.7, 25.6, 37.0, 49.4, 59.7, 63.7, 115.2, 122.9, 123.2, 125.1, 126.7, 127.4, 127.6, 128.5 (2C), 133.7, 134.3, 136.0, 139.2, 145.9, 161.4, 169.8, 178.6. HRMS (DART) calcd for C23H21ClNO4 [M + H]+, 410.1154; found, 410.1153; HPLC analysis (column, Daicel Chiralpak IB-3, 2-propanol/hexane = 2:98, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 13.4 (major), 15.8 (minor). Compound γ-trans-4h was determined to have 90% ee.
Ethyl (1S,5R)-1′-acetyl-5-(naphthalene-1-yl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4i)11a,c
White solid (93%, γ/α = >20:1): [α]25D = −373 (c = 0.215 in CHCl3) (lit.: ref. 11a, [α]25D = −445 (c = 1.3 in CHCl3) for (1S,5R)-4i, >99% ee, γ/α = >95:5; ref. 11c, [α]25D = +211.9 (c = 0.5 in CHCl3) for 98% ee with use of (R)-SITCP). IR 3055, 2941, 1749, 1715, 1653, 1636 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.04 (t, 3H, J = 7.2 Hz), 2.58 (s, 3H), 3.19 (ddd, 1H, J = 3.0 8.4, 18.6 Hz), 3.43 (ddd, 1H, J = 1.8, 9.6, 18.6 Hz), 4.00 (q, 2H, J = 7.2 Hz), 5.22 (dd, 1H, J = 8.4, 9.0 Hz), 6.94 (d, 2H, J = 4.2 Hz), 6.98–7.01 (m, 1H), 7.17 (t, 1H, J = 7.8 Hz), 7.31 (d, 1H, J = 7.2 Hz), 7.37 (t, 1H, J = 7.2 Hz), 7.41–7.45 (m, 2H), 7.54 (d, 1H, J = 7.8 Hz), 7.69 (dd, 1H, J = 1.2, 8.4 Hz), 7.76 (d, 1H, J = 8.4 Hz), 8.03 (d, 1H, J = 9.0 Hz). 13C NMR (150 MHz, CDCl3): δ 12.7, 25.5, 37.2, 49.0, 59.7, 64.2, 115.1, 121.6, 123.09, 123.13, 123.3, 124.5, 125.06, 125.08, 126.9, 127.1, 127.4, 127.9, 130.9, 131.4, 132.5, 136.5, 138.4, 146.0, 161.5, 169.3, 179.4. HRMS (DART) calcd for C27H24NO4 [M + H]+, 426.1700; found, 426.1702; HPLC analysis (column, Daicel Chiralpak IA-3, 2-propanol/hexane = 2:98, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 18.3 (major), 23.9 (minor). Compound γ-trans-4i was determined to have 86% ee.
Ethyl (1S,5S)-1′-acetyl-5-(furan-2-yl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4j)11a
Yellow oil (99%, γ/α = 5.3:1): [α]25D = –67.2 (c = 1.12 in CHCl3). IR 3032, 2987, 1751, 1708, 1635 cm−1. 1H NMR (CDCl3, 600 MHz): δ 1.06 (t, 3H, J = 7.2 Hz), 2.75 (s, 3H), 3.10 (ddd, 1H, J = 3.0, 9.0, 18.0 Hz), 3.16 (ddd, 1H, J = 2.4, 10.2, 18.0 Hz), 3.98 (q, 2H, J = 7.2 Hz), 4.32 (dd, 1H, J = 9.0, 9.6 Hz), 5.90 (dd, 1H, J = 1.2, 2.4 Hz), 6.01 (dd, 1H, J = 2.4, 3.0 Hz), 6.88 (dd, 1H, J = 1.2, 7.2 Hz), 6.95–6.98 (m, 2H), 7.18 (dt, 1H, J = 1.2, 7.8 Hz), 7.24 (t, 1H, J = 2.4 Hz), 8.11 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.7, 35.6, 49.0, 60.8, 63.8, 106.9, 109.8, 116.1, 123.6, 124.4, 127.6, 128.7, 138.3, 139.9, 142.0, 145.8, 151.7, 162.2, 170.9, 179.7. HRMS (DART) calcd for C21H20NO5 [M + H]+, 366.1336; found, 366.1338; HPLC analysis (column, Daicel Chiralpak IC-3, 2-propanol/hexane = 10:90, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 11.9 (major), 25.8 (minor). Compound γ-trans-4j was determined to have 85% ee.
Ethyl (1S,5S)-1′-acetyl-2′-oxo-5-(quinolin-2-yl)spiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4k)11a
Yellowish oil (99%, γ/α = 16:1): [α]25D = −37.0 (c = 1.27 in CHCl3) (lit.: [α]25D = −15 (c = 0.6 in CHCl3) for (1S,5S)-4k, 97% ee, γ/α = 90:10). IR 3020, 2989, 1750, 1705, 1635, 1600 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.09 (t, 3H, J = 7.0 Hz), 2.85 (s, 3H), 3.21 (ddd, 1H, J = 3.6, 8.4, 18.6 Hz), 3.81 (ddd, 1H, J = 1.8, 10.2, 18.6 Hz), 4.02 (q, 2H, J = 7.2 Hz), 4.70 (dd, 1H, J = 8.4, 10.2 Hz), 6.74 (dt, 1H, J = 1.2, 7.8 Hz), 6.81 (dd, 1H, J = 1.2, 7.2 Hz), 6.95–6.98 (m, 2H), 7.37–7.41 (m, 2H), 7.58 (dt, 1H, J = 1.2, 8.4 Hz), 7.61 (d, 1H, J = 8.4 Hz), 7.74 (d, 1H, J = 8.4 Hz), 7.85 (d, 1H, J = 8.4 Hz), 8.02 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.8, 34.8, 57.3, 60.8, 64.1, 115.9, 119.3, 123.6, 124.2, 126.1, 126.5, 127.3, 128.2, 128.3, 129.0, 129.5, 136.2, 138.4, 140.1, 146.5, 147.3, 157.1, 162.4, 171.1, 180.5. HRMS (DART) calcd for C26H23N2O4 [M + H]+, 427.1652; found, 427.1650; HPLC analysis (column, Daicel Chiralpak IC-3, 2-propanol/hexane = 10:90, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 17.0 (major), 41.5 (minor). Compound γ-trans-4k was determined to have 90% ee.
Ethyl (1S,5S)-1′-acetyl-2′-oxo-5-(3-phenylpropyl)spiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4l)
Yellow oil: [α]24D = −48.2 (c = 0.106 in CHCl3). IR 3030, 2938, 1755, 1707, 1604 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.06 (t, 3H, J = 7.2 Hz), 1.40 (m, 1H), 1.55 (m, 1H), 2.25 (ddd, 1H, J = 5.4, 10.8, 13.8 Hz), 2.43 (ddd, 1H, J = 5.4, 10.8, 13.2 Hz), 2.51 (ddd, 1H, J = 1.8, 9.6, 18.0 Hz), 2.71 (s, 3H), 2.93 (ddd, 1H, J = 3.0, 8.4, 18.0 Hz), 3.06 (m, 1H), 3.98 (q, 2H, J = 7.2 Hz), 6.88 (d, 2H, J = 7.2 Hz), 7.01 (dd, 1H, J = 0.6, 7.2 Hz), 7.12 (t, 1H, J = 7.8 Hz), 7.15–7.19 (m, 4H), 7.34 (dt, 1H, J = 1.2, 7.8 Hz), 8.28 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 14.2, 26.9, 31.2, 34.6, 44.8, 55.8, 56.9, 60.6, 116.4, 121.4, 125.89, 125.92, 128.29 (2C), 128.31, 128.5, 135.5, 138.1, 138.2, 140.4 (2C), 141.3, 163.9, 170.8, 179.3. HRMS (DART) calcd for C25H26NO4 [M + H]+, 404.1856; found, 404.1855; HPLC analysis (column, Daicel Chiralpak IB-3, 2-propanol/hexane = 1.5:98.5, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 12.4 (major), 17.9 (minor). Compound γ-trans-4l was determined to have 91% ee.
γ-cis-4l.
Yellow oil: IR 3030, 2938, 1753, 1711, 1634, 1605 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.32 (t, 3H, J = 7.2 Hz), 1.52–1.59 (m, 1H), 1.86 (ddd, 1H, J = 4.8, 12.0, 13.2 Hz), 2.15 (ddd, 1H, J = 6.6, 7.2, 13.2 Hz), 2.35 (m, 1H), 2.68 (s, 3H), 2.77 (td, 1H, J = 2.4, 18.6 Hz), 3.12 (td, 1H, J = 2.4, 18.6 Hz), 3.59 (m, 1H), 4.18–4.27 (m, 2H), 6.72 (d, 2H, J = 7.8 Hz), 6.82 (q, 1H, J = 2.4 Hz), 7.08 (t, 1H, J = 7.8 Hz), 7.13 (t, 2H, J = 7.8 Hz), 7.23 (dt, 1H, J = 0.6, 7.2 Hz), 7.32 (dd 1H, J = 0.6, 7.2 Hz), 7.41 (dt, 1H, J = 1.2, 7.8 Hz), 8.31 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 14.3, 26.7, 31.1, 33.8, 45.4, 53.9, 57.0, 60.6, 116.7, 123.8, 125.2, 125.9, 128.16 (2C), 128.24 (2C), 128.8, 130.8, 138.1, 139.5, 140.0, 141.1, 164.2, 170.9, 181.7. HRMS (DART) calcd for C25H26NO4 [M + H]+, 404.1856; found, 404.1855.
α-trans-4l.
Yellow oil: IR 3026, 2928, 1753, 1710, 1639, 1604 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.30 (t, 3H, J = 7.2 Hz), 2.12–2.19 (m, 2H), 2.36–2.41 (m, 2H), 2.72 (s, 3H), 2.76 (dd, 1H, J = 3.0, 18.0 Hz), 3.25 (td, 1H, J = 3.0, 18.0 Hz), 3.27–3.29 (m, 1H), 4.19–4.26 (m, 2H), 6.90 (m, 1H), 7.00 (d, 2H, J = 7.8 Hz), 7.12 (t, 1H, J = 7.8 Hz), 7.16–7.23 (m, 4H), 7.33 (dt, 1H, J = 1.8, 7.8 Hz), 8.24 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 14.2, 26.9, 31.2, 34.6, 44.8, 55.8, 56.9, 60.6, 116.4, 121.4, 125.89, 125.93, 128.29 (2C), 128.32 (2C), 128.5, 135.5, 138.1, 138.2, 140.4, 141.3, 163.9, 170.8, 179.3. HRMS (DART) calcd for C25H26NO4 [M + H]+, 404.1856; found, 404.1857.
Ethyl (1S,5S)-1′-acetyl-5-hexyl-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4m)
Yellow oil: [α]24D = −72.1 (c = 0.876 in CHCl3). IR 3032, 2930, 1755, 1707 cm−1. 1H NMR (600 MHz, CDCl3): δ 0.79 (t, 3H, J = 7.2 Hz), 0.95–1.17 (m, 13H), 2.47 (ddd, 1H, J = 2.4, 10.2, 18.6 Hz), 2.71 (s, 3H), 2.89 (ddd, 1H, J = 3.0, 7.8, 18.6 Hz), 2.99 (quint, 1H, J = 7.8 Hz), 3.97 (q, 2H, J = 7.2 Hz), 6.98 (dd, 1H, J = 1.2, 7.8 Hz), 7.14 (dt, 1H, J = 1.2, 7.8 Hz), 7.18 (t, 1H, J = 2.4 Hz), 7.32 (dt, 1H, J = 1.2, 7.8 Hz), 8.26 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.7, 13.9, 22.3, 26.6, 27.6, 29.0, 30.6, 31.4, 38.6, 50.4, 60.6, 63.0, 116.6, 123.7, 124.6, 128.3, 128.7, 139.0, 139.9, 146.7, 162.5, 171.0, 180.5. HRMS (DART) calcd for C23H30NO4 [M + H]+, 384.2169; found, 384.2169; HPLC analysis (column, Daicel Chiralpak IA-3, 2-propanol/hexane = 1:99, flow rate = 1.0 mL min−1): λ = 220 nm, tR (min) = 11.3 (major), 19.1 (minor). Compound γ-trans-4m was determined to have 92% ee.
Ethyl (1S,5R)-1′-acetyl-5-(hept-1-yn-1-yl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4n)11a
Yellow oil: [α]24D = −23.1 (c = 0.609 in CHCl3) (lit.: [α]25D = −19 (c = 1.7 in CHCl3) for (1S,5R)-4n, 86% ee, γ/α = 80:20). IR 2934, 1756, 1708, 1635 cm−1. 1H NMR (CDCl3, 600 MHz): δ 0.80 (t, 3H, J = 7.2 Hz), 0.95–0.99 (m, 2H), 1.02–1.08 (m, 5H), 1.11–1.15 (m, 2H), 1.82–1.84 (m, 2H), 2.71 (s, 3H), 2.86 (ddd, 1H, J = 1.8, 10.2, 18.0 Hz), 3.07 (ddd, 1H, J = 3.0, 8.4, 18.0 Hz), 3.77 (m, 1H), 3.95–4.10 (m, 2H), 7.06 (dd, 1H, J = 1.2, 7.8 Hz), 7.12–7.18 (m, 2H), 7.31 (dt, 1H, J = 1.2, 7.8 Hz), 8.26 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 13.9, 18.3, 22.1, 26.7, 28.0, 30.5, 39.6, 41.7, 60.8, 64.0, 76.4, 86.2, 116.4, 123.7, 124.6, 128.5, 128.8, 138.1, 140.0, 145.7, 162.2, 170.9, 179.4. HRMS (DART) calcd for C24H28NO4 [M + H]+, 394.2013; found, 394.2013; HPLC analysis (column, Daicel Chiralpak IC-3, 2-propanol/hexane = 1:99, flow rate = 1.0 mL min−1): λ = 205 nm, tR (min) = 16.4 (major), 39.8 (minor). Compound γ-trans-4n was determined to have 92% ee.
Ethyl (1S,5S)-1′-acetyl-5-(3-methyl-3-nitrobutyl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4o)
Yellow oil: [α]24D = −89.5 (c = 0.74 in CHCl3). IR 3031, 2934, 1756, 1706, 1635, 1541 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.07 (t, 3H, J = 7.2 Hz), 1.09–1.36 (m, 9H), 1.85 (dt, 1H, J = 4.2, 13.2 Hz), 2.49 (m, 1H), 2.72 (s, 3H), 2.90–2.98 (m, 2H), 3.98 (q, 2H, J = 7.2 Hz), 6.97 (d, 1H, J = 7.2 Hz), 7.15–7.18 (m, 2H), 7.35 (dt, 1H, J = 1.2, 7.8 Hz), 8.27 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 24.4, 25.3, 26.6, 31.3, 38.6, 38.7, 49.4, 60.8, 62.8, 87.4, 116.7, 123.8, 124.9, 127.6, 129.1, 139.0, 139.9, 146.1, 162.3, 170.9, 180.2. HRMS (DART) calcd for C22H27N2O6 [M + H]+, 415.1864; found, 415.1865; HPLC analysis (column, Daicel Chiralpak IB-3, 2-propanol/hexane = 1.5:98.5, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 15.2 (major), 23.9 (minor). Compound γ-trans-4o was determined to have 90% ee.
Ethyl (1S,5R)-1′-acetyl-2′-oxo-5-((E)-styryl)spiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4p)
Yellow oil (99%, trans/cis = 5:1): [α]24D = 22.5 (c = 0.584 in CHCl3). IR 3032, 2986, 1750, 1708, 1635 cm−1. 1H NMR (600 MHz, CDCl3): δ 1.08 (t, 3H, J = 7.2 Hz), 2.70 (s, 3H), 2.86 (ddd, 1H, J = 1.8, 9.6, 18.6 Hz), 2.97 (ddd, 1H, J = 2.4, 7.8, 18.0 Hz), 3.79 (q, 1H, J = 9.0 Hz), 4.00 (m, 2H), 5.71 (dd, 1H, J = 9.0, 15.6 Hz), 6.38 (d, 1H, J = 15.6 Hz), 7.02 (d, 2H, J = 6.6 Hz), 7.08 (dd, 1H, J = 1.2, 7.8 Hz), 7.15–7.20 (m, 4H), 7.24 (t, 1H, J = 2.4 Hz), 7.31 (dt, 1H, J = 1.2, 7.8 Hz), 8.22 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 26.7, 38.5, 53.9, 60.8, 64.2, 116.8, 123.3, 124.8, 126.27, 126.34, 126.4, 127.7, 128.5, 129.0, 132.7, 136.4, 138.8, 139.8, 146.4, 162.3, 170.8, 179.7. HRMS (DART) calcd for C25H24NO4 [M + H]+, 402.1700; found, 402.1699; HPLC analysis (column, Daicel Chiralpak IA-3, 2-propanol/hexane = 1.5:98.5, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 15.2 (major), 22.7 (minor). Compound γ-trans-4p was determined to have 85% ee.
Ethyl (1S,5S)-1′-acetyl-5-((Z)-hex-3-en-1-yl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4q)
Yellow oil: [α]24D = −89.8 (c = 0.299 in CHCl3). IR 3019, 1749, 1706, 1653, 1636 cm−1. 1H NMR (600 MHz, CDCl3): δ 0.84 (t, 3H, J = 7.2 Hz), 1.06 (t, 3H, J = 7.2 Hz), 1.08–1.14 (m, 1H), 1.24–1.30 (m, 1H), 1.72–1.84 (m, 4H), 2.50 (ddd, 1H, J = 2.4, 10.2, 18.6 Hz), 2.71 (s, 3H), 2.91 (ddd, 1H, J = 3.0, 7.8, 18.6 Hz), 3.02 (m, 1H), 3.98 (q, 2H, J = 7.2 Hz), 5.10 (m, 1H), 5.27 (m, 1H), 6.99 (dd, 1H, J = 1.2, 7.8 Hz), 7.15 (dt, 1H, J = 1.2, 7.2 Hz), 7.18 (t, 1H, J = 2.4 Hz), 7.32 (dt, 1H, J = 1.2, 7.8 Hz), 8.27 (d, 1H, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3): δ 13.8, 14.2, 20.2, 25.3, 26.7, 30.9, 38.6, 50.1, 60.7, 63.0, 116.6, 123.7, 124.7, 127.5, 128.2, 128.8, 132.4, 139.0, 140.0, 146.6, 162.4, 171.0, 180.4. HRMS (DART) calcd for C23H28NO4 [M + H]+, 382.2013; found, 382.2012; HPLC analysis (column, Daicel Chiralpak IB-3, 2-propanol/hexane = 1.5:98.5, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 7.6 (major), 13.5 (minor). Compound γ-trans-4q was determined to have 92% ee.
Diethyl (1S,5S)-1′-acetyl-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2,5-dicarboxylate (γ-trans-4r)
White solid (90%, trans/cis = 2.3:1): [α]26D = −59.2 (c = 0.603 in CHCl3). IR 3033, 2986, 1714, 1636 cm−1. 1H NMR (600 MHz, CDCl3): δ 0.71 (t, 3H, J = 7.2 Hz), 1.08 (t, 3H, J = 7.2 Hz), 2.74 (s, 3H), 2.92 (ddd, 1H, J = 3.0, 9.0, 19.2 Hz), 3.33 (ddd, 1H, J = 2.4, 9.6, 19.2 Hz), 3.60–3.71 (m, 2H), 3.93 (t, 1H, J = 9.6 Hz), 3.99 (q, 2H, J = 7.2 Hz), 6.99 (dd, 1H, J = 1.2, 7.8 Hz), 7.11 (dt, 1H, J = 1.2, 7.8 Hz), 7.16 (t, 1H, J = 2.4 Hz), 7.31 (dt, 1H, J = 1.2, 7.8 Hz), 8.23 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.3, 13.8, 26.6, 33.5, 54.0, 60.8, 60.9, 61.0, 116.4, 122.9, 125.0, 129.2, 129.3, 137.3, 140.4, 145.8, 161.9, 169.6, 170.9, 179.3. HRMS (DART) calcd for C20H22NO6 [M + H]+, 372.1442; found, 372.1443; HPLC analysis (column, Daicel Chiralpak IC-3, 2-propanol/hexane = 3:97, flow rate = 0.5 mL min−1): λ = 230 nm, tR (min) = 13.1 (major), 14.6 (minor). Compound γ-trans-4r was determined to have 91% ee.
Ethyl (1S,5R)-1′-acetyl-5-cyclohexyl-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4s)
Yellow oil: [α]23D = −98.3 (c = 0.596 in CHCl3). IR 3030, 2933, 1756, 1706, 1635 cm−1. 1H NMR (600 MHz, CDCl3): δ 0.66–0.75 (m, 2H), 0.92–1.12 (m, 7H), 1.22 (m, 1H), 1.40 (m, 1H), 1.49 (m, 1H), 1.64–1.70 (m, 2H), 2.56 (ddd, 1H, J = 2.4, 9.6, 17.4 Hz), 2.71 (s, 3H), 2.78–2.89 (m, 2H), 3.96 (dq, 2H, J = 1.2, 7.2 Hz), 7.02 (dd, 1H, J = 1.2, 7.8 Hz), 7.13–7.16 (m, 2H), 7.32 (dt, 1H, J = 1.2, 7.8 Hz), 8.26 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.7, 25.4, 25.7, 25.9, 26.7, 30.7, 31.6, 37.2, 38.8, 56.2, 60.6, 62.0, 116.5, 123.8, 124.6, 128.3, 128.7, 139.6, 140.0, 146.0, 162.4, 171.2, 180.6. HRMS (DART) calcd for C23H28NO4 [M + H]+, 382.2013; found, 382.2015; HPLC analysis (column, Daicel Chiralpak IA-3, 2-propanol/hexane = 1.5:98.5, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 8.5 (major), 12.7 (minor). Compound γ-trans-4s was determined to have 90% ee.
Ethyl (1S,5R)-1′-acetyl-5-(tert-butyl)-2′-oxospiro[cyclopentane-1,3′-indolin]-2-ene-2-carboxylate (γ-trans-4t)
Yellow oil: [α]25D = −71.7 (c = 0.165 in CHCl3). IR 3024, 2951, 1758, 1705, 1603 cm−1. 1H NMR (600 MHz, CDCl3): δ 0.73 (s, 9H), 1.03 (t, 3H, J = 7.2 Hz), 2.71 (s, 3H), 2.74 (ddd, 1H, J = 3.0, 7.8, 18.0 Hz), 2.82 (ddd, 1H, J = 2.4, 11.4, 18.0 Hz), 2.99 (dd, 1H, J = 7.8, 11.4 Hz), 3.94 (dq, 2H, J = 2.4, 7.2 Hz), 7.05 (dd, 1H, J = 1.2, 7.2 Hz), 7.09 (dt, 1H, J = 1.2, 7.2 Hz), 7.23 (t, 1H, J = 2.4 Hz), 7.31 (dt, 1H, J = 1.8, 7.8 Hz), 8.26 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 13.9, 26.9, 28.8 (3C), 33.4, 33.9, 60.7, 61.8, 62.2, 116.8, 124.4, 125.7, 128.8, 129.0, 139.8, 140.1, 146.2, 162.4, 171.2, 181.1. HRMS (DART) calcd for C21H26NO4 [M + H]+, 356.1856; found, 356.1857; HPLC analysis (column, Daicel Chiralpak IA-3, 2-propanol/hexane = 1.5:98.5, flow rate = 1.0 mL min−1): λ = 230 nm, tR (min) = 8.42 (major), 12.4 (minor). Compound γ-trans-4t was determined to have 80% ee.
Preparation of racemic γ-trans-4a and α-trans-4a for X-ray crystallographic analysis
The following reaction was carried out under N2. Compound 3 (0.410 mL, 3.56 mmol) was added to a stirred solution of 2a (500 mg, 1.90 mmol) and PPh3 (100 mg, 0.380 mmol) in toluene (5.9 mL). After stirring at room temperature for 63 h, the mixture was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel twice (EtOAc/hexane, 1:10) and preparative thin-layer chromatography (CH2Cl2) to provide γ-trans-4a (368 mg, 52%) as slightly yellow crystals and α-trans-4a (24.6 mg, 3%) as slightly yellow crystals.
Ethyl (1R*,2R*)-1′-acetyl-2′-oxo-2-phenylspiro[cyclopentane-1,3′-indolin]-3-ene-3-carboxylate (α-trans-4a)
IR 3031, 2933, 1749, 1714, 1647 cm−1. 1H NMR (600 MHz, acetone-d6): δ 1.12 (t, 3H, J = 7.2 Hz), 2.65 (s, 3H), 3.00 (dt, 1H, J = 2.4, 18.6 Hz), 3.16 (ddd, 1H, J = 0.6, 3.0, 18.6 Hz), 4.03–4.13 (m, 2H), 4.64 (s, 1H), 6.42 (dd, 1H, J = 1.2, 7.8 Hz), 6.75 (dt, 1H, J = 1.2, 7.8 Hz), 6.93 (m, 2H), 7.09–7.14 (m, 4H), 7.20 (q, 1H, J = 2.4 Hz), 8.05 (d, 1H, J = 8.4 Hz). 13C NMR (150 MHz, CDCl3): δ 14.0, 26.7, 43.1, 58.2, 60.0, 60.5, 115.8, 124.39, 124.42, 127.4, 128.0, 128.2, 128.3, 129.1, 137.0, 137.3, 139.3, 142.3, 163.8, 170.8, 181.3. HRMS (DART) calcd for C23H22NO4 [M + H]+, 376.1543; found, 376.1544.
Data availability
The data supporting this article have been included as part of the ESI:† Experimental details, 1H, 1H–1H COSY and 13C NMR spectra, HPLC data, computational methods, and X-ray data. CCDC 2343919, 2343921, and 2343914.†Xyz cartesian coordinates format. For ESI and crystallographic data in CIF or other electronic format, see.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported in part by JSPS KAKENHI Grant Number 15K07875 and the Hoansha Foundation. We also acknowledge We thank Ms M. Kawamura and Y. Sugiura for their technical assistance.
References
- For selected reviews:
(a) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049 CAS;
(b) S. B. Nallapati and S.-C. Chuang, Asian J. Org. Chem., 2018, 7, 1743 CAS.
-
(a) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CAS;
(b) Y. Du, X. Lu and C. Zhang, Angew. Chem., Int. Ed., 2003, 42, 1035 CAS.
- For selected reviews:
(a) Y. Xiao, Z. Sun, H. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089 Search PubMed;
(b) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344 CAS.
- S. Gladiali, E. Alberico, K. Junge and M. Beller, Chem. Soc. Rev., 2011, 40, 3744 CAS.
- For selected examples:
(a) D. Wang, Z.-F. Song, W.-J. Wang and T. Xu, Org. Lett., 2019, 21, 3963 CAS;
(b) D. Wang, W. Liu, Y. Hong and X. Tong, Org. Lett., 2018, 20, 5002 CAS.
- For selected examples:
(a) A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030 CAS;
(b) N. Pinto, M. Neel, A. Panossian, P. Retailleau, G. Frison, A. Voituriez and A. Marinetti, Chem. – Eur. J., 2010, 16, 1033 CAS.
- For selected examples of amino acid-derived phosphine catalysts:
(a) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369 CAS;
(b) B. J. Cowen and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 10988 CAS;
(c) H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467 CAS For other phosphine catalysts, see ref. 3.
- For selected reviews:
(a) S. S. Panda, A. S. Girgis, M. N. Aziz and M. S. Bekheit, Molecules, 2023, 28, 618 CAS;
(b) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CAS.
- For selected reviews:
(a) M. Ganesh and S. Suraj, Org. Biomol. Chem., 2022, 20, 5651 CAS;
(b) Y. Wang, A. A. Cobo and A. K. Franz, Org. Chem. Front., 2021, 8, 4315 CAS;
(c) A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026 CAS;
(d) P.-W. Xu, J.-S. Yu, C. Chen, Z.-Y. Cao, F. Zhou and J. Zhou, ACS Catal., 2019, 9, 1820 CAS;
(e) G.-J. Mei and F. Shi, Chem. Commun., 2018, 54, 6607 CAS;
(f) A. Ding, M. Meazza, H. Guo, J. W. Yang and R. Rios, Chem. Soc. Rev., 2018, 47, 5946 CAS;
(g) L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023 CAS.
- For selected reviews:
(a) Q.-F. Li, J. Ma, J. Meng and E.-Q. Li, Adv. Synth. Catal., 2023, 365, 4412 CAS;
(b) R. Saeed, A. P. Sakla and N. Shankaraiah, Org. Biomol. Chem., 2021, 19, 7768 CAS;
(c) G. Molteni and A. Silvani, Eur. J. Org. Chem., 2021, 1653 CAS;
(d) A. Voituriez, A. Marinetti and M. Gicquel, Synlett, 2015, 142 CAS;
(e) H. Li and Y. Lu, Asian J. Org. Chem., 2017, 6, 1130 CAS.
-
(a) A. Voituriez, N. Pinto, M. Neel, P. Retailleau and A. Marinetti, Chem. – Eur. J., 2010, 16, 12541 CAS;
(b) C. Gomez, M. Gicquel, J.-C. Carry, L. Schio, P. Retailleau, A. Voituriez and A. Marinetti, J. Org. Chem., 2013, 78, 1488 CAS;
(c) D. Wang, G.-P. Wang, Y.-L. Sun, S.-F. Zhu, Y. Wei, Q.-L. Zhou and M. Shi, Chem. Sci., 2015, 6, 7319 CAS;
(d) M. Gicquel, C. Gomez, M. C. G. Alvarez, O. Pamlard, V. Guérineau, E. Jacquet, J. Bignon, A. Voituriez and A. Marinetti, J. Med. Chem., 2018, 61, 9386 CAS For reactions of (2-oxoalkylidene)indolinones:
(e) X.-C. Zhang, S.-H. Cao, Y. Wei and M. Shi, Chem. Commun., 2011, 47, 1548 CAS For reactions of bis(alkoxycarbonyl)methyleneindolinones:
(f) X.-C. Zhang, S.-H. Cao, Y. Wei and M. Shi, Org. Lett., 2011, 13, 1142 CAS For reactions of dicyanomethylideneindolinones:
(g) S. Guo, R. Wang, J. Li, C. Li, H. Deng and X. Jia, Synlett, 2011, 2256 CAS For reactions of pyrazoloneyldiene oxindoles:
(h) W. Luo, B. Shao, J. Li, X. Xiao, D. Song, F. Ling and W. Zhong, Org. Chem. Front., 2020, 7, 1016 CAS For reactions of isoindigos:
(i) W.-L. Chan, X. Tang, F. Zhang, G. Quek, G.-J. Mei and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 6260 CAS.
-
(a) B. Tan, N. R. Candeias and C. F. Barbas III, J. Am. Chem. Soc., 2011, 133, 4672 CAS;
(b) Z. Lu, Y. Jia, X. Chen and P. Li, J. Org. Chem., 2022, 87, 3184 CAS For reactions of dicyanomethylideneindolinone and its analogues:
(c) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CAS;
(d) H.-P. Deng, Y. Wei and M. Shi, Org. Lett., 2011, 13, 3348 CAS;
(e) S. Kitagaki, M. Tsuji, H. Teramoto, N. Takenaga and K. Yoshida, Heterocycles, 2020, 101, 127 CAS For reactions of pyrazoloneyldiene oxindoles:
(f) J. Zhang, W.-L. Chan, L. Chen, N. Ullah and Y. Lu, Org. Chem. Front., 2019, 6, 2210 CAS.
- J. Zhang, C. Cheng, D. Wang and Z. Miao, J. Org. Chem., 2017, 82, 10121 CAS.
- C. Hu, Q. Zhang and Y. Huang, Chem. – Asian J., 2013, 8, 1981 CAS.
- The vinylogous reactions using 3-alkylideneoxindole as a nucleophile have been reported. For selected examples:
(a) Y.-J. Tsou, N. Sathishkumar, I.-T. Chen, T.-A. Lee, H.-T. Chen and J.-L. Han, J. Org. Chem., 2022, 87, 2520 CAS;
(b) J.-X. Xu, Y.-S. Jiang, C.-H. Chen, N. Sathishkumar, K.-T. Chu, M.-H. Chiang, H.-T. Chen and J.-L. Han, J. Org. Chem., 2022, 87, 197 CAS;
(c) N. D. Iorio, P. Righi, S. Ranieri, A. Mazzanti, R. G. Margutta and G. Bencivenni, J. Org. Chem., 2015, 80, 7158 CrossRef PubMed.
-
(a) One example of reaction of 3-methylenebenzofuranone bearing an alkyl substituent on the methylene carbon has been reported. See ref. 11c;
(b) Chiral phosphine-catalyzed (4 + 2) cyclization of γ-benzyl-substituted allenoates also includes only use of benzylideneindolinones. S. Jia, M. Ma, E.-Q. Li, Z. Duan and F. Mathey, Org. Lett., 2021, 23, 3337 CrossRef CAS.
-
(a) S. E. Gibson and J. D. Knight, Org. Biomol. Chem., 2003, 1, 1256 RSC;
(b)
V. Rosenberg, E. Sergeeva and H. Hopf, in Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, Germany, 2004, p. 435 Search PubMed;
(c) A. A. Aly and A. B. Brown, Tetrahedron, 2009, 65, 8055 CrossRef CAS;
(d) J. Paradies, Synthesis, 2011, 3749 CrossRef CAS;
(e) Special Issue: Cyclophanes, Isr. J. Chem., 2012, 52, 1;
(f) Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947 RSC.
- S. Felder, S. Wu, J. Brom, L. Micouin and E. Benedetti, Chirality, 2021, 33, 506 CrossRef CAS PubMed.
- Phosphines:
(a) T. G. Driver, J. R. Harris and K. A. Woerpel, J. Am. Chem. Soc., 2007, 129, 3836 CrossRef CAS;
(b) T.-Z. Zhang, L.-X. Dai and X.-L. Hou, Tetrahedron: Asymmetry, 2007, 18, 1990 CrossRef CAS Phosphoric acids:
(c) E. Xie, S. Huang and X. Lin, Org. Lett., 2019, 21, 3682 CrossRef CAS PubMed Thioureas:
(d) Y. Lu, Y. Ma, S. Yang, M. Ma, H. Chu and C. Song, Tetrahedron: Asymmetry, 2013, 24, 1082 CrossRef CAS;
(e) S. Kitagaki, T. Ueda and C. Mukai, Chem. Commun., 2013, 49, 4030 RSC;
(f) S. Kitagaki, E. Shimo, S. Takeda, R. Fukai, N. Kojima, S. Yoshioka, N. Takenaga and K. Yoshida, Heterocycles, 2021, 103, 678 CrossRef CAS N-Heterocyclic carbenes:
(g) L. Wang, Z. Chen, M. Ma, W. Duan, C. Song and Y. Ma, Org. Biomol. Chem., 2015, 13, 10691 RSC;
(h) P. An, Y. Huo, Z. Chen, C. Song and Y. Ma, Org. Biomol. Chem., 2017, 15, 3202 RSC Imines:
(i) M. Negru, D. Schollmeyer and H. Kunz, Angew. Chem., Int. Ed., 2007, 46, 9339 CrossRef CAS Hypervalent iodines:
(j) Y. Wang, H. Yuan, H. Lu and W.-H. Zheng, Org. Lett., 2018, 20, 2555 CrossRef CAS;
(k) Y. Wang, C.-Y. Zhao, Y.-P. Wang and W.-H. Zheng, Synthesis, 2019, 3675 CAS NAD(P)H models:
(l) Z.-H. Zhu, Y.-X. Ding, B. Wu and Y.-G. Zhou, Chem. Sci., 2020, 11, 10220 RSC;
(m) Z.-B. Zhao, J. Wang, Z.-H. Zhu, M.-W. Chen and Y.-G. Zhou, Org. Lett., 2021, 23, 9112 CrossRef CAS;
(n) Z.-H. Zhu, Y.-X. Ding and Y.-G. Zhou, Chem. Commun., 2022, 58, 3973 RSC Isothioureas:
(o) S. Yuan, C. Liao and W.-H. Zheng, Org. Lett., 2021, 23, 4142 CrossRef CAS.
-
(a) S. Kitagaki, Y. Ohta, R. Takahashi, M. Komizu and C. Mukai, Tetrahedron Lett., 2013, 54, 384 CrossRef CAS;
(b) N. Takenaga, S. Adachi, A. Furusawa, K. Nakamura, N. Suzuki, Y. Ohta, M. Komizu, C. Mukai and S. Kitagaki, Tetrahedron, 2016, 72, 6892 CrossRef CAS.
- S. Kitagaki, K. Nakamura, C. Kawabata, A. Ishikawa, N. Takenaga and K. Yoshida, Org. Biomol. Chem., 2018, 16, 1770 RSC.
-
(a) CCDC 2343919.†;
(b) CCDC 2343921.†.
- For mechanistic studies:
(a) T. Dudding, O. Kwon and E. Mercier, Org. Lett., 2006, 8, 3643 CrossRef CAS;
(b) E. Mercier, B. Fonovic, C. Henry, O. Kwon and T. Dudding, Tetrahedron Lett., 2007, 48, 3617 CrossRef CAS;
(c) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS;
(d) X.-F. Zhu, C. E. Henry and O. Kwon, J. Am. Chem. Soc., 2007, 129, 6722 CrossRef CAS PubMed;
(e) Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem. – Eur. J., 2008, 14, 4361 CrossRef CAS PubMed;
(f) Y. Liang, S. Liu and Z.-X. Yu, Synlett, 2009, 905 CAS;
(g) G.-T. Huang, T. Lankau and C.-H. Yu, J. Org. Chem., 2014, 79, 1700 CrossRef CAS;
(h) G.-T. Huang, T. Lankau and C.-H. Yu, Org. Biomol. Chem., 2014, 12, 7297 RSC;
(i) M. C. Holland, R. Gilmour and K. N. Houk, Angew. Chem., Int. Ed., 2016, 55, 2022 CrossRef CAS PubMed;
(j) H. Ni, Z. Yu, W. Yao, Y. Lan, N. Ullah and Y. Lu, Chem. Sci., 2017, 8, 5699 RSC;
(k) C.-X. Cui, C. Shan, Y.-P. Zhang, X.-L. Chen, L.-B. Qu and Y. Lan, Chem. – Asian J., 2018, 13, 1076 CrossRef CAS PubMed;
(l) Z. Yu, Z. Jin, M. Duan, R. Bai, Y. Lu and Y. Lan, J. Org. Chem., 2018, 83, 9729 CrossRef CAS PubMed;
(m) S. Anwar, L.-T. Lin, V. Srinivasadesikan, V. B. Gudise and K. Chen, RSC Adv., 2021, 11, 38648 RSC.
- See the ESI† for details of computational methods.
- CCDC 2343914.†.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, 1H, 1H–1H COSY and 13C NMR spectra, HPLC data, computational methods, and X-ray data. CCDC 2343919, 2343921, and 2343914. Xyz cartesian coordinates format. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00831f |
| ‡ Current address: Faculty of Pharmacy, Juntendo University, Urayasu, Chiba 279-0013, Japan. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Tomoki
Nakayoshi
*a,
Tomoki
Nakayoshi