
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in thiochromene chemistry
Solai
Murugappan
a,
Pranali Vijaykumar
Kuthe
a,
Kondapalli Venkata Gowri
Chandra Sekhar
 b and
Murugesan
Sankaranarayanan
*a
b and
Murugesan
Sankaranarayanan
*a
aDepartment of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Vidya Vihar, Pilani-333031, Rajasthan, India. E-mail: murugesan@pilani.bits-pilani.ac.in
bDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Hyderabad Campus, Jawahar Nagar, Kapra Mandal, Hyderabad-500078, Telangana, India
First published on 11th July 2024
Abstract
Thiochromenes are versatile sulfur-containing heterocyclic compounds that have received considerable interest in drug discovery because of their ability to act as crucial building blocks for synthesizing bioactive compounds. In particular, these scaffolds have found utility in the design of anticancer, anti-HIV, antioxidant, and antimicrobial agents, among others. Despite their pharmacological potential, the synthesis of these scaffolds is less explored in contrast to their oxygen-containing counterparts. This review classifies the synthetic processes into Michael addition, cycloaddition, ring-opening, coupling, cyclization and Diels–Alder reactions, and others. Reaction mechanisms, circumstances, and important instances are thoroughly discussed in each area. For instance, chiral catalysts and substrates like mercaptobenzaldehyde and cinnamaldehyde are used in Michael addition processes to achieve excellent enantioselectivity. In cycloaddition reactions, readily available substrates such as thioisatins and alkynes achieve regioselectivity and product production. Thiochromenes are also synthesized by ring-opening reactions with epoxides or aziridines. These reactions demonstrate the importance of catalysts and solvents in reaction control, particularly palladium catalysts for aryl halides and thiol coupling processes. Another major class discussed is cyclization reactions with alkynyl thiols and alkynes under regulated temperature and pressure conditions to efficiently synthesize thiochromenes. With the use of chiral substrates and catalysts, Diels–Alder processes increase yields and selectivity and enhance the variety of thiochromene compounds. This review emphasizes the versatility of thiochromenes in drug discovery and consolidates the existing literature on thiochromenes, scrutinizing the gaps and opportunities for synthesizing novel thiochromene-containing lead molecules.
 Solai Murugappan | Ms Solai Murugappan completed her MS (Pharm) in Medicinal Chemistry from the National Institute of Pharmaceutical Education and Research, Hyderabad, Telangana, India, in 2023. She is pursuing her PhD in Pharmacy at Birla Institute of Technology and Science (BITS) Pilani, Pilani Campus, India, under the guidance of Dr S. Murugesan. Her current interest mainly focuses on the synthesis of novel thiochromene derivatives. |
 Pranali Vijaykumar Kuthe | Ms Pranali Vijaykumar Kuthe completed her M. Pharm in Pharmaceutical Chemistry from All India Shri Shivaji Memorial Society's College of Pharmacy, Pune, Maharashtra, India, in 2022. Currently, she is a Ph.D. scholar under the supervision of Dr S. Murugesan at Birla Institute of Technology and Science Pilani, Pilani Campus, Rajasthan, India. Her research endeavors revolve around synthesizing novel inhibitors to combat the challenges of cancer and malaria (epigenetics). |
 Kondapalli Venkata Gowri Chandra Sekhar | Prof. K. V. G. Chandra Sekhar, a distinguished academic, obtained his Ph.D. in Medicinal Chemistry (2009), M. Pharm (2003), and B. Pharm (Hons.) (1999) from Birla Institute of Technology & Science, Pilani. With rich teaching experience spanning over two decades at BITS Pilani, Hyderabad, and Rajasthan campuses, he currently serves as a Professor at BITS Pilani, Hyderabad Campus. Prof. KVG's research interests encompass diverse areas of medicinal chemistry, focusing on the synthesis of heterocyclic compounds targeting various therapeutic applications and the development of compounds such as quorum sensing inhibitors, anti-leishmanial agents, anticancer agents, and treatments for neuropsychiatric disorders. |
 Murugesan Sankaranarayanan | Prof. S. Murugesan is an accomplished academician who joined the Department of Pharmacy at BITS, Pilani in 2010 as an Assistant Professor. He holds a Ph.D. from BIT, Mesra (2009) in synthetic medicinal chemistry, has published over 200 research papers, and has presented 100 posters at national and international forums. With expertise in computer aided drug design and synthetic medicinal chemistry, Dr Murugesan is actively involved in the development of non-nucleoside reverse transcriptase inhibitors for combating opportunistic infections like HIV, TB, malaria, and leishmaniasis as well as addressing rare disorders such as sickle cell disease, Gaucher's disease and lifestyle disorders such as NAFLD. |
1. Introduction
Heteroatoms such as oxygen, sulfur, and nitrogen are essential structural elements in several pharmaceutically active ingredients. The widespread use of these atoms as a part of cyclic or open chain fragments is due to their abundance in natural products and nucleic acids. Among them, several scientists have primarily explored nitrogen and oxygen over the years due to their ability to modify and improve potency, selectivity and ability to act as bioisosteric replacements to alter the pharmacokinetic properties of drug molecules.1 In the midst of this is the sulfur atom, which has comparable functional characteristics, including higher volatility and improved reactivity, but has yet to be explored as thoroughly as the other two heteroatoms.Among the sulfur-based compounds, heterocyclic derivatives like thiazole, thiopyran, benzothiopyran, thiophene, and thioazolidinone are frequently found as a part of several bioactive scaffolds.2,3 In particular, these mono/bi-cyclic scaffolds with a thio atom serve as building blocks for developing drugs and agricultural products.4,5 As of 2018, over 285 sulfur-containing compounds were identified in the list of FDA-approved drugs for treating various ailments, and sulfonamides constitute the largest group in this series.6
However, despite their success in drug discovery, certain sulfur pharmacophores still need to be explored compared to well-established ones like sulfoxides and thioethers.7 They include sulfoximine, sulfondiimine, sulfonyl fluoride, and thienopentathiepine, among others. For instance, sulfoximines discovered in the 1970s still represent a class of underrated pharmacophores despite their chemical stability, dual nature as hydrogen bond donors/acceptors, and promising pharmacological activities.7 Consistent with this, the other sulfur functionalities classified under the less-explored ones, as shown in Fig. 1, demonstrate excellent potential in the development of newer leads in medicinal chemistry. Under this category, thiochromenes or benzothiopyrans have recently established themselves as distinguished scaffolds owing to the increased attention garnered by these pharmacophores over the years. These scaffolds are the thio-analogs of chromenes and exhibit promising biological activities, ranging from anti-inflammatory to anticancer effects.8–10
 | ||
| Fig. 1 Representative examples of sulfur-containing scaffolds of biological importance. | ||
The pharmaceutical importance of bicyclic ring systems with a thio-atom as the pharmacophore is emphasized by the presence of FDA-approved drugs, namely tazarotene, flupentixol, and meticrane (Fig. 2).11–14 Besides their application in medicinal chemistry, thiochromenes also find use in materials science and optoelectronics due to their characteristic photo-physical behavior, such as slow photodegradation and efficient biomolecular photoreaction in solution. This distinguishing trait facilitates their use in developing laser dyes, organic light-emitting diodes (OLEDs), and fluorescent probes.15–17
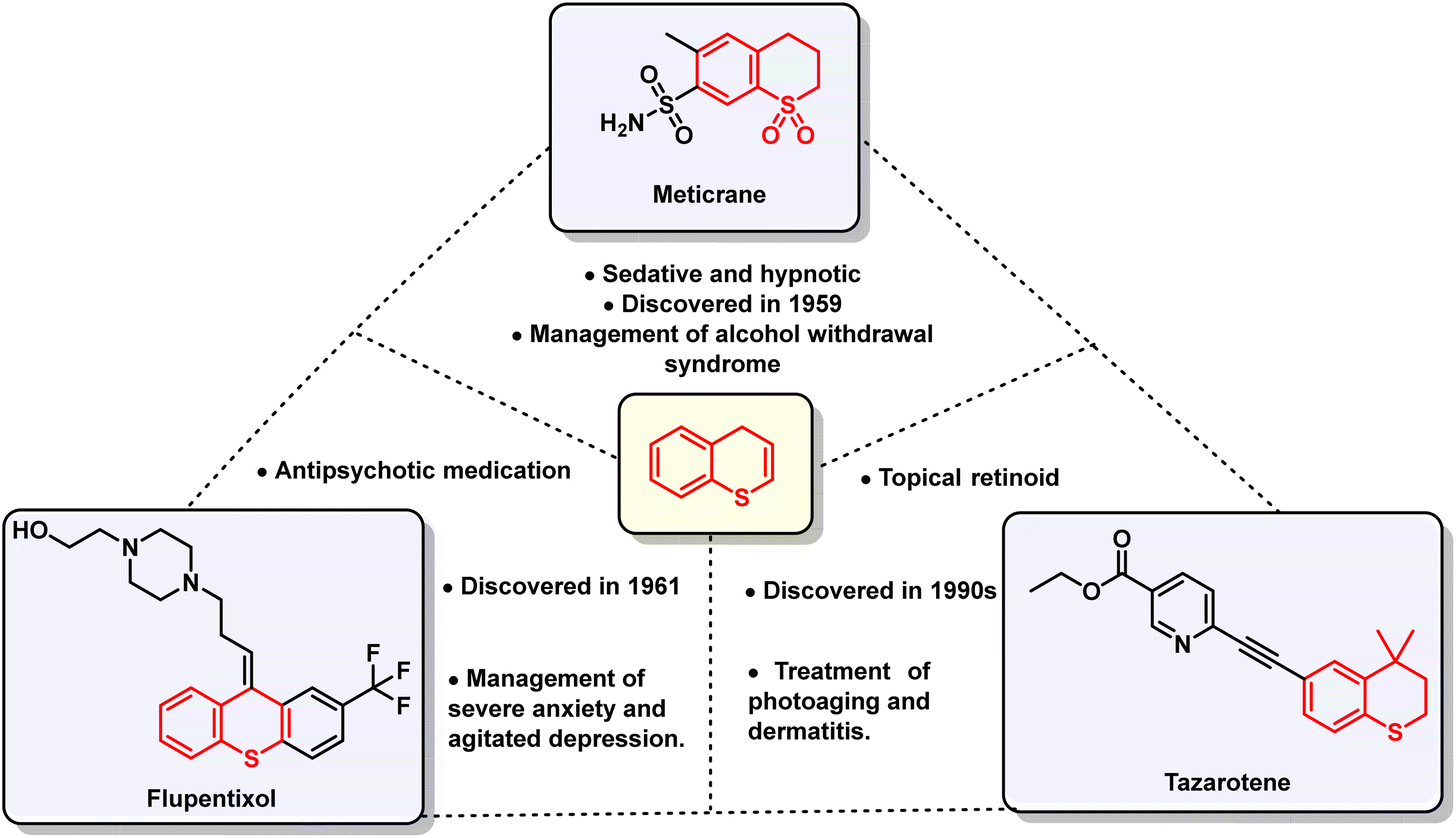 | ||
| Fig. 2 Structure of FDA-approved drugs containing a thiochromane/thiochromene nucleus. | ||
Despite its structural similarity to chromenes, the synthesis of this sulfur analog is hindered by several challenges due to the inherent nature of sulfur and its structural complexity. This is mainly due to the multiple oxidation states and tendency shown by sulfur atoms to form diverse bonding patterns because of their ability to expand their valence shell to occupy more than eight electrons.7,18 Additionally, highly reactive sulfur atoms tend to alter the structural integrity of molecules with more than one reactive site.19 Such changes are often witnessed by the development of undesired by-products or side reactions, which ultimately complicate the purification and isolation of the pure compound. These challenges have emphasized the need for novel synthetic approaches and streamlined reaction sequences to attain thiochromenes with better purity and yield.
The methods used for synthesizing thiochromenes can be transition metal-catalyzed reactions or organocatalytic strategies. In the case of the former, transition metal complexes are leveraged to catalyze the critical bond-forming step to aid in synthesizing thiochromenes. One promising protocol emerging under this category is the combination of cyanuric chloride (2,4,6-trichloro-1,3,5-triazine) with dimethylformamide (DMF) in the presence of metal catalysts to yield substituted thiochromenes from readily available starting materials.20 The most significant appeal of these approaches is their high selectivity and faster reaction times, making them admissible thiochromene synthesis options. However, their utility is constrained because of their cost and inability to generate chiral thiochromenes under more simplified reaction conditions, making it challenging to develop a library of desired compounds.
Organocatalytic approaches act as promising alternatives for thiochromene synthesis. These reactions offer enhanced stereoselectivity and functional group compatibility, countering metal-catalyzed reactions’ cons.21,22 Organocatalysts initiate key transformations by activating specific functional groups, which guide the reaction toward forming the final products. Strategies such as Lewis acid catalysis and intramolecular tandem Michael addition-type reactions work on this principle and assist in generating structurally diverse thiochromenes.23,24 In addition to this, such approaches also open avenues for further functionalization at the stereogenic center, broadening these sulfur-containing heterocycles’ applicability in medicinal chemistry and materials science.
This comprehensive review endeavors to provide an in-depth analysis of the recent advancements over the decade in synthesizing thiochromenes by categorizing them based on their reaction type. By exploring the reaction compatibility with the substrate and mechanistic insights offered by these methodologies, this work aims to provide an overview of the current landscape in thiochromene-related chemistry.
The subsequent sections will cover the various synthetic strategies employed to generate thiochromenes by categorizing them into seven distinct classes as follows:
(a) Michael addition
(b) Cycloaddition reactions
(c) Ring-opening reactions
(d) Coupling reactions
(e) Cyclization reactions
(f) Diels–Alder reactions
(g) Miscellaneous reactions
1.1. Michael addition
 | ||
| Scheme 1 Pyrrolidine-catalyzed synthesis of thiochromen-3-carbaldehyde using cinnamaldehyde. | ||
 | ||
| Scheme 2 (a) Stereoselective synthesis of thiochromene-3-carbaldehyde; (b) preferred conformation of the transition state; (c) select examples. | ||
After establishing a sulfa-Michael cascade reaction for the synthesis of 2H-thiochromenes from mercaptobenzaldehyde and vinyl phosphonates in their earlier studies,28 Simlandy's group in the year 2017 tried to control the initial Michael step to facilitate Julia–Kocienski olefination for the enantioselective formation of 3,4-unsubstituted thiochromenes.29 This attempt proved to be successful by furnishing the desired products in yields of 28–80% with good enantiomeric ratios (er = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3–99:1). The reaction proceeded via the formation of an iminium intermediate (8a), which is bound to the amine catalyst and gets attacked by the vinyl phenyl tetrazole sulfone (vinyl PT sulfone or 9) in a Si-face selective manner. This study further explored the synthetic feasibility of thiochromenes to obtain a thiochromane ring (11 by reduction) and a thioflavone (12 by oxidation) (Scheme 3).
:3–99:1). The reaction proceeded via the formation of an iminium intermediate (8a), which is bound to the amine catalyst and gets attacked by the vinyl phenyl tetrazole sulfone (vinyl PT sulfone or 9) in a Si-face selective manner. This study further explored the synthetic feasibility of thiochromenes to obtain a thiochromane ring (11 by reduction) and a thioflavone (12 by oxidation) (Scheme 3).
 | ||
| Scheme 3 (a) Julia–Kocienski olefination for synthesizing thiochromenes; (b) mechanism of the Michael addition reaction; (c) select examples; (d) synthetic application of the protocol. | ||
Thiols comprise one of the most frequently used substrates for synthesizing thiochromene analogs. Despite their extensive application in this domain, thiols suffer from unpleasant odour and toxicity and potentiate the formation of multiple by-products.30 To overcome this, Wu et al., in 2023, realized the replacement of thiols with their oxidized products, disulfides, which are more stable and act as thiol surrogates under the reaction conditions.31 On this note, dithiobenzaldehyde (14) was treated with aromatic bromoenal (13) in the presence of a reducing agent, PPh3, and an N-heterocyclic carbine (NHC) catalyst. Control experiments established the importance of water in this reaction as it took part in the generation of the sulfur anion (14a) from dithiobenzaldehyde (14) in a manner resembling the Corey–Nicolaou macro-lactonization pathway. Moreover, the addition of this anion to the acyl azolium intermediate (13a) is the probable mechanistic pathway that was identified to be the enantio-determining step (sulfa-Michael addition), resulting in the generation of products (15) with high enantioselectivities with an enantiomeric ratio as high as 97:3 (Scheme 4).
 | ||
| Scheme 4 (a) N-Heterocyclic carbine (NHC) catalyzed synthesis of thiochromenes using disulfides as the sulfur source; (b) mechanism of the Michael addition reaction; (c) select examples. | ||
 | ||
| Scheme 5 (a) Thio-Michael addition reaction for the synthesis of thiochromene-3-carboxylate ester; (b) mechanism of the Michael addition reaction; (c) select examples. | ||
A similar approach was undertaken by Sangeetha et al. in 2019 to actualize a one-pot synthetic approach for obtaining thiochromenones by the reaction of halogenated chalcones (19) and xanthates.33 The uniqueness of this reaction lies in its ability to generate iodine in situ to yield a halogenated thiochromane intermediate (20), which, on reduction, gives the final thiochromenone product. Interestingly, this reaction uses the waste by-product (potassium iodide) to create a facile protocol with minimal waste generation. The same conditions with the addition of iodine during the reduction step result in the formation of bis-thiochromenones (22) in considerable yields (52–62%), depicting a simple yet novel route for the generation of such compounds (Scheme 6).
 | ||
| Scheme 6 (a) One-pot reaction to synthesize thiochromenes using xanthate as the sulfur source; (b) mechanism of the Michael addition reaction; (c) synthetic application; (d) select examples of thiochromene derivatives. | ||
In 2023, Saini and co-workers developed a one-pot Knoevenagel–Michael cascade reaction to synthesize thiochromenes bearing a spirocyclic framework (26).34 In this reaction, the dehydrated derivative of ninhydrin (23) reacts with naphthalene thiol (24) to generate a Knoevenagel condensation intermediate (23a), which then undergoes an addition reaction with 1,3-dicarbonyl compounds (25) to release the desired spiroindene-thiochromene derivatives (26) via removal of a molecule of water (Scheme 7).
 | ||
| Scheme 7 (a) Knoevenagel–Michael cascade reaction to synthesize thiochromenes; (b) mechanism of the Michael addition reaction; (c) select examples. | ||
:1.0. The mixture did not undergo diastereomeric salt crystallization using methanol, but proceeded to yield the final thiochromene products (30a and 30b) by sequential oxidation, deprotonation cum Henry intramolecular cyclization by a carbanion and dehydration at the C3–C4 bond of the thiochromene ring (29b) (Scheme 8). In the same year, Muthupandi et al. attempted a similar reaction by preceding it with aryl C–S bond formation to yield thiochromenes (34).36 This protocol demonstrated good tolerability with a range of substrates to furnish the corresponding products in 66–98% yields. Furthermore, this work revealed that the copper catalyst was essential for the C(aryl)–S bond formation and subsequent in situ generation of formylbenzenethiolate (31d), which then undergoes Michael-aldol elimination (Scheme 9).
 | ||
| Scheme 8 Synthesis of diastereomeric mixtures of thiochromenes from mercaptobenzoic acid. | ||
 | ||
| Scheme 9 (a) Xanthate promoted C–S aryl bond formation to synthesize thiochromenes; (b) mechanism of the Michael addition reaction; (c) select examples. | ||
Le et al., in the same year, devised a novel and efficient method for attaining thiochromenes by the in situ generation of mercaptobenzaldehyde from the reaction of bromobenzaldehyde (35) and nitroalkene (36).37 This protocol proceeded by a thiol-Michael addition tandem intramolecular aldol condensation of o-formyl thiophenolate to styrene, followed by the dehydration of the ensuing intermediate to achieve the desired thiochromene (37). This reaction demonstrated better yields with the use of chalcone (38a), cinnamaldehyde (38b), and its esters (38c) in place of styrene, as the former group furnished the product with one equivalent. At the same time, the latter required 2.5 equivalents (Scheme 10).
 | ||
| Scheme 10 (a) Thio-Michael addition reaction using bromobenzaldehyde to synthesize thiochromenes from nitroalkenes; (b) chalcones and their esters; (c) select examples. | ||
In the subsequent year, Sundaravelu and Sekar came out with a double hetero-Michael addition reaction encompassing halo-benzaldehydes (38) and substituted chalcones (39 or 42) in the presence of a copper catalyst using xanthate (A) as the sulfur source and chemoselective reducing agent.38 This group noted that this reaction resulted in the generation of an amino/bromo-benzoyl-substituted thiochromene ring (40 or 43) subjected to intramolecular Michael addition without the formation of by-products. The amine-bearing substrate's subsequent base-catalyzed reaction led to quinolinone ring formation (41). In contrast, the progression of the reaction involving the bromo-bearing substrate at increased temperature resulted in the generation of a thioflavone ring (44) (Scheme 11).
 | ||
| Scheme 11 (a) Chemoselective synthesis of thiochromenes by the sulfa-Michael addition reaction; (b) mechanism of the Michael addition reaction; (c) select examples. | ||
Control experiments and literature analysis were extensively conducted to further comprehend this reaction's mechanistic intricacies. Through these approaches, the group noted a common pathway that bifurcates into two distinct mechanisms depending on the substrate employed for the cyclization. The copper complexed halo-benzaldehyde (38a) initially exchanges ligands with xanthate (A), generating potassium iodide as the by-product. The intermediate (38c) formed then undergoes sulfa-Michael addition cum aldol condensation to yield an intermediate (38e). In the next stage, two different mechanisms are observed. The bromo-substituted chalcones (43) correspond to the formation of chromene-isoflavones (44) by intramolecular sulfa-Michael addition, while nitro-bearing chalcones (40) undergo the reaction to generate quinolinone rings (41) by aza-Michael addition. Intriguingly, both these parallel pathways proceed via an oxidation step catalyzed by the iodine generated in situ in the reaction of potassium iodide in the initial steps (Scheme 11).
 | ||
| Scheme 12 (a) 1,8-Diazabicyclo-[5.4.0]-undec-7-ene (DBU) mediated cyclization to synthesize thiochromenes; (b) select examples; (c) PTSA mediated synthesis of thiochromenes; (d) select examples. | ||
In the same year, another highly diastereoselective method was reported by Wang and co-workers for the synthesis of annulene fused thiochromenes (56) from bridged biaryl-enones (54) and mercaptoacetophenones (55) using a chiral phosphoric acid catalyst.40 The reaction demonstrated good tolerance towards electron donating and withdrawing functionalities, yielding a single diastereomer (56) with 89–99% enantiomeric excess (ee) and diastereomeric ratio (dr) > 20:1. The catalyst employed in this reaction initially reacts with the thiol group to generate a nucleophile that attacks the enone via the Re face through Michael addition. In the next step, an intramolecular aldol condensation occurs, followed by protonation to produce the desired product (Scheme 13).
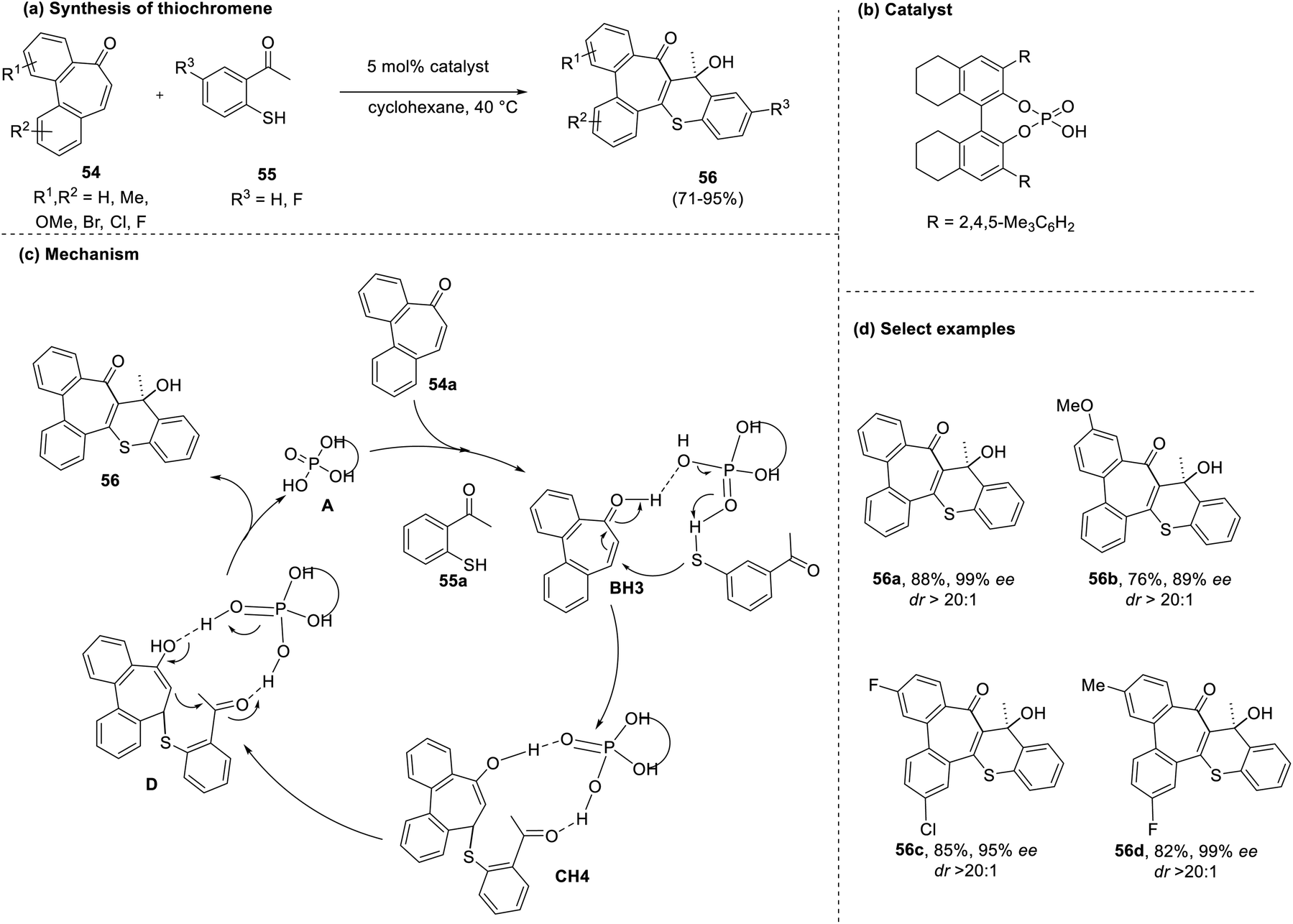 | ||
| Scheme 13 (a) Chiral phosphoric acid-catalyzed synthesis of thiochromenes; (b) chiral phosphoric acid catalyst; (c) mechanism for the synthesis of thiochromenes; (d) select examples. | ||
1.2. Cycloaddition reactions
Inami et al. in 2014 put forth a facile approach for synthesizing thiochromones by reacting thioisatins (57) with alkynes (58).41 While the reaction involved a decarbonylative cycloaddition to furnish the final product (59) as a single regioisomer, specific substrates resulted in a mixture of regioisomers. Using octyne, methyl or phenyl cyclopropyl acetylene, and terminal alkynes gave rise to a mixture of isomers (Scheme 14).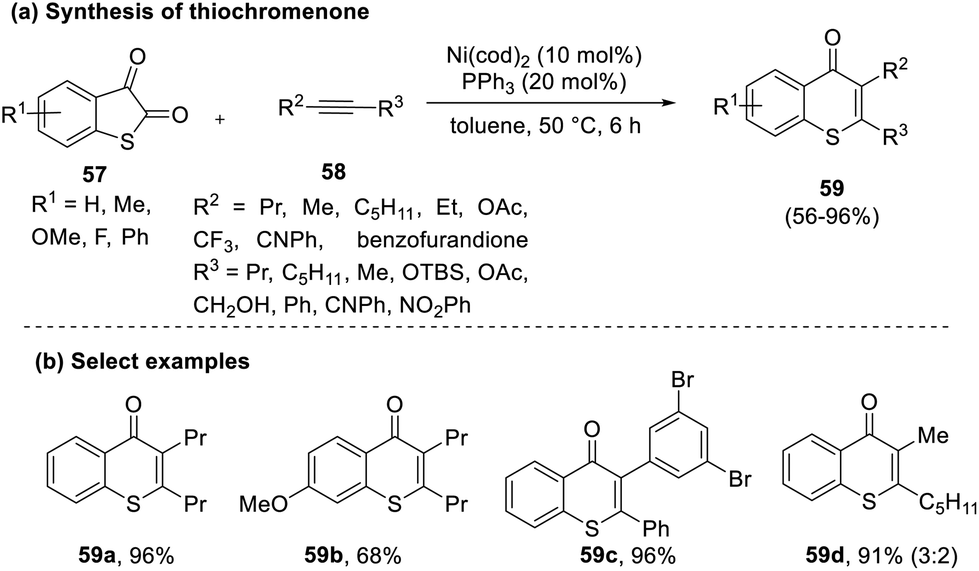 | ||
| Scheme 14 (a) Nickel-catalyzed decarbonylative cycloaddition to synthesize thiochromenones; (b) select examples. | ||
Continuing with this approach, Zhu et al., in 2018, revamped the previous work by establishing a [3 + 2 + 1] cyclization protocol for achieving thiochromenones (63) by the use of aromatic sulfides (60) and alkynes (62) by a rhodium-catalyzed reaction.42 This reaction tolerated electron donating and withdrawing functionalities but failed to generate the products with internal alkynes. The other recalcitrant alkynes for this reaction were cyclohexyl acetylene and octyne (Scheme 15).
 | ||
| Scheme 15 (a) Rhodium-catalyzed [3 + 2 + 1] cycloaddition to synthesize thiochromenones; (b) mechanism of cycloaddition; (c) select examples. | ||
In 2023, Pan et al. further exploited this area by developing a [3 + 2 + 1] transannulation protocol whereby thiadiazoles (64) reacted with alkynes (65) using a de-nitrogenative carbonylation approach.43 In this method, carbon monoxide (CO) acted as the C1 synthon, and PPh3 was used to convert the rhodium catalyst into a highly reactive species. With the help of mechanistic investigations, the reaction was noted to proceed by forming a cyclorhodium intermediate that undergoes CO insertion and furnishes the thiochromenone product (66) via reductive elimination (Scheme 16).
 | ||
| Scheme 16 (a) Rhodium-catalyzed de-nitrogenative transannulation to synthesize thiochromenones; (b) select examples. | ||
1.3. Ring-opening reactions
The chemical versatility of chromenes and thiochromenes has established them as significant building blocks for synthesizing promising therapeutic agents. In this context, synthetic protocols, mainly catalyst-aided reactions, have been primarily explored for these motifs of interest. While these methods offer thiochromenes in good yields, their economic feasibility hinders their widespread utility. In a bid to tackle this, Roy et al., in 2014, devised a simple methodology utilizing a mild chlorinating agent, 2,4,6-trichloro-1,3,5-triazine (TCT) (68), to furnish thiochromenes (69) via ring opening of hydroxyl thiochromane possessing a cyclopropane moiety (67).20 This protocol, carried out under anhydrous conditions, resulted in excellent yields of E-configuration thiochromenes (69) (84–88%). Mechanistic studies revealed the concurrence of cyclopropane ring opening to release the angle strain and subsequent dehydrogenation at the C3–C4 position to form a diene by the release of the leaving group. The synthetic applicability of this reaction was tested for the generation of cannabinoid derivatives (71) by the Diels–Alder reaction wherein a benzodioxole-containing substrate (69c) was reacted with N-phenyl maleimide (70) (Scheme 17). | ||
| Scheme 17 (a) 2,4,6-Trichloro-1,3,5-triazine (TCT)-mediated ring opening to synthesize thiochromenes; (b) synthetic application; (c) select examples; (d) mechanism of ring opening. | ||
In 2020, Ponra and co-workers explored the ring opening of cyclopropyl thioethers (72) using palladium as the metal catalyst.44 In continuation of their interest in the ring opening of cyclopropanes, this group tested the reactivity of the thioether analog (72) in the presence of various palladium catalysts. However, it was observed that the use of palladium (0) catalysts, particularly palladium acetate, was the best choice. An investigation of the substrate scope revealed that the reaction tolerated electron-donating functionalities but generated poor yields using chloro-substituted thioethers. Furthermore, isomerization of the double bond of the thiochromene (73), resulting in regioisomers with an isomeric ratio of 2.5:1, was noticed in the presence of substrates bearing acyl groups. In the mechanistic inspection, the principal intermediate formed is a seven-membered palladacycle (72c), which undergoes subsequent elimination to generate the final product (73). The oxidative addition of the Pd(0) catalyst to the thioether is the only sluggish step in this reaction, and this is attributed to the presence of the sulfur atom ortho to the halogen, which results in a rise in the electron density of the aromatic ring, thereby hampering the oxidative addition (Scheme 18).
 | ||
| Scheme 18 (a) Palladium-catalyzed ring opening of cyclopropyl thioether to synthesize thiochromenes; (b) mechanism of ring opening; (c) select examples. | ||
1.4. Coupling reactions
Coupling reactions have been of paramount importance in generating C–S bonds. However, such reactions often face limitations due to catalyst poisoning and using thiol as the precursor. Considering this, Shen and co-workers in 2016 envisaged a one-pot technique utilizing a reagent capsule to generate a thiochromenone (76) by circumventing the catalyst poisoning and undesired side products usually encountered in these cases.45 To facilitate this, the reaction was carried out between halogenated benzenes (74), aromatic acetylenes (75), and sodium sulfide nonahydrate in the presence of a palladium catalyst. While sodium sulfide served as a conducive sulfur source, it was also identified to contribute towards catalytic poisoning and side-product formation. Shen and his group tackled this by encapsulating anhydrous sodium sulfide in paraffin wax capsules and controlling their release with time to obtain the final product (76) in good yields (Scheme 19).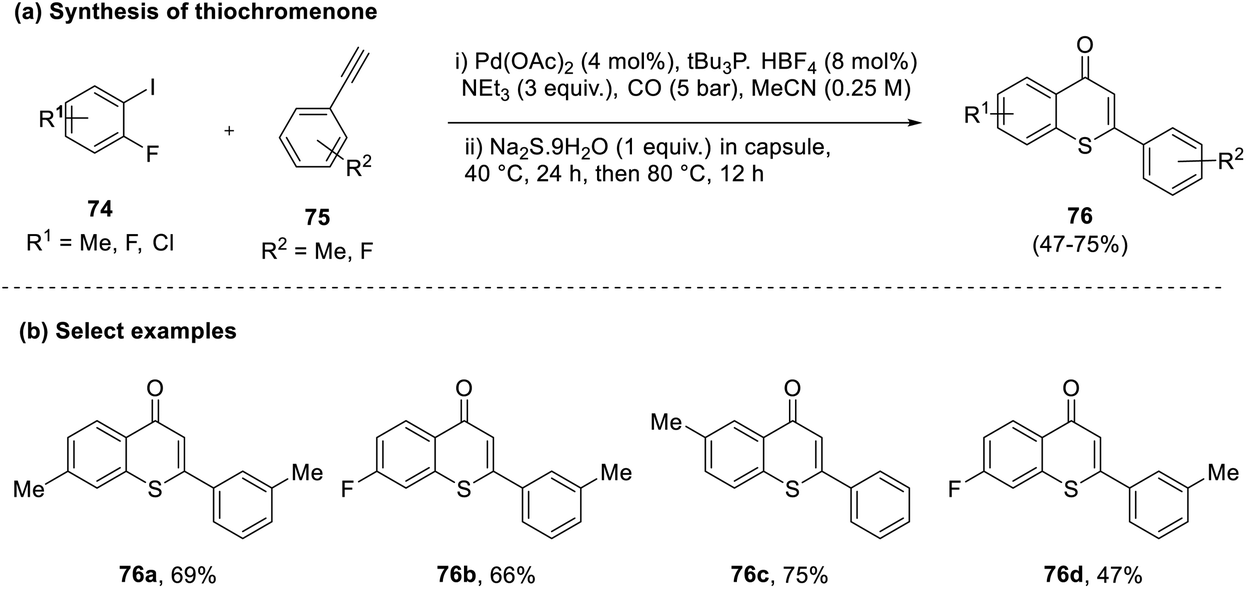 | ||
| Scheme 19 (a) Reagent capsule-based approach to selectively synthesize thiochromenes; (b) select examples. | ||
Sundaravelu et al. devised a greener approach for C–S coupling to achieve thiochromene derivatives. This protocol employed xanthate (78) as the sulfur surrogate and proceeded under visible light conditions, which is crucial for the initial C–S bond formation between the xanthate (78) and halogenated benzaldehydes (77). This step facilitated intermolecular charge transfer, followed by inter/intramolecular Michael addition to attain the final product (79).46 Interestingly, the use of crotonaldehyde resulted in the formation of the thiochromene ring (81) under the same reaction conditions, while chalcones led to the corresponding thiochromanol (dr: 77:23 to 83:17) (83) (Scheme 20).
 | ||
| Scheme 20 (a) Visible light-mediated synthesis of thiochromenes; (b) synthetic application; (c) select examples. | ||
The existing literature for synthesizing thiochromene-4-imines (86) showed that an improvement is required regarding the regioselectivity and higher loading of catalysts and ligands. Singh and co-workers proposed a greener approach using D-glucosamine as the ligand under Cu(I)-catalyzed conditions to address these challenges.47 This reaction proceeded through the formation of a copper complex (84b), which transitions to form a thiolate anion (84c) that ultimately generates a thietane ring (84e) by C–S bond formation. This four-membered cycle then undergoes cyclative rearrangement induced by the attack of the alkynyl group to yield the desired product (72–86%) (86) (Scheme 21).
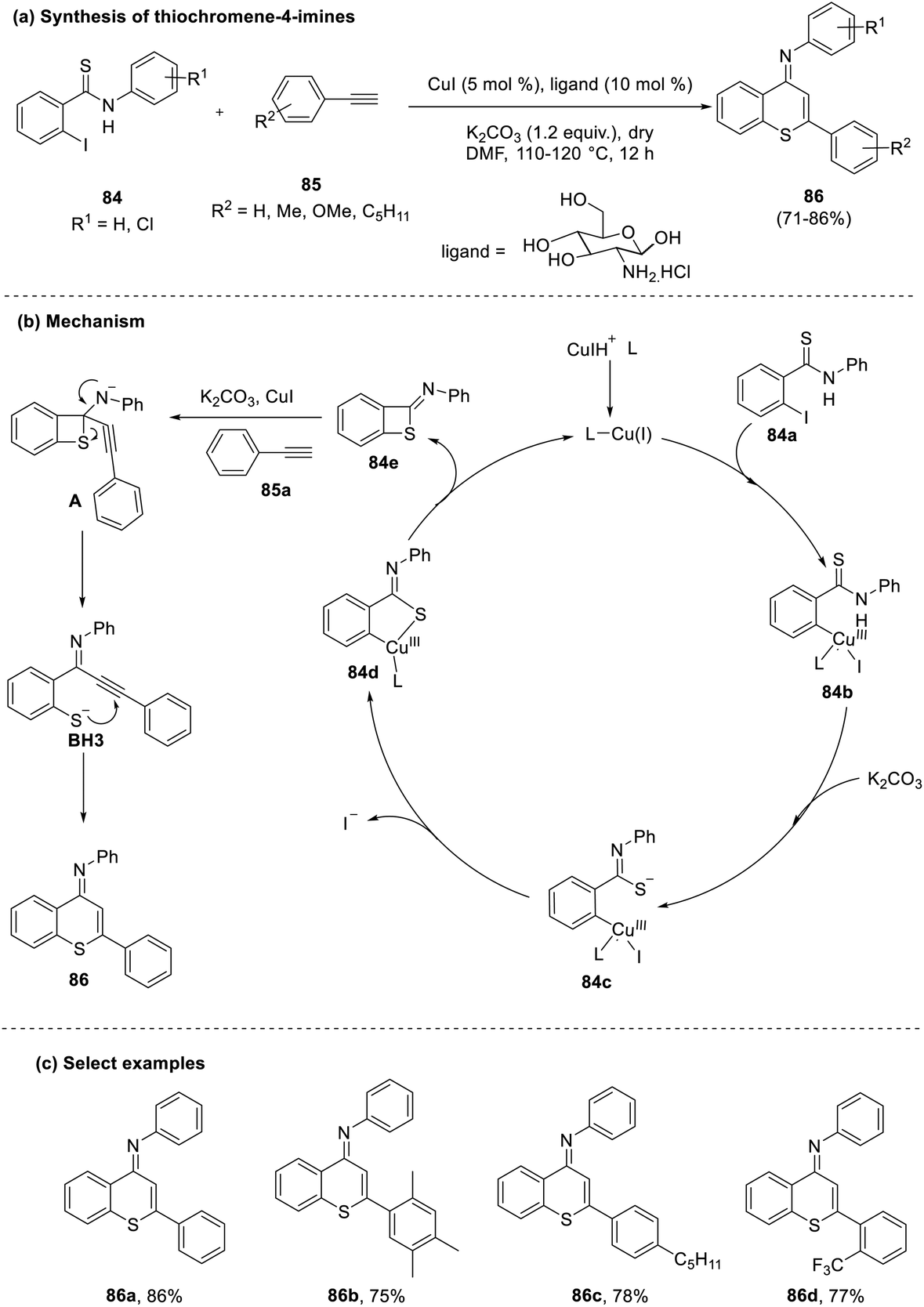 | ||
| Scheme 21 (a) Glucosamine-mediated cyclization to synthesize thiochromenes; (b) mechanism of coupling; (c) select examples. | ||
In 2023, Deepika et al. reported a novel de-nitrative C–S coupling methodology for synthesizing pyrazole-tethered thiochromenes (89) using elemental sulfur (88) as the sulfur source.48 This environmentally benign protocol leveraged 1,4-diazabicyclo-[2.2.2]-octane (DABCO) for activating the sulfur source and aided in regioselective C–H bond formation, simultaneously removing the nitro motif from the arene ring. Following control experiments, two possible mechanistic pathways were identified for this reaction. In path A, the reaction proceeds through the Michael addition of a trisulfur anion to the double bond of the chalcone, which subsequently undergoes S–S bond cleavage and an intramolecular nucleophilic substitution reaction to generate the final product (89). Contrarily, the other pathway proceeds by generating a polysulfide zwitterion. In both pathways, DMSO was crucial as it stabilized the intermediates and radicals generated during the reaction mechanism (Scheme 22).
 | ||
| Scheme 22 (a) 1,4-Diazabicyclo-[2.2.2]-octane (DABCO) mediated regioselective synthesis of thiochromenes; (b) mechanism involved in the formation of thiochromene-4-imines; (c) select examples. | ||
1.5. Cyclization reactions
In furtherance of the work exploring cyclization reactions for thiochromene synthesis, Yugandar and co-workers in the year 2017 reported a one-pot palladium-catalyzed intramolecular cyclization of haloarenes (90) with a thieno-fused thiodiketone (91).49 This reaction proceeded via two steps, forming an open chain thiovinyl ketone (A) in the first step, which is then cyclized to afford a thiochromene derivative (92) using 10 mol% palladium acetate. While thienyl- and furanyl-substitutions at the thiocarbonyl portion of diketones were well tolerated in this reaction, the introduction of a pyrrole ring or thienyl ring with a dimethylamino substituent at this end caused a deviation from the desired product, leading to the formation of the corresponding benzothiophene product (Scheme 23). | ||
| Scheme 23 (a) Palladium-catalyzed direct C–H alkylation to synthesize thiochromenes; (b) select examples. | ||
The reactions of sulfur-containing allenes (93) under acidic conditions with halogens have been studied by several groups like Zhou et al. and Ma et al.50,51 However, despite the promising results obtained from these studies, the electrophilic activation reactions entailing these molecules have been unchartered. To bridge this gap, Lozovskiy's group, in 2018, investigated the transformation of sulfur-containing allenes (93) under the influence of different Brønsted acids and observed the formation of a thiochromene system (94) with the use of trifluoroacetic acid at high temperature.52 Using NMR and DFT calculations, the ortho-carbon of the phenyl ring was discerned to be a highly reactive electrophilic center, which aided in intramolecular cyclization towards heterocycle formation. Furthermore, allenes with a p-methyl phenyl ring resulted in the formation of a 7-methyl substituted product as opposed to the 6-methyl substitution due to a [6,7]-methyl shift, which occurs due to the action of the superacid at high temperatures (Scheme 24).
 | ||
| Scheme 24 (a) Brønsted acid-mediated formation of thiochromenes; (b) mechanism of cyclization; (c) select examples. | ||
In the same year, Wang's group unveiled a novel protocol for the synthesis of halogenated thiochromenes conjugated with aliphatic amines (101) like piperidine, N-methyl piperazine, diethyl amine, and pyrrolidine.53 Friedel–Crafts reaction conditions were employed to facilitate the intramolecular cyclization of thiophenols (95) with simple chiral carboxylic acids to furnish thiochromanones (97), which were then subjected to the Vilsmeier–Haack protocol to achieve the thiochromene scaffold (98). These compounds were then substituted with aliphatic amines under alkaline conditions to obtain the final thiochromene rings substituted with heterocyclic rings (101) in yields of 46–82% (Scheme 25).
 | ||
| Scheme 25 (a) Intramolecular cyclization of thiophenol to synthesize thiochromenes; (b) select examples. | ||
Ma et al. envisaged a Ullmann-type coupling of aryl thioamides (102) with phenylacetylene (103) for the synthesis of thiochromene-4-imines (104) using L-proline as the ligand. This actualized reaction preferentially generated (E)-configuration products.54 A plausible mechanism was identified through X-ray analysis and literature findings, as shown in Scheme 26. The reaction proceeds through coupling with a copper catalyst to form a complex (A), which, under redox conditions, furnishes a 2-imino-benzothietane intermediate (E). In the subsequent step, the thietane ring opens, and a nucleophilic attack occurs, generating the desired product.
 | ||
| Scheme 26 (a) Proline-catalyzed cyclization to synthesize thiochromenes; (b) mechanism of cyclization; (c) select examples. | ||
Shigeno and co-workers in 2018 proposed a two-step peri-selective methodology for the synthesis of benzo thiochromenes (108) from the reaction of a methyl thioether derivative of naphthalene (105) with aromatic aldehydes (106) in the presence of a rhodium catalyst.55 The first step of the reaction was not compatible with aliphatic alkynes. In contrast, the use of diphenyl or unsymmetrical alkynes gave a mixture of products with an E/Z ratio of up to 11:89. In the final step, a 6-endo-trig pattern of annulation was observed to yield the final product in good yields (40–69%) (Scheme 27).
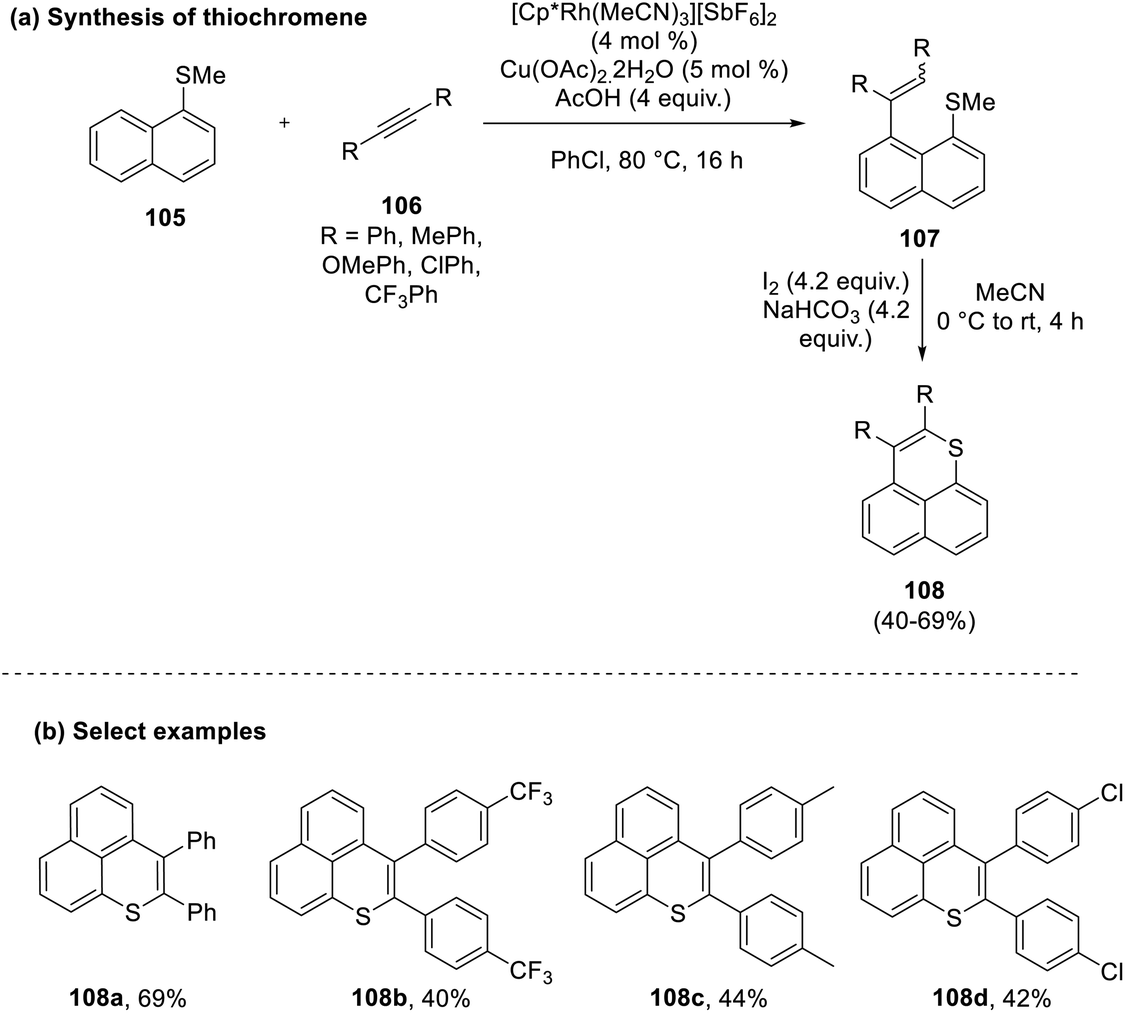 | ||
| Scheme 27 (a) Peri-selective cyclization of bicyclic systems to synthesize thiochromenes; (b) select examples. | ||
Intrigued by the importance of sulfur derivatives, Ali's group in 2019 sought to explore the chemical reactivity of enaminones (109) against phosphorus sulfides. This approach resulted in the formation of trichrome-4-thione (112) via intramolecular annulation of the thio-analog of an enaminone.56 Interestingly, when the same reaction was carried out using Lawesson's reagent as the thionation reagent, a [4 + 2] cycloaddition reaction took place, resulting in the formation of an oxathiaphosphine ring in place of the anticipated thiochromene ring (Scheme 28).
 | ||
| Scheme 28 Lawesson's reagent mediated generation of thiochromenes. | ||
In the same year, Zhou et al. devised a novel strategy employing carbene chemistry to generate a thiochromenone (117) from the hydroacylation cum cyclization of a thiadiazole (113) and propionaldehyde (114) in the presence of a rhodium catalyst.57 While the reaction was well tolerated with most of the substrates, propionaldehydes with iodophenyl, dimethyl pentanyl, and cyclopropyl substitutions were identified to be un-reactive substrates, possibly due to the steric effects of these groups. Expansion of the substrate scope demonstrated that alkenyl aldehydes are suitable substrates that are annulated in the presence of low catalyst levels. Further investigations revealed that the reactions proceeded only when the electronic properties of both substrates complemented each other (Scheme 29).
 | ||
| Scheme 29 (a) Ring expansion of thiadiazoles to synthesize thiochromenones; (b) select examples. | ||
Yang et al. also put forth a single electron transfer (SET) cyclization method for the synthesis of similar thiochromene molecules (120) from 1-thiomethyl naphthalene (118) in a peri-selective manner.58 The actualized reaction differed intrinsically from the conventional methods owing to the selective heteroarylation at the peri-position rather than at the C2 position, which is usually involved due to the steric effect. This approach gave rise to good yields with various substrates but failed to discern the by-products formed, excluding the heteroaryl sulfides and unreacted starting materials. Mechanistically, it was observed that 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) was essential for the cyclization as it was postulated to have a stabilizing effect on the cation-radical intermediate (118a) to aid in the single electron transfer cum annulation approach (Scheme 30).
 | ||
| Scheme 30 (a) HFIP-mediated single electron transfer annulation to synthesize thiochromenes; (b) mechanism of cyclization; (c) select examples. | ||
Velasco and co-workers explored the possibility of achieving thiochromene rings (122) by intramolecular cyclization of S-aryl propargyl thioethers (121) with N-iodosuccinimide (NIS) as the electrophilic reagent.59 Concomitantly, reactions using other catalysts that might act as iodinium agents were also attempted, with silver triflate identified as the most appropriate one on account of its ability to engender good coordinating counterions in comparison with other silver salts. This protocol was, however, incompatible with electron-withdrawing functionalities. meta-Substitution of the thioether led to a mixture of regioisomers in a ratio of 2:1 by 6-endo cyclization. In contrast, using the naphthol derivative of thioether corresponded to the generation of linear and angular thiochromenes in a ratio of 1.2:1 (Scheme 31).
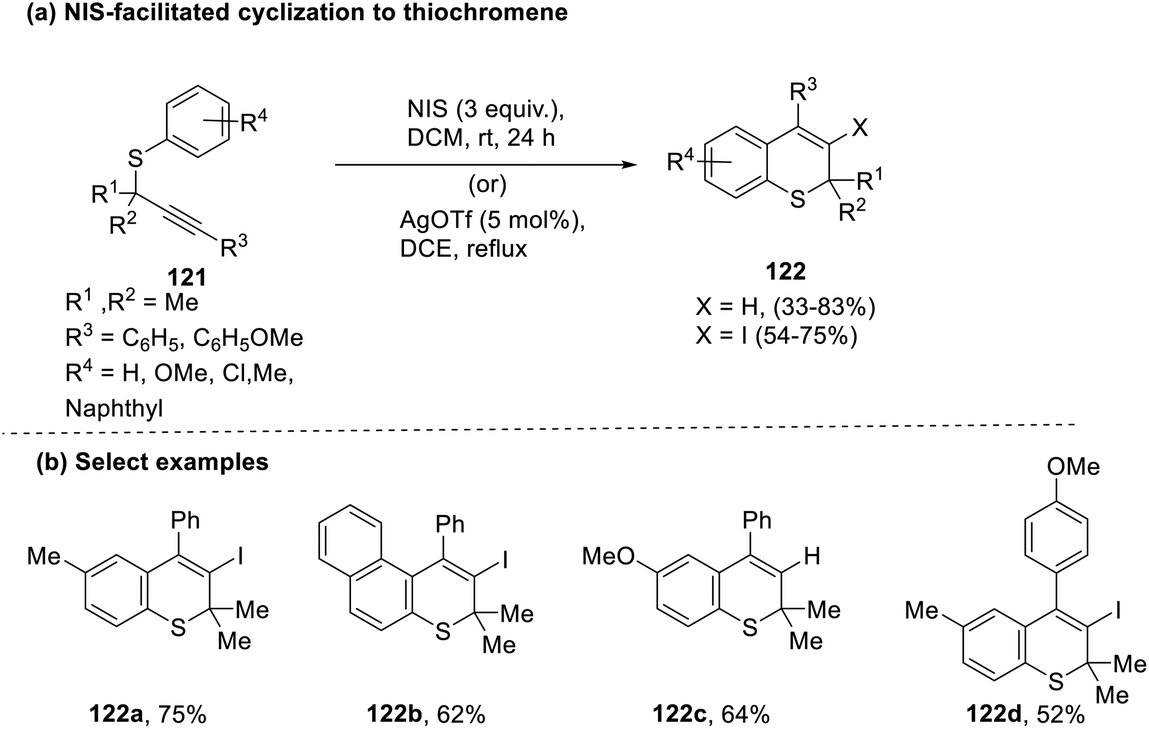 | ||
| Scheme 31 (a) N-Iodosuccinimide (NIS)-facilitated cyclization to synthesize thiochromenes; (b) select examples. | ||
In 2021, Wang et al. impelled the synthesis of thiochromenones (125) by developing a carbonylative reaction utilizing aromatic sulfonyl chlorides (123) and alkynes (124) in the presence of a nickel catalyst. This one-pot reaction used sulfonyl chloride as the sulfur surrogate and proceeded through a 6-endo-dig cyclization pathway.60 Interestingly, this reaction generated lower yields with o-substituted alkynes compared with m- or p-substituted alkynes, which is ascribed to the steric effects seen in the case of the former. Through control experiments, two mechanistic cycles were postulated bearing a common intermediate II as the starting point. This intermediate is generated by the migratory insertion of CO into nickel complex I and then reacts with terminal alkynes to produce alkynones that, on cyclization, generate thiochromenones (125a); similarly, when the same intermediate reacted with internal alkynes, ring expansion followed by reductive elimination furnished the final product (125) (Scheme 32).
 | ||
| Scheme 32 (a) 6-Endo-dig cyclization of sulfonyl chloride to synthesize thiochromenes; (b) mechanism of cyclization; (c) select examples. | ||
Ahlemeyer et al., in 2021, attempted the cascade cyclization of cinnamic acid thioesters (126), which proceeded via aldol cyclization cum lactonization to generate thiochromenes (130) under neat reaction conditions.61 The asymmetric synthetic protocol envisaged by this group demonstrated good yields (up to 79%) with an ee of 96% using electron-rich amidine-based catalysts (Scheme 33).
 | ||
| Scheme 33 (a) Amidine catalyst mediated synthesis of thiochromenes; (b) select examples. | ||
The conventional synthesis of thiochromene-containing polycyclic systems often necessitates transition metal catalysts and proceeds through multiple steps.62,63 These approaches, while feasible, are constrained by restricted substrate compatibility and reliance on environmentally harmful catalysts. Prompted by this challenge, Deng and co-workers (2021) streamlined the synthetic approach to present a sustainable base-catalyzed domino reaction of thioisatin (131) with bromoketone (132) to achieve thiochromene-fused furan scaffolds (20–80%) (133) (Scheme 34).64
 | ||
| Scheme 34 (a) Base-catalyzed intermolecular cyclization of thiochromenes; (b) synthetic application; (c) select examples. | ||
Song et al. (2021) adopted mild reaction conditions to actualize the synthesis of highly stereoselective (>19:1 Z/E ratio) thiochromene dioxides (137) through visible light-mediated photocatalysis.65 This approach utilized sodium metabisulfite as the sulfur dioxide surrogate and reacted it with terminal alkynes (136) in the presence of a phenyl ethynyl derivative of a diazonium coupling agent (135). Examination of the substrate scope revealed that the reaction was impeded when strong electron-withdrawing groups were substituted in the aryl rings attached to the coupling agent. This was ascribed to the formation of an unstable intermediate (alkenyl radical) in situ (Scheme 35).
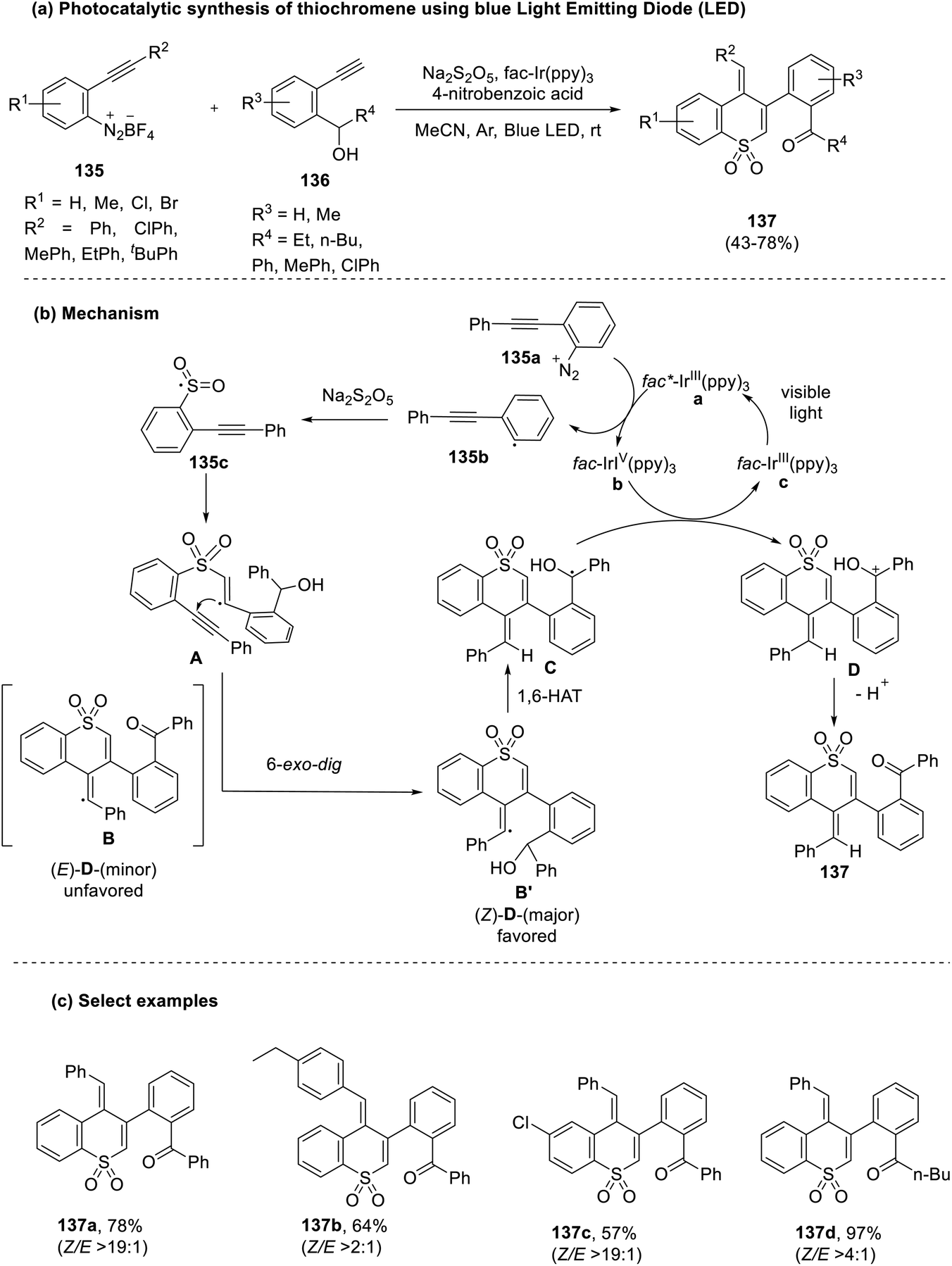 | ||
| Scheme 35 (a) Photocatalytic synthesis of thiochromenes using a blue Light Emitting Diode (LED); (b) mechanism of cyclization; (c) select examples. | ||
In 2023, Davoine and co-workers synthesized carboxy-(thio)-chromenes (142) by the cyclization of substituted mercaptobenzaldehyde (138) under optimum reaction conditions.66 This reaction was facilitated by the formation of an α,β-unsaturated ketone (140) or a dioxane-dione intermediate (139) that undergoes annulation to give the desired product (Scheme 36).
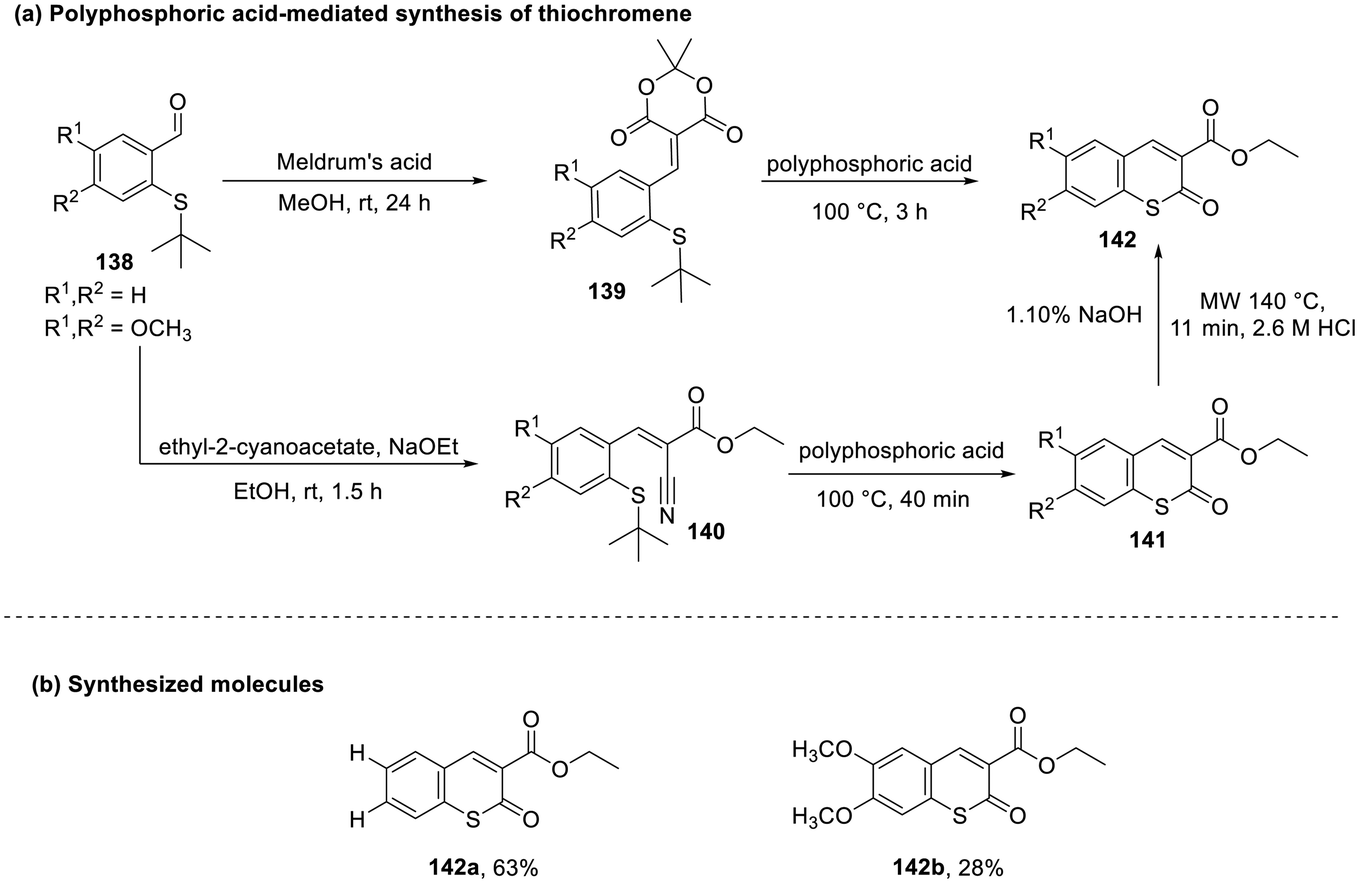 | ||
| Scheme 36 (a) Polyphosphoric acid-mediated synthesis of thiochromenes; (b) synthesized molecules. | ||
In the same context, Ortiz et al. (2023) proposed three novel methodologies for the synthesis of thiochromenes from thiophenol (143) and bromopropionic acid (144) or crotonic acid (145).67 The reaction with bromopropionic acid was catalyzed by the use of a basic buffer solution. In contrast, the reaction with crotonic acid was facilitated by a phase transfer catalyst, tetra-n-butylammonium fluoride (TBAF) (Scheme 37).
 | ||
| Scheme 37 (a) Mild base-catalyzed reaction of thiophenol to synthesize thiochromenones; (b) select examples. | ||
Taking into account the widespread utility of thiochromenes, Bartz et al., in 2023, devised a sustainable protocol for achieving sulfenyl thiochromenes (154) using thiols (153) and thioaryl nones (150) in the presence of visible light.68 This work sought to improve the protocols reported by Xu's group (2019) and Ai's group (2020) in this area, which demanded oxidant species and excess solvent.69,70 Besides offering sustainable reaction conditions, the recently reported method commenced with light irradiation to generate an intramolecular complex (B), which, in the presence of a thiol, triggers Hydrogen Atom Transfer (HAT) to give a thiyl radical (C). This radical initiates chain propagation and undergoes intramolecular ring cyclization to afford the final product (154) (Scheme 38).
 | ||
| Scheme 38 (a) Hydrogen Atom Transfer (HAT)-mediated synthesis of thiochromenes; (b) mechanism of cyclization; (c) select examples. | ||
1.6. Diels–Alder reactions
Wang's group laid the foundation for the synthesis of 3-substituted thiochromenes, which was then explored by Rios et al.71,72 These asymmetric catalytic methods activated by iminium ions or thiourea facilitated the substitution of an electron-withdrawing functionality at the C3 position of the thiochromene, thereby limiting its further extension. Inspired by this, Ahlemeyer et al. sought to design 3-unsubstituted thiochromenes (156) from thioesters of mercaptobenzaldehyde (155) by using a homobenzotetramisole derivative (HBTM-2), an amidine-based catalyst.73 This protocol demonstrated an enormous substrate scope and offered products with high enantioselectivity (>99% ee), forming only carbon dioxide as the byproduct. Using transition state models, they proposed a hetero-Diels–Alder reaction, wherein C–S and C–C bond formation happens simultaneously, which is responsible for the absolute configuration of the thiochromene derivatives (Scheme 39). | ||
| Scheme 39 Homobenzotetramisole (HBTM-2) catalyzed synthesis of thiochromenes. | ||
Mlostoń and co-workers in 2018 devised a thia-Diels–Alder protocol for attaining novel 4H-thiochromenes (159 and 161) from thiochalcones (157) and anthroquinones (158 and 160).74 In this reaction, a highly regioselective [4 + 2] cycloaddition was observed between the two substrates to yield the desired product in 89–94% yields. Further investigation into the substrate scope revealed that the reaction was not bench-feasible with benzoquinones, as the pure synthesized product underwent decomposition under ambient conditions, posing a challenge for practical application. Besides this, the reaction showed compatibility issues with menadione as well owing to the extended timeframe needed and the formation of significant amounts of [4 + 2] decomposed products along with the product (Scheme 40).
 | ||
| Scheme 40 (a) Thia-Diels–Alder approach to thiochromene synthesis; (b) select examples. | ||
Expanding further on this domain, An et al. leveraged aryne chemistry and C–S bond formation to streamline the synthesis of benzothiochromenes (166).75 In particular, they addressed the requirement of excess base, harsh reaction conditions, and the use of costly catalysts through the use of Kobayashi's reagent, an aryne precursor (163) and a thionoester (162). This reaction employed a relatively inexpensive phase transfer catalyst (18-crown-ether-6) and made use of 4 Å (Angstrom) molecular sieves (MS) to selectively yield benzothiochromenes (166) (Scheme 41).
 | ||
| Scheme 41 (a) Kobayashi's reagent catalyzed synthesis of thiochromenes; (b) select examples. | ||
1.7. Miscellaneous reactions
Chanda et al., in 2021, reported a straightforward approach for the synthesis of benzothiochromenes (169) via carboarylation of alkynes (167) with activated benzylic alcohols (168).76 This reaction promoted the use of a cheaper and greener iron catalyst for actualizing the desired product with high chemo- and regio-selectivity. In accordance with the previous reports and control experiments, a plausible mechanism was outlined. Initially, a highly reactive benzylic carbocation (B) is generated by the activation of the alcohol (168) by the iron catalyst. This then transitions to form a stable aryl vinyl carbocation (C), which cyclizes and forms the final product by following the Friedel–Crafts alkylation pathway (Scheme 42).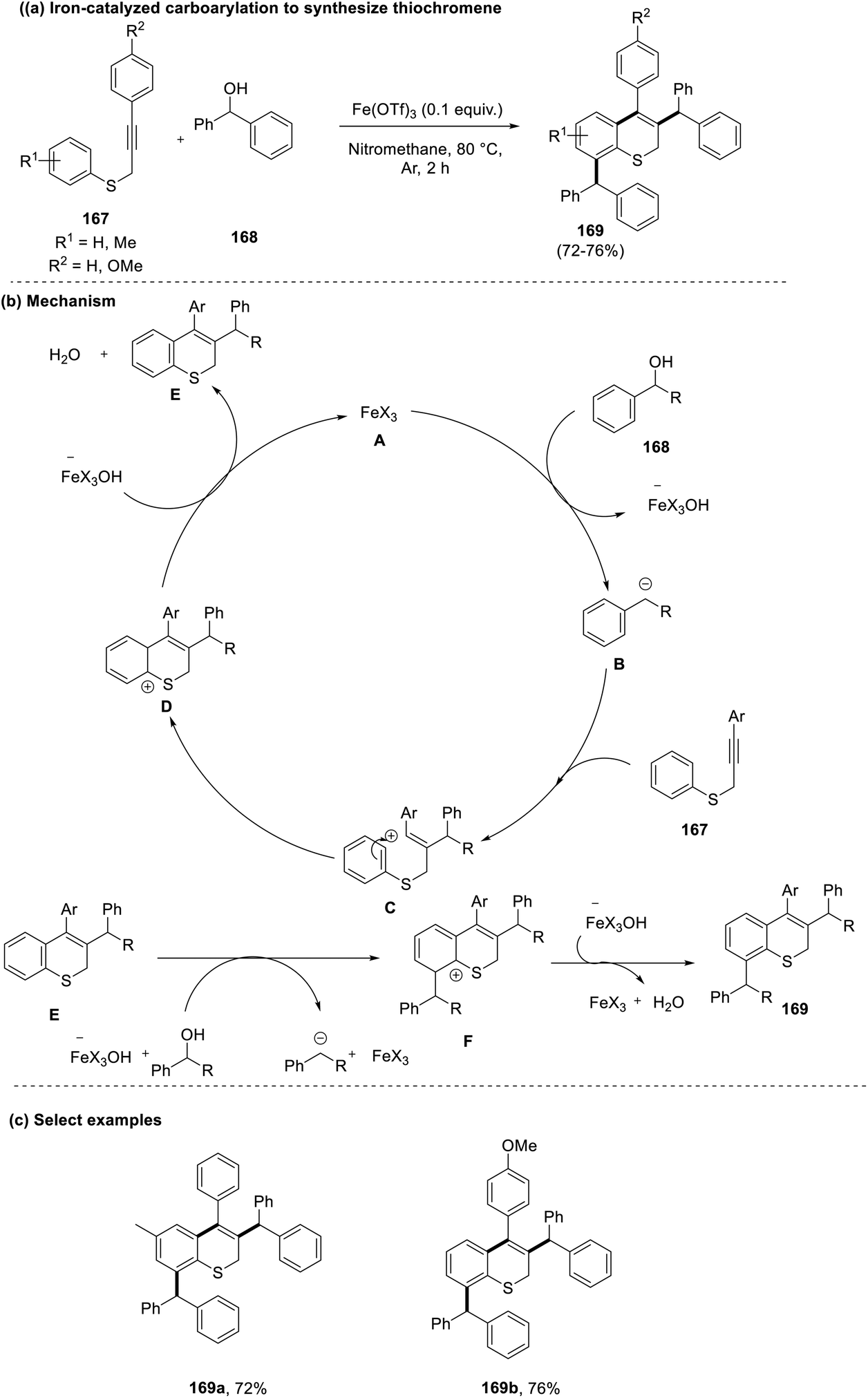 | ||
| Scheme 42 (a) Iron-catalyzed carboarylation to synthesize thiochromenes; (b) mechanism involved; (c) select examples. | ||
In the same year, Shibata's group put forth an atom-economical protocol for the synthesis of indenothiochromene (171) by a Ni-catalyzed carbothiolation reaction of symmetric phenyl-substituted diynes (170).77 This reaction took place in the presence of a non-activated C–S bond, which underwent cleavage in the presence of a catalyst and was subsequently difunctionalized to generate the thiochromene scaffold (171). In contrast, the use of unsymmetric diynes led to the generation of a regioisomeric mixture of the product in lower yields (34%). Exploration of the synthetic scope of this work revealed the uniqueness of this substrate in undergoing self-coupling to produce thiochromene entities (172) when the employed Ni-catalyst was reduced in situ during the reaction (Scheme 43).
 | ||
| Scheme 43 (a) Nickel-catalyzed carbothiolation to synthesize thiochromenes; (b) mechanism involved; (c) synthetic application; (d) select examples. | ||
Virumbrales et al., in 2022, also focussed on the development of indenothiochromenes (174) by the use of a cyclopropyl gold catalyst.78 The thio analog of (E)-(alkynyl)-styrene (173), used as a substrate for this reaction, participated in a cascade process wherein the styrene underwent intramolecular rearrangement and subsequent cyclization by following the Friedel–Crafts pathway. The use of the cyclopropyl gold catalyst with the Z-configuration of the substrate, however, led to the formation of diastereoisomers (dr value: 1:1.6 to 4:1) (Scheme 44).
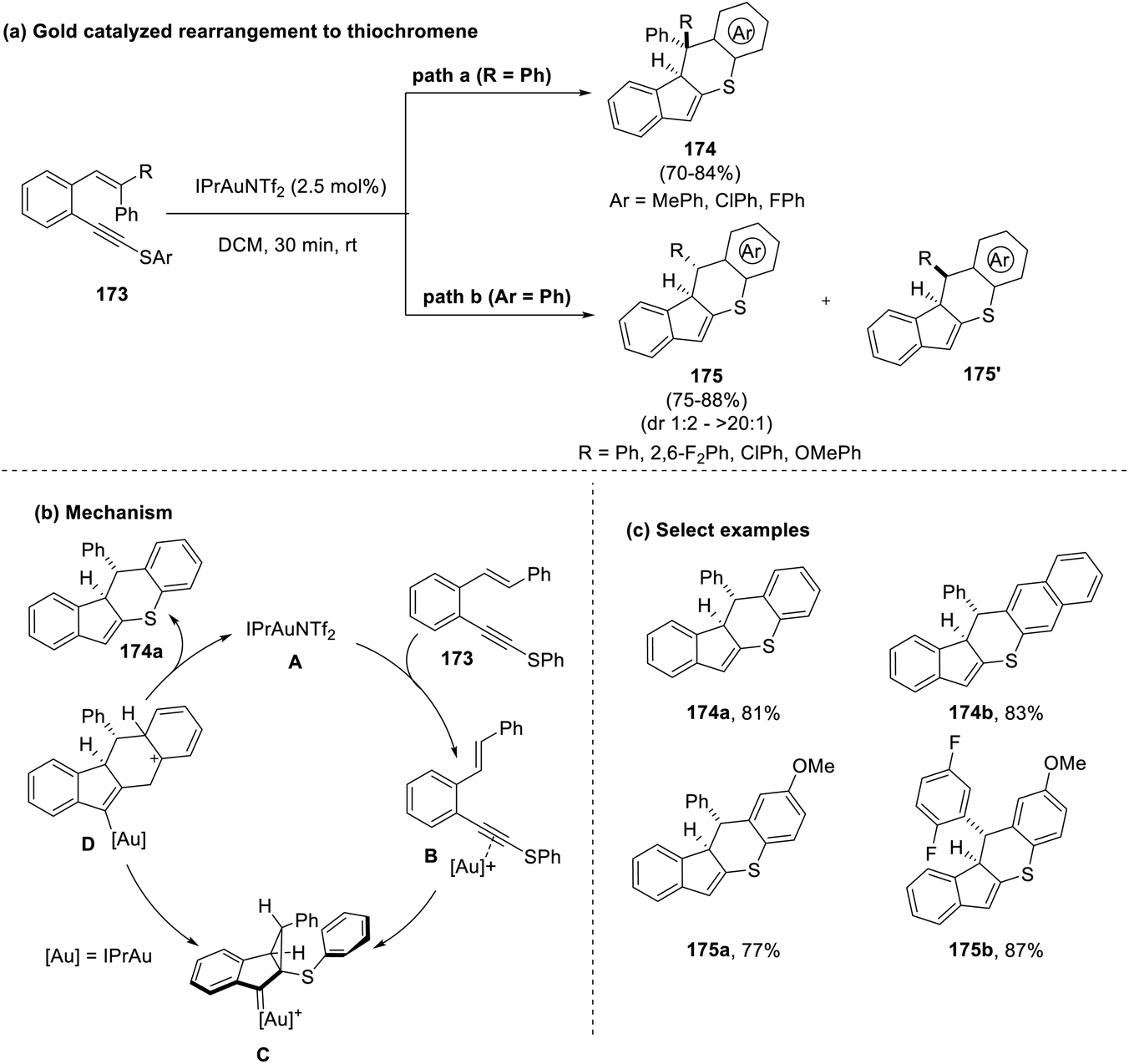 | ||
| Scheme 44 (a) Gold-catalyzed rearrangement to synthesize thiochromenes; (b) mechanism involved; (c) select examples. | ||
2. Functionalization of thiochromenes
Mahato et al., in 2017, reported a mild [3 + 2] cycloaddition reaction for the synthesis of thiochromenones bearing an oxime functionality (178) from thiocoumarin (176) and styrenes (177).79 The reaction tolerated a wide range of substituents in both substrates and proceeded via a regioselective annulation as sulfur serves as a better nucleophile in contrast to its oxygen counterpart, as the former has a larger size and better polarizability. Furthermore, the final product was utilized as the synthon for Beckmann rearrangement and Semmler–Wolf type reactions to generate the corresponding products (179 and 180), as shown in Scheme 45. | ||
| Scheme 45 (a) [3 + 2] Cycloaddition to synthesize thiochromenones under basic conditions; (b) synthetic application; (c) select examples. | ||
The late-stage functionalization of thiochromenones attempted by Barakat's group in 2021 further explored the synthetic utility of these reactions.80 In particular, the synthetic transformation of a natural compound, D-10-camphorsulfonyl chloride (183), was facilitated using the previously optimized reaction conditions to generate a thiochromenone-bearing derivative (185), as shown in Scheme 46.
 | ||
| Scheme 46 Late-stage functionalization of D-10-camphorsulfonyl chloride. | ||
Barakat et al., in 2021, devised a 1,3-dipolar cycloaddition approach for the synthesis of thiochromenones tethered with a spiroxindole motif (188) in a wholly stereoselective and regioselective manner.81 This reaction used L-proline as the catalyst, which initially reacts with isatin (187) to yield a cyclic lactone (A), which then undergoes cycloaddition with the chalcone (186) in a trans-selective manner to minimize the steric repulsion that usually arises when the carbonyl groups of isatin are cis to the chalcone moiety. The molecular electron density theory study also validated this unique ortho-/endo-selective nature of the reaction carried out by the same group in the year 2023 (Scheme 47).81
 | ||
| Scheme 47 (a) Stereoselective synthesis of spiroxindole tethered thiochromenes using proline; (b) select examples. | ||
Das et al., in 2022, ventured into the development of novel protocols for actualizing double intramolecular cyclization in a one-pot, atom-economical manner to attain polycyclic benzo-fused systems (190).82 The use of thiobutanal with boron trifluoride ethereate (189) smoothly delivered the products in good yields with the exception of substrates bearing halogens in the ortho position, as the inductive effect of the latter reduced the reactivity of the S-aryl ring for double cyclization. In addition, the presence of the m-methoxy functionality also served to highlight an anomaly as this substrate gave rise to a mixture of regioisomers that could not be separated (10:1). Mechanistic investigation revealed the formation of a carbenium ion, which depicted three possible conformational isomers, one trans and two cis forms; despite the existence of three conformers, the cis-fused axial conformer (D′) with enhanced hyperconjugative stabilization triumphed over the other two due to its minimal energy requirement for cyclization and the absence of the 1,3-allylic strain witnessed in the trans- and cis-conformers (equatorial) (Scheme 48).
 | ||
| Scheme 48 (a) Double intramolecular annulation to obtain polycyclic thiochromenes; (b) select examples; (c) mechanism involved. | ||
Etsè and co-workers in 2021 designed a series of thiochromane dioxides (195 and 196), which were envisioned to be the bio-isosteric replacement of nitrogen in the thiadiazine ring to explore the bioactive potential of this scaffold.83 To attain this, thiochromanone (192) was reacted with a Grignard reagent, dehydrated, and subsequently oxidized to generate the final product (195 and 196). During this reaction, palladium on carbon was used for hydrogenation in the final step to generate the final product. However, this Pd/C hydrogenation condition rendered the reaction with the use of the chloro-substituted substrate, aiding in removing the chlorine atom (Scheme 49).
 | ||
| Scheme 49 (a) Grignard reagent catalyzed synthesis of thiochromane dioxides; (b) select examples. | ||
Recently, in 2024, Ali et al. explored the thia-Michael addition reaction of hydroxyl thiocoumarin (197) with cinnamaldehyde (198) using an organocatalyst (L-proline) to synthesize novel thiopyranothiochromenone products (199) in a one-pot reaction.84 This reaction proceeded in the presence of protic solvents while using aprotic non-polar solvents hindered the reaction. Furthermore, the reaction showed excellent compatibility with cinnamaldehyde substituted with electron-donating functionalities yet failed to proceed when they were replaced with electron-withdrawing groups. Mechanistic investigation proposed the abstraction of a proton from hydroxythiocoumarin by the zwitterionic form of proline to form a nucleophile (B) that tautomerizes to form B′ and reacts with cinnamaldehyde (198) to form a Mannich intermediate (D) which finally undergoes ring cyclization to yield the product with the removal of proline (Scheme 50).
 | ||
| Scheme 50 (a) Proline catalyzed synthesis of thiopyranothiochromene from thiocoumarin; (b) mechanism involved in ring cyclization; (c) select examples. | ||
3. Conclusion
Various approaches have been explored, ranging from catalytic asymmetric reactions to aryne-chemistry-based reactions, for constructing a diverse portfolio of thiochromene derivatives. In the past decade, thiol surrogates have been extensively explored with newer alternatives like disulfides, thionoesters, and thioamides, aiding in developing cleaner reaction outcomes with minimal by-product generation, as discussed earlier. Concurrently, several greener methodologies have also been accomplished, highlighting greener catalysts like iron catalysts for the chemoselective synthesis of benzothiochromenes or D-glucosamine to drive the sustainable synthesis of thiochromene-4-imines. In addition to these noteworthy improvements, chalcones might be involved in improving atom economy as reactions employing these substrates have resulted in higher yields of up to 95%. Furthermore, this review also emphasizes that while the use of expensive metal catalysts like gold and rhodium is beneficial, relatively more straightforward reaction conditions employing p-toluene sulfonic acid, tetramethyl guanidine, PPh3, or potassium iodide offer a more practical approach for synthesizing the desired thiochromene derivatives.Despite the advancement in synthetic chemistry, the synthesis of thiochromenes can be streamlined and optimized even further. Given the importance of heterocyclic molecules in developing phytopharmaceuticals, synthetic methods to obtain enantioenriched thiochromenes as intermediates during complex natural product development require further work, as only one such work has been reported. Moreover, despite the exploration of metal catalysts and phase transfer catalysts, the use of enzyme catalysis, sonochemical methods, ionic liquids, and electrochemical catalysis still needs to be explored and, therefore, needs more attention. In addition, while several protocols attempted in this area show good substrate compatibility, specific methods suffer from limited substrate scope, emphasizing the need for newer reactions to diverge this aspect.
While the synthesis of thiochromenes has made strides in the recent decade, ample opportunities for refining and optimizing synthetic methodologies still exist. Therefore, the continued exploration of the design of eco-friendly and diverse catalytic approaches holds the potential to advance the synthetic methods of thiochromenes and their derivatives.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review titled “Recent Developments in Thiochromene Chemistry”.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We sincerely express our gratitude to BITS-Pilani, Pilani Campus, and BITS-Pilani, Hyderabad Campus, for providing the necessary facilities to do the work. The authors SM and KVGC acknowledge the funding received from the SERB-CRG projects (Ref. No. CRG/2022/005290 and CRG/2022/001889) under the Department of Science and Technology (DST), New Delhi.References
- J. Jampilek, Molecules, 2019, 24(21), 3839 CrossRef CAS.
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- M. Mustafa and J. Y. Winum, Expert Opin. Drug Discovery, 2022, 17, 501–512 CrossRef CAS PubMed.
- S. Pathania, R. K. Narang and R. K. Rawal, Eur. J. Med. Chem., 2019, 180, 486–508 CrossRef CAS PubMed.
- K. Laxmikeshav, P. Kumari and N. Shankaraiah, Med. Res. Rev., 2022, 42, 513–575 CrossRef CAS PubMed.
- K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- M. J. Tilby and M. C. Willis, Expert Opin. Drug Discovery, 2021, 16, 1227–1231 CrossRef.
- S. W. Schneller, Adv. Heterocycl. Chem., 1975, 18, 59–97 CrossRef CAS.
- J. Song, L. M. Jones, G. E. Chavarria, A. K. Charlton-Sevcik, A. Jantz, A. Johansen, L. Bayeh, V. Soeung, L. K. Snyder, S. D. Lade Jr., D. J. Chaplin, M. L. Trawick and K. G. Pinney, Bioorg. Med. Chem. Lett., 2013, 23, 2801–2807 CrossRef CAS PubMed.
- E. J. Choi, J. I. Lee and G. H. Kim, Int. J. Mol. Med., 2012, 29, 252–256 CAS.
- A. Roeder, M. Schaller, M. Schäfer-Korting and H. C. Korting, Skin Pharmacol. Appl. Skin Physiol., 2004, 17, 111–118 CrossRef CAS PubMed.
- S. Frigoli, C. Fuganti, L. Malpezzi and S. Serra, Org. Process Res. Dev., 2005, 9, 646–650 CrossRef CAS.
- N. A. Abdallah, Int. J. Electrochem. Sci., 2016, 11, 10715–10731 CrossRef CAS.
- I. V. Ukrainets and N. L. Bereznyakova, Chem. Heterocycl. Compd., 2012, 48, 155–165 CrossRef CAS.
- R. Feringa, H. S. Siebe, N. Klement, J. D. Steen and W. R. Browne, Adv. Mater., 2022, 3, 282–289 RSC.
- N. Kang, J. M. Lee, A. Jeon, H. Bin Oh and B. Moon, Tetrahedron, 2016, 72, 5612–5619 CrossRef CAS.
- A. Kumar, V. Hüch and V. J. Ram, CrystEngComm, 2013, 15, 7019–7019 RSC.
- U. Lücking, Org. Chem. Front., 2019, 6, 1319–1324 RSC.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
- R. Roy, S. Rakshit, T. Bhowmik, S. Khan, A. Ghatak and S. Bhar, J. Org. Chem., 2014, 79, 6603–6614 CrossRef CAS PubMed.
- Q.-L. Zhou and Q.-L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352–5353 CrossRef CAS.
- B. M. Sahoo and B. K. Banik, Curr. Organocatal., 2019, 6, 92–105 CrossRef CAS.
- C. Bhanja, S. Jena, S. Nayak and S. Mohapatra, Beilstein J. Org. Chem., 2012, 8, 1668–1694 CrossRef CAS.
- S. Saha and C. Schneider, Chem. – Eur. J., 2015, 21, 2348–2352 CrossRef CAS.
- W. Wang, H. Li, J. Wang and L. Zu, J. Am. Chem. Soc., 2006, 128, 10354–10355 CrossRef CAS PubMed.
- R. Rios, H. Sundén, I. Ibrahem, G.-L. Zhao, L. Eriksson and A. Córdova, Tetrahedron Lett., 2006, 47, 8547–8551 CrossRef CAS.
- H. H. Kinfe, F. M. Mebrahtu, F. L. Makolo, P. T. Moshapo and M. M. Manana, J. Org. Chem., 2014, 79, 3111–3118 CrossRef CAS PubMed.
- A. R. Choudhury and S. Mukherjee, Adv. Synth. Catal., 2013, 355, 1989–1995 CrossRef CAS.
- A. K. Simlandy and S. Mukherjee, J. Org. Chem., 2017, 82, 4851–4858 CrossRef CAS.
- C. Hoyle, A. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- Q. Wu, S. Wu, J. Zou, Q. Wang, C. Mou, P. Zheng and Y. R. Chi, Org. Lett., 2023, 25, 3971 Search PubMed.
- T. T. H. Nguyen, T. X. Nguyen, T. T. T. Cao, T. H. Dinh, H. H. Nguyen, T. T. T. Bui, V. P. Pham and D. H. Mac, Synlett, 2017, 429–432 CAS.
- S. Sangeetha and G. Sekar, Org. Lett., 2019, 21, 75–79 CrossRef.
- M. Saini, K. Raigar and A. K. Guleria, Polycyclic Aromat. Compd., 2023, 1–17 CAS.
- V. Luque-Agudo, J. Albarrán-Velo, J. G. Fernández-Bolaños, O. López, M. E. Light, J. M. Padrón, I. Lagunes, E. Román, J. A. Serranoa and M. V. Gil, New J. Chem., 2017, 41, 3154–3162 RSC.
- P. Muthupandi, N. Sundaravelu and G. Sekar, J. Org. Chem., 2017, 82, 1936–1942 CrossRef CAS.
- T. T. H. Le, Y. Youhei, Q. H. Le, T. B. Nguyen and D. H. Mac, Org. Biomol. Chem., 2019, 17, 6355–6358 RSC.
- N. Sundaravelu and G. Sekar, Chem. Commun., 2020, 56, 8826–8829 RSC.
- C. Ortiz, F. Echeverri, S. Robledo, D. Lanari, M. Curini, W. Quiñones and E. Vargas, Molecules, 2020, 25, 800 CrossRef CAS.
- X. Wang, Y. Luo, J. Zhao and S. Luo, Org. Biomol. Chem., 2023, 21, 6697–6701 RSC.
- T. Inami, T. Kurahashi and S. Matsubara, Org. Lett., 2014, 16, 5660–5662 CrossRef CAS PubMed.
- F. Zhu and X.-F. Wu, J. Org. Chem., 2018, 83, 13612–13617 CrossRef CAS.
- J. Pan, Z. Zhang, L. Chen and N. Jiao, Chin. J. Chem., 2023, 41, 509–513 CrossRef CAS.
- S. Ponra, A. Nyadanu, N. Pan, E. Martinand-Lurin, A. Savy, M. Vitale, L. E. Kaim and L. Grimaud, Org. Process Res. Dev., 2020, 24, 827–834 CrossRef CAS.
- C. Shen, A. Spannenberg and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 5067–5070 CrossRef CAS PubMed.
- N. Sundaravelu, A. Nandy and G. Sekar, Org. Lett., 2021, 23, 3115–3119 CrossRef CAS PubMed.
- S. K. Singh, M. S. Yadav, A. S. Singh, A. K. Agrahari, N. Mishra, S. Kumar and V. K. Tiwari, ACS Omega, 2021, 6, 21125–21138 CrossRef CAS PubMed.
- D. Deepika, A. K. Paul, C. C. Malakar, A. Bansal and V. Singh, Asian J. Org. Chem., 2023, 12, e202300289 CrossRef.
- S. Yugandar, S. Konda and H. Ila, Org. Lett., 2017, 19, 1512–1515 CrossRef CAS.
- C. Zhou, C. Fu and S. Ma, Tetrahedron, 2007, 63, 7612–7616 CrossRef CAS.
- S. Ma, H. Ren and Q. Wei, J. Am. Chem. Soc., 2003, 125, 4817–4830 CrossRef CAS PubMed.
- S. V. Lozovskiy, A. Y. Ivanov, O. V. Khoroshilova and A. V. Vasilyev, Beilstein J. Org. Chem., 2018, 14, 2897–2906 CrossRef CAS PubMed.
- D. J. Wang, Z. Hou, H. Xu, R. An, X. Su and C. Guo, Bioorg. Med. Chem. Lett., 2018, 28, 3574–3578 CrossRef CAS.
- Y. G. Ma, M. Q. Huang, Z. Liu, J. Q. Liu and X. S. Wang, J. Org. Chem., 2018, 83, 9504–9509 CrossRef CAS PubMed.
- M. Shigeno, Y. Nishii, T. Satoh and M. Miura, Asian J. Org. Chem., 2018, 7, 1334–1337 CrossRef CAS.
- T. E. Ali, M. A. Assiri, I. S. Yahia and H. Y. Zahran, Synlett, 2019, 550–557 CAS.
- B. Zhou, Q. Wu, Z. Dong, J. Xu and Z. Yang, Org. Lett., 2019, 21, 3594–3599 CrossRef CAS PubMed.
- S. Yang, R. Cheng, M. Zhang, Z. Bin and J. You, ACS Catal., 2019, 9, 6188–6193 CrossRef CAS.
- N. Velasco, A. Suarez, F. Martinez-Lara, M. A. Fernandez-Rodriguez, R. Sanz and S. Suarez-Pantiga, J. Org. Chem., 2021, 86, 7078–7091 CrossRef CAS PubMed.
- W. Wang, Z. P. Bao, X. Qi and X.-F. Wu, Org. Lett., 2021, 23, 6589–6593 CrossRef CAS PubMed.
- N. A. Ahlemeyer, M. R. Straub, D. M. Leace, B. A. Matz and V. B. Birman, J. Org. Chem., 2021, 86, 1191–1197 CrossRef CAS PubMed.
- T. Arai and Y. Yamamoto, Org. Lett., 2014, 16, 1700–1703 CrossRef CAS.
- K. C. Majumdar and B. Roy, Synlett, 2003, 133–142 CAS.
- Q. Deng, A. Yu, S. Zhang and X. Meng, Org. Chem. Front., 2021, 8, 936–940 RSC.
- K. X. Song, X. Y. Qin, Z. X. Ma, F. Z. Geng, W. J. Hao, S. J. Tu and B. Jiang, Org. Chem. Front., 2021, 8, 5681–5686 RSC.
- C. Davoine, A. Traina, J. Evrard, S. Lanners, M. Fillet and L. Pochet, Eur. J. Med. Chem., 2023, 259, 115636 CrossRef CAS.
- C. Ortiz, M. Breuning, S. Robledo, F. Echeverri, E. Vargas and W. Quiñones, Heliyon, 2023, 9, e17801 CrossRef CAS.
- R. H. Bartz, K. B. Silva, R. R. S. A. Santos, T. J. Peglow, E. J. Lenardao, B. A. Iglesias, F. Penteado, G. Perin and R. G. Jacob, ChemCatChem, 2023, 15, e202201557 CrossRef CAS.
- J. Xu, F. Zhang, S. Zhang, L. Zhang, X. Yu, J. Yan and Q. Song, Org. Lett., 2019, 21, 1112–1115 CrossRef CAS PubMed.
- Z. Ai, J. Xiao, Y. Li, B. Guo, Y. Du and K. Zhao, Org. Chem. Front., 2020, 7, 3935–3940 RSC.
- W. Wang, H. Li, J. Wang and L. Zu, J. Am. Chem. Soc., 2006, 128, 10354–10355 CrossRef CAS.
- R. Rios, H. Sundén, I. Ibrahem, G.-L. Zhao, L. Eriksson and A. Cordova, Tetrahedron Lett., 2006, 47, 8547–8551 CrossRef CAS.
- N. A. Ahlemeyer and V. B. Birman, Org. Lett., 2016, 18, 3454–3457 CrossRef CAS.
- G. Mlostoń, K. Urbaniak, P. Urbaniak, A. Marko, A. Linden and H. Heimgartner, Beilstein J. Org. Chem., 2018, 14, 1834–1839 CrossRef.
- Y. An, F. Zhang, G. Du, Z. Cai and L. He, Org. Chem. Front., 2021, 8, 6979–6984 RSC.
- R. Chanda, A. Kar, A. Das, B. Chakraborty and U. Jana, Org. Biomol. Chem., 2021, 19, 5155–5160 RSC.
- T. Shibata, A. Sekine, M. Akino and M. Ito, Chem. Commun., 2021, 57, 9048–9051 RSC.
- C. Virumbrales, M. El-Remaily, S. Suarez-Pantiga, M. A. Fernandez-Rodriguez, F. Rodriguez and R. Sanz, Org. Lett., 2022, 24, 8077–8082 CrossRef CAS PubMed.
- K. Mahato, P. R. Bagdi and A. T. Khan, Org. Biomol. Chem., 2017, 15, 5625–5634 RSC.
- A. Barakat, M. S. Islam, M. Ali, A. M. Al-Majid, S. Alshahrani, A. S. Alamary, S. Yousuf and M. I. Choudhury, Symmetry, 2021, 13, 1426 CrossRef CAS.
- M. S. Islam, A. M. Al-Majid, M. Haukka, Z. Parveen, N. Ravaiz, A. Wadood, A. U. Rehman, M. Ríos-Gutiérrez, L. R. Domingo and A. Barakat, Chem. Biol. Drug Des., 2023, 102, 972–995 CrossRef CAS.
- A. J. Das and S. K. Das, J. Org. Chem., 2022, 87, 5085–5096 CrossRef CAS.
- K. S. Etsè, J. Dorosz, K. McLain Christensen, J. Y. Thomas, I. Botez Pop, E. Goffin, T. Colson, P. Lestage, L. Danober, B. Pirotte, J. S. Kastrup and P. Francotte, ACS Chem. Neurosci., 2021, 12, 2679–2692 CrossRef.
- A. Ali, S. Faraz and A. T. Khan, Org. Biomol. Chem., 2024, 22, 1426–1433 RSC.
| This journal is © The Royal Society of Chemistry 2024 |