
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of cytochalasan analogues with aryl substituents at position 10†
Žaneta
Javorská
 a,
Silvie
Rimpelová
b,
Magdaléna
Labíková
a and
Pavla
Perlíková
*ac
a,
Silvie
Rimpelová
b,
Magdaléna
Labíková
a and
Pavla
Perlíková
*ac
aDepartment of Organic Chemistry, Faculty of Chemical Technology, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague, Czech Republic. E-mail: perlikop@vscht.cz
bDepartment of Biochemistry and Microbiology, Faculty of Food and Biochemical Technology, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague, Czech Republic
cInstitute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo nám. 2, 160 00 Prague, Czech Republic
First published on 13th May 2024
Abstract
Cytochalasans are fungal metabolites that are known to inhibit actin polymerization. Despite their remarkable bioactivity, there are few studies on the structure–activity relationship (SAR) of the cytochalasan scaffold. The full potential of structural modifications remains largely unexplored. The substituent at position 10 of the cytochalasan scaffold is derived from an amino acid incorporated into the cytochalasan core, thus limiting the structural variability at this position in natural products. Additionally, modifications at this position have only been achieved through semisynthetic or mutasynthetic approaches using modified amino acids. This paper introduces a modular approach for late-stage modifications at position 10 of the cytochalasan scaffold. Iron-mediated cross-coupling reactions with corresponding Grignard reagents were used to introduce aryl or benzyl groups in position 10, resulting in the synthesis of six new cytochalasan analogues bearing non-natural aromatic residues. This methodology enables further exploration of modifications at this position and SAR studies among cytochalasan analogues.
Introduction
Cytochalasans are an important class of natural compounds that affect the dynamics of the actin cytoskeleton.1–3 They bind specifically the barbed end of F-actin, thus inhibiting the actin polymerization.4,5 As a result, cytochalasans exhibit cytotoxic effects and/or migrastatic activities by inhibiting cancer cell migration.6–9 The concept of migrastatics,10–12i.e., drugs that inhibit cancer cell invasion and metastasis, shows potential in developing new cancer therapies. Existing anticancer agents often fail to inhibit metastasis formation, which is a major contributor to cancer-related deaths.13,14 Therefore, cytochalasans and their analogues are promising candidates for medicinal chemists seeking to address this critical aspect of cancer treatment. At the same time, knowledge of the structure–activity relationship (SAR) of this class of compounds is limited15–21 and the full potential of structural modification has not been explored, yet.Cytochalasans are hybrid fungal metabolites that combine polyketides and amino acids.22 They exhibit a characteristic structure consisting of a perhydroisoindolone core and a complex macrocycle. Cytochalasans can be classified based on the specific amino acid incorporated into the perhydroisoindolone core, which may include aromatic amino acids such as phenylalanine in cytochalasins (e.g., cytochalasin B, 1), tryptophan in chaetoglobosins (e.g., chaetoglobosin A, 2), O-methyltyrosine in pyrichalasins (e.g., pyrichalasin H, 3), and aliphatic amino acids such as leucine in aspochalasins, valine in trichalasins, and alanine in alachalasins (Fig. 1). Consequently, the cytochalasan scaffold exhibits limited structural variability in position 10 among natural cytochalasans. So far, analogues with modifications at position 10 have only been provided through a semisynthetic and mutasynthetic approach using non-natural amino acids, particularly substituted L-phenylalanines and L-tyrosines.23,24 Hence, there is untapped potential for further structural modifications at position 10 and investigations into the structure–activity relationship of cytochalasans.
 | ||
| Fig. 1 Structures of natural cytochalasans with aromatic substituents in position 10 and cytochalasan analogues from this work. Amino acids incorporated into the cytochalasan core are shown in blue. | ||
Since their discovery, chemists have been pursuing the total synthesis of cytochalasans due to their intricate structure, which poses a persistent challenge.25–29 However, none of the total syntheses have involved the late-stage introduction of substituents into position 10, so far. Only a synthesis of cytochalasan analogues based on desymmetrization reactions of succinimide derivatives has been reported.18 Early attempts to introduce phenyl groups into the cytochalasan scaffold through cuprate reagents in the 1990s were unsuccessful.30,31 To date, all syntheses of cytochalasans have relied on the use of amino acids as starting materials. However, this approach is impractical for achieving modification at position 10, as each multistep synthesis would need to be optimized according to the amino acid being used. Therefore, a modular approach for introducing modifications to position 10 is necessary.
Although the first attempts for arylation in position 10 were unsuccessful thirty years ago, significant advancements have been made in synthetic methodology for sp2–sp3 C–C bond formation. The transition metal-catalyzed couplings predominantly encompass arylations catalyzed by palladium,32 nickel,33 cobalt,34,35 or iron.35,36 In this study, we investigated these reactions to achieve a modular approach for introducing aryl groups of different bulkiness in position 10 of the cytochalasan core. This approach facilitates the synthesis of derivatives modified at this position and enables more thorough SAR studies for cytochalasan analogues.
Results and discussion
Our investigation aimed to develop a streamlined method for introducing aryl groups at position 10 of the cytochalasan core. Due to the complexity of the cytochalasan core, the arylation conditions were first screened using simple model compounds, specifically derivatives of 4-methyl-2-pyrrolidone. A transition-metal-catalyzed approach was chosen, such as Suzuki cross-coupling reactions with organoboron reagents,37 or copper(I)-38 and palladium(II)-mediated39 reactions with corresponding Grignard reagents. However, none of these conditions provided the arylation. Consequently, our attention shifted towards cobalt(III)-40 and iron(III)-catalyzed41 reactions.The 2-pyrrolidone ring contains an amide group with an acidic hydrogen, susceptible to interaction with a base, such as a Grignard reagent. Therefore, the consideration of an N-protecting group, such as benzoyl (Bz), tert-butyloxycarbonyl (Boc), or methoxymethyl (MOM) was paramount. In the cytochalasan synthesis, the N-benzoyl group is commonly employed, however, its instability under basic reaction conditions42 precludes its use in this approach. The Boc group, previously used in arylations of pyrrolidines,40 is also unsuitable due to its propensity to enhance the electrophilicity of the 2-pyrrolidone carbonyl group, leading to lactam ring opening upon treatment with a strong nucleophile (data not shown). Thus, the MOM group was identified as the most appropriate choice, mitigating both of the issues.
The initial optimization step involved the selection of an appropriate leaving group for arylations. Bromide 4 and tosylate 5 were synthesized using established procedures or in analogy with known procedures.30,43–45 (see ESI for details, Scheme S1†). The reactions of the MOM-protected 4-methyl-2-pyrrolidone derivatives with phenylmagnesium bromide in the presence of Co(acac)3 (0.05 eq.) as a catalyst and N,N,N′,N′-tetramethylethylendiamine (TMEDA) as a ligand were carried out in THF at room temperature under an argon atmosphere (Scheme 1). In the case of tosylate 5, arylation product 6 was not observed at all. Instead, bromide 4 was formed (entry 1, Table 1). On the other hand, bromide 4 provided the arylation in 5 h (entry 2, Table 1). Full conversion was achieved after 24 h and phenyl derivative 6 was isolated in 88% yield (entry 3, Table 1), indicating that bromine is a good leaving group for arylations.
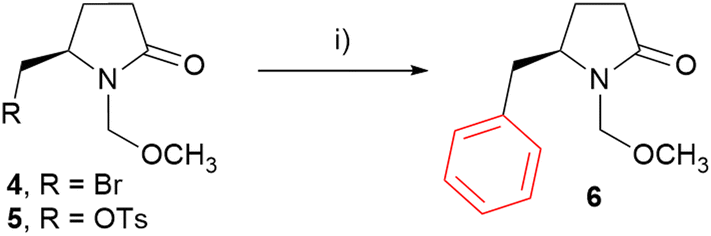 | ||
| Scheme 1 Leaving group optimization for arylations of starting materials with 2-pyrrolidone moiety. Reagents and conditions: (i) PhMgBr (1.3 eq.), Co(acac)3 (0.05 eq.), TMEDA, THF, RT, see Table 1. | ||
Encouraged by the results, our interest extended to exploring the applicability of the reaction conditions to the arylations of unprotected 4-bromomethyl-2-pyrrolidone (7) (Scheme 2 and Table 2). In light of the anticipated interaction between the free NH group and the Grignard reagent, the quantity of phenylmagnesium bromide was elevated to 2.6 equivalents (Table 2, entry 1). The arylated pyrrolidone 8 was formed but full conversion was not achieved after 24 h. The formation of 4-methyl-2-pyrrolidone 9, a debrominated byproduct, was also observed, likely due to a radical mechanism of the reaction. A reduction in the amount of TMEDA to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with the catalyst increased conversion and improved the ratio of phenyl derivative 8 to debrominated byproduct 9 (entry 2, Table 2). Further improvements in conversion and selectivity were achieved by elevating the reaction temperature to 55 °C (entry 3, Table 2). On the other hand, in situ protection of the amide group with N,O-bis(trimethylsilyl)acetamide (BSA)46 only led to poor conversion (entry 4, Table 2). Replacement of the catalyst by Co(PPh3)Cl2 provided better or equal conversion compared to Co(acac)3 but with a preference for the formation of debrominated byproduct 9 (entries 5 and 6, Table 2).
:1 ratio with the catalyst increased conversion and improved the ratio of phenyl derivative 8 to debrominated byproduct 9 (entry 2, Table 2). Further improvements in conversion and selectivity were achieved by elevating the reaction temperature to 55 °C (entry 3, Table 2). On the other hand, in situ protection of the amide group with N,O-bis(trimethylsilyl)acetamide (BSA)46 only led to poor conversion (entry 4, Table 2). Replacement of the catalyst by Co(PPh3)Cl2 provided better or equal conversion compared to Co(acac)3 but with a preference for the formation of debrominated byproduct 9 (entries 5 and 6, Table 2).
 | ||
| Scheme 2 Optimization of the cross-coupling reaction using unprotected pyrrolidone-type bromide 7. Reagents and conditions: (i) PhMgBr, catalyst, TMEDA, THF, see Table 2. | ||
| Entry | PhMgBr (eq.) | Catalyst (equivalents) | TMEDA (eq.) | Temperature | Time | NMR ratio 7:8:9 |
|---|---|---|---|---|---|---|
|
a Isolated yield 13%.
b (1) BSA, THF, 50 °C, 3 h (2) PhMgBr, catalyst, TMEDA, THF.
c Degassed THF.
d Compound 9 observed in 1H NMR, ratio could not be determined, ratio 7:8 is 40:60.
e Isolated yield 36%.
f Isolated yield 59%.
|
||||||
| 1 | 2.6 | Co(acac)3 (0.05) | 0.20 | RT | 24 h | 49:18a:33 |
| 2 | 2.6 | Co(acac)3 (0.05) | 0.05 | RT | 24 h | 30:41:29 |
| 3 | 2.6 | Co(acac)3 (0.05) | 0.05 | 55 °C | 24 h | 17:63:20 |
| 4b | 2.6 | Co(acac)3 (0.05) | 0.05 | RT | 18 h | 92:8:0 |
| 5 | 2.6 | Co(PPh3)2Cl2 (0.05) | 0.05 | RT | 48 h | 21:35:44 |
| 6 | 2.6 | Co(PPh3)2Cl2 (0.05) | 0.05 | 55 °C | 24 h | 19:38:43 |
| 7c | 2.6 | Fe(acac)3 (0.05) | 0.05 | 0 °C to RT | 4.5 h | 88:12:0 |
| 8c | 2.6 | Fe(acac)3 (0.05) | 0.05 | 0 °C to RT | 24 h | 83:17:0 |
| 9c | 7.7 | Fe(acac)3 (0.05) | 0.05 | 0 °C to RT | 24 h | ndd |
| 10 | 7.7 | Fe(acac)3 (1.00) | 1.00 | 0 °C to RT | 18 h | 0:100e:0 |
| 11c | 7.7 | Fe(acac)3 (1.00) | 1.00 | 0 °C to RT | 18 h | 0:100f:0 |
Due to unsatisfactory results with cobalt catalysts, Fe(acac)3 was explored as a catalyst41,47 (Scheme 2 and Table 2). In these experiments, the solvent was carefully degassed and the Grignard reagent was added at 0 °C. The full conversion was not achieved after 4.5 or 24 h, however, the debrominated byproduct 9 was not observed (entries 7 and 8, Table 2). Increasing the equivalents of the Grignard reagent resulted in an increased conversion, however, the debrominated byproduct 9 was also detected by 1H NMR. In this case, the NMR ratio of the phenyl derivative 8 and debrominated byproduct 9 could not be accurately determined due to overlapping peaks (entry 9, Table 2). The reaction was also carried out using stoichiometric amounts of both Fe(acac)3 and TMEDA in degassed or undegassed THF. In both cases, full conversion was achieved without the formation of debrominated byproduct 9. However, the isolated yield of the phenylated product 8 was higher when using degassed solvent (entries 10 and 11, Table 2). In this case, the reaction mixture contained fewer impurities, making the isolation of product 8 much easier.
Following the initial screening of reaction conditions with pyrrolidone derivatives, we focused on the synthesis of the starting material 16 with a cytochalasan scaffold and a phenylalkyl side chain instead of the macrocycle (Scheme 3). This structural analogue was chosen based on our previous work showing that cytochalasan analogues lacking the macrocyclic moiety still exhibit biological activities.21 The key step of the synthesis is the formation of the perhydroisoindolone core by the Diels–Alder reaction. First, we considered N-MOM protection of a dienophile but the approach failed due to dienophile instability (data not shown). The presence of an electron-withdrawing protecting group, such as the benzoyl group, is required. The first step of the synthesis of dienophile 14 was a benzoylation of O-silylated pyrrolidone 10 using NaH and benzoyl chloride (Scheme 3) yielding N-benzoyl pyrrolidone 11 (86%).48 An enolate was then generated in situ using lithium bis(trimethylsilyl)amide (LiHMDS) and reacted with methyl chloroformate to give acylated pyrrolidone 12 (88%).27 Following a similar procedure, the phenylselenyl group was introduced using LiHMDS and PhSeCl as an electrophile, yielding selenide 13 (91%).21 Both intermediates 12 and 13 were obtained as mixtures of diastereoisomers. While the diastereoisomers of acylated pyrrolidones 12 are inseparable, the selenides 13 can be separated by flash chromatography. However, separation was not necessary as the subsequent reactions led to the formation of the same dienophile. The oxidation and subsequent spontaneous elimination of selenide 13 with hydrogen peroxide produced the dienophile 14, which had to be used immediately in the subsequent Diels–Alder reaction due to its limited stability. The Diels–Alder reaction was performed with either DCM or 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) as a solvent. In both cases, a mixture of two diastereoisomers 16 (the intended endo-diastereoisomer) and 16′ (exo-diastereoisomer) was formed. The reaction in HFIP gave a higher yield (85% over two steps) and better diastereoselectivity (16/16′ ratio: 5.8/1) compared to the reaction in DCM (56% over two steps, 16/16′ ratio: 4.5/1). Separation of the diastereoisomers was achieved by flash chromatography using an excess of silica.
 | ||
| Scheme 3 Reagents and conditions: (i) NaH, BzCl, THF, 0 °C to RT, 24 h; (ii) LiHMDS, ClCO2Me, THF, −78 °C, 5.5 h; (iii) LiHMDS, PhSeCl, THF, −78 °C, 4 h; (iv) H2O2, DCM, RT, 2 h; (v) HFIP, 35 °C, 18 h. | ||
The aliphatic side chain was introduced using our previously published procedure (Scheme 4).21 The hydroxy group of compound 16 was alkylated with a triflate that was generated in situ from (2-iodoethyl)benzene using silver triflate and 2,6-di-tert-butylpyridine. Derivative 17 was synthesized with a high yield of 90%. Subsequent steps involved the removal of the O- and N-protecting groups in derivative 17. Optimal outcomes were achieved by employing Zemplén's conditions for the initial removal of the N-Bz protecting group. Following this step, the TBS group in the debenzoylated intermediate 18 was efficiently cleaved using (HF)3·NEt3, yielding the unprotected intermediate 19 (Scheme 4). It is noteworthy that the deprotection sequence can be reversed (O-deprotection followed by N-deprotection), however, in such case, a partial migration of the N-Bz group to the hydroxy group was observed (Scheme S2†). Finally, an Appel reaction of intermediate 19 with CBr4 and PPh3 smoothly led to the key bromide 20 (81%, Scheme 4).
 | ||
| Scheme 4 Reagents and conditions: (i) (2-iodoethyl)benzene, AgOTf, 2,6-di-tert-butylpyridine, DCM, RT to 35 °C, 72 h; (ii) Na, MeOH, RT, 2 h; (iii) (HF)3·NEt3, CH3CN, RT, 5 h; (iv) CBr4, PPh3, DCM, RT, 2.5 h. | ||
Bromide 20 is more complex than pyrrolidone 7, so conclusions drawn from arylations with a simple model compound may not be applicable. Therefore, both cobalt and iron catalysis were considered. We first investigated its reactivity in cobalt-mediated cross-coupling reactions with phenylmagnesium bromide (Scheme 5). However, the cobalt catalysts proved to be inefficient. Indeed, rapid screening of the reaction conditions (temperature, phenylmagnesium bromide equivalents, catalyst and ligand; entries 1–4, Table 3) resulted in poor conversion and the formation of the debrominated byproduct 22 at the expense of the target cross-coupling product 21.
 | ||
| Scheme 5 Optimization of the cross-coupling reaction using cytochalasan bromide 20. Reagents and conditions: (i) PhMgBr, catalyst (0.05 eq.), ligand, THF (non-degassed), see Table 3. | ||
| Entry | PhMgBr (eq.) | Catalyst | Ligand (eq.) | Temperature | Time | NMR ratio 20:21:22 |
|---|---|---|---|---|---|---|
| a Isolated yield 36%. b Et2O was used as a solvent. c 1.00 eq. | ||||||
| 1 | 2.6 | Co(acac)3 | TMEDA (0.36) | 55 °C | 24 h | 62:13:25 |
| 2 | 4.0 | Co(acac)3 | TMEDA (0.05) | RT | 22 h | 67:11:22 |
| 3 | 2.6 | Co(PPh3)2Cl2 | TMEDA (0.05) | RT | 22 h | 88:4:8 |
| 4 | 2.6 | Co(acac)3 | XPhos (0.1) | RT | 24 h | 93:7:0 |
| 5 | 7.7 | Fe(acac)3 | TMEDA (0.05) | 0 °C to RT | 4.5 h | 0:62a:38 |
| 6 | 7.7 | Fe(acac)3 | TMEDA (0.05) | 0 °C to 50 °C | 4.5 h | 36:23:41 |
| 7 | 7.7 | FeCl3 | TMEDA (0.05) | 0 °C to RT | 4.5 h | 38:35:27 |
| 8b | 7.7 | Fe(acac)3 | TMEDA (0.05) | 0 °C to RT | 4.5 h | 42:24:34 |
| 9 | 7.7 | Fe(acac)3 | TMEDA (1.2) | 0 °C to RT | 4.5 h | 43:26:31 |
| 10 | 7.7 | Fe(acac)3 | dppe (0.05) | 0 °C to RT | 4.5 h | 49:20:31 |
| 11 | 7.7 | Fe(acac)3 | SIPr (0.05) | 0 °C to RT | 4.5 h | 0:41:59 |
| 12 | 7.7 | Fe(acac)3c |
TMEDA (1.00) | 0 °C to RT | 4.5 h | 0:70:30 |
In contrast, our experiments with iron catalyst proved more promising. Building on the successful conditions for arylation of the model compound 7 with a substantial excess of Grignard reagent, subsequent attempts with the cytochalasan starting material 20 followed the same approach. Full conversion was achieved within 4.5 h (entry 5, Table 3). However, a mixture of products was formed: the desired phenylated compound 21 and a debrominated byproduct 22 (1H NMR ratio of 62/38; isolated yield of 21: 36%; entry 5, Table 3). 1H NMR yields were determined according to the chemical shifts of the signals that did not overlap (signal of H-13 of compound 20: δ 3.35–3.25 ppm, compound 21: δ 3.23 ppm, compound 22: δ 3.15 ppm; see ESI†). It was crucial to achieve full conversion as the similar retention factors of bromide 20 and phenyl derivative 21 made their separation very difficult (compounds 20 and 21 are only separable by preparative HPLC) and precluded the possibility of monitoring the reaction by TLC. The reaction conditions were further optimized. However, changes in the reaction temperature, solvent, catalyst or amount and type of a ligand led to incomplete conversion and a decrease in the ratio of product 21 and byproduct 22 (entries 7–10, Table 3). Only the use of the SIPr ligand led to full conversion but the preferential formation of the debrominated byproduct 22 was observed (1H NMR ratio of 21/22: 41/59; entry 11, Table 3). Some alternative ligands were tested without improved results (for details, see ESI, Table S1†). Finally, stoichiometric amounts of both Fe(acac)3 and TMEDA were used, leading to complete conversion and a 21/22 ratio of 70/30 (entry 12, Table 3). These conditions were found to be the most efficient for the arylation.
A series of cytochalasan analogues was synthesized by the arylation of bromide 20 with aryl Grignard reagents of various sizes and substitutions (Scheme 6). Because the iron-mediated cross-coupling reactions with benzyl Grignard reagents have been reported,49 a reaction with benzylmagnesium chloride was also included. Experiments conducted in both degassed and non-degassed THF yielded similar outcomes. The aromatic cytochalasan analogues 23–27 were isolated in modest yields (8–28%), along with the benzylic analogue 28 (7%). The reactions of bromide 20 with 2-pyridylmagnesium bromide or 1-naphthylmagnesium bromide did not yield the desired derivatives 29 and 30, respectively. The low yields were attributed to incomplete conversion, formation of the debrominated byproduct 22, and the challenging purification of the final products, requiring multiple rounds of flash chromatography and preparative HPLC. The inconvenience of incomplete conversion and difficult separation can be partially solved by treating the crude reaction product with sodium methoxide that substitutes the bromine of the unreacted starting material 20. The substitution product can be easily removed from the mixture by chromatography. Despite the challenges of achieving optimal yields and purifications, our approach marks a significant milestone as the first successful introduction of a non-natural aryl moiety onto the cytochalasan scaffold.
 | ||
| Scheme 6 Reagents and conditions: (i) R2MgBr or R2MgCl (7.7 eq.), TMEDA (1 eq.), Fe(acac)3 (1 eq.), THF, 0 °C to RT, 4.5–24 h. | ||
Compounds 22–28 were screened for their cytotoxic activities against human melanoma BLM cell line and MRC-5 fibroblasts (Table 4) after 72 h treatment. None of the compounds showed selectivity towards the cancer cell line. Compounds 23, 25, 27, and 28 showed moderate activity similar to or lower than the cytotoxic effect of previously reported derivative 21.21 On the other hand, after 72 h treatment, methyl derivative 22, phenoxyphenyl 24 and phenanthrenyl 26 derivatives did not show sufficient activity (IC50 > 50 μM), indicating that the presence of an aryl moiety improves the cytotoxic effect, however, the bulkiness, and/or sterical factors need to be considered to maintain the activity.
| Cell line | BLM | MRC-5 |
|---|---|---|
| Compound | IC50 ± SEM [μM] | |
| Values represent the mean that originates from at least three independent experiments and the standard error of the mean.a Values from ref. 21. | ||
| 21 | 27.07 ± 1.24 | 30.50 ± 0.38 |
| 22 | 50 | 50 |
| 23 | 27.06 ± 1.61 | 30.76 ± 0.33 |
| 24 | 50 | 50 |
| 25 | 32.68 ± 3.09 | 39.30 ± 0.78 |
| 26 | 50 | 50 |
| 27 | 37.64 ± 3.39 | 42.13 ± 3.63 |
| 28 | 31.85 ± 4.30 | 39.41 ± 4.59 |
Experimentals
General remarks
Solvents and chemicals were either purchased from commercial suppliers or purified using standard techniques. tert-Butyldimethylsilylpyrrolidone 10,30 dienol 15 (ref. 50) and (2-iodoethyl)benzene51 were synthesized using previously published protocols. Flash chromatography was performed by using silica gel Silicycle – Siliaflash® P 60 (particle size 40–63 μm, pore diameter 60 Å). Thin-layer chromatography (TLC) was conducted using silica gel plates Merck 60 F254.1H, 13C, 19F NMR spectra were measured on Agilent 400-MR DDR2 or JEOL-ECZL400G (400.0 MHz for 1H, 100.6 MHz for 13C, 376.0 MHz for 19F). The complete assignment of all NMR signals was done by a combination of H,H-COSY, H,H-ROESY, H,C-HSQC, and H,C-HMBC experiments. The spectra were recorded in CDCl3. It served as an internal standard (δCDCl3 = 7.26 ppm) for 1H NMR and (δCDCl3 = 77.0 ppm) for 13C NMR, CFCl3 (δCFCl3 = 0.00 ppm) was used as an external standard for 19F NMR. Cytochalasan atom numbering was used for the assignment of the NMR signals.52 Low- and high-resolution mass spectroscopic data were obtained on LTQ Orbitrap XL (Thermo Fisher Scientific) using ESI at the Laboratory of Mass spectrometry, IOCB Prague. Optical rotations were measured at 20 °C, and [α]D values are given in 10−1 deg cm2 g−1. Flash chromatography (FC) was performed using the Büchi Pure C-815 Flash system on CHROMABOND Flash empty cartridges filled with silica gel Silicycle – Siliaflash® P 60 (particle size 40–63 μm, pore diameter 60 Å). Purification of some compounds was done by HPLC Büchi Pure C-850 FlashPrep system on a column packed with 5 μm normal phase (ProntoSIL 60-5-Si 150 × 20 mm, BISCHOFF Chromatography). The purity of all final compounds was determined by clean NMR spectra and by HPLC.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–Bz), 134.98 (C–i-Bz), 131.80 (CH–p-Bz), 128.71 (CH–o-Bz), 127.85 (CH–m-Bz), 63.70 (CH2–OSi), 58.63 (CH-5), 32.71 (CH2-3), 25.87 (SiCCH3), 21.25 (CH2-4), 18.21 (SiCCH3), −5.47 (SiCH3), −5.60 (SiCH3) ppm. HRMS (ESI) m/z calcd for C18H28O3NSi [M + H]+ 334.1833; found 334.1835.
O–Bz min.), 170.51 (CO–Bz/C-2 maj.), 170.39 (CO–Bz/C-2 maj.), 170.34 (C-2 min.), 169.84 (COOCH3 maj.), 169.31 (COOCH3 min.), 134.30 (C–i-Bz maj.), 133.97 (C–i-Bz min.), 132.67 (CH–p-Bz, min.), 131.97 (CH–p-Bz, maj.), 129.52 (CH–o-Bz min.), 128.61 (CH–o-Bz maj.), 127.98 (CH–m-Bz min.), 127.92 (CH–m-Bz maj.), 64.07 (CH2–OSi maj.), 61.29 (CH2–OSi min.), 57.08 (CH-5 maj.), 57.00 (CH-5 min.), 52.90 (COOCH3 min.), 52.85 (COOCH3 maj.), 50.09 (CH-3 maj.), 49.04 (CH-3 min.), 26.04 (CH2-4 maj.), 25.86 (SiCCH3 maj.), 25.77 (SiCCH3 min.), 23.93 (CH2-4 min.), 18.22 (SiCCH3 min.), 18.19 (SiCCH3 maj.), −5.50 (SiCH3 maj.), −5.54 (SiCH3 min.), −5.61 (SiCH3 min.), −5.63 (SiCH3 maj.) ppm. HRMS (ESI) m/z calcd for C20H30O5NSi [M + H]+ 392.1888; found 392.1885.
O–Bz), 170.07 (C-2), 169.32 (COOCH3), 137.76 (CH–o-PhSe), 134.04 (C–i-Bz), 132.37 (CH–p-Bz), 130.22 (CH–p-PhSe), 129.29 (CH–o-Bz/CH–m-PhSe), 129.19 (CH–o-Bz/CH–m-PhSe), 127.85 (CH–m-Bz), 126.05 (C–i-PhSe), 61.05 (CH2–OSi), 55.64 (CH-5), 53.96 (COOCH3), 53.38 (C-3), 31.92 (CH2-4), 25.72 (SiCCH3), 18.20 (SiCCH3), −5.62 (SiCH3), −5.70 (SiCH3) ppm. HRMS (ESI) m/z calcd for C26H34O5N80SeSi [M + H]+ 548.1366; found 548.1363. Minor:1H NMR (400 MHz, CDCl3) δ 7.73 (dd, JH–o-PhSe,H–m-PhSe = 8.2, JH–o-PhSe,H–p-PhSe = 1.3 Hz, 2H, H–o-PhSe), 7.59 (m, ∑J = 8.4 Hz, 2H, H–o-Bz), 7.54 (m, ∑J = 17.5 Hz, 1H, H–p-Bz), 7.47–7.32 (m, 5H, 2× H–m-Bz, 2× H–m-PhSe, H–p-PhSe), 4.42 (tdd, J5,4a,b = 7.6, J5,OCH2a = 5.0, J5,OCH2b = 2.6 Hz, 1H, H-5), 3.95 (dd, Jgem = 10.4, JOCH2a,5 = 5.0 Hz, 1H, OCH2a), 3.72–3.64 (m, 4H, OCH3 + OCH2b), 2.78 (dd, Jgem = 13.9, J4a,5 = 7.4 Hz, 1H, H-4a), 2.53 (dd, Jgem = 14.0, J4b,5 = 7.9 Hz, 1H, H-4b), 0.85 (s, 9H, C(CH3)3), −0.02 (s, 3H, SiCH3), −0.08 (s, 3H, SiCH3) ppm. 13C NMR (101 MHz, CDCl3) δ 171.07 (CO–Bz), 170.04 (C-2), 169.35 (COOCH3), 137.95 (CH–o-PhSe), 133.84 (C–i-Bz), 132.67 (CH–p-Bz), 129.98 (CH–p-PhSe), 129.57 (CH–o-Bz), 129.12 (CH–m-PhSe), 127.88 (CH–m-Bz), 126.27 (C–i-PhSe), 60.72 (CH2–OSi), 56.51 (CH-5), 54.36 (C-3), 53.53 (COOCH3), 32.24 (CH2-4), 25.87 (SiCCH3), 18.25 (SiCCH3), −5.46 (SiCH3), −5.60 (SiCH3) ppm. HRMS (ESI) m/z calcd for C26H34O5N80SeSi [M + H]+ 548.1366; found 548.1363.
O–Bz), 140.81 (C-6), 134.31 (C–i-Bz), 132.32 (C–p-Bz), 129.07 (C–o-Bz), 127.92 (C–m-Bz), 126.36 (C-7), 62.68 (C-9), 62.14 (C-10), 61.48 (C-14), 58.03 (C-3), 53.00 (COOCH3), 46.25 (C-4), 37.69 (C-8), 34.39 (C-5), 33.16 (C-13), 25.83 (SiCCH3), 20.08 (C-12), 18.34 (SiCCH3), 13.78 (C-11), −5.54 (SiCH3), −5.63 (SiCH3) ppm. HRMS (ESI) m/z calcd for C28H41O6NNaSi [M + Na]+ 538.2595; found 538.2598. 16′: 1H NMR (400 MHz, CDCl3) δ 7.72 (m, ∑J = 8.0 Hz, 2H, H–o-Bz), 7.53 (m, ∑J = 20.0 Hz, 1H, H–p-Bz), 7.41 (ddd, Jm,p = Jm,o = 7.6, Jm,m = 1.8 Hz, 2H, H–m-Bz), 5.48 (m, ∑J = 10.0 Hz, 1H, H-7), 4.21 (ddd, J3,4 = 6.0, J3,10a = 5.0, J3,10b = 3.0 Hz, 1H, H-3), 3.92 (dd, Jgem = 10.5, J10a,3 = 5.0 Hz, 1H, H-10a), 3.78–3.67 (m, 5H, OCH3, H-10b,14a), 3.58 (ddd, Jgem = 11.0, J14b,13a = 8.5, J14b,13b = 5.3 Hz, 1H, H-14b), 2.71 (dd, J4,3 = J4,5 = 5.9 Hz, 1H, H-4), 2.47 (m, ∑J = 20.0 Hz, 1H, H-8), 2.15–2.01 (m, 2H, H-5,13a), 1.84–1.66 (m, 4H, H-12,13b), 1.18 (d, J11,5 = 7.1 Hz, 3H, H-11), 0.87 (s, 9H, C(CH3)3), 0.02 (s, 3H, SiCH3), −0.04 (s, 3H, SiCH3) ppm. 13C NMR (101 MHz, CDCl3) δ 173.63 (C-1), 171.28 (COOCH3), 171.02 (CO–Bz), 136.91 (C-6), 134.09 (C–i-Bz), 132.59 (C–p-Bz), 129.45 (C–o-Bz), 127.93 (C–m-Bz), 124.17 (C-7), 61.11 (C-3), 60.95 (C-14), 60.25 (C-10), 58.69 (C-9), 52.36 (COOCH3), 45.57 (C-4), 34.56 (C-5), 34.19 (C-8), 34.06 (C-13), 25.73 (SiCCH3), 21.10 (C-12), 18.20 (SiCCH3), 17.93 (C-11), −5.58 (SiCH3), −5.66 (SiCH3) ppm. HRMS (ESI) m/z calcd for C28H41O6NNaSi [M + Na]+ 538.2595; found 538.2591.
O–Bz), 140.35 (C-6), 139.20 (C–i-Ph), 134.41 (C–i-Bz), 132.20 (C–p-Bz), 129.03 (C–o-Bz), 128.91 (C–o-Ph), 128.18 (C–m-Ph), 127.89 (C–m-Bz), 126.41 (C-7), 125.98 (C–p-Ph), 71.34 (C-15), 69.40 (C-14), 62.79 (C-10), 62.54 (C-9), 57.69 (C-3), 52.80 (COOCH3), 46.06 (C-4), 38.32 (C-8), 36.28 (C-16), 34.36 (C-5), 29.55 (C-13), 25.83 (SiCCH3), 20.03 (C-12), 18.34 (SiCCH3), 13.63 (C-11), −5.55 (SiCH3), −5.62 (SiCH3) ppm. HRMS (ESI) m/z calcd for C36H49O6NNaSi [M + Na]+ 642.3221; found 642.3223.
O–Bz), 134.98 (C–i-Bz), 131.80 (CH–p-Bz), 128.71 (CH–o-Bz), 127.85 (CH–m-Bz), 63.70 (CH2–OSi), 58.63 (CH-5), 32.71 (CH2-3), 25.87 (SiCCH3), 21.25 (CH2-4), 18.21 (SiCCH3), −5.47 (SiCH3), −5.60 (SiCH3) ppm. HRMS (ESI) m/z calcd for C18H28O3NSi [M + H]+ 334.1833; found 334.1835.
O–Bz min.), 170.51 (CO–Bz/C-2 maj.), 170.39 (CO–Bz/C-2 maj.), 170.34 (C-2 min.), 169.84 (COOCH3 maj.), 169.31 (COOCH3 min.), 134.30 (C–i-Bz maj.), 133.97 (C–i-Bz min.), 132.67 (CH–p-Bz, min.), 131.97 (CH–p-Bz, maj.), 129.52 (CH–o-Bz min.), 128.61 (CH–o-Bz maj.), 127.98 (CH–m-Bz min.), 127.92 (CH–m-Bz maj.), 64.07 (CH2–OSi maj.), 61.29 (CH2–OSi min.), 57.08 (CH-5 maj.), 57.00 (CH-5 min.), 52.90 (COOCH3 min.), 52.85 (COOCH3 maj.), 50.09 (CH-3 maj.), 49.04 (CH-3 min.), 26.04 (CH2-4 maj.), 25.86 (SiCCH3 maj.), 25.77 (SiCCH3 min.), 23.93 (CH2-4 min.), 18.22 (SiCCH3 min.), 18.19 (SiCCH3 maj.), −5.50 (SiCH3 maj.), −5.54 (SiCH3 min.), −5.61 (SiCH3 min.), −5.63 (SiCH3 maj.) ppm. HRMS (ESI) m/z calcd for C20H30O5NSi [M + H]+ 392.1888; found 392.1885.
O–Bz), 170.07 (C-2), 169.32 (COOCH3), 137.76 (CH–o-PhSe), 134.04 (C–i-Bz), 132.37 (CH–p-Bz), 130.22 (CH–p-PhSe), 129.29 (CH–o-Bz/CH–m-PhSe), 129.19 (CH–o-Bz/CH–m-PhSe), 127.85 (CH–m-Bz), 126.05 (C–i-PhSe), 61.05 (CH2–OSi), 55.64 (CH-5), 53.96 (COOCH3), 53.38 (C-3), 31.92 (CH2-4), 25.72 (SiCCH3), 18.20 (SiCCH3), −5.62 (SiCH3), −5.70 (SiCH3) ppm. HRMS (ESI) m/z calcd for C26H34O5N80SeSi [M + H]+ 548.1366; found 548.1363. Minor:1H NMR (400 MHz, CDCl3) δ 7.73 (dd, JH–o-PhSe,H–m-PhSe = 8.2, JH–o-PhSe,H–p-PhSe = 1.3 Hz, 2H, H–o-PhSe), 7.59 (m, ∑J = 8.4 Hz, 2H, H–o-Bz), 7.54 (m, ∑J = 17.5 Hz, 1H, H–p-Bz), 7.47–7.32 (m, 5H, 2× H–m-Bz, 2× H–m-PhSe, H–p-PhSe), 4.42 (tdd, J5,4a,b = 7.6, J5,OCH2a = 5.0, J5,OCH2b = 2.6 Hz, 1H, H-5), 3.95 (dd, Jgem = 10.4, JOCH2a,5 = 5.0 Hz, 1H, OCH2a), 3.72–3.64 (m, 4H, OCH3 + OCH2b), 2.78 (dd, Jgem = 13.9, J4a,5 = 7.4 Hz, 1H, H-4a), 2.53 (dd, Jgem = 14.0, J4b,5 = 7.9 Hz, 1H, H-4b), 0.85 (s, 9H, C(CH3)3), −0.02 (s, 3H, SiCH3), −0.08 (s, 3H, SiCH3) ppm. 13C NMR (101 MHz, CDCl3) δ 171.07 (CO–Bz), 170.04 (C-2), 169.35 (COOCH3), 137.95 (CH–o-PhSe), 133.84 (C–i-Bz), 132.67 (CH–p-Bz), 129.98 (CH–p-PhSe), 129.57 (CH–o-Bz), 129.12 (CH–m-PhSe), 127.88 (CH–m-Bz), 126.27 (C–i-PhSe), 60.72 (CH2–OSi), 56.51 (CH-5), 54.36 (C-3), 53.53 (COOCH3), 32.24 (CH2-4), 25.87 (SiCCH3), 18.25 (SiCCH3), −5.46 (SiCH3), −5.60 (SiCH3) ppm. HRMS (ESI) m/z calcd for C26H34O5N80SeSi [M + H]+ 548.1366; found 548.1363.
O–Bz), 140.81 (C-6), 134.31 (C–i-Bz), 132.32 (C–p-Bz), 129.07 (C–o-Bz), 127.92 (C–m-Bz), 126.36 (C-7), 62.68 (C-9), 62.14 (C-10), 61.48 (C-14), 58.03 (C-3), 53.00 (COOCH3), 46.25 (C-4), 37.69 (C-8), 34.39 (C-5), 33.16 (C-13), 25.83 (SiCCH3), 20.08 (C-12), 18.34 (SiCCH3), 13.78 (C-11), −5.54 (SiCH3), −5.63 (SiCH3) ppm. HRMS (ESI) m/z calcd for C28H41O6NNaSi [M + Na]+ 538.2595; found 538.2598. 16′: 1H NMR (400 MHz, CDCl3) δ 7.72 (m, ∑J = 8.0 Hz, 2H, H–o-Bz), 7.53 (m, ∑J = 20.0 Hz, 1H, H–p-Bz), 7.41 (ddd, Jm,p = Jm,o = 7.6, Jm,m = 1.8 Hz, 2H, H–m-Bz), 5.48 (m, ∑J = 10.0 Hz, 1H, H-7), 4.21 (ddd, J3,4 = 6.0, J3,10a = 5.0, J3,10b = 3.0 Hz, 1H, H-3), 3.92 (dd, Jgem = 10.5, J10a,3 = 5.0 Hz, 1H, H-10a), 3.78–3.67 (m, 5H, OCH3, H-10b,14a), 3.58 (ddd, Jgem = 11.0, J14b,13a = 8.5, J14b,13b = 5.3 Hz, 1H, H-14b), 2.71 (dd, J4,3 = J4,5 = 5.9 Hz, 1H, H-4), 2.47 (m, ∑J = 20.0 Hz, 1H, H-8), 2.15–2.01 (m, 2H, H-5,13a), 1.84–1.66 (m, 4H, H-12,13b), 1.18 (d, J11,5 = 7.1 Hz, 3H, H-11), 0.87 (s, 9H, C(CH3)3), 0.02 (s, 3H, SiCH3), −0.04 (s, 3H, SiCH3) ppm. 13C NMR (101 MHz, CDCl3) δ 173.63 (C-1), 171.28 (COOCH3), 171.02 (CO–Bz), 136.91 (C-6), 134.09 (C–i-Bz), 132.59 (C–p-Bz), 129.45 (C–o-Bz), 127.93 (C–m-Bz), 124.17 (C-7), 61.11 (C-3), 60.95 (C-14), 60.25 (C-10), 58.69 (C-9), 52.36 (COOCH3), 45.57 (C-4), 34.56 (C-5), 34.19 (C-8), 34.06 (C-13), 25.73 (SiCCH3), 21.10 (C-12), 18.20 (SiCCH3), 17.93 (C-11), −5.58 (SiCH3), −5.66 (SiCH3) ppm. HRMS (ESI) m/z calcd for C28H41O6NNaSi [M + Na]+ 538.2595; found 538.2591.
O–Bz), 140.35 (C-6), 139.20 (C–i-Ph), 134.41 (C–i-Bz), 132.20 (C–p-Bz), 129.03 (C–o-Bz), 128.91 (C–o-Ph), 128.18 (C–m-Ph), 127.89 (C–m-Bz), 126.41 (C-7), 125.98 (C–p-Ph), 71.34 (C-15), 69.40 (C-14), 62.79 (C-10), 62.54 (C-9), 57.69 (C-3), 52.80 (COOCH3), 46.06 (C-4), 38.32 (C-8), 36.28 (C-16), 34.36 (C-5), 29.55 (C-13), 25.83 (SiCCH3), 20.03 (C-12), 18.34 (SiCCH3), 13.63 (C-11), −5.55 (SiCH3), −5.62 (SiCH3) ppm. HRMS (ESI) m/z calcd for C36H49O6NNaSi [M + Na]+ 642.3221; found 642.3223.
General procedure for cross-coupling reactions
Fe(acac)3 (1.00 eq.) was added to a dry Schlenk flask that was then evacuated and filled with argon (3×). A solution of compound 19 (1.00 eq.) in anhydrous THF (0.5–1 mL), TMEDA (1.00 eq.) and THF (0.5–1 mL) were added and the resulting mixture was cooled to 0 °C. Then, a Grignard reagent (7.7 eq.) was slowly added dropwise and the mixture was stirred at RT under argon atmosphere for 4.5–24 h. A solution of NH4Cl (5 mL) and EtOAc (5 mL) were added, phases were separated and aqueous phase was extracted with EtOAc (2 × 5 mL). The combined organic layers were washed with a solution of NaHCO3 and EDTA (4 × 5 mL) and brine (1 × 5 mL) and was dried (MgSO4) and the solvent was evaporated in vacuo. The crude products were purified by flash chromatography and preparative HPLC on silica gel (eluent: EtOAc in hexane 0% to 80%). The debrominated compound 22 was formed as a byproduct.Cultivation of cell lines
Human BLM (derived from melanoma) and MRC-5 (lung fibroblasts; Merck, USA) cells were utilized in this study to assess the cytotoxicity of the prepared compounds in vitro. Both cell lines were kept at the exponential phase of growth and regularly passaged using trypsin–EDTA solution. BLM cells were cultured in high-glucose DMEM supplemented with 10% (v/v) fetal bovine serum (FBS; Merck, USA) and stable L-glutamine. MRC-5 were maintained in MEM + 10% (v/v) FBS, stable L-glutamine, and 1% (v/v) non-essential amino acids. The cells were incubated at 37 °C, 5% CO2 in the atmosphere and 95% humidity.Compound cytotoxicity measurement
Cytotoxicity of the evaluated compounds was determined using a colorimetric WST-1 viability assay (Merck, USA) similarly as reported in ref. 21. The amount of 5000 BLM and MRC-5 cells were seeded in 100 μL of cell media into wells of flat-bottom 96-well plates and incubated at standard conditions for 24 h. Then, the cells were treated with a concentration series of the measured compounds diluted in 100 μL of cell media. This was added into the wells with cells and incubated for 72 h. After that, the viability of BLM and MRC-5 cells was determined using WST-1 (4% v/v solution in FluoroBrite DMEM) after 2 h incubation, at which formazan absorption was assessed spectrophotometrically at 450 nm (reference wavelength at 650 nm) by a UV-vis spectrophotometer (Bio-Rad, USA). Untreated cells and cells treated only with a vehicle (DMSO) served as controls. The experiment occurred in at least three independent measurements each of which consisted of three technical replicates. The data were plotted as dose–response curves from which the half-maximal inhibitory concentrations (IC50) were calculated by AAT Bioquest.Conclusions
This study explores the potential of transition metal-mediated reactions for introducing aryl groups at position 10 of the cytochalasan scaffold as a tool for modular modifications at this position. The methodology was optimised using 2-pyrrolidones as model starting materials. An excellent yield was obtained using a Co(acac)3-catalyzed reaction with PhMgBr and TMEDA as a ligand when using N-MOM protected 5-(bromomethyl)-2-pyrrolidone 4 as the starting material. However, unprotected 5-(bromomethyl)-2-pyrrolidone 7 was only successfully arylated with PhMgBr using stoichiometric amounts of Fe(acac)3 and TMEDA. Although arylations with the corresponding cytochalasan-like bromide suffered from partial debromination of the starting material, we identified conditions that predominantly led to the formation of arylated products. This methodology was applied to the synthesis of six new aryl- and benzyl-substituted cytochalasan analogues 23–28. These compounds exhibited comparable or lower cytotoxic activity compared to known cytochalasan analogues lacking the macrocyclic moiety.Our results demonstrate a viable method for introducing aryl groups of different sizes and substitutions at position 10 during the late stages of cytochalasan analogue synthesis, more than 30 years after the arylation was first proposed. It is the first example of a late-stage modification in this position and due to its modular character, it holds promise for conducting further SAR studies on cytochalasans. It could contribute to a deeper understanding of the role of aryl groups in their interaction with actin and other protein targets.
Conflicts of interest
No conflict of interest to declare.Acknowledgements
This work was supported by the Czech Science Foundation (23-05336M, Ž. J., S. R. and P. P.), by institutional support of the Dagmar Procházková Fund (P. P.), a grant of Specific university research (reg. no. A2_FCHT_2022_064; Ž. J.), and by IOCB Tech. The authors acknowledge Prof. Andrea Brancale for critical reading of the manuscript.References
- K. Scherlach, D. Boettger, N. Remme and C. Hertweck, Nat. Prod. Rep., 2010, 27, 869–886 RSC.
- H. Zhu, C. Chen, Q. Tong, Y. Zhou, Y. Ye, L. Gu and Y. Zhang, in Progress in the chemistry of organic natural products 114, ed. A. Douglas Kinghorn, H. Falk, S. Gibbons, J. Kobayashi, Y. Asakawa and J.-K. Liu, Springer International Publishing, Cham, 2021, pp. 1–134 Search PubMed.
- C. Lambert, K. Schmidt, M. Karger, M. Stadler, T. E. B. Stradal and K. Rottner, Biomolecules, 2023, 13, 1247 CrossRef CAS PubMed.
- M. D. Flanagan and S. Lin, J. Biol. Chem., 1980, 255, 835–838 CrossRef CAS PubMed.
- S. S. Brown and J. A. Spudich, J. Cell Biol., 1981, 88, 487–491 CrossRef CAS PubMed.
- B. Hagmar and W. Ryd, Int. J. Cancer, 1977, 19, 576–580 CrossRef CAS PubMed.
- I. R. Hart, A. Raz and I. J. Fidler, JNCI, J. Natl. Cancer Inst., 1980, 64, 891–900 CAS.
- D. Murray, G. Horgan, P. Macmathuna and P. Doran, Br. J. Cancer, 2008, 99, 1322–1329 CrossRef CAS PubMed.
- W. Sun, C. T. Lim and N. A. Kurniawan, J. R. Soc., Interface, 2014, 11, 20140638 CrossRef PubMed.
- J. Solomon, M. Raškova, D. Rösel, J. Brábek and H. Gil-Henn, Cells, 2021, 10, 1845 CrossRef PubMed.
- D. Rosel, M. Fernandes, V. Sanz-Moreno and J. Brábek, Trends Cancer, 2019, 5, 755–756 CrossRef CAS PubMed.
- A. Gandalovičová, D. Rosel, M. Fernandes, P. Veselý, P. Heneberg, V. Čermák, L. Petruželka, S. Kumar, V. Sanz-Moreno and J. Brábek, Trends Cancer, 2017, 3, 391–406 CrossRef PubMed.
- M. B. Sporn, Lancet, 1996, 347, 1377–1381 CrossRef CAS PubMed.
- H. Dillekås, M. S. Rogers and O. Straume, Cancer Med., 2019, 8, 5574–5576 CrossRef PubMed.
- H. Minato, T. Katayama, M. Matsumoto, K. Katagiri and S. Matsuura, Chem. Pharm. Bull., 1973, 21, 2268–2277 CrossRef CAS PubMed.
- I. Yahara, F. Harada, S. Sekita, K. Yoshihira and S. Natori, J. Cell Biol., 1982, 92, 69–78 CrossRef CAS PubMed.
- S. Sekita, K. Yoshihira, S. Natori, F. Harada, K. Iida and I. Yahara, J. Pharmacobio-Dyn., 1985, 8, 906–916 CrossRef CAS PubMed.
- M. Sellstedt, M. Schwalfenberg, S. Ziegler, A. P. Antonchick and H. Waldmann, Org. Biomol. Chem., 2016, 14, 50–54 RSC.
- M. Zaghouani, O. Gayraud, V. Jactel, S. Prevost, A. Dezaire, M. Sabbah, A. Escargueil, T. L. Lai, C. Le Clainche, N. Rocques, S. Romero, A. Gautreau, F. Blanchard, G. Frison and B. Nay, Chem. – Eur. J., 2018, 24, 16686–16691 CrossRef CAS PubMed.
- R. Kretz, L. Wendt, S. Wongkanoun, J. J. Luangsa-Ard, F. Surup, S. E. Helaly, S. R. Noumeur, M. Stadler and T. E. B. Stradal, Biomolecules, 2019, 9, 73 CrossRef CAS PubMed.
- B. Formánek, D. Dupommier, T. Volfová, S. Rimpelová, A. Škarková, J. Herciková, D. Rösel, J. Brábek and P. Perlíková, RSC Med. Chem., 2024, 15, 322–343 RSC.
- E. Skellam, Nat. Prod. Rep., 2017, 34, 1252–1263 RSC.
- C. Wang, V. Hantke, R. J. Cox and E. Skellam, Org. Lett., 2019, 21, 4163–4167 CrossRef CAS PubMed.
- C. Wang, C. Lambert, M. Hauser, A. Deuschmann, C. Zeilinger, K. Rottner, T. E. B. Stradal, M. Stadler, E. J. Skellam and R. J. Cox, Chem. – Eur. J., 2020, 26, 13578–13583 CrossRef CAS PubMed.
- Y. Zhao, X. Long, H. Wu and J. Deng, Org. Chem. Front., 2022, 9, 6979–6998 RSC.
- O. Gayraud, B. Laroche, N. Casaretto and B. Nay, Org. Lett., 2021, 23, 5755–5760 CrossRef CAS PubMed.
- X. Long, Y. Ding and J. Deng, Angew. Chem., Int. Ed., 2018, 57, 14221–14224 CrossRef CAS PubMed.
- J. R. Reyes, N. Winter, L. Spessert and D. Trauner, Angew. Chem., Int. Ed., 2018, 57, 15587–15591 CrossRef CAS PubMed.
- R. Bao, C. Tian, H. Zhang, Z. Wang, Z. Dong, Y. Li, M. Gao, H. Zhang, G. Liu and Y. Tang, Angew. Chem., Int. Ed., 2018, 57, 14216–14220 CrossRef CAS PubMed.
- J. Ackermann, M. Matthes and C. Tamm, Helv. Chim. Acta, 1990, 73, 122–132 CrossRef CAS.
- M. Matthes and C. Tamm, Helv. Chim. Acta, 1991, 74, 1585–1590 CrossRef CAS.
- N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC.
- T. Iwasaki and N. Kambe, in Ni- and Fe-based cross-coupling reactions, ed. A. Correa, Springer International Publishing, Cham, 2017, pp. 1–36 Search PubMed.
- A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351–1363 CrossRef PubMed.
- S. H. Kyne, G. Lefèvre, C. Ollivier, M. Petit, V.-A. Ramis Cladera and L. Fensterbank, Chem. Soc. Rev., 2020, 49, 8501–8542 RSC.
- B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed.
- C.-T. Yang, Z.-Q. Zhang, Y.-C. Liu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 3904–3907 CrossRef CAS PubMed.
- K. Nakata, C. Feng, T. Tojo and Y. Kobayashi, Tetrahedron Lett., 2014, 55, 5774–5777 CrossRef CAS.
- A. C. Frisch, N. Shaikh, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2002, 41, 4056–4059 CrossRef CAS PubMed.
- S.-F. Hsu, C.-W. Ko and Y.-T. Wu, Adv. Synth. Catal., 2011, 353, 1756–1762 CrossRef CAS.
- M. Nakamura, K. Matsuo, S. Ito and E. Nakamura, J. Am. Chem. Soc., 2004, 126, 3686–3687 CrossRef CAS PubMed.
- E. Vedejs and S. M. Duncan, J. Org. Chem., 2000, 65, 6073–6081 CrossRef CAS PubMed.
- M. A. Berliner and K. Belecki, J. Org. Chem., 2005, 70, 9618–9621 CrossRef CAS PubMed.
- J. Gao, G. Liao, L. Wang and Z. Guo, Org. Lett., 2014, 16, 988–991 CrossRef CAS PubMed.
- N. Kotoku, S. Fujioka, C. Nakata, M. Yamada, Y. Sumii, T. Kawachi, M. Arai and M. Kobayashi, Tetrahedron, 2011, 67, 6673–6678 CrossRef CAS.
- J. F. Klebe, H. Finkbeiner and D. M. White, J. Am. Chem. Soc., 1966, 88, 3390–3395 CrossRef CAS.
- T. Nagano and T. Hayashi, Org. Lett., 2004, 6, 1297–1299 CrossRef CAS PubMed.
- S. A. Harkin, O. Singh and E. J. Thomas, J. Chem. Soc., Perkin Trans. 1, 1984, 1489–1499 RSC.
- R. B. Bedford, P. B. Brenner, E. Carter, P. M. Cogswell, M. F. Haddow, J. N. Harvey, D. M. Murphy, J. Nunn and C. H. Woodall, Angew. Chem., Int. Ed., 2014, 53, 1804–1808 CrossRef CAS PubMed.
- J. Wu, X. Jiang, J. Xu and W.-M. Dai, Tetrahedron, 2011, 67, 179–192 CrossRef CAS.
- S. Rieckhoff, J. Meisner, J. Kästner, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2018, 57, 1404–1408 CrossRef CAS PubMed.
- M. Binder, C. Tamm, W. B. Turner and H. Minato, J. Chem. Soc., Perkin Trans. 1, 1973, 1146–1147 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00634h |
| This journal is © The Royal Society of Chemistry 2024 |