
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of chalcogen-bonding interactions and their role in the trans–trans conformation of thiourea†
Takumi
Inoue
a,
Nami
Morita
a,
Yui
Amijima
a,
Rika
Sakai
a,
Shohei
Hamada
 a,
Seikou
Nakamura
b,
Yusuke
Kobayashi
a and
Takumi
Furuta
*a
a,
Seikou
Nakamura
b,
Yusuke
Kobayashi
a and
Takumi
Furuta
*a
aLaboratory of Pharmaceutical Chemistry, Kyoto Pharmaceutical University, Yamashina-ku, Kyoto 607-8414, Japan. E-mail: furuta@mb.kyoto-phu.ac.jp
bLaboratory of Pharmacognosy, Kyoto Pharmaceutical University, Yamashina-ku, Kyoto 607-8414, Japan
First published on 14th June 2024
Abstract
The chalcogen-bonding interactions formed at both sides of the thiocarbonyl group in thiourea were investigated. In particular, the role of these chalcogen-bonding interactions in the trans–trans conformation of thiourea was evaluated via single-crystal X-ray diffraction analysis and DFT calculations. The obtained results indicate that the Se⋯S⋯Se dual chalcogen-bonding interactions play a stronger role in controlling the planar structure than the S⋯S⋯S interactions.
Thiourea derivatives have been recognized as privileged organocatalysts and ligands for a variety of organic transformations.1 For the development of thiourea-based organocatalysts with high catalytic performance and stereoselectivity, their stereostructures are of crucial importance.
Thiourea derivatives such as N,N′-diaryl thiourea, possess four rotatable C–N single bonds (a, a′, b, and b′ in Fig. 1A), which results in a rich conformational variety. For example, rotation of the b and b′ bonds leads to trans–trans, cis–trans, and cis–cis conformations,2,3 while rotation around the a and a′ bonds in the case of derivatives that bear R substituents causes a different substituent geometry as typically shown in the trans–trans conformation.
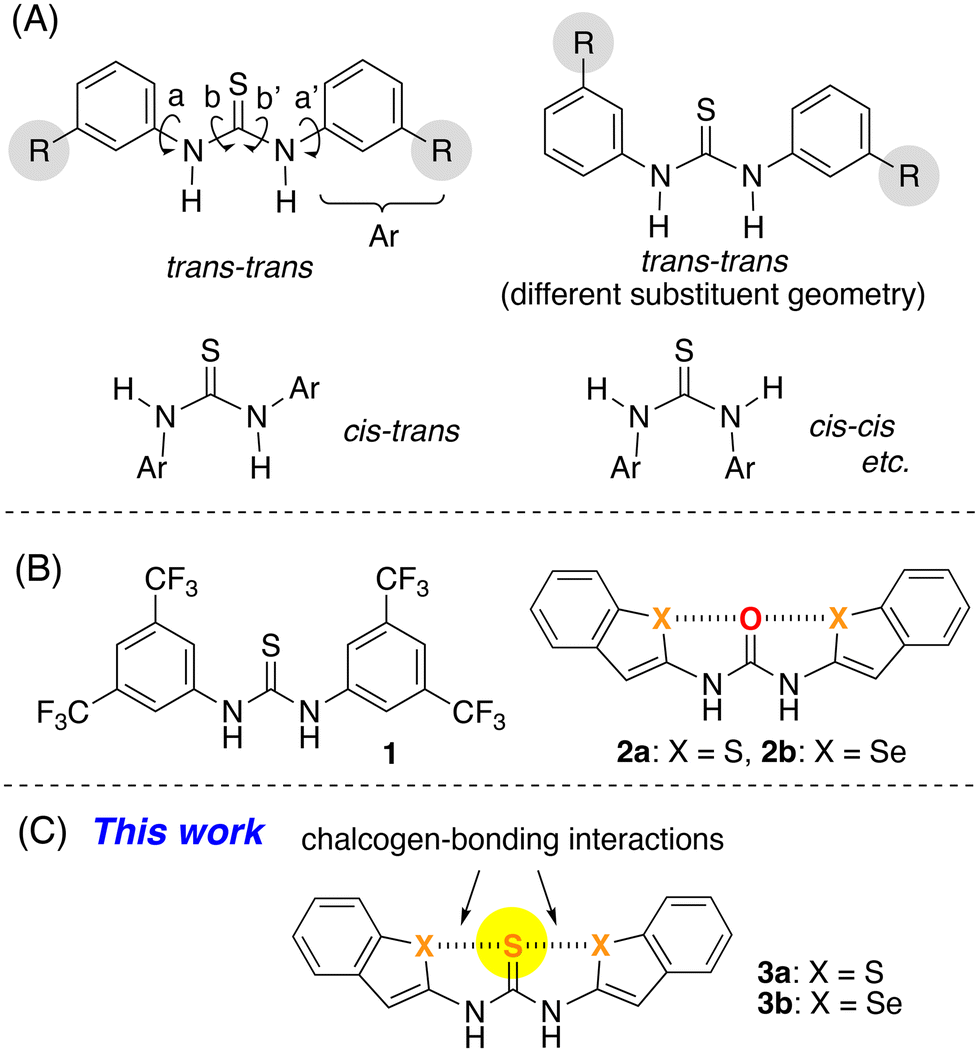 | ||
| Fig. 1 (A) Conformations of N,N′-diaryl thiourea derivatives bearing substituents (R). (B) Previously reported urea and thiourea derivatives with trans–trans conformation. (C) This work. | ||
Among the conformations of thiourea, the trans–trans conformation is expected to show superior catalytic activity and molecular recognition ability by virtue of the hydrogen-bonding interactions involving both N–H groups. Therefore, establishing conformational control over thiourea to obtain the trans–trans conformation has attracted considerable attention. Wittkopp and Schreiner have developed a series of N,N′-diaryl thiourea derivatives including 1, which bears electron-withdrawing CF3 groups (Fig. 1B).4 It has been proposed that thiourea 1 adopts the trans–trans conformation due to the formation of intramolecular hydrogen-bonding interactions between the sulfur atom of the thiocarbonyl group and the acidic C–H groups at the ortho-position of the aniline moieties.
However, to the best of our knowledge, controlling the conformation of thiourea to favor the trans–trans conformation remains largely unexplored, apart from the aforementioned examples.
Chalcogen-bonding interactions represent attractive noncovalent interactions between chalcogen atoms (sulfur, selenium, and tellurium) and heteroatoms including oxygen and nitrogen.5 We have previously reported strategies to establish conformational control over urea derivatives 2a and 2b, which bear benzothiophene and benzoselenophene moieties, to obtain the trans–trans conformation with S⋯O⋯S and Se⋯O⋯Se arrangements by virtue of intramolecular chalcogen-bonding interactions (Fig. 1B).6 With this approach to establish conformational control in hand, we envisioned that the stereostructure of the corresponding thioureas 3a and 3b could also be controlled to favor the trans–trans conformation (Fig. 1C). However, it can be expected that the conformational flexibility is increased in the N,N′-diaryl thiourea derivative relative to the N,N′-diaryl urea derivative.2c Furthermore, the electronic repulsion between the heavy chalcogen atoms might induce adverse effects toward the formation of chalcogen-bonding interactions. Thus, in an initial step, we investigated the formation of chalcogen-bonding interactions at both sides of the thiocarbonyl group in the trans–trans conformation. Herein, the formation of chalcogen-bonding interactions in 3a and 3b and the evaluation of their role in the formation of the planar structure of the trans–trans conformation are described.
Thioureas 3a and 3b were prepared via benzothiophene and benzoselenophene isothiocyanate intermediates (Scheme 1). First, benzochalcogenophene carboxylic acids 4a and 4b, which bear sulfur and selenium atoms, respectively, were converted into the corresponding N-Boc derivatives (5a and 5b) via a Curtius rearrangement. Deprotection of the Boc groups under acidic conditions led to hydrochloride salts 6a and 6b, which were subjected to treatment with thiophosgene to give isothiocyanates 7a and 7b. Upon treatment of 7a and 7b with 1.5 equivalent of 6a and 6b in the presence of iPr2EtN in CH2Cl2 at −20 °C, the corresponding thioureas (3a and 3b) were obtained in 35% and 33% yield, respectively.
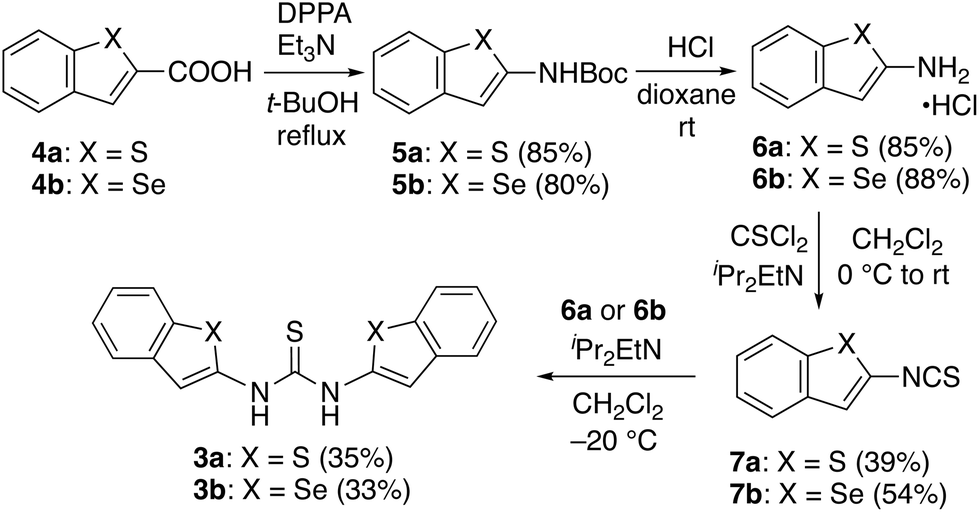 | ||
| Scheme 1 Synthesis of thioureas 3a and 3b. | ||
The stereostructure of 3a was determined unambiguously by an X-ray diffraction (XRD) analysis of a single crystal obtained from acetone.7 As shown in Fig. 2A, the three sulfur atoms are aligned on the same side of the molecule. The distances between the sulfur atoms of the benzothiophene and the thiocarbonyl moieties (3.0429(9) and 3.0810(9) Å, respectively) are both shorter than the sum of the van der Waals radii (3.6 Å) of two sulfur atoms. These short contacts indicate that chalcogen-bonding interactions are formed at both sides of the thiocarbonyl group, resulting in an S⋯S⋯S arrangement (red dotted lines). Due to these chalcogen-bonding interactions, the benzothiophene rings and the thiocarbonyl group are located almost on the same plane, albeit that a small twist of the thiocarbonyl group was observed (ϕ S’,C′–C,S = 18.6(1)°; ϕ S′′,C′′–C,S = −19.5(1)°) (Fig. 2A, top view).
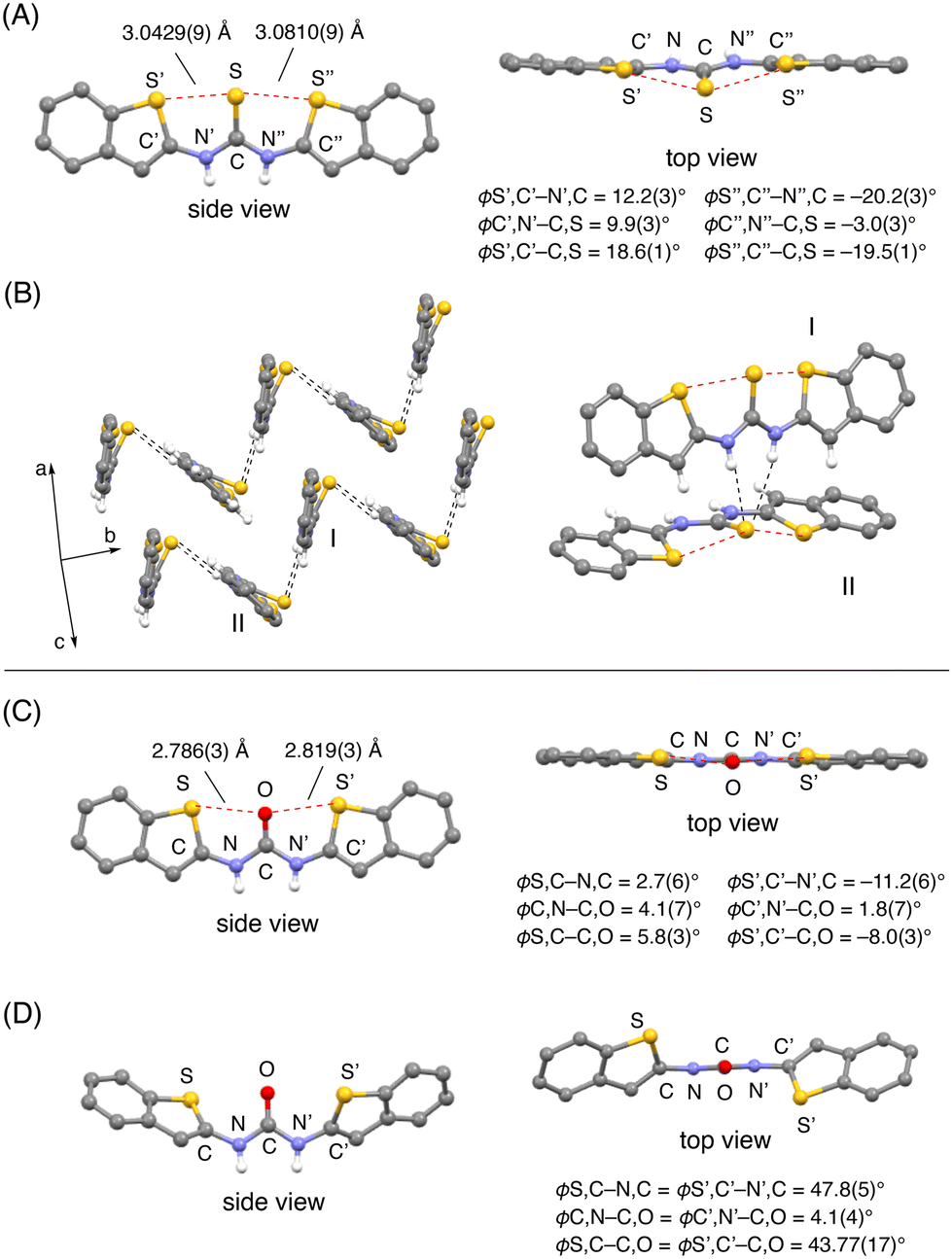 | ||
| Fig. 2 (A) Crystal structure of 3a. (B) Packing of 3a and associated molecules I and II. (C) Crystal structure of a single crystal of 2a obtained from acetone, exhibiting chalcogen-bonding interactions. (D) Crystal structure of a single crystal of 2a obtained from THF without chalcogen-bonding interactions. Only the hydrogen atoms of the N–H groups are depicted for clarity. The red and black dotted lines indicate chalcogen-bonding interactions and hydrogen-bonding interactions, respectively. | ||
This planar structure of 3a is a typical feature for the urea derivatives bearing the chalcogen-bonding interactions. In our previous study, the crystal structures of urea 2a with and without chalcogen-bonding interactions were determined.8 In the structure of 2a without chalcogen-bonding interactions, the dihedral angles between the carbonyl group and the benzothiophene moieties were significantly higher (ϕ S,C–C,O = ϕ S’,C′–C,O = 43.77(17)°) (Fig. 2D). On the other hand, 2a with chalcogen-bonding interactions exhibited the planar structure (Fig. 2C).
Accordingly, the planar structure with the small twist of the thiocarbonyl group in 3a supports the formation of chalcogen-bonding interactions.
It is also interesting to compare the dihedral angles of the thiocarbonyl group in 3a (ϕ S’,C′–C,S = 18.6(1)°; ϕ S′′,C′′–C,S = −19.5(1)°) with those in 2a with chalcogen-bonding interactions (ϕ S,C–C,O = 5.8(3)°; ϕ S’,C′–C,O = −8.0(3)°) (Fig. 2C). The larger dihedral angles in 3a might be due to (i) the presence of a repulsive interaction between the sulfur atoms in the thiocarbonyl group and the aromatic rings as initially expected, and/or (ii) the formation of intermolecular hydrogen-bonding interactions in the crystal. As shown in Fig. 2B, 3a assembles through intermolecular hydrogen-bonding interactions between the sulfur atom of the thiocarbonyl group and the N–H groups (black dotted lines) as shown for the representative molecules I and II (Fig. 2B).
To investigate the conformations of 3a and 3b in solution, nuclear Overhauser effect (NOE) studies were carried out in THF-d8 and in DMSO-d6, respectively. In both cases, NOE correlations were observed between the N–H protons and the protons at the 3,3′-positions (Fig. 3).9 These results suggest that both compounds adopt the trans–trans conformation with S⋯S⋯S and Se⋯S⋯Se arrangement in solution.
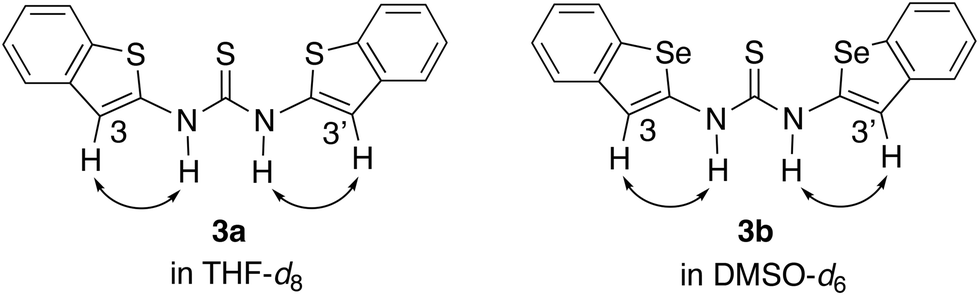 | ||
| Fig. 3 NOE correlations of 3a (in THF-d8) and 3b (in DMSO-d6). | ||
To gain further insight into the chalcogen-bonding interactions, the crystal structure of 3a and the DFT-optimized structure were subjected to an NBO analysis10 at the ωB97XD/6-311G(d,p) level11 under SMD(DMSO) conditions (Fig. 4A). The analysis of the crystal structure of 3a supports the presence of the chalcogen-bonding interactions at both sides of the sulfur atom of the thiocarbonyl group, indicating orbital overlap between the sulfur s-type lone pair (LP1)12 and the antibonding orbitals of the C–S bonds (σ*C–S), which were calculated to have second-order perturbation energies of 1.01 and 1.16 kcal mol−1, respectively. The overlap between the sulfur p-type lone pair (LP2)12 and the antibonding orbitals of the C–S bonds (σ*C–S) with energies of 3.81 and 4.37 kcal mol−1, respectively, was also confirmed (Fig. 4A). The NBO analysis of the DFT-optimized structure revealed similar stabilization energies (shown in parenthesis in Fig. 4A).
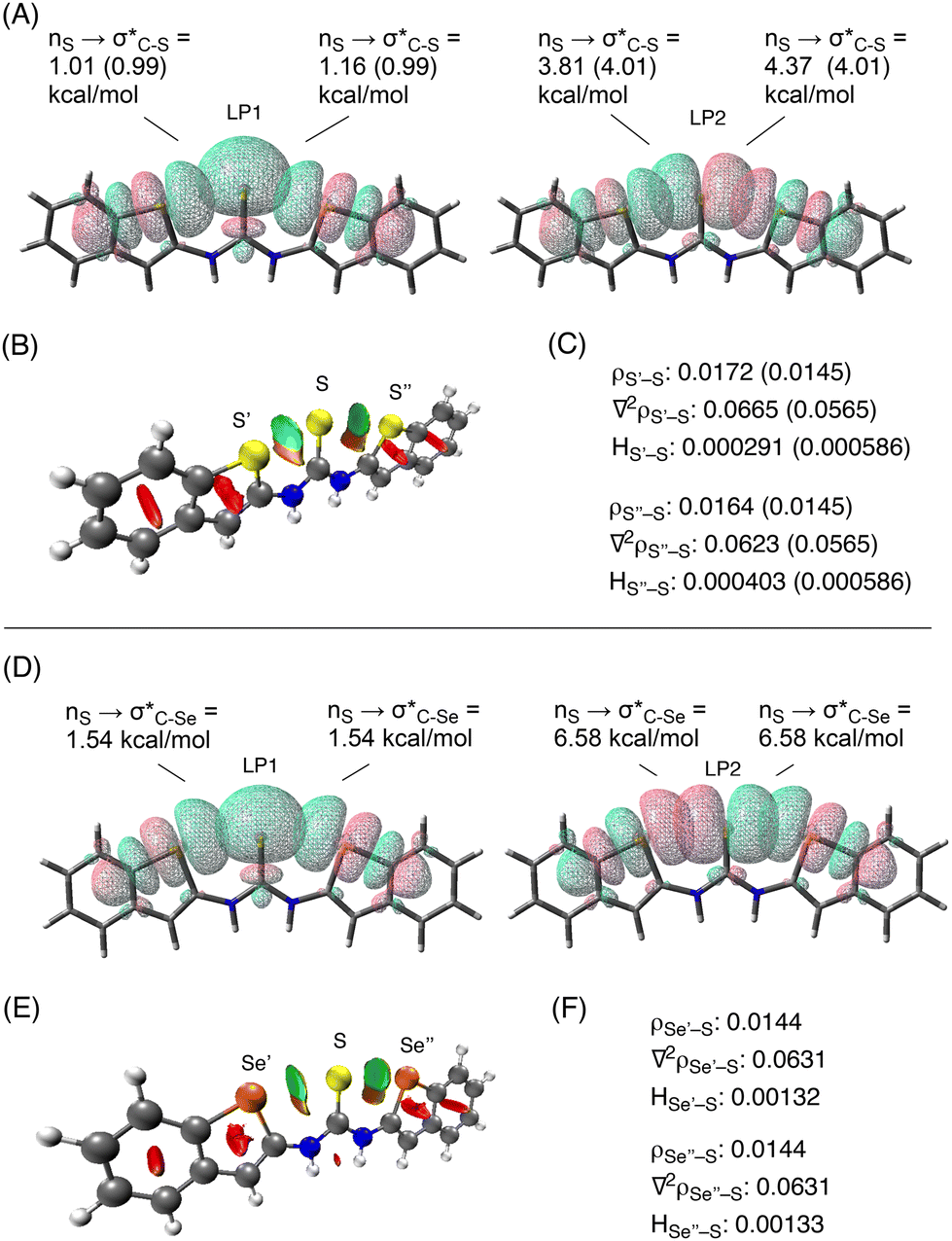 | ||
| Fig. 4 (A) NBO analysis of the crystal structure of 3a. Data in parenthesis refer to the values of the optimized structure. (B) NCI plot isosurfaces of the crystal structure of 3a. (C) Results of the QTAIM analysis of the crystal structure of 3a. Data in parenthesis refer to the values of the optimized structure. (D) NBO analysis of the optimized structure of 3b. (E) NCI plot isosurfaces of the optimized structure of 3b. (F) Results of the QTAIM analysis of the optimized structure of 3b. The calculations were performed at the ωB97XD/6-311G(d,p) level under SMD(DMSO) conditions. | ||
The noncovalent interaction (NCI) plot index analysis13 of the crystal structure of 3a also supports the presence of the chalcogen-bonding interactions at both sides of the thiocarbonyl group (indicated by the green areas between S’ and S and between S and S′′ in Fig. 4B). Furthermore, the quantum theory of atoms in molecules (QTAIM) analysis14 of the crystal structure showed electron-density values (ρ = 0.0172 and 0.0164 a.u.) at the bond critical points (BCPs), which is comparable to those previously reported for chalcogen-bonding interactions (Fig. 4C).15 The depletion of electron density at the BCPs, which suggests that the interactions are noncovalent, is also supported by positive Laplacian values (∇2ρS′–S: 0.0665, ∇2ρS′′–S: 0.0623). Moreover, the positive values of HS′–S (0.000291) and HS′′–S (0.000403) indicate that the interactions are electrostatic in nature. In their entirety, these results confirm the formation of chalcogen-bonding interactions between the sulfur atoms in 3a.
To evaluate the chalcogen-bonding interactions between the selenium and the sulfur atoms in 3b, the structure was optimized and analyzed at the ωB97XD/6-311G(d,p) level11 under SMD(DMSO) conditions. The NBO, NCI, and QTAIM data supported the formation of chalcogen-bonding interactions (Fig. 4D–F). It is worth noting that the chalcogen-bonding interactions calculated via NBO analysis for 3b are stronger than those calculated for 3a (1.54 kcal mol−1 for LP1 and 6.58 kcal mol−1 for LP2 in 3bvs. 0.99 kcal mol−1 for LP1 and 4.01 kcal mol−1 for LP2 in optimized 3a).16
Therefore, the population of the trans–trans conformation of 3b with the Se⋯S⋯Se arrangement can be expected to be larger than that of 3a with the S⋯S⋯S arrangement.
This prediction was supported by the conformational analysis of trans–trans3a and 3b on the basis of the calculated S,C–N,C and S′,C′–N′,C dihedral angles and the Se,C–N,C and Se′,C′–N′,C dihedral angles (Fig. 5). In the case of 3a, the trans–trans planar conformation I with the S⋯S⋯S arrangement should be present, as indicated by the NOE study (Fig. 3). However, even in the presence of chalcogen-bonding interactions, conformation I was calculated to be less stable (+0.69 kcal mol−1 at the ωB97XD/6-311G(d,p) level under SMD(DMSO) conditions) than conformation II, wherein the benzothiophene ring adopts a perpendicular arrangement relative to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S group (Fig. 5A). The Boltzmann population for conformations I, II, and IV was calculated to be 22.9, 73.6, and 3.5% under SMD(DMSO) conditions. In the calculations under SMD(THF) conditions, the conformations exhibit the same order of stability.
S group (Fig. 5A). The Boltzmann population for conformations I, II, and IV was calculated to be 22.9, 73.6, and 3.5% under SMD(DMSO) conditions. In the calculations under SMD(THF) conditions, the conformations exhibit the same order of stability.
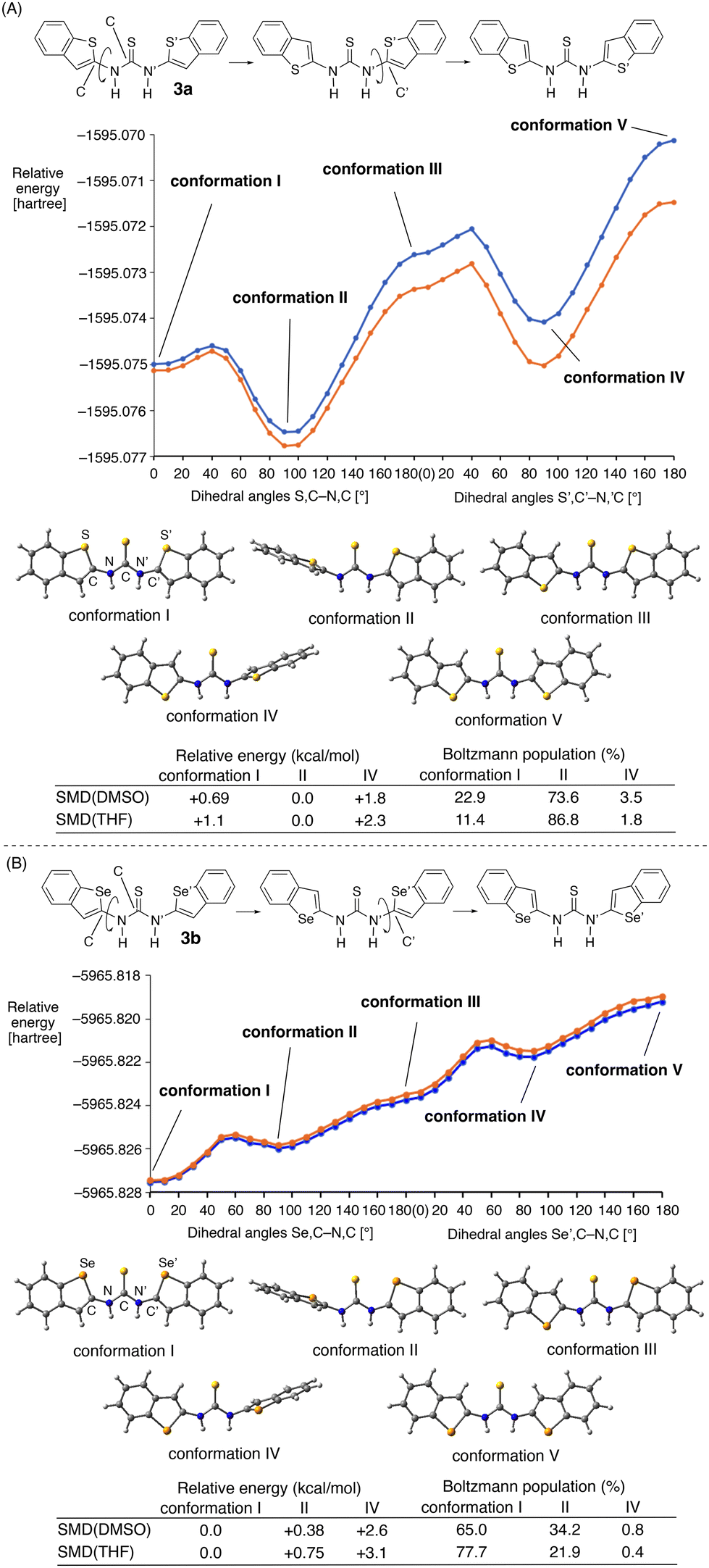 | ||
| Fig. 5 (A) Conformational analysis of 3a on the basis of the S,C–N,C and S’,C′–N′,C dihedral angles (blue dotted line: SMD(DMSO), orange dotted line: SMD(THF)), structures obtained under SMD(DMSO) conditions, as well as relative energies and Boltzmann population of the conformations. (B) Conformational analysis of 3b on the basis of the Se,C–N,C and the Se′,C′–N′,C dihedral angles (blue dotted line: SMD(DMSO), orange dotted line: SMD(THF)), structures obtained under SMD(DMSO) conditions, as well as relative energies and Boltzmann population of the conformations. The calculations were performed at the ωB97XD/6-311G(d,p) level. | ||
In contrast, the trans–trans conformation I of 3b with the Se⋯S⋯Se arrangement was by 0.38 and 0.75 kcal mol−1 more stable than conformation II under SMD(DMSO) and SMD(THF) conditions, respectively (Fig. 5B). The Boltzmann populations were calculated to be 65.0% and 77.7% under SMD(DMSO) and SMD(THF) conditions, respectively. This reversal of the order of stability between conformations I and II could potentially be attributed to the formation of stronger chalcogen-bonding interactions between the selenium and the sulfur atoms than between the sulfur atoms in 3a. These results confirm the formation of chalcogen-bonding interactions at both sides of the thiocarbonyl group in the trans–trans conformation, and, in particular, that the sulfur–selenium interactions contribute more strongly to the control of the planar structure of the thiourea moiety.
Conclusions
The formation of chalcogen-bonding interactions in the trans–trans conformation of thiourea derivatives was demonstrated by performing experimental X-ray diffraction and theoretical analyses. The Se⋯S⋯Se interaction in 3b was found to be stronger than the S⋯S⋯S interaction in 3a. A conformational analysis revealed that the population of the trans–trans planar conformation of 3b is higher than that of 3a. This conformational preference could provide a theoretical basis for the development of advanced thiourea-based catalysts. Further studies on the conformational analysis including cis–trans and cis–cis conformations, as well as the derivatization of these chalcogen-containing thioureas, including their application as organocatalysts are currently in progress in our laboratory.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. I.: investigation, writing of the original manuscript draft. N. M., Y. A., and R. S.: investigation. S. H., S. N., and Y. K.: investigation, formal analysis. T. F.: supervision, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a Grant-in-Aid for Scientific Research (B) (21H02611 and 24K02158). This work was the result of using shared research equipment(s) (Bruker/Avance NEO 500 MHz spectrometer and JEOL/JMS-GCmateII) supported by MEXT and Kyoto Pharmaceutical University.References
- (a) M. S. Sigmann and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901 CrossRef; (b) A. G. Wenzel and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 12964 CrossRef CAS PubMed; (c) P. R. Schreiner and A. Wittkopp, Org. Lett., 2002, 4, 217 CrossRef CAS PubMed; (d) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRef CAS PubMed.
- (a) Á. Madarász, Z. Dósa, S. Varga, T. Soós, A. Csámpai and I. Pápai, ACS Catal., 2016, 6, 4379 CrossRef; (b) A. Supady, S. Hecht and C. Baldauf, Org. Lett., 2017, 4, 4199 CrossRef PubMed; (c) G. Luchini, D. M. H. Ascough, J. V. Alegre-Requena, V. Gourverneur and R. S. Paton, Tetrahedron, 2019, 75, 697 CrossRef CAS; (d) A. A. Ehrhard, L. Gunkel, S. Jäger, A. C. Sell, Y. Nagata and J. Hunger, ACS Catal., 2022, 12, 12689 CrossRef CAS PubMed; L. Rummel, M. H. J. Domanski, H. Hausmann, J. Becker and P. R. Schreiner, Angew. Chem., Int. Ed., 2022, 61, e202204393 Search PubMed.
- In this paper, ‘trans’ refers to the case in which the NHAr group is oriented in opposing direction relative to the aryl group across the partial CN bond of the thioamide moiety.
- A. Wittkopp and P. R. Schreiner, Chem. – Eur. J., 2003, 407 CrossRef CAS PubMed.
- For selected reviews, see: (a) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383 CrossRef CAS PubMed; (b) H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291 CrossRef CAS PubMed; (c) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121 RSC; (d) M. Breugst, D. von der Heiden and J. Schmauck, Synthesis, 2017, 49, 3224 CrossRef CAS; (e) L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880 CrossRef CAS PubMed; (f) K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977 CrossRef CAS PubMed.
- T. Inoue, M. Ota, Y. Amijima, H. Takahashi, S. Hamada, S. Nakamura, Y. Kobayashi, T. Sasamori and T. Furuta, Chem. – Eur. J., 2023, e202302139 CrossRef CAS PubMed.
- The crystallographic data for 3a has been deposited at the Cambridge Crystallographic Data Center under reference number CCDC 2348537.†.
- The crystal structures of 2a with and without chalcogen-bonding interactions were obtained from acetone and THF, respectively. For details, see: ref. 6.
- Decomposition of 3b was observed in THF-d8. Therefore, the NOE study of 3b was carried out in DMSO-d6.
- (a) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS; (b) F. Weinhold, J. Comput. Chem., 2012, 33, 2363 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- (a) K. B. Wiberg, M. Marquez and H. Castejon, J. Org. Chem., 1994, 59, 6817 CrossRef CAS; (b) G. J. Bartlett, A. Choughary, R. T. Raines and D. N. Woolfson, Nat. Chem. Biol., 2010, 6, 615 CrossRef CAS PubMed.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed; (b) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef PubMed.
- (a) R. F. W. Bader, Atoms in Molecules, A Quantum Theory, Oxford University Press, Oxford, 1990 CrossRef; (b) T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- (a) S. Biswal, A. K. Sahu, B. Galmés, A. Frontera and D. Chopra, ChemBioChem, 2022, 23, e202100498 CrossRef PubMed; (b) Y. Lu, Z.-X. Wang and X.-Y. Chen, Angew. Chem., Int. Ed., 2022, 61, e202116071 CrossRef CAS PubMed ; see also: ref. 6.
- This is in accord with the order of strength of the chalcogen-bonding interactions (S < Se < Te).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectroscopic data, computational details. CCDC 2348537. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00723a |
| This journal is © The Royal Society of Chemistry 2024 |