
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organometallic copper(II) complex of meso–meso N-methyl N-confused pyrrole-bridged doubly N-methyl N-confused hexaphyrin†
Manik
Jana
a,
Daniel
Blasco
b,
Dage
Sundholm
 *c and
Harapriya
Rath
*a
*c and
Harapriya
Rath
*a
aSchool of Chemical Sciences, Indian Association for the Cultivation of Science, 2A/2B Raja S.C Mullick Road, Jadavpur, Kolkata, West Bengal 700 032, India. E-mail: ichr@iacs.res.in
bDepartamento de Química, Instituto de Investigación en Química (IQUR), Universidad de La Rioja, Madre de Dios 53, 26006, Logroño, Spain
cDepartment of Chemistry, Faculty of Science, University of Helsinki, P. O. Box 55 (A. I. Virtasen aukio 1), FIN-00014, Helsinki, Finland. E-mail: sundholm@chem.helsinki.fi
First published on 15th May 2024
Abstract
Synthesis, spectroscopic and theoretical characterization of a hitherto unknown meso–meso N-confused N-methylpyrrole-bridged doubly N-confused hexaphyrin (molecule 5) and its organometallic copper(II) complex (molecule 6) are reported herein. The absence of Q-type bands in the UV-Vis spectrum and the high chemical shifts of the inner proton signals of 5 suggest its globally non-aromaticity. The spectroscopic evidence of non-aromaticity for 5 and the paramagnetic nature of 6, are fully supported by density functional theory (DFT) calculations of the UV-Vis spectra, electron paramagnetic resonance (EPR) g-tensor parameters, and the magnetically induced current density strengths obtained with the gauge-including magnetically induced currents (GIMIC) method.
The bicycloaromaticity hypothesis that two aromatic π-electron circuits can exist independently of each other in nonplanar molecules was introduced by Goldstein and Hoffmann.1 Two connected molecular rings with a common conjugation pathway in one part of them share π electrons that sustain independent ring currents, which can lead to a more complex bicyclic ring with dual-potential electronic networks.2 Such bicyclic ring systems exhibit the potential to operate as switching functional motifs and may have three-dimensionally extended aromaticity and/or antiaromaticity. Thus, recent years have witnessed an upsurge in demand for [4n]/[4n + 2] annuleno [4n]/[4n + 2] annulene-type expanded porphyrinoids3 to unravel bicycloaromaticity. The meso–meso bridging approach, i.e. the linking of two meso positions of a porphyrinoid with a conjugated organic group, was first introduced by Osuka et al., who obtained an almost planar structure for decaphyrin using a para-phenylene bridge.3a The same group also reported that vinylene and pyrrole-bridged hexaphyrins I and II (Chart 1) adopt a major [26π] electronic conjugation pathway, whereas thiophene-bridged III exhibits dual [18π] and [26π] aromaticity, and meta-phenylene-bridged IV exhibits a predominant contribution from the [18π] porphyrin network.3c–e Depending on whether the chalcogen present in a thiophene-bridged doubly-substituted hexaphyrin is sulfur(V) or selenium(VI), a dual [18π] and [26π] conjugation pathway or a major contribution from the [26π] aromatic circuit are revealed, respectively.3f,g The meso–meso azulene bridged hexaphyrin VII exhibits a single [26π] electron pathway.4a These reports confirm that meso–meso bridged moieties adopting different orientations with respect to the mean meso plane of the macrocycle may trigger bicycloaromaticity instead of monocyclic aromaticity.3,4
 | ||
| Chart 1 Schematic presentation of meso–meso bridged hexaphyrins. | ||
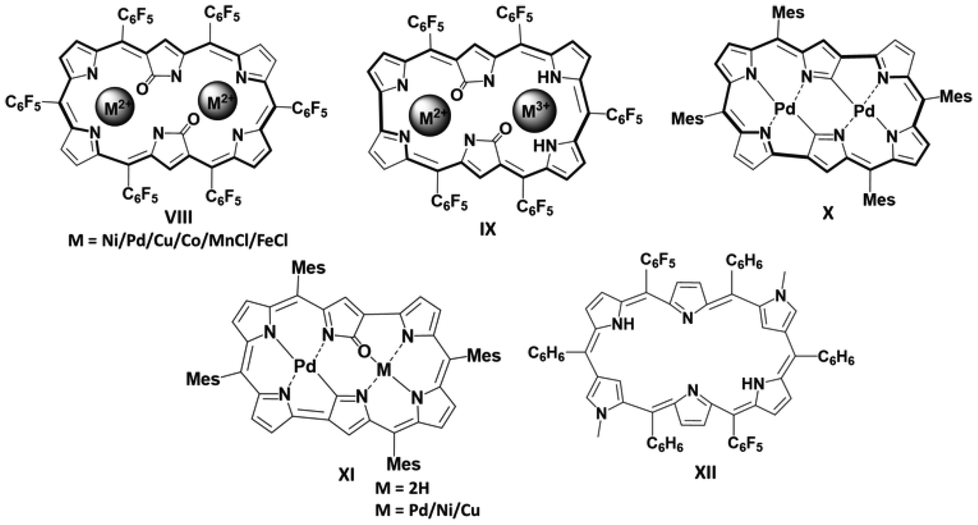
In the chemistry of expanded porphyrins, N-confused expanded porphyrins (NCPs)5a are important newcomers, which contribute to highlight the potential of expanded porphyrinoids. Chart 2 summarizes progress in single macrocyclic π-conjugated (anti)aromatic doubly N-confused hexaphyrins,5b–h each of which exhibited distinct properties upon metal ion complexation. For example, the metal (CuII, NiII, CoII, PdII, MnIII, FeIII ions) complexes of aromatic doubly N-confused [26]dioxohexaphyrin(1.1.1.1.1.1) VIII are attractive near-infrared-absorbing and/or emitting dyes,5b,f while the doubly N-confused and ring-contracted aromatic [26]dioxohexaphyrin(1.1.1.1.1.0) IX led to the formation of π-radical species upon complexation with palladium(II) cations.5d Ring-contracted [24]hexaphyrin PdII complexes (X and XI) happen to be stable antiaromatic,5g while nonaromatic figure-eight cross-conjugated doubly N-methyl N-confused hexaphyrin XII exhibited antiaromaticity upon protonation.5h From these, it is apparent that competitive mono vs. dual macrocyclic π-conjugation via meso–meso bridging strategy remained so far unexplored in case of N-confused expanded porphyrinoids. Herein, we report the synthesis, electronic properties, and aromaticity studies of a new-generation doubly N-confused hexaphyrin with a N-confused N-methylpyrrole moiety as the meso–meso bridging unit. Furthermore, this novel hexaphyrin has the potential to act as a tetradentate ligand for transition metal complexes, as demonstrated here for copper(II).
 | ||
| Chart 2 Progress in doubly N-confused hexaphyrins. | ||
As outlined in Scheme 1,4b the targeted doubly N-methyl N-confused hexaphyrin 5 is obtained by a [3 + 1] acid-catalyzed MacDonald-type oxidative condensation i.e., the reaction of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of tripyrrane 3, bearing bulky meso-2,6-difluorophenyl rings (to confer rigidity to the π-extended porphyrinoid), with the 1,3-bis-N-methylpyrrole aldehyde 4, in dichloromethane. Trifluoroacetic acid (TFA) was used as the catalyst. The reaction is followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4 benzoquinone (DDQ), which led to the exclusive formation of meso–meso bridged doubly N-methyl N-confused hexaphyrin 5. Column chromatographic separation followed by repeated preparative thin layer chromatography (PTLC) purification led to the isolation of 5 in 13% yield as a blue solid. 5 is stable against ambient temperature, light, and air both as solid as well as in solution. The new macrocycle has been thoroughly characterized via various spectroscopic and in-depth DFT level theoretical analyses. The positive-mode ESI-TOF mass spectrometry showed the parent ion peak at m/z of 1080.2544 Da for 5 (Fig. S2†) supporting the proposed structure.
:1 ratio of tripyrrane 3, bearing bulky meso-2,6-difluorophenyl rings (to confer rigidity to the π-extended porphyrinoid), with the 1,3-bis-N-methylpyrrole aldehyde 4, in dichloromethane. Trifluoroacetic acid (TFA) was used as the catalyst. The reaction is followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4 benzoquinone (DDQ), which led to the exclusive formation of meso–meso bridged doubly N-methyl N-confused hexaphyrin 5. Column chromatographic separation followed by repeated preparative thin layer chromatography (PTLC) purification led to the isolation of 5 in 13% yield as a blue solid. 5 is stable against ambient temperature, light, and air both as solid as well as in solution. The new macrocycle has been thoroughly characterized via various spectroscopic and in-depth DFT level theoretical analyses. The positive-mode ESI-TOF mass spectrometry showed the parent ion peak at m/z of 1080.2544 Da for 5 (Fig. S2†) supporting the proposed structure.
 | ||
| Scheme 1 Rational syntheses of meso–meso bridged doubly N-confused hexaphyrin 5 and its CuII complex 6. | ||
The electronic absorption spectral pattern of 5 (Fig. 1A) shows a strong Soret band at 544 nm while lacking the typical Q-type bands of porphyrins,6 along with a very weak band at 800 nm stretching up to 900 nm that could be expected for a disrupted π-conjugation of cross-conjugated system.5h The broken conjugation pathway through the oxo groups of the N-confused N-methylpyrrole rings due to the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups is confirmed by the IR absorption band at 1696 cm−1 for 5 (Fig. S4†). The low temperature 1H NMR spectral pattern of 5 (Fig. S10†) revealed structural rigidity with the macrocyclic framework lacking conformational fluxionality. Based on the 2D HSQC (heteronuclear single quantum coherence) spectra (Fig. S14†), the broad signals at 13.34 and 11.43 ppm are assigned to NH peaks (c and i in Fig. 1A′). The doublets a–b and g–h (δ = 7.9, 8.31 ppm and 7.62, 8.101 ppm, respectively) have been assigned to the β-CH protons of the amine type pyrrole rings owing to the COSY (correlated spectroscopy) correlations with NH signals (Fig. S11†). The singlets at 5.95 ppm and 6.62 ppm assigned to α-CH and β-CH respectively of the meso–meso bridging N-methyl N-confused pyrrole ring, is based on the dipolar coupling with the methyl peak at 2.87 ppm while exhibiting dipolar coupling with NH signals c and i in the 2D ROESY (rotating frame Overhauser enhancement spectroscopy, Fig. S12†). The doublets at δ = 7.06, 6.54 ppm and 6.96, 6.40 ppm, respectively, have been unambiguously assigned to the β-CH protons e–f and l–k of the imine-type pyrrole rings based on COSY (Fig. S11†). The sharp singlet at 2.64 ppm exhibiting no scalar or dipolar coupling with any other signals, concludes as methyl peaks of N-confused N-methylpyrrole rings possessing oxo groups. The resonances of the individual NH and CH peaks in the 1H NMR spectrum of 5 suggest that it is weakly antiaromatic7 or nonaromatic through the major 28π macrocyclic conjugation (5′, Scheme 1). Our observation is in line with the reduced (anti)aromaticity which has been obtained in most of the N-substituted N-confused porphyrinoids reported to date.8,5h
O groups is confirmed by the IR absorption band at 1696 cm−1 for 5 (Fig. S4†). The low temperature 1H NMR spectral pattern of 5 (Fig. S10†) revealed structural rigidity with the macrocyclic framework lacking conformational fluxionality. Based on the 2D HSQC (heteronuclear single quantum coherence) spectra (Fig. S14†), the broad signals at 13.34 and 11.43 ppm are assigned to NH peaks (c and i in Fig. 1A′). The doublets a–b and g–h (δ = 7.9, 8.31 ppm and 7.62, 8.101 ppm, respectively) have been assigned to the β-CH protons of the amine type pyrrole rings owing to the COSY (correlated spectroscopy) correlations with NH signals (Fig. S11†). The singlets at 5.95 ppm and 6.62 ppm assigned to α-CH and β-CH respectively of the meso–meso bridging N-methyl N-confused pyrrole ring, is based on the dipolar coupling with the methyl peak at 2.87 ppm while exhibiting dipolar coupling with NH signals c and i in the 2D ROESY (rotating frame Overhauser enhancement spectroscopy, Fig. S12†). The doublets at δ = 7.06, 6.54 ppm and 6.96, 6.40 ppm, respectively, have been unambiguously assigned to the β-CH protons e–f and l–k of the imine-type pyrrole rings based on COSY (Fig. S11†). The sharp singlet at 2.64 ppm exhibiting no scalar or dipolar coupling with any other signals, concludes as methyl peaks of N-confused N-methylpyrrole rings possessing oxo groups. The resonances of the individual NH and CH peaks in the 1H NMR spectrum of 5 suggest that it is weakly antiaromatic7 or nonaromatic through the major 28π macrocyclic conjugation (5′, Scheme 1). Our observation is in line with the reduced (anti)aromaticity which has been obtained in most of the N-substituted N-confused porphyrinoids reported to date.8,5h
 | ||
| Fig. 1 UV-Vis absorption spectra of (A) 5 and (B) 6 in CH2Cl2 at 298 K; (A′) completely assigned 1H NMR spectra of 5 in CDCl3 at 298 K; (B′) EPR spectrum of 6 in CH2Cl2 at 77 K; DFT optimised geometries of (A′′) 5 and (B′′) 6. Colour code: C, grey; H, white; Cu, brown; F, green; N, blue; O, red. | ||
In our next attempt, we have synthesized the organometallic copper(II) complex 6 following an adapted literature procedure.5 Based on the observation of the molecular ion peak at m/z of 1140.1608 Da (Fig. S3†), the elemental composition of 6 was confirmed. In the electronic absorption spectrum, complex 6 exhibited a Soret band at 402 nm with Q-type bands at 615 and 709 nm (Fig. 1B), supporting π-conjugation upon metalation. The paramagnetic behaviour of complex 6 is obvious by the absence of 1H NMR resonances, thus supporting an open-shell nature for 6. Electron paramagnetic resonance (EPR) spectrum recorded for 6 in dichloromethane at liquid nitrogen temperature exhibits a pattern which is typical of that expected for the CuII ion in tetragonal environment (Fig. 1B′).9 Of the four parallel lines, two are very well resolved and, in the perpendicular region, the superhyperfine interactions between the CuII ion and coordinated N atoms are seen. The EPR parameters do not differ much from those of copper(II) 5,10,15,20-(tetraphenyl) porphyrin (CuTPP; g∥ = 2.19; g⊥ = 2.05; A∥Cu = 202.0 × 10−4 cm−1; A⊥Cu = 33 × 10−4 cm−1; α2Cu = 0.82), suggesting that the meso–meso bridging ring has little effect on the electronic structure of the CuII ion. The g-tensor components have been calculated for an optimized model of 6 (vide infra) at the DFT level of theory within a relativistic spin-orbit exact-two-component framework.10,11Table 1 summarizes the experimental EPR parameters together with the calculated ones. The calculated g-tensor components are of the same size as the experimental ones, thus supporting the proposed tetragonal C, N, N, O coordination environment for CuII in complex 6.
| Experimental | Calculated | |
|---|---|---|
| g ∥ | 2.14 | 2.125 |
| g ⊥ | 2.004 | 2.031, 2.037 |
| A ∥ Cu | 201.3 × 10−4 | — |
| A ⊥ Cu | 35 × 10−4 | — |
| A ⊥ N | 16.32 × 10−4 | — |
| α 2 Cu | 0.738 | — |
The monocyclic vs. bicyclic conjugation pathways and optical properties of hexaphyrins 5 and 6 were computationally studied at the DFT level of theory. The optimized structures of 5 and 6 are depicted in Fig. 1A′′ and B′′, respectively. The N-confused hexaphyrin scaffold of 5 is roughly planar, except for the fact that the ring strain imposed by the short length of the protruding N-methylpyrrole bridge tilts the inner carbonyl groups out of the mean plane. Besides, the bridging moiety is almost perpendicular to the hexaphyrin plane. CuII coordination in 6 induces planarization of one of the N-confused porphyrin subcycles, leading to a folded structure. The possible bicyclic π-conjugation pathway of hexaphyrin 5 and its CuII complex 6 was evaluated with the gauge-including magnetically induced current (GIMIC) method and by visualization of the delocalization pathways.12–15 We have integrated the magnetically induced current (MIC) density passing through planes that cut the bonds shown in Fig. S17.† If 5 and 6 are globally (anti)aromatic, a strong ring current must flow along these bonds. Table S1† summarizes the calculated ring-current strengths. The obtained values of <3.0 nA T−1 are too small to indicate any global (anti)aromaticity for these macrocycles, thus precluding bicycloaromatic character. The computational study suggesting nonaromaticity is in line with 1H NMR and UV-Vis spectral observation for 5. The non-aromatic behavior of 5 and 6 is further studied by visualizing the delocalization pathways of the MIC as 3D streamlines. This is a successful strategy for revealing independent ring currents, as demonstrated for dithienothiophene-bridged [34]octaphyrins,2 lemniscular molecules such as [12]infinitene,16,17 and figure-eight octaphyrins.18 The ring-current pathways of 5 and 6 are depicted in Fig. 2. The weak ring current in 5 mainly follows the expected route along the hexaphyrin scaffold. It is complicated by loops on the pyrrole subunits and diversions towards the meso 2,6-difluorophenyl groups that cancel the net ring current. Notably, almost no current density circulates through the meso–meso bridged N-confused N-methylpyrrole ring, supporting a preferred single macrocyclic π-conjugation pathway for 5. Conversely, the metalation induced planarization of the meso–meso bridging N-confused N-methylpyrrole ring in complex 6 leads to a major delocalization pathway on the copper(II) N-methyl N-confused porphyrin subcycle. This agrees with the observation of Q-type bands in the UV-Vis-NIR spectrum of 6, which are absent in the spectrum of free-base 5. The simulated spectrum of 5 by means of time-dependent DFT (TD-DFT; Fig. S18 and Tables S2 and S3†) calculations features an intense excitation at 506 nm, which is in good agreement with the position of the Soret band at 554 nm. It corresponds to the S2 ← S0 transition and consists of a mixture of transitions from the HOMO, HOMO−1 and HOMO−2 to the LUMO. These orbitals are not located on specific parts but on the whole molecule. The interpretation of the spectrum of complex 6 is not straightforward due to its open-shell character, that leads to numerous small contributions from the low-lying d states of CuII. More interestingly, the low-energy band edge of the spectrum is red shifted with respect to that of 5, which agrees with the experimental measurements.
 | ||
| Fig. 2 The magnetically induced current density delocalization pathways of molecules 5 (left) and 6 (right) as 3D streamlines. The increasing MIC strength is represented with a black-red-orange-yellow-white colour scale. Colour code: C, grey; H, white; Cu, brown; F, green; N, blue; O, red. | ||
Conclusions
In conclusion, we reported syntheses of a meso–meso N-confused N-methylpyrrole bridged doubly N-methyl N-confused hexaphyrin (5) exhibiting major single macrocyclic conjugation through charge separated canonical resonance structure (5′) while its CuII complex (6) exhibits dual macrocyclic conjugation pathways. The α- and β-carbon oxidation hampered in the global macrocyclic π-conjugation pathways leading to nonaromaticity as evident from calculated magnetically induced current density. The experimentally observed g-tensor confirms that CuII has a tetragonal coordination environment. Further, such thriving scientific investigations are currently under progress in our laboratory for expanded N-confused porphyrinoid analogues beyond six pyrrole (heterocycle) subunits.Author contributions
HR designed the complete scientific project. MJ synthesized all the macrocycles, purified the macrocycles, and plotted all spectroscopic data. DB and DS performed the theoretical studies. HR, DB, and DS wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MJ thanks UGC for senior research fellowship. HR thanks SERB (SPG/2021/002173) New Delhi, India for a research grant. DS thanks the Academy of Finland for a research grant (project 340583). DB and DS acknowledge CSC – IT Center for Science, Finland for computational resources. DB thanks Universidad de La Rioja for a Margarita Salas post-doc scholarship financed by the Spanish Ministerio de Universidades and the European Union-NextGenerationEU program.References
- (a) M. J. Goldstein and R. Hoffmann, J. Am. Chem. Soc., 1971, 93, 6193–6204 CrossRef CAS; (b) M. J. Goldstein, J. Am. Chem. Soc., 1967, 89, 6357–6359 CrossRef CAS; (c) J. B. Grutzner and S. Winstein, J. Am. Chem. Soc., 1970, 92, 3186–3187 CrossRef CAS; (d) J. B. Grutzner and W. L. Jorgensen, J. Am. Chem. Soc., 1981, 103, 1372–1375 CrossRef CAS.
- R. R. Valiev, H. Fliegl and D. Sundholm, Phys. Chem. Chem. Phys., 2018, 20, 17705–17713 RSC.
- (a) V. G. Anand, S. Saito, S. Shimizu and A. Osuka, Angew. Chem., Int. Ed., 2005, 44, 7244–7248 CrossRef CAS PubMed; (b) M. Suzuki and A. Osuka, J. Am. Chem. Soc., 2007, 129, 464–465 CrossRef CAS PubMed; (c) M.-C. Yoon, S. Cho, M. Suzuki, A. Osuka and D. Kim, J. Am. Chem. Soc., 2009, 131, 7360–7367 CrossRef CAS PubMed; (d) H. Mori, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 12997–13001 CrossRef CAS PubMed; (e) G. Karthik, M. Sneha, V. P. Raja, J. M. Lim, D. Kim, A. Srinivasan and T. K. Chandrashekar, Chem. – Eur. J., 2013, 19, 1886–1890 CrossRef CAS PubMed; (f) J. M. Lim, G. Karthik, Y. M. Sung, A. Srinivasan, T. K. Chandrashekar and D. Kim, Chem. Commun., 2014, 50, 4358–4360 RSC; (g) G. Karthik, W.-Y. Cha, A. Ghosh, T. Kim, A. Srinivasan, D. Kim and T. K. Chandrashekar, Chem. – Asian J., 2016, 11, 1447–1453 CrossRef CAS PubMed.
- (a) M. J. Białek and L. Latos-Grażyński, Chem. Commun., 2018, 54, 1837–1840 RSC; (b) S. Sahoo, G. Velmurugan, P. Comba and H. Rath, Org. Chem. Front., 2023, 10, 5601–5609 RSC.
- (a) M. Togano and H. Furuta, Chem. Rev., 2022, 122, 8313–8437 CrossRef PubMed; (b) A. Srinivasan, T. Ishizuka, A. Osuka and H. Furuta, J. Am. Chem. Soc., 2003, 125, 878–879 CrossRef CAS PubMed; (c) M. Suzuki, M.-C. Yoon, D. Y. Kim, J. H. Kwon, H. Furuta, D. Kim and A. Osuka, Chem. – Eur. J., 2006, 12, 1754–1759 CrossRef CAS PubMed; (d) Y. Hisamune, K. Nishimura, K. Isakari, M. Ishida, S. Mori, S. Karasawa, T. Kato, S. Lee, D. Kim and H. Furuta, Angew. Chem., Int. Ed., 2015, 54, 7323–7327 CrossRef CAS PubMed; (e) K. Yamasumi, K. Nishimura, Y. Hisamune, Y. Nagae, T. Uchiyama, K. Kamitani, T. Hirai, M. Nishibori, S. Mori, S. Karasawa, T. Kato, K. Furukawa, M. Ishida and H. Furuta, Chem. – Eur. J., 2017, 23, 15322–15326 CrossRef CAS PubMed; (f) Y. Wang, K. Ogasahara, D. Tomihama, R. Mysliborski, M. Ishida, Y. Hong, Y. Notsuka, Y. Yamaoka, T. Murayama, A. Muranaka, M. Uchiyama, S. Mori, Y. Yasutake, S. Fukatsu, D. Kim and H. Furuta, Angew. Chem., 2020, 132, 16295–16300 CrossRef; (g) F. Luo, L. Liu, H. Wu, L. Xu, Y. Rao, M. Zhou, A. Osuka and J. Song, Nat. Commun., 2023, 14, 5028 CrossRef CAS PubMed; (h) A. Mallick, J. Oh, D. Kim, M. Ishida, H. Furuta and H. Rath, Chem. – Eur. J., 2016, 22, 5504–5508 CrossRef CAS PubMed.
- M. Gouterman, G. H. Wagnière and L. C. Snyder, J. Mol. Spectrosc., 1963, 11, 108–127 CrossRef CAS.
- J. A. Pople and K. G. Untch, J. Am. Chem. Soc., 1966, 88, 4811–4815 CrossRef CAS.
- (a) J. L. Sessler, D.-G. Cho, M. Stepien, V. Lynch, J. Waluk, Z. S. Yoon and D. Kim, J. Am. Chem. Soc., 2006, 128, 12640–12641 CrossRef CAS PubMed; (b) P. J. Chmielewski and L. Latos-Grażyński, J. Chem. Soc., Perkin Trans. 2, 1995, 503–509 RSC.
- F. A. Wlker, in Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 1999, vol. V, ch. 36 Search PubMed.
- Y. J. Franzke and J. M. Yu, J. Chem. Theory Comput., 2022, 18, 2246–2266 CrossRef CAS PubMed.
- Y. J. Franzke and J. M. Yu, J. Chem. Theory Comput., 2022, 18, 323–343 CrossRef CAS PubMed.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- D. Sundholm, M. Dimitrova and R. J. F. Berger, Chem. Commun., 2021, 57, 12362–12378 RSC.
- M. Orozco-Ic, R. R. Valiev and D. Sundholm, Phys. Chem. Chem. Phys., 2022, 24, 6404–6409 RSC.
- Q. Wang, M. Orozco-Ic and D. Sundholm, Phys. Chem. Chem. Phys., 2023, 25, 19207–19213 RSC.
- Q. Wang, J. Pyykkö, M. Dimitrova, S. Taubert and D. Sundholm, Phys. Chem. Chem. Phys., 2023, 25, 12469–12478 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00691g |
| This journal is © The Royal Society of Chemistry 2024 |