
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of fluorinated curcumin derivatives for detecting amyloid plaques by 19F-MRI†
Sebastiano
Micocci‡
a,
Rachele
Stefania‡
 *b,
Francesca
Garello
a,
Umberto
Fasoglio
a,
Ivan
Hawala
a,
Lorenzo
Tei
b,
Simonetta
Geninatti Crich
a and
Giuseppe
Digilio
b
*b,
Francesca
Garello
a,
Umberto
Fasoglio
a,
Ivan
Hawala
a,
Lorenzo
Tei
b,
Simonetta
Geninatti Crich
a and
Giuseppe
Digilio
b
aDepartment of Molecular Biotechnology and Health Sciences, University of Turin, Via Nizza 52, 10126, Torino, Italy
bDepartment of Science and Technological Innovation, University of Eastern Piedmont “Amedeo Avogadro”, Viale Teresa Michel 11, 15120, Alessandria, Italy. E-mail: rachele.stefania@uniupo.it
First published on 3rd July 2024
Abstract
The most prominent pathophysiological hallmark of Alzheimer's disease is the aggregation of amyloid-β (Aβ) peptides into senile plaques. Curcumin and its derivatives exhibit a high affinity for binding to Aβ fibrils, effectively inhibiting their growth. This property holds promise for both therapeutic applications and diagnostic molecular imaging. In this study, curcumin was functionalized with perfluoro-tert-butyl groups to create candidate molecular probes specifically targeted to Aβ fibrils for use in 19F-magnetic resonance imaging. Two types of fluorinated derivatives were considered: mono-substituted (containing nine fluorine atoms per molecule) and disubstituted (containing eighteen fluorine atoms). The linker connecting the perfluoro moiety with the curcumin scaffold was evaluated for its impact on binding affinity and water solubility. All mono-substituted compounds and one disubstituted compound exhibited a binding affinity toward Aβ fibrils on the same order of magnitude as reference curcumin. The insertion of a charged carboxylate group into the linker enhanced the water solubility of the probes. Compound Curc-Glu-F9 (with one L-glutamyl moiety and a perfluoro-tert-butyl group), showed the best properties in terms of binding affinity towards Aβ fibrils, water solubility, and intensity of the 19F-NMR signal in the Aβ oligomer bound form.
Introduction
Alzheimer's disease (AD) is characterized by a progression from episodic memory problems to a slow global decline in cognitive function. The scientific research in the field is focused on the investigation of a simple and accurate way to detect Alzheimer's before these devastating symptoms begin. Cerebral amyloid-beta (Aβ) accumulation and aggregation is the primary event in AD pathogenesis.1a,b It has been proposed that the rest of the disease process, including the formation of neurofibrillary tangles containing tau protein, results from an imbalance between Aβ production and Aβ clearance.2a,b Accordingly, estimating the level of Aβ deposition in the brain would be informative for early diagnosis of AD and for evaluating AD progression. Among the various aggregates that Aβ can generate, oligomers gather during the early phases of Aβ aggregation and have the most neurotoxic effects.2a,3 Larger aggregates, such as protofibrils and fibrils, can fragmentate, releasing them and acting as sinks. In order to achieve early detection of Aβ oligomers and protofibrils, many researchers have tried to develop chemical probes that have a specific affinity for Aβ aggregates. Compounds able to bind selectively with high affinity the Aβ aggregates in vitro and in vivo are derivatives based on highly conjugated aromatic systems, such as thioflavin T, Congo red, chrysamine G, benzoxazoles, curcumin, and stilbenes.4 Among them, curcumin is of great interest because it is food-derived and shows a superior safety profile. Curcumin is a low molecular weight yellow-orange pigment derived from the turmeric plant with numerous pharmacological properties including anti-tumor, anti-oxidative, anti-inflammatory, hepatoprotective, nephroprotective, and anti-amyloid effects. Several studies have reported that curcumin has a high binding affinity to Aβ aggregates and inhibits the aggregation.5 Structurally, curcumin contains two methoxyphenol rings linked by a conjugated diene-β-dicarbonyl backbone. Curcumin is a potentially great scaffold to develop probes for Aβ diagnostic imaging and/or therapy because of its blood–brain barrier (BBB) permeability, high-affinity binding to senile plaques, and low toxicity.6 Curcumin is also reported to reduce Aβ aggregates in Alzheimer's transgenic mice.7 Considerable progress in Aβ imaging has been achieved in recent years.8 Currently, positron emission tomography (PET) is the most efficient imaging modality to detect Aβ deposition because of its high sensitivity and the ability to quantify the accumulated probe. Pittsburgh compound B ([11C]PiB), which is a derivative of thioflavin (Th-T), was the first successful Aβ-selective PET radioligand;9 then, different 18F-labeled Aβ-targeting derivatives emerged to overcome the 20 min radioactive decay half-life limitation of 11C-PiB.10,11a,b Magnetic resonance imaging (MRI) is another important and widely clinically used diagnostic technique that provides detailed anatomical information with excellent soft tissue contrast, and it is amenable to molecular imaging applications provided that suitable MRI probes are available. MRI probes are typically based on paramagnetic contrast agents, but diamagnetic probes carrying the MR active 19F nuclei are attracting a growing interest because of several advantages: first, the 19F nucleus has a high gyromagnetic ratio and a natural isotopic abundance of 100%, hence an MR sensitivity approaching that of 1H. Second, biological tissues contain essentially no 19F: only negligible amounts of endogenous fluorine are embedded in the teeth and bone matrix of the human body. Therefore, the introduction of exogenous 19F signals in vivo will yield background-free images. Third, 19F MRI exhibits relatively high spatial resolution. In addition, it is worth noting that 19F is a naturally occurring halogen and a stable, non-radioactive isotope of fluorine. Thus, unlike the radioactive isotope 18F commonly used in PET imaging, incorporating fluorine into a probe is synthetically smoother. Yanagisawa et al. developed the first perfluoro curcumin analogue, FMeC1,12 for 19F MRI to facilitate visualization of Aβ in vivo. FMeC1, named then Shiga-Y5, containing two trifluoromethoxy groups in place of the methoxy ones and a methylpropanoate moiety on the C4 position (Fig. 1), could cross the blood–brain barrier and bind to Aβ plaques in a transgenic mouse model of AD after injection via the tail vein. They also developed and investigated several 19F-containing curcumin analogue, called the Shiga-Y series, with different moieties at the C4 position;13 among them, Shiga-Y25 (Fig. 1) with a short PEG chain ending with a trifluoromethoxy group successfully detected Aβ depositions in the brain of a living mouse.14 All developed probes contained a limited number of 19F atoms/molecule, typically six/molecule, except the case of Shiga-Y25 which contains nine 19F atoms. | ||
| Fig. 1 Structure of curcumin, Shiga-Y5, Shiga Y51, Shiga-Y25. | ||
This work aims at synthesizing a series of novel 19F-containing curcumins with a high number of equivalent 19F nuclei, suitably spaced from the aromatic part of the molecule. Furthermore, we present a novel synthetic approach that involves direct modification of the OH group attached to the phenol moiety of curcumin to produce novel 19F curcumin derivatives (Curc-C3-F9, Curc-C6-F9, Curc-Glu-F9, Curc-C6-F18, Curc-Glu-F18). The affinity of the novel derivatives for Aβ fibrils was evaluated in vitro by a fluorescence-based assay, and the 19F-NMR properties in the fibril-bound state were investigated in vitro.
Results and discussion
Design of the 19F-curcumin imaging probes
Several aspects must be kept into account when designing curcumin-based 19F-MRI probes targeted to Aβ fibrils. The enol form of these compounds must be preserved as it shows preference for binding to Aβ fibrils,15 whereas the keto form favours the binding to Aβ oligomers.16 Another important parameter to consider is the hydrophilicity/hydrophobicity balance of the fluorinated probe for an efficient crossing of the BBB.17 Finally, the highest achievable sensitivity to 19F-MRI detection must be pursued by introducing in the molecule as many as possible magnetically equivalent fluorine atoms. A common drawback of fluorinated molecular probes is that their 19F-NMR linewidth can be significantly affected by binding interactions. If the re-orientational motions of the perfluoroalkyl moieties are restricted in the bound state, a significant line broadening of the 19F-NMR signal would arise, leading ultimately to a dramatic loss of the 19F-MRI signal.Based on these observations, we have designed and synthesized a range of novel curcumin derivatives that contain nine or eighteen equivalent 19F atoms. To counteract potentially detrimental line-broadening effects, the perfluorinated alkyl groups were linked to the curcumin structure through flexible linkers, such to preserve local re-orientational freedom also in the bound-state. These linkers varied in length and hydrophilicity. Specifically, we have developed three monosubstituted curcumin derivatives, one with a short aliphatic chain (Curc-C3-F9), one with a longer aliphatic chain (Curc-C6-F9), and one with a spacer containing a carboxylic group suitable to improve the solubility (Curc-Glu-F9, Fig. 2). The conjugation reactions occur on the hydroxyl groups (4-OH) attached to the phenyl rings of natural product of curcumin. The monosubstituted compounds maintain one curcumin phenolic group, which is known to be important for interaction with fibrils.18 We also synthesized bis-functionalized derivatives to maximize the number of 19F atoms per molecule. These probes were assessed for the best compromise between water solubility, fibril targeting ability, and sensitivity to 19F-MRI detection.
 | ||
| Fig. 2 Structure of 19F MRI curcumin-based probes synthesized in this work. | ||
Synthesis of the fluorinated curcumin imaging probes
The synthesis of the probes started by preparing the perfluorinated amines, characterized by the presence of the nonafluoro-tert-butyloxy tail, using the condensation under Mitsunobu conditions19 of the readily available 6-(Boc-amino)-1-hexanol or 3-(Boc-amino)-1-propanol with perfluoro-tert-butanol (Scheme 1). The corresponding perfluoro-tert-butyl ethers were obtained, from which the Boc group was removed in acidic conditions, using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of trifluoroacetic acid (TFA) in dichloromethane (CH2Cl2). In order to improve the solubility of the final perfluoro curcumin derivative, we also designed a perfluorinated amine containing a carboxylate moiety: thus, compound F9-C3-NH2 with the shorter chain was conjugated to Boc-L-glutamic acid 5-tert-butyl ester (Boc-Glu(OtBu)-OH) using DCC/DMAP approach. Then, compound F9-Glu-NH2 was obtained as a trifluoroacetate salt after the deprotection of the Boc group with TFA.
:1 mixture of trifluoroacetic acid (TFA) in dichloromethane (CH2Cl2). In order to improve the solubility of the final perfluoro curcumin derivative, we also designed a perfluorinated amine containing a carboxylate moiety: thus, compound F9-C3-NH2 with the shorter chain was conjugated to Boc-L-glutamic acid 5-tert-butyl ester (Boc-Glu(OtBu)-OH) using DCC/DMAP approach. Then, compound F9-Glu-NH2 was obtained as a trifluoroacetate salt after the deprotection of the Boc group with TFA.
 | ||
| Scheme 1 Synthesis of perfluoroamine derivatives: (i) nonafluoro-tert-butyl alcohol, PPh3, DIAD, Et2O, (ii) TFA, CH2Cl2, (iii) Boc-Glu(OtBu)-OH, DCC, DMAP, CH2Cl2, (iv) TFA, CH2Cl2 (1:1). | ||
While curcumin is naturally derived, its derivatives like those containing 19F in its structure are generally produced by a chemical reaction between by acetylacetone and its derivatives with appropriate aryl-aldehydes. This assembly method can yield multiple chemical analogues, such as compounds with trifluoromethoxy groups on the benzene ring and alkyl substituents on the middle carbon of the linker (C4 position).15,20 The functionalization at C4 can be performed by a Michael addition starting from curcumin.21 Here, the synthesis of perfluoro curcumin derivatives MRI probes was carried out in four steps starting from commercial Curcuma longa powder (C1386, Merck), which is composed of curcumin (77%), demethoxycurcumin (17%), and bisdemethoxycurcumin (3%). Conjugation reactions take place on one or two hydroxyl groups of the phenyl rings of curcumin to synthesize mono- or bifunctional nonafluoro derivatives in a facile synthetic route. The reaction of curcumin with 0.5 equivalents of t-butyl bromoacetate in the presence of potassium carbonate as base led to the formation of mono-tBu ester derivative and bis-tBu ester derivative as by-product (compound 1a and compound 1b, Scheme 2). The monofunctionalized curcumin was isolated, after purification by column chromatography on silica gel, with about 30% yield. The bis-functionalized derivative was also collected (10% yield) and used for further functionalization to evaluate its binding to the Aβ aggregates and to compare with the mono-functionalized derivatives. Then, the t-Bu esters were deprotected with TFA and the mono and bis-carboxylic acid curcumin derivatives (compound 2a and 2b, respectively) were reacted with EDC/NHS in order to obtain the mono and bis-N-hydroxysuccinimide esters (Curc-mono-NHS and Curc-bis-NHS, compounds 3a and 3b). The final 19F-MRI probes were obtained by amide coupling reaction between the NHS-activated esters of curcumin and the perfluorinated amine (F9-C3-NH2, F9-C6-NH2 and F9-Glu-NH2) in a mixture of CH3CN and a phosphate buffer at pH 7.5, at room temperature (Scheme 2). The bis-NHS curcumin 3b was also conjugated to F9-C6-NH2 and F9-Glu-NH2 to afford final bis-perfluorinated derivatives with a higher number of 19F nuclei (Curc-C6-F18 and Curc-Glu-F18) (Scheme 2). The compounds were then purified by RP-HPLC and characterized by UPLC-UV-MS(ESI+) and NMR spectroscopy. The characteristic 1H, 19F, 2D 1H, 1H-COSY, 2D 1H,13C-HSQC, 2D 1H,13C-HMBC NMR (600 MHz, DMSO-d6, 300 K) and 19F NMR (500 MHz, ethanol, 300 K) of all 19F-curcumin imaging probes reported are presented as ESI,† as well as the UPLC-UV-MS(ESI+) chromatogram. The complete 1H NMR chemical shift assignment of Curc-C3-F9 dissolved in DMSO-d6 is shown in Fig. 3. The peak at 6.13 ppm corresponds to the proton on the α-carbon in the keto–enol tautomer (H1 in Fig. 3). Moreover, the methoxy, the aromatic and the conjugated methyne protons of the two sides of the molecule are not magnetically equivalent confirming the asymmetry of the structure. For all compounds, the purity was found to be between 96 and 98% as measured by UPLC at λ = 220 nm and λ = 413 nm.
 | ||
| Scheme 2 Synthesis of mono and bis-perfluorinated curcumin derivatives: (i) t-butyl bromoacetate, K2CO3 in CH3CN; (ii) TFA/CH2Cl2 (1:1); (iii) EDC/NHS, 5 mol% DMAP in NMP; (iv) F9-C3-NH2 or F9-C6-NH2 or F9-Glu-NH2, buffer phosphate (0.1 M, pH = 7.5), CH3CN. | ||
 | ||
| Fig. 3 1H-NMR spectrum of compound Curc-C3-F9 (DMSO-d6, 300 K). | ||
Binding activity of curcumin derivatives to the amyloid-beta (Aβ) fibrils
Aβ aggregates in protofibrillar and fibrillar state were prepared by incubating Aβ 1–42 peptide (50 μM) in phosphate buffer 10 mM (pH 7.4) containing 11 mM NaCl, for 4 days at 37 °C under stirring.22 The formation of Aβ aggregates was checked by measuring Thioflavin-T (ThT) fluorescence enhancement during fibril formation at 37 °C under stirring at 600 rpm (see Fig. 4A). ThT exhibits a significant shift in the excitation maximum (from 385 nm to 450 nm) and the emission maximum (from 445 nm to 482 nm) due to its binding to Aβ.23 ThT is an effective indicator of fibrillization, as confirmed by morphological analysis using field emission scanning electron microscopy (FESEM) (Fig. 4B). These experiments showed that aggregation and maturity of the fibrils was optimal after 5 days incubation under our experimental conditions. | ||
| Fig. 4 (A) ThT fluorescence enhancement at different times of incubation (37 °C, under stirring, [Aβ] 300 nM, [ThT] 50 nM, ex. 450 nm, em. 476 nm). (B) FESEM images of Aβ aggregates (5 days incubation) at 15k× and 100k× magnification, probe set at 100 pA and the electron beam energy at 5 keV. | ||
Curcumin and its fluorinated derivatives, at the concentrations used, generate negligible fluorescence emission in the range 488–497 nm. In the absence of fibrils (Fig. 5, dashed line, λex = 450 nm). The binding of curcumin and its derivatives to mature Aβ fibrils is known to be characterised by a steep increase of their fluorescence emission24a,b and also by a red shift of the absorption maximum.25a,b In our conditions, the maximum fluorescence emission of curcumin derivatives in the presence of Aβ fibrils was found using an excitation wavelength of 450 nm. This excitation wavelength was used in binding titrations, where a fixed amount of a curcumin derivative was incubated with increasing amounts of Aβ fibrils. The fluorescence emission at around 490 nm was plotted as a function of the fibril concentration to obtain binding isotherms (Fig. 6), from which the binding affinity Ka for Aβ could be extracted by computer aided best fitting to eqn (1) (Experimental section). The obtained thermodynamic association constants Ka, assuming a 1:1 interaction with Aβ, are listed in Table 1.
 | ||
| Fig. 5 Fluorescence emission spectra (λex = 450 nm) as a function of aggregated Aβ: (A) Curc-C3-F9, 22.0 nM; (B) Curc-C6-F9, 25.6 nM; (C) Curc-Glu-F9, 20.8 nM; (D) Curc-Glu-F18, 28.3 nM; (E) Curc-C6-F18, 25.0 nM; (F) curcumin, 21.0 nM. | ||
 | ||
| Fig. 6 Plot of the fluorescence emission (λex = 450 nm) of curcumin and its derivatives: (A) Curc-C3-F9, λem = 497 nm, R2 = 0.99957; (B) Curc-C6-F9, λem = 495 nm, R2 = 0.99836; (C) Curc-Glu-F9, λex = 495 nm, R2 = 0.99334; (D) Curc-Glu-F18, λem = 488 nm, R2 = 0.992; (E) Curc-C6-F18, λex = 488 nm; (F) curcumin, λem = 495 nm, R2 = 0.91. Titrations were done in triplicate. Error bars correspond to ±SD of the mean values. | ||
| Sample | F b ± SD (CPS mA−1 M−1) | K a ± SD (M−1) |
|---|---|---|
| Curc-C3-F9 | 5.5 × 1013 ± 1.1 × 1013 | 3.8 × 105 ± 0.9 × 105 |
| Curc-C6-F9 | 3.2 × 1013 ± 3.2 × 1013 | 3.9 × 105 ± 0.8 × 105 |
| Curc-Glu-F9 | 1.7 × 1014 ± 0.4 × 1014 | 1.1 × 105 ± 0.9 × 105 |
| Curc-Glu-F18 | 3.5 × 1013 ± 2.9 × 1013 | 2.1 × 105 ± 0.2 × 105 |
| Curc-C6-F18 | n.d. | n.d. |
| Curcumin | 9.7 × 1014 ± 7.4 × 1014 | 2.6 × 105 ± 1.3 × 105 |
The affinity constant Ka for curcumin obtained by this assay is in line with that reported in the literature.26 All fluorinated compounds showed a good affinity for fibrils, with Ka in the range (1.1–3.9 × 105 M−1), except Cur-C6-F18. The latter compound showed a very low fluorescence increase which prevented the calculation of its affinity constant. This could be likely due to the very poor solubility of the compound in aqueous medium (see below).
While the extension of the spacer between curcumin and the perfluoro-tert-butyl ether from C3 to C6 does not affect the affinity, the presence of a carboxylic group, which is negatively charged at neutral pH, slightly reduces the Ka of Curc-Glu-F9 derivative.
Although it is known that at least one phenolic group in the aromatic portion of the structure is needed to preserve binding to the fibril beta-sheets layer, we found that the disubstituted Curc-Glu-F18 (having no such phenolic group) had a binding affinity in the same order of magnitude of the monosubstituted Curc-Glu-F9 counterpart. We might speculate that the two amide groups within each of the linkers of Curc-Glu-F18 may provide suitable hydrogen bonding capabilities to form a suitable hydrogen bond network with the fibril.
Binding to human serum albumin (HSA)
For an initial assessment of the binding selectivity, we chose serum albumin as a model protein offering multiple potential binding sites for the fluorinated compounds, each site having different binding properties. It has been reported that the curcumin fluorescence emission in the 450–490 nm range (with λex at 430 nm) strongly increases when bound to the hydrophobic pockets of HSA.26 To this purpose the fluorescence emission at 485 nm was plotted as a function of the HSA concentration, assuming a 1:1 interaction (see ESI, section 3†). As expected, reference curcumin showed specific binding to HSA with an affinity constant Ka of 5.9 × 105 M−1, in line with the value reported in the literature.27 The monosubstituted Curc-C6-F9 and Curc-Glu-F9 showed a very low binding to HSA as demonstrated by the low fluorescence increase compared to the autofluorescence of HSA alone at the same concentrations. The only compound showing a binding affinity for HSA was Curc-Glu-F18, with a binding affinity constant higher than that of reference curcumin (Ka = 4.2 × 106 M−1).
Solubility of fluorinated curcumin derivatives
All fluorinated curcumin derivatives exhibit pronounced hydrophobicity, especially the bis-functionalized Curc-C6-F18 derivative, which does not dissolve in any of the potentially injectable formulations prepared for in vivo use. The very low solubility in aqueous medium is due to the presence of the central curcuminoid body, consisting of a conjugated alkenyl and aromatic components, as well as the transformation of the curcumin –OH groups into alkyl ethers terminating with three bulky and hydrophobic –CF3 groups. To achieve a suitable solubility and stability of the solutions in water, addition of CREMOPHOR EL, a non-ionic surfactant derived from castor oil, was necessary. The solubility properties of the compounds at 10 mg mL−1 in DMSO, MeOH, and HEPES buffer with the addition of 10% and 20% CREMOPHOR are shown in Table 2. The curcumin derivatives that gain a negative charge at physiological pH (Curc-Glu-F9 and Curc-Glu-F18) have been shown to effectively enhance water solubility.19F-NMR study
Compounds Curc-C6-F9, Curc-Glu-F9, and Curc-Glu-F18, which showed an acceptable solubility in the hepes/cremophor biocompatible medium, were selected for 19F-MRI sensitivity assessment. All these compounds show a single 19F-NMR singlet falling 4–5 ppm upfield relative to TFA (see ESI section 2.14†). In MRI signal acquisition, the relaxation times T1 and T2 of 19F nuclei play a crucial role, as they determine both the signal-to-noise ratio achievable in the image and the acquisition times of the imaging experiment. Indeed, high values of T1 (greater than 1 s) require long acquisition times, while too small values of T2 (less than 100 ms) result in signal loss due to both line-broadening and dead times of the acquisition sequence. The T1 and T2 values for the single 19F resonance of the synthesized products are reported in Table 3.| T 1 (ms) | T 2 (ms) | SNR | |
|---|---|---|---|
| Curc-C6-F9 HEPES + cremophor 20% | 610 | 126 | 13.7 |
| Curc-Glu-F9 HEPES + cremophor 10% | 558 | 139 | 11.4 |
| Curc-Glu-F18 HEPES + cremophor 10% | 523 | 102 | 22.0 |
The relaxation times are within the suitable range for 19F-MRI. Indeed, T1 values around 500 ms allow for the acquisition of a reasonable number of images in a short time without saturating the signal, while T2 values slightly above 100 ms are enough to avoid appreciable signal loss during spin-echo trains. Accordingly, 19F MR-images show a good signal-to-noise and the expected proportionality of the 19F-MRI signal intensity to the number of fluorine atoms present in each compound (Table 3 and Fig. 7).
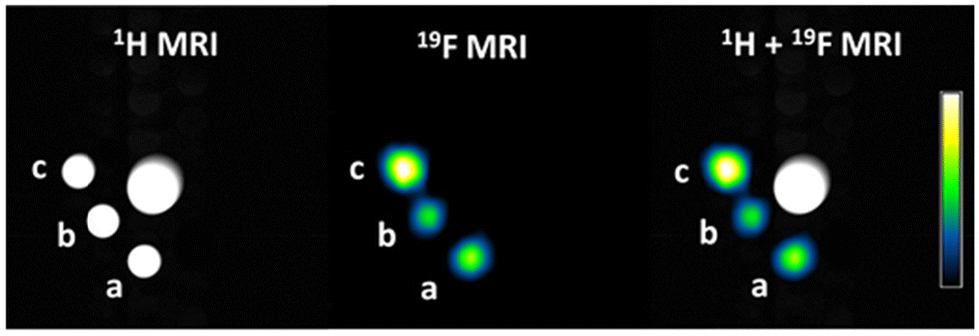 | ||
| Fig. 7 1H, 19F, and merged MRI of (a) Curc-C6-F9, (b) Curc-Glu-F9, (c) and Curc-Glu-F18 solubilized at 4.5 mM in HEPES/NaCl buffer + cremophor. The normalized 19F signal intensity scale is reported in the calibration bar. The central cone is to provide an appropriate volume for shimming. | ||
Next, we tried to acquire the 19F-NMR signal in the presence of amyloid fibrils. The critical issue in this kind of measurement is that our model of amyloid fibrils is incompatible with the surfactant needed to solubilize the probe. Therefore, experiments were carried out without the surfactant with compound Curc-Glu-F9, which showed the best compromise between binding affinity to fibrils, selectivity and water solubility. In phosphate buffer (without any surfactants) Curc-Glu-F9 (50 μM) yielded a barely detectable signal (Fig. 8A), because the compound aggregates into supramolecular structures having a heterogeneous distribution of molecular sizes and leading to a substantial line-broadening. In the presence of monomers or oligomers at 50 μM concentration, a 19F-NMR signal at −70.6 ppm (linewidth about 25 Hz) became clearly detectable (Fig. 8B and C). In such experimental conditions, the theoretically expected fraction of fibril-bound probe would be 80%. In the presence of mature fibrils, the signal disappears (Fig. 8D). The increase of 19F-NMR signal intensity in the presence of monomers/oligomers is explained in terms of the establishment of a binding equilibrium between the aggregated, NMR invisible form of the fluorinated compound and an NMR detectable form where Curc-Glu-F9 is bound to amyloid monomers/oligomers.
 | ||
| Fig. 8 19F-NMR spectrum of Curc-Glu-F9: (A) without Aβ, (B) with Aβ monomers, (C) with soluble Aβ oligomers and (D) with mature fibrils. Asterisks denote TFA (*) and hexafluoro-2-propanol (HFIP, **) as contaminants. | ||
In the presence of mature fibrils, such adducts have a large molecular size, leading to massive 19F-NMR line broadening which in turn hampers the detection of the 19F-NMR signal. These results indicate that, under the in vitro assay conditions, Curc-Glu-F9 can detect Aβ monomers/oligomers rather than mature fibrils. Soluble Aβ oligomers are toxic to neurons and are believed to be major contributors to synaptic dysfunction and neuronal death observed in Alzheimer's disease. Detection and quantification of soluble Aβ and tau oligomers in cerebrospinal fluid (CSF) may serve as pre-symptomatic biomarkers.29a,b This approach to study the binding interaction has limitations, as the in vitro conditions may not mimic properly those likely met in a physiological environment. For instance, fibril-bound forms may exist in equilibrium with forms bound to the hydrophobic components of the ECM rather than in equilibrium with self-aggregated forms. In vivo studies with murine models of brain deposition of amyloid fibrils are awaited to gain further insights about the NMR properties of the 19F-NMR signal, and to assess whether the high number of fluorine atoms is paralleled by a proportional increase of signal intensity. As a matter of fact, such kind of studies for the compounds of the Shiga-family were carried out directly by in vivo measurements.12,14,16
Conclusions
The use of 19F-MRI to detect amyloid fibrils in the brain presents several challenges. The main hurdle is the limited sensitivity of the 19F-MRI technique, eventually requiring a high concentration of fluorine-19 nuclei in the imaging voxel (in the order of millimoles per litre), under a realistic clinical scenario,30 and a suitably small signal linewidth. We successfully synthesized new curcumin derivatives substituted with one or two perfluoro-tert-butyl groups, containing 9 or 18 equivalent fluorine atoms respectively. Amongst these compounds, the monosubstituted Curc-Glu-F9 (i) retained a high binding affinity towards Aβ fibrils; (ii) had an acceptable water solubility in formulation with surfactants; and (iii) showed a detectable 19F-NMR signal in the form bound to Aβ oligomers in a preliminary in vitro assay. In vivo studies are needed to determine if the large number of equivalent fluorine atoms per molecule will result in a corresponding increase in 19F-MRI signal intensity in a more realistic brain imaging setting.Experimental
General synthetic methods
All reagents were purchased by Sigma Aldrich (Darmstad, Germany) and solvents by VWR International (Radnor, USA) and were used without further purifications. Column chromatographic separations were performed using silica gel (VWR International) with a particle size of 0.040–0.063 mm. Preparative HPLC-MS were carried out on a Waters AutoPurification system (3100 Mass Detector, 2545 Pump Gradient Module, 2767 Sample Manager, and 2998 PDA detector). UPLC analysis was performed using a UPLC Acquity H-Class coupled with the QDa and TUV detectors, using Kinetex® F5 column, 1.7 μm, 2.1 × 100 mm, applying a gradient of CH3CN (0.05% TFA) in H2O (0.05% TFA) from 50% to 100% in 8 min and 100% of B in 4 min (0.2 mL min−1), peak area revealed at 210 nm and 430 nm (method 1). All compounds are >95% pure by HPLC. NMR spectra were recorded at 310 K on a Bruker AVANCE 600 MHz and a Bruker Avance Neo 500 MHz spectrometer.Synthesis of mono-NHS and bis-NHS esters of curcumin
:2 to 1:1) to give 1a (0.34 g, yield 30%) and 1b (0.53 g, yield 19%) as a yellow oil. Compound 1a: 1H NMR (DMSO-d6), δ (ppm): 9.70 (s, 1H, phenolic-OH), 7.60 (d, 2H, J = 15.8 Hz, H4/H4′), 7.41 (s, 1H, H6), 7.41 (s, 1H, H6′), 7.27 (d, 1H, J = 8.0 Hz, H10), 7.20 (d, 1H, J = 8.2 Hz, H10′), 6.93 (d, 1H, J = 8.3 Hz, H9), 6.88 (d, 1H, J = 15.8 Hz, H3), 6.86 (d, 1H, J = 8.3 Hz, H9′), 6.80 (d, 1H, J = 15.9 Hz, H3′), 6.13 (s, 1H, H1), 4.76 (s, 2H, H12), 3.89 (s, 3H, H20), 3.88 (s, 3H, H20′), 1.47 (s, 9H, tBu) (see ESI section 2.1†). Direct infusion mass analysis with methanol/water 9:1 v:v at 0.2 mg mL−1: ESI-MS (m/z) calcd: for C27H30O8 [M + H]+ 483.19 found: 483.35. Compound 1b: 1H NMR (DMSO-d6), δ (ppm): 7.61 (d, 2H, J = 15.9 Hz, H4/H4′), 7.30 (s, 2H, H6/H6′), 7.27 (d, 2H, J = 8.4 Hz, H10/H10′), 6.94 (d, 2H, J = 8.4 Hz, H9, H9′), 6.87 (d, 2H, J = 15.9 Hz, H3/H3′), 6.16 (s, 1H, H1), 4.72 (s, 4H, H12/H12′), 3.88 (s, 6H, H20/H20′), 1.46 (s, 18H, tBu). Direct infusion mass analysis with methanol/water 9:1 v:v at 0.2 mg mL−1: ESI-MS (m/z): calcd: for C33H40O10 [M + H]+ 597.26 found: 597.37.
Compound 2a: UPLC-UV (λ = 220 nm, 430 nm): tR 2.05 min, 98% purity. 1H-NMR (DMSO-d6), δ (ppm): 16.33 (exch br s, 1H, H11), 13.06 (exch br s, 1H, COOH), 9.67 (s, 1H, phenolic-OH), 7.60 (d, 2H, J = 15.9 Hz, H4/H4′), 7.41 (s, 1H, H6), 7.36 (s, 1H, H6′), 7.26 (d, 1H, J = 8.1 Hz, H10), 7.20 (d, 1H, J = 8.3 Hz, H10′), 6.94 (d, 1H, J = 8.1 Hz, H9), 6.87 (d, 1H, J = 15.9 Hz, H3), 6.86 (d, 1H, J = 8.3 Hz, H9′), 6.80 (d, 1H, J = 15.9 Hz, H3′), 6.13 (s, 1H, H1), 4.78 (s, 2H, H12), 3.88 (s, 3H, H20), 3.87 (s, 3H, H20′). 13C NMR (DMSO-d6), δ (ppm): 184.4, 183.2, 170.5, 150.0, 149.8, 149.7, 148.6, 141.6, 140.7, 128.8, 126.9, 123.8, 123.0, 122.9, 121.7, 116.3, 113.5, 112.0, 111.6, 101.5, 65.5, 56.3. ESI-MS (m/z): calcd: for C23H22O8 [M + H]+ 427.13 found: 427.33; [M + Na]+ 449.12 found: 449.34 (see ESI section 2.2†).
Compound 2b: UPLC-UV (λ = 220 nm, 254 nm): tR 2.50 min, 93% purity.1 HNMR (DMSO-d6), δ (ppm): 7.62 (d, 2H, J = 15.9 Hz, H4/H4′), 7.41 (s, 2H, H6/H6′), 7.27 (d, 2H, J = 8.4 Hz, H10/H10′), 6.94 (d, 2H, J = 8.4 Hz, H9, H9′), 6.88 (d, 2H, J = 15.9 Hz, H3/H3′), 6.16 (s, 1H, H1), 4.77 (s, 4H, H12/H12′), 3.88 (s, 6H, H20/H20′). 13C NMR (DMSO), δ (ppm): 184.9, 172.6, 151.5, 151.2, 141.8, 131.2, 123.9, 123.8, 115.4, 112.5, 67.0, 56.8. ESI-MS (m/z): calcd: For C25H24O10 [M + H]+ 485.14 found: 485.26 (see ESI section 2.3†).
:1 v:v at 0.2 mg mL−1: Compound 3a: ESI-MS (m/z): calcd: for C27H25NO10 [M + H]+ 524.15 found: 524.26; [M + Na]+ 546.49; Compound 3b: ESI-MS (m/z): calcd: for C33H30N2O14 [M + H]+ 679.17 found: 679.37; [M + Na]+ 701.61.
Synthesis of F9-C3-NH2 and F9-C6-NH2
3-(Boc-amino)-1-propanol (0.37 g, 2.1 mmol) or 6-(Boc-amino)-1-hexanol (0.46 g, 2.1 mmol) and triphenylphosphine (0.66 g, 2.52 mmol) in Et2O (16 mL) were added to an ice cooled and stirred solution of nonafluoro-tert-butyl alcohol (0.5 g, 2.1 mmol) in Et2O (8 mL). After 5 min, a solution of diisopropyl azodicarboxylate (DIAD, 0.55 g, 2.73 mmol) in Et2O (5 mL) was added during 15 min. Then, the ice bath was removed and the mixture stirred at RT for 24 h. The solid precipitate was removed by filtration and the filtrate evaporated. The crude product was purified by chromatography (silica gel column, DCM/MeOH 98:2) to afford Boc-HN-C3-F9 or Boc-HN-C6-F9 as a yellow oil (52% and 32%, respectively). For the deprotection of the Boc-group, 0.3 g of Boc-perfluoroamine were dissolved in CH2Cl2 (2 mL) and cooled to 0 °C. 2 mL of TFA were added and the solution was allowed to warm to room temperature. After stirring at room temperature until starting material was consumed (TLC monitoring) the solution was concentrated in vacuo (ca. 80%). Boc-NH-C3-F9: 1H-NMR (CDCl3, 600 MHz) δ 1.48 (s, 9H), 1.94 (t, 2H, J = 5.92), 3.29 (m, 2H), 4.13 (t, 2H, J = 5.96). ESI-MS (m/z): calcd: for C12H16F9NO3 [M + H]+ 394.10 found: 394.14. F9-C3-NH2: 1H-NMR (MeOD, 600 MHz) δ 2.12 (m, 2H), 3.10 (m, 2H), 4.28 (t, 2H, J = 5.93), 6.87 (m, 3H) (see ESI section 2.4†). ESI-MS (m/z): calcd: for C7H8F9NO [M + H]+ 294.05 found: 294.13. Boc-NH-C6-F9: 1H-NMR (CDCl3, 600 MHz) δ 1.39 (m, 2H), 1.46 (m, 2H), 1.50 (s, 9H), 1.54 (m, 2H), 1.73 (m, 2H), 3.16 (t, 2H), 4.05 (t, 2H). 13C-NMR (CDCl3, 600 MHz) δ 24.32, 25.60, 27.67, 28.88, 29.22, 39.71, 68.99, 116.76, 118.72, 120.66, 122.58, 155.26. ESI-MS (m/z): calcd: for C15H22F9NO3 [M + H]+ 436.15 found: 436.15. F9-C6-NH2: 1H-NMR (MeOD 600 MHz) δ 1.51(m, 4H), 1.71 (m, 2H), 1.79 (m, 2H), 2.97 (m, 2H), 4.15 (t, 2H, J = 5.94) (see ESI section 2.5†). 13C-NMR (MeOD, 600 MHz) δ 23.91, 24.92, 26.30, 28.43, 38.50, 69.17, 116.80, 118.72, 120.67, 122.62, 159.03. ESI-MS (m/z): calcd: for C10H14F9NO [M + H]+ 336.09 found: 336.27.
Synthesis of F9-Glu-NH2
Boc-L-glutamic acid 5-tert-butyl ester (0.18 g, 0.6 mmol) was dissolved in 10 mL of CH3CN and DIPEA (0.18 g, 1.4 mmol) and HATU (0.23 g, 0.7 mmol) were added. After 5 min, F9-C3-NH2 (0.2 g, 0.7 mmol) dissolved in CH3CN (1 mL) was added dropwise and then the mixture was stirred at room temperature for 6 h under N2 atmosphere. The solvent was then removed under vacuum and the residue was dissolved in CH2Cl2 (10 mL) and washed three times with brine (2 × 10 mL) and water (2 × 10 mL) and the separated organic phases were dried with anhydrous Na2SO4. Then, the crude product obtained by evaporation of the solvent was purified by chromatography (silica gel column, petroleum ether/ethyl acetate 7:3) to give F9-C3-Glu(OtBu)-NH-Boc as a pale yellow oil (55%). 1H-NMR (CDCl3, 600 MHz) δ 1.49 (s, 9H), 1.51 (s, 9H), 1.88 (m, 1H), 1.98 (m, 2H), 2.19 (m, 1H), 2.30 (m, 2H), 3.43 (m, 2H), 4.16 (m, 3H), 5.31 (s, 1H), 6.63 (s, 1H). 13C-NMR (CDCl3, 600 MHz) δ 27.16, 27.56, 28.85, 29.21, 31.85, 35.78, 52.64, 67.62, 79.05, 81.67, 116.33, 118.58, 120.64, 122.51, 155.43, 170.70, 171.73. ESI-MS (m/z): calcd: for C21H31F9N2O6 [M + H]+ 579.20 found: 579.36. For the deprotection of the Boc and tert-butyl groups, 0.2 g of F9-C3-Glu(OtBu)-NH-Boc were dissolved in CH2Cl2 (2 mL) and cooled to 0 °C; then, 2 mL of TFA were added and the solution was allowed to warm to room temperature. After stirring at room temperature overnight the solution was concentrated in vacuo to obtain F9-C3-Glu-NH2 in 92% yield. 1H-NMR (DMSO-d6, 600 MHz) δ 1.85 (m, 2H), 2.02 (m, 2H), 2.32 (m, 2H), 3.18 (m, 2H), 3.97 (m, 1H), 4.13 (t, 2H, J = 6.41 Hz), 8.03 (t. 1H, J = 5.33 Hz), 8.27 (s, 2 H) (see ESI section 2.6†). 13C-NMR (CDCl3, 600 MHz) δ 28.05, 30.36, 32.86, 36.58, 54.74, 64.52, 116.24, 118.42, 172.40, 174.83. ESI-MS (m/z): calcd: for C12H15F9N2O4 [M + H]+ 423.09 found: 423.18.
Synthesis of monosubstituted curcumin derivatives (Curc-C3-F9, Curc-C6-F9, Curc-Glu-F9)
A solution of mono-NHS ester of curcumin (compound 3a, 0.38 mmol) in acetonitrile (4 mL) was slowly added at room temperature to a solution of perfluoroamine (F9-C3-NH2, F9-C6-NH2, F9-Glu-NH2) (0.38 mmol) dissolved in sodium phosphate buffer (0.1 M, pH 7.5, 4 mL) and CH3CN (4 mL). The biphasic mixture was allowed to stir vigorously for 2 h. Then the acetonitrile was evaporated under reduced pressure and the aqueous phase was washed with dichloromethane (3 × 100 mL). The organic phase was dried with anhydrous Na2SO4, and, after filtration, the solvent was evaporated to give yellow solids. The solids were then purified by preparative HPLC by using a Water XTerra™ Prep RPdC8 19/100 column, applying a gradient of CH3CN (0.1% TFA) in H2O (0.1% TFA) from 50% to 100% in 15 min (20 mL min−1). The pure products were obtained as yellow powders. The purity of the compounds was determined by UPLC using method 1.
Curc-C3-F9: (0.17 g). Yield: 62%, tR 5.80 min, 98% purity. 1H-NMR (600 MHz, DMSO-d6, 300 K), δ ppm: 9.69 (s, 1H, phenolic-OH), 8.08 (t, J = 5.6 Hz, 1H, H14), 7.61 (d, J = 15.9 Hz, 2H, overlapping H4/H4′), 7.43 (d, J = 1.9 Hz, 1H, H6), 7.37 (d, J = 1.9 Hz, 1H, H6′), 7.27 (dd, J = 8.3 and 1.9 Hz, 1H, H10), 7.20 (dd, J = 8.2 and 1.9 Hz, 1H, H10′), 6.97 (d, J = 8.3 Hz, 1H, H9), 6.88 (d, 15.9 Hz, 1H, H3), 6.87 (d, J = 8.2 Hz, 1H, H9′), 6.81 (d, J = 15.9 Hz, 1H, H3′), 6.13 (s, 1H, H1), 4.57 (s, 2H, H12), 4.13 (t, J = 6.2 Hz, 2H, H17), 3.90 (s, 3H, H20), 3.88 (s, 3H, H20′), 3.27 (q, J = 6.2 Hz, 2H, H15), 1.88 (m, J = 6.2 Hz, 2H, H16) (see ESI section 2.7† for details and NMR assignment). 19F-NMR (470 MHz, ethanol/DMSO-d6 550:50, 298 K), δ ppm (relative to TFA −76.55 ppm): −71.71 (s). 13C-NMR (150 MHz, δ ppm from 2D HSQC and 2D HMBC, DMSO-d6, 300 K): 184.4 (C2′), 182.8 (C2), 168.0 (C13), 150.0 (overlapping C7, C8, C8′), 148.5 (C7′), 141.4 (C4′), 140.3 (C4), 129.2 (C5), 126.7 (C5′), 123.7 (C10′), 123.0 (C3), 122.7 (C10), 121.6 (C3′), 116.3 (C9′), 114.3 (C9), 111.8 (C6′), 111.5 (C6), 101.3 (C1), 68.8 (C17), 68.6 (C12), 56.2 (overlapping C20, C20′), 35.2 (C15), 29.8 (C16). ESI-MS (m/z): calcd: for C30H28F9NO8 [M + H]+ 702.17 found: 702.19; [M + Na]+ 724.33.
Curc-C6-F9: (0.15 g). Yield: 52%, tR 6.58 min, 98% purity. 1H-NMR (600 MHz, DMSO-d6, 300 K), δ ppm: 9.70 (s, 1H, phenolic-OH), 7.95 (t, J = 5.7 Hz, 1H, H14), 7.60 (d, J = 15.8 Hz, 2H, overlapping H4/H4′), 7.43 (d, J = 1.9 Hz, 1H, H6), 7.36 (d, J = 1.9 Hz, 1H, H6′), 7.27 (dd, J = 8.4 and 1.9 Hz, 1H, H10), 7.20 (dd, J = 8.4 and 1.9 Hz, 1H, H10′), 6.97 (d, J = 8.4 Hz, 1H, H9), 6.88 (d, J = 15.8 Hz, 1H, H3), 6.86 (d, J = 8.4 Hz, 1H, H9′), 6.80 (d, J = 15.8 Hz, 1H, H3′), 6.12 (s, 1H, H1), 4.56 (s, 2H, H12), 4.09 (t, J = 6.2 Hz, 2H, H20), 3.90 (s, 3H, H23), 3.88 (s, 3H, H23′), 3.15 (q, J = 6.2 Hz, 2H, H15), 1.66 (m, J = 6.4 Hz, 2H, H19), 1.46 (m, J = 7.4 Hz, 2H, H16), 1.37 (m, J = 7.4 Hz, 2H, H18), 1.295 (m, J = 7.4 Hz, 2H, H17) (see ESI section 2.8† for details and NMR assignment). 19F-NMR (470 MHz, ethanol/DMSO-d6 550:50, 298 K), δ ppm (relative to TFA −76.55 ppm): −71.79 (s). 13C-NMR (150 MHz, δ ppm from 2D HSQC and 2D HMBC, DMSO-d6, 300 K): 184.4 (C2′), 182.7 (C2), 167.7 (C13), 149.8 (overlapping C7, C8, C8′), 148.7 (C7′), 141.5 (C4′), 140.2 (C4), 129.2 (C5), 126.7 (C5′), 123.7 (C10′), 123.0 (C3), 122.9 (C10), 121.6 (C3′), 116.2 (C9′), 114.4 (C9), 111.8 (C6′), 111.5 (C6), 101.4 (C1), 70.7 (C20), 68.5 (C12), 56.15 (overlapping C23, C23′), 38.6 (C15), 29.55 (C19), 29.3 (C16), 26.3 (C17), 24.9 (C16). ESI-MS (m/z): calcd: for C33H34F9NO8 [M + H]+ 744.21 found: 744.28; [M + Na]+ 766.25.
Curc-Glu-F9: (0.08 g). Yield: 34%, tR 4.13 min, 97% purity. 1H-NMR (600 MHz, DMSO-d6, 300 K), δ ppm: 9.70 (s, 1H, phenolic-OH), 8.24 (br, 1H, H14), 7.92 (t, J = 5.6 Hz, 1H, H19), 7.60 (d, J = 15.8 Hz, 2H, overlapping H4/H4′), 7.43 (d, J = 1.6 Hz, 1H, H6), 7.36 (d, J = 1.6 Hz, 1H, H6′), 7.26 (dd, J = 8.4 and 1.6 Hz, 1H, H10), 7.20 (dd, J = 8.4 and 1.6 Hz, 1H, H10′), 7.01 (d, J = 8.3 Hz, 1H, H9), 6.88 (d, J = 15.8 Hz, 1H, H3), 6.86 (d, J = 8.4 Hz, 1H, H9′), 6.80 (d, J = 15.8 Hz, 1H, H3′), 6.13 (s, 1H, H1), 4.64 (AB system, 2H, H12), 4.28 (m, br, 1H, H15), 4.11 (t, J = 6.2 Hz, 2H, H22), 3.90 (s, 3H, H26), 3.88 (s, 3H, H26′), 3.15 (q, J = 6.6 Hz, 2H, H20), 2.16 (m, 2H, H17), 2.06 (m, 1H, H16a), 1.87 (m, 1H, H16b), 1.81 (m, J = 6.5 Hz, 2H, H21) (see ESI section 2.9† for details and NMR assignment). 19F-NMR (470 MHz, ethanol/DMSO-d6 550:50, 298 K), δ ppm (relative to TFA −76.55 ppm): −71.80 (s). 13C-NMR (150 MHz, δ ppm from 2D HSQC and 2D HMBC, DMSO-d6, 300 K): 184.3 (C2′), 182.8 (C2), 171.7 (C18), 168.0 (C13), 149.8 (overlapping C7, C8, C8′), 148.5 (C7′), 141.4 (C4′), 140.2 (C4), 129.4 (C5), 126.8 (C5′), 123.8 (C10′), 123.1 (C3), 122.9 (C10), 121.6 (C3′), 116.2 (C9′), 114.5 (C9), 111.8 (C6′), 111.6 (C6), 101.3 (C1), 68.9 (C22), 68.2 (C12), 56.3 (overlapping C26, C26′), 52.3 (C15), 35.3 (C20), 32.03 (C17), 29.9 (C21), 27.7 (C16). ESI-MS (m/z): calcd: for C35H35F9N2O11 [M + H]+ 831.21 found: 831.24; [M + Na]+ 753.14, [M + 2H]+ 416.32.
Synthesis of disubstituted curcumin derivatives (Curc-C6-F18, Curc-Glu-F18)
Compound Curc-C6-F18 and Curc-Glu-F18 were obtained by performing the same procedure, starting from bis-NHS ester of curcumin (compound 3b, 0.7 g, 0.11 mmol) and perfluoroamine derivatives (0.25 mmol).
Curc-C6-F18: (0.06 g). Yield: 48%, tR 8.73 min, 96% purity. 1H-NMR (CD3CN, 600 MHz, 310 K), δ ppm: 7.60 (d, J = 15.9 Hz, 2H, H4), 7.30 (s, 2H, H6), 7.19 (d, J = 8.0 Hz, 2H, H10), 7.01 (t, br, 2H, H14), 6.95 (d, J = 8.0 Hz, 2H, H9), 6.75 (d, J = 15.9 Hz, 2H, H3), 5.95 (s, 1H, H1), 4.50 (s, 4H, H12), 4.05 (t, J = 5.6 Hz, 4H, H20), 3.91 (s, 6H, H23), 3.22 (q, J = 6.6 Hz, 4H, H15), 1.64 (m, 4H, H19), 1.48 (m, 4H, H16), 1.37 (m, 4H, H18), 1.29 (m, 4H, H17) (see ESI section 2.10† for details and NMR assignment). 19F-NMR (470 MHz, ethanol/DMSO-d6 550:50, 298 K), δ ppm (relative to TFA −76.55 ppm): −71.84 (s). ESI-MS (m/z): calcd: for C45H48F18N2O10 [M + 2H]2+ 560.19, found: 560.25.
Curc-Glu-F18: (0.06 g). Yield: 32%, tR 5.08 min, 98% purity. 1H-NMR (600 MHz, DMSO-d6, 300 K), δ ppm: 8.29 (d, J = 7.7 Hz, 2H, H14), 7.92 (t, J = 5.6 Hz, 2H, H19), 7.62 (d, J = 15.8 Hz, 2H, H4), 7.43 (d, J = 1.5 Hz, 2H, H6), 7.27 (dd, J = 8.4 and 1.5 Hz, 2H, H10), 7.01 (d, J = 8.4 Hz, 2H, H9), 6.90 (d, J = 15.8 Hz, 2H, H3), 6.15 (s, 1H, H1), 4.65 (AB system, 4H, H12), 4.30 (m, 2H, H15), 4.11 (t, J = 6.2 Hz, 4H, H22), 3.91 (s, 6H, H26), 3.15 (m, 4H, H20), 2.17 (m, 4H, H17), 2.06 (m, 2H, H16a), 1.88 (m, 2H, H16b), 1.82 (m, J = 6.5 Hz, 4H, H21) (see ESI section 2.11† for details and NMR assignment). 19F-NMR (470 MHz, ethanol/DMSO-d6 550:50, 298 K), δ ppm (relative to TFA −76.55 ppm): −71.72 (s). ESI-MS (m/z): calcd: for C49H50F18N4O16 [M + 2H]2+ 647.14, found: 647.39.
In vitro assays
Stock solutions of the 19F-labelled curcumin probes were prepared by dissolving the required mass of the probe into 5 mL of analytical grade ethanol to obtain a concentration in the 0.5–3.0 mM range. The exact concentration of stock solutions was assessed by quantitative 19F-NMR spectroscopy (qNMR, see ESI section 1.2†).Preparation of amyloid-beta (Aβ) aggregates
5 mg of Aβ (1–42) (Anaspec) lyophilized powder were solubilized in 7 mL HFIP (hexafluoro-2-propanol), portioned, evaporated ON at room temperature and stored at −20 °C.An Aβ aliquot (45 μg) was solubilized in 20 μL of a freshly prepared mixture consisting of CH3CN/300 μM Na2CO3/250 mM NaOH (48.3/48.3/3.4, v/v/v) to obtain a 500 μM stock solution (pH 13). The stock solution was sonicated in a water bath to remove preformed aggregates. The Aβ peptide solution was diluted 1:10 (v/v) in phosphate buffer 10 mM pH 7.4 containing 11 mM NaCl22 and then incubated at 4 °C, no stirring for 24 hours to have Aβ oligomers,31 or at 37 °C, 600 rpm for 4 days to have Aβ aggregates in protofibrillar and fibrillar state.
Field emission scanning electron microscopy (FESEM)
Morphological analysis of Aβ aggregates was performed by FESEM. After 5 days of incubation, the samples are diluted to remove excess salts, centrifuged and the supernatant removed. The pellet was resuspended in pure water and 10 μL of this solution was spotted onto a gold-coated glass coverslip. To assess fibril morphology, images of the samples are acquired using a Tescan FEG-SEM S9000. Measurements are acquired with a Schotty emitter, the probe set at 100 pA and the electron beam energy at 5 keV. The analysis was performed with an in-beam SE detector. Microanalysis was performed using OXFORD – Ultim Max detector – AZTEC software.Binding assay to amyloid-beta (Aβ) aggregates
The fluorometric assays were performed with a Horiba Jobin Yvon spectrofluorometer Fluoromax-4 (Kyoto, Japan). Stock solution (3 mM) of commercial curcumin, Curc-C3-F9, Curc-C6-F9, Curc-Glu-F9, Curc-C6-F18, Curc-Glu-F18 were prepared in 99% (v/v) ethanol. The assay was performed using a fixed concentration of reporter ligand (between 20–30 nM) and varying concentrations of Aβ aggregates (0–3.3 μM). After 4 days of incubation, the Aβ solutions (50 μM) were diluted with phosphate buffer 10 mM pH 7.4 plus 11 mM NaCl and 0.5% ethanol, containing the reporter ligand, up to a final volume of 2.0 ml. The fluorescence emission signal was monitored at 495, 497 nm, or 488 nm (λex = 450 nm) with excitation and emission slits of 5 nm bandwidth. An emission calibration curve for each compound was prepared to determine their molar fluorescence emission observed when the compound is unbound. From the plot of the S1/R1 fluorescent emission at the respective wavelength, fitted by using [eqn (1)], it was possible to obtain a thermodynamic dissociation constant assuming a 1:1 interaction with Aβ. | (1) |
K a = association constant, Ct = compound fixed concentration, n = number of binding sites, x = Aβ concentration, Fb = molar fluorescence intensity observed at the peak of the emission spectra when the compound is bound to fibrils, Ff = molar fluorescence intensity observed at the peak of the emission spectra when the compound is free, Fpr = molar fluorescence emission observed at the peak of the emission spectra for Aβ aggregates, BKG = background fluorescence (buffer plus 0.5% ethanol).
Human serum albumin (HSA) binding assay
The fluorometric assays were performed with a Shimadzu RF-6000. Stock solution (3 mM) of commercial curcumin, Curc-C3-F9, Curc-C6-F9, Curc-Glu-F9, Curc-C6-F18, Curc-Glu-F18 were prepared in 99% (v/v) ethanol. The assay was performed using a fixed concentration of reporter ligand (about 30 nM) and varying concentrations of HSA (0–27 μM) (lyophilized powder, ≥96% (agarose gel electrophoresis), purchased from Sigma-Aldrich). The HSA stock solution (2 mg mL−1) was diluted with phosphate buffer 10 mM pH 7.4 plus 11 mM NaCl and 0.5% ethanol, containing the reporter ligand, up to a final volume of 2.0 ml. The fluorescence emission signal was monitored at 485 nm (λex = 430 nm) with excitation and emission slits of 5 nm bandwidth. The plot of the S1 fluorescent emission at 485 nm vs. HSA concentration was fitted by using [eqn (1)] substituting HSA to Aβ thus obtaining the thermodynamic association constant assuming a 1:1 interaction with the protein.
Solubility test in physiological buffer
Solubility tests were carried out by solubilizing the compound in NaCl/HEPES buffer (4,2-hydroxyethyl-1-piperazineethanesulfonic acid) at 300 mOsm with a concentration of 4.5 mM of each compound. It was decided to operate at a surfactant concentration of CREMOPHOR EL ranging from 10% to 20% v/v, depending on the compound's degree of lipophilicity and behavior during the test. To aid solubilization, the solutions were sonicated (40 kHz) for 5 minutes at 30 °C between each buffer addition. Finally, the pH of the mixture was monitored using a pH meter and maintained at a value of 7.4 (physiological pH).Longitudinal and transverse (T1 and T2) relaxation times measurements
To measure the longitudinal (T1) and transverse (T2) 19F-NMR relaxation times, Curc-Glu-F9 and Curc-Glu-F18 were solubilized in NaCl/HEPES buffer (300 mOsm) + 10% v/v CREMOPHOR EL, while Curc-C6-F9 in NaCl/HEPES buffer (300 mOsm) + 20% v/v CREMOPHOR EL. Measurements were conducted at room temperature by means of a Bruker Avance microimaging scanner operating at 7 T, corresponding to 300 and 282 MHz proton and 19F Larmor frequency, respectively. The scanner was equipped with a 40 mm 1H/19F volume transmit-receive probe. T1 was measured by inversion recovery with repetition time = 5 s, number of averages = 10, number of variable delays = 16. T2 was measured by means of the CPMG pulse sequence, with the same parameters as above and an echo time of 0.0002 s.MRI image acquisition
Experiments were conducted at room temperature using the Bruker Avance 300 MHz magnet (7 T), equipped with a 40 mm 1H/19F volume transmit-receive probe. Samples were solubilized as previously reported at a concentration of 4.5 mM. First, a T2-weighted 1H image was acquired to localize the samples with the following parameters: echo time (ms): 72.79, repetition time (s): 4, rare factor: 32, matrix: 256 × 256, FOV (cm): 3.5 × 3.5, slice thickness (mm): 3, acquisition time (min): 1.40. Then a 19F MR image was acquired with the following parameters: SFO1 (MHz) 282.3903307, echo time (ms): 3, repetition time (s): 1, rare factor: 24, matrix: 32 × 32, FOV (cm): 3.5 × 3.5, slice thickness (mm): 3: number of averages: 630, acquisition time (min): 10.30. The signal to noise ratio (SNR) for each sample was calculated using the formula:
Here, Ssample represents the average 19F signal measured within a specific region of interest (ROI) for each sample, SH2O is the average 19F signal measured in a control sample without fluorine, and SDnoise denotes the standard deviation of the 19F signal measured in the background of the image, capturing the statistical intensity distribution of the noise.
19F-NMR spectroscopy
19F-NMR spectroscopy to study the interaction between the fluorinated curcumin derivatives and Aβ aggregates was performed with a Bruker AvanceNeo 9.4 T NMR spectrometer (376 MHz, 19F Larmor frequency). A stock solution (3 mM) of Curc-Glu-F9 was prepared in 99% (v/v) ethanol. The Aβ aggregates samples (monomers, oligomers and fibrils) were diluted with phosphate buffer 10 mM pH 7.4 plus 11 mM NaCl, 16% D2O and 3.5% ethanol, containing the fluorinated compound, up to a final volume of 0.3 ml. The NMR spectra were acquired at 298 K using the following parameters: spectral width (sw) 30120.482 Hz, relaxation delay (d1) 2.5 s, number of scans (ns) 256–512.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included in the text and in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 964934, Nectar project. The authors acknowledge the Italian Ministry of Research for FOE contribution to the EuroBioImaging MultiModal Molecular Imaging Italian Node (https://www.mmmi.unito.it). G. D. acknowledges MUR PRIN 2022 funding (project 2022598YAX, OPTIMA). F. G. acknowledges financial support from PON Ricerca e Innovazione 2014–2020, Action IV.6. Maria Carmen Valsania and Erica Rebba are acknowledge for the acquisition of FESEM images at the Chemistry Department of the University of Turin.References
- (a) R. U. Haquea and A. I. Levey, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(52), 26224–26229 CrossRef PubMed; (b) A. Serrano-Pozo, M. P. Frosch, E. Masliah and B. T. Hyman, Cold Spring Harbor Perspect. Med., 2011, 1(1), a006189 Search PubMed.
- (a) J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed; (b) J. Lewis, D. W. Dickson, W. L. Lin, L. Chisholm, A. Corral, G. Jones, S. H. Yen, N. Sahara, L. Skipper, D. Yager, C. Eckman, J. Hardy, M. Hutton and E. McGowan, Science, 2001, 293(5534), 1487–1491 CrossRef CAS PubMed.
- (a) W. F. Xue, A. L. Hellewell, E. W. Hewitt and S. E. Radford, Prion, 2010, 4, 20–25 CrossRef CAS PubMed; (b) J. C. Lee, S. J. Kim, S. Hong and Y. Kim, Exp. Mol. Med., 2019, 51, 1–10 Search PubMed.
- X. Zhang and C. Ran, Curr. Org. Chem., 2013, 17(6), 580–593 CrossRef CAS PubMed.
- F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, C. Pingping, K. Rakez, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS PubMed.
- G. Si, S. Zhou, G. Xu, J. Wang, B. Wu and S. Zhou, Dyes Pigm., 2019, 163, 509–515 CrossRef CAS.
- G. P. Lim, T. Chu, F. Yang, W. Beech, S. A. Frautschy and G. M. Cole, J. Neurosci., 2001, 21(21), 8370–8377 CrossRef CAS PubMed.
- V. L. Villemagne, V. Dore, P. Bourgeat, S. C. Burnham, S. Laws, O. Salvado, C. L. Masters and C. C. Rowe, Semin. Nucl. Med., 2017, 47, 75–88 CrossRef PubMed.
- W. E. Klunk, H. Engler, A. Nordberg, Y. Wang, G. Blomqvist, D. P. Holt, M. Bergström, I. Savitcheva, G. F. Huang, S. Estrada, B. Ausén, M. L. Debnath, J. Barletta, J. C. Price, J.- Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis and B. Långström, Ann. Neurol., 2004, 55(3), 306–319 CrossRef CAS PubMed.
- W. Zhang, S. Oya, M. P. Kung, C. Hou, D. L. Maier and H. F. Kung, J. Med. Chem., 2005, 48(19), 5980–5988 CrossRef CAS PubMed.
- (a) E. K. Degenhardt, M. M. Witte, M. G. Case, P. Yu, D. B. Henley, H. M. Hochstetler, D. N. D'Souza and P. T. Trzepacz, Psychosomatics, 2016, 57, 208–216 CrossRef PubMed; (b) L. Filippi, A. Chiaravalloti, O. Bagni and O. Schillaci, 18F-labeled radiopharmaceuticals for the molecular neuroimaging of amyloid plaques in Alzheimer's disease, Am. J. Nucl. Med. Mol. Imaging, 2018, 8, 268–281 CAS.
- D. Yanagisawa, T. Amatsubo, S. Morikawa, H. Taguchi, M. Urushitani, N. Shirai, K. Hirao, A. Shiino, T. Inubushi and I. Tooyama, Neuroscience, 2011, 184, 120–127 CrossRef CAS PubMed.
- D. Yanagisawa, H. Taguchi, S. Morikawa, T. Kato, K. Hirao, N. Shirai and I. Tooyama, Biochem. Biophys. Rep., 2015, 4, 357–368 Search PubMed.
- I. Tooyama, D. Yanagisawa, H. Taguchi, T. Kato, K. Hirao, N. Shirai, T. Sogabe, N. F. Ibrahim, T. Inubushi and S. Morikawa, Ageing Res. Rev., 2016, 30, 85–94 CrossRef CAS PubMed.
- D. Yanagisawa, N. Shirai, T. Amatsubo, H. Taguchi, K. Hirao, M. Urushitani, S. Morikawa, T. Inubushi, M. Kato, F. Kato, K. Morino, H. Kimura, I. Nakano, C. Yoshida, T. Okada, M. Sano, Y. Wada, K. Wada, A. Yamamoto and I. Tooyama, Biomaterials, 2010, 31(14), 4179–4185 CrossRef CAS PubMed.
- D. Yanagisawa, N. F. Ibrahim, H. Taguchi, S. Morikawa, T. Tomiyama and I. Tooyama, Molecules, 2021, 26(5), 1362 CrossRef CAS PubMed.
- T. Amatsubo, S. Morikawa, T. Inubushi, M. Urushitani, H. Taguchi, N. Shirai, K. Hirao, M. Kato, K. Morino, H. Kimura, I. Nakano, C. Yoshida, T. Okada, M. Sano and I. Tooyama, Neurosci. Res., 2009, 63(1), 76–81 CrossRef CAS PubMed.
- E. Chainoglou and D. H. Litina, Int. J. Mol. Sci., 2020, 21(6), 1975 CrossRef CAS PubMed.
- G. Schitter, A. J. Steiner, G. Pototschnig, E. Scheucher, M. Thonhofer, C. A. Tarling, S. G. Withers, K. Fantur, E. Paschke, D. J. Mahuran, B. A. Rigat, M. B. Tropak, C. Illaszewicz, R. Saf, A. E. Stütz and T. M. Wrodnigg, ChemBioChem, 2010, 11(14), 2026–2033 CrossRef CAS PubMed.
- S. Nasir Abbas Bukhari and I. Jantan, Mini-Rev. Med. Chem., 2015, 15(13), 1110–1121 CrossRef PubMed.
- B. Laha, A. R. Tiwari, E. Gravel, E. Doris and I. N. N. Namboothiri, Org. Biomol. Chem., 2024, 22(7), 1346–1359 RSC.
- A. De Simone, M. Naldi, D. Tedesco, A. Milelli, M. Bartolini, L. Davani, D. Widera, M. L. Dallas and V. Andrisano, ACS Omega, 2019, 4(7), 12308–12318 CrossRef CAS PubMed.
- M. Groenning, J. Chem. Biol., 2010, 3(1), 1–18 CrossRef PubMed.
- (a) G. Si, S. Zhou, G. Xu, J. Wang, B. Wu and S. Zhou, Dyes Pigm., 2019, 163, 509–515 CrossRef CAS; (b) J. Den Haan, T. H. J. Morrema, A. J. Rozemuller, F. H. Bouwman and J. J. Hoozemans, Acta Neuropathol. Commun., 2018, 6(1), 75 CrossRef PubMed.
- (a) K. I. Priyadarsini, Photophysics, J. Photochem. Photobiol., C, 2009, 10(2), 81–95 CrossRef CAS; (b) A. Barik, K. I. Priyadarsini and H. Mohan, Photochem. Photobiol., 2007, 77(6), 597–603 CrossRef.
- Z. Qia, M. Wua, Y. Fua, T. Huanga, T. Wanga, Y. Suna, Z. Fengc and C. Lia, Cell. Physiol. Biochem., 2017, 44, 618–633 CrossRef PubMed.
- A. Barik, B. Mishra, A. Kunwar and K. I. Priyadarsini, Chem. Phys. Lett., 2007, 436, 239–243 CrossRef CAS.
- Y. W. Kim, K. N. Chung, H. S. Kang and Y. Y. Sheen, Biomol. Ther., 2008, 16(1), 40–45 CrossRef CAS.
- (a) Y. C. Youn, B. S. Lee, G. J. Kim, J. S. Ryu, K. Lim, R. Lee, J. Suh, Y. H. Park, J. M. Pyun, N. Ryu, M. J. Kang, H. R. Kim, S. Kang, S. S. A. An and S. Kim, J. Alzheimer's Dis., 2020, 75, 493–499 CAS; (b) S. M. Wang, D. W. Kang, Y. H. Um, S. Kim, C. U. Lee, P. Scheltens and H. K. Li, Alzheimer's Res. Ther., 2024, 16(1), 55 CrossRef CAS PubMed.
- H. Amiri, M. Srinivas, A. Veltien, M. J. van Uden, J. M. de Vries and A. Heerschap, Eur. Radiol., 2015, 25, 726–735 CrossRef PubMed.
- W. B. Stine, K. N. Dahlgren, G. A. Krafft and M. J. LaDu, J. Biol. Chem., 2003, 278, 11612–11622 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00730a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |