
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium catalyzed stereoselective intramolecular [3 + 2] cycloaddition reactions of (E) & (Z)-ene-vinylidenecyclopropanes†
Chao
Ning
a,
Zi-Qi
Yu
 a,
Yin
Wei
*b and
Min
Shi
*ab
a,
Yin
Wei
*b and
Min
Shi
*ab
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling-Ling Lu, Shanghai, 200032, China. E-mail: weiyin@sioc.ac.cn; mshi@mail.sioc.ac.cn
First published on 10th May 2024
Abstract
A palladium-catalyzed ring-opening cyclization of (E) & (Z)-ene-vinylidenecyclopropanes has been developed via an intramolecular [3 + 2] cycloaddition process in the presence of a sterically bulky biaryl phosphine ligand, stereoselectively affording fused cis- & trans-bicyclo[4.3.0] skeletal products in good yields with a broad substrate scope and good functional tolerance. A plausible reaction mechanism was proposed on the basis of previous work and the DFT calculations.
Bicyclo[4.3.0] skeletons are widely found in natural products and are important backbones commonly observed in natural compounds with important biological activities.1 For example, globostellatic acid is extracted from the marine sponge Rhabdastrella globostellata.2 This novel triterpenoidal compound can be used as selective anti-proliferative agents against human umbilical vein endothelial cells. Chiloscyphone is a natural product with the bicyclo[4.3.0] framework that is extracted from liverworts and shows multiple antimicrobial activities.3 Moreover, 7-epi-pinguisone exhibits insect anti-feedant activity (Scheme 1).4
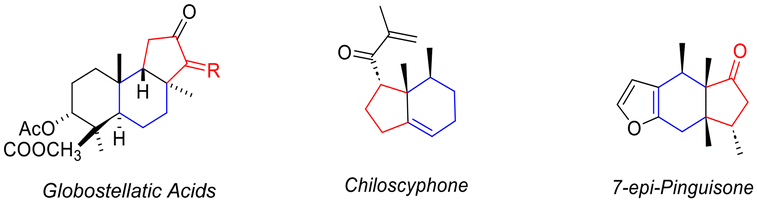 | ||
| Scheme 1 Bioactive compounds containing bicyclo[4.3.0] cores. | ||
Among the synthetic methodologies reported in the past, transition metal (TM)-catalyzed C–C bond activation and cycloaddition reactions have been characterized by good atom economy and mild reaction conditions for the rapid construction of bicyclic compounds.5 Therefore, transition metal catalysis provides a very powerful strategy to construct bicyclic compounds. Methylenecyclopropanes (MCPs) and vinylidenecyclopropanes (VDCPs) play important roles as three-carbon partners in the construction of bicyclic compounds.6,7 Generally, for MCP and VDCP moieties (Scheme 2A), the oxidative addition of a TM to the distal C–C bond of a cyclopropane has been shown to produce the metallocyclobutane species Int-A, which is accompanied by the migratory insertion of an unsaturated bond to produce the metallocyclohexane species Int-B. The metallocyclohexane species Int-B undergoes reductive elimination to produce the bicyclic products. Recently, several significant examples of [3 + 2] cycloaddition reactions of MCPs and VDCPs using Pd,8 Rh,9 Co,10 and Ni11 as catalysts have been disclosed.
 | ||
| Scheme 2 Previous work and this work. | ||
Our group has recently reported an asymmetric cycloaddition reaction of ene-VDCPs catalyzed by cationic rhodium and chiral phosphine ligands, resulting in the production of bicyclic products in high yields along with good ee values. By varying the reaction temperature, the selective construction of two different bicyclic products can be realized (Scheme 2B).12
In this work, we report a novel palladium-catalyzed [3 + 2] cycloaddition of (E) & (Z)-ene-VDCPs to stereoselectively obtain fused cis- & trans-bicyclic products in good yields (Scheme 2C, this work).
At the start of our studies, (E)-ene-VDCP 1a was used as the model substrate to examine the reaction outcomes. We found that the reaction successfully took place in the presence of 6.0 mol% of Pd2dba3 and 12 mol% of tBuXPhos in anhydrous toluene at 100 °C after 8 hours and the cis-bicyclic product 2a was obtained in 96% isolated yield (Table 1, entry 1). Phosphine-RuPhos, another bulky biaryl, can also promote the reaction, affording the corresponding product 2a in yields as low as 60% (Table 1, entry 2). Using dppf, dppb, BINAP and L2 as ligands, the reaction did not occur (Table 1, entries 3–5, & 8). In addition, upon using dtbpf, L1 and PtBu3 as ligands, this [3 + 2] cycloaddition reaction did not take place smoothly and the desired product was not obtained; the reaction system became complex and substrate 1a decomposed and only a small amount of 1a was recovered (Table 1, entries 6, 7, & 9). The examination of solvents showed that toluene is the best choice for this reaction (entry 3 vs. entries 10–13). On lowering the reaction temperature to 80 °C, no reaction occurred (Table 1, entry 14). It is worth noting that we did not find other products based on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond migration on the cyclopentane ring.
C bond migration on the cyclopentane ring.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | L | Solvent | T (°C) | Yieldb/% |
| a Reaction conditions: substrate 1a (0.10 mmol), Pd2(dba)3 (6 mol%) and L (12 mol%) in 0.1 mL anhydrous toluene under an argon atmosphere for 8 h. b Isolated yield. c K2CO3 (12 mol%) was added. d NR = no reaction. | |||||
| 1 | Pd 2 (dba) 3 | t BuXPhos | Toluene | 100 | 96 |
| 2 | Pd2(dba)3 | RuPhos | Toluene | 100 | 60 |
| 3 | Pd2(dba)3 | dppf | Toluene | 100 | NRd |
| 4 | Pd2(dba)3 | dppb | Toluene | 100 | NRd |
| 5 | Pd2(dba)3 | (rac)-BINAP | Toluene | 100 | NRd |
| 6 | Pd2(dba)3 | dtbpf | Toluene | 100 | — |
| 7 | Pd2(dba)3 | L1 | Toluene | 100 | — |
| 8c | Pd2(dba)3 | L2 | Toluene | 100 | NRd |
| 9c | Pd2(dba)3 | HPtBu3BF4 | Toluene | 100 | — |
| 10 | Pd2(dba)3 | t BuXPhos | Dioxane | 100 | 80 |
| 11 | Pd2(dba)3 | t BuXPhos | DCE | 100 | 76 |
| 12 | Pd2(dba)3 | t BuXPhos | PhCl | 100 | 82 |
| 13 | Pd2(dba)3 | t BuXPhos | MeCN | 100 | NR |
| 14 | Pd2(dba)3 | t BuXPhos | Toluene | 80 | NR |

|
|||||

Having determined the optimal reaction conditions, we next explored the substrate scope of (E)-ene-VDCPs 1 and the results are summarized in Scheme 3. It was found that R1 can be an alkyl, a cycloalkyl or a benzyl group, and the desired products 2a–2j were obtained in good yields ranging from 88% to 96%. The structure of 2c was determined by X-ray diffraction and its ORTEP drawing is shown in Scheme 3. The (E)-ene-VDCP 1k having an acetal group gave the desired product 2k in 88% yield. We also investigated the aryl substitution in this reaction and for the electron-donating groups found at the para- and meta-positions of the benzene ring, the reaction proceeded smoothly and the target products 2l–2o were obtained in good yields ranging from 80% to 84%. We also attempted to synthesize substrates bearing electron-withdrawing substituents such as OMe, F, Cl at the benzene ring. However, we only obtained the by-products instead of the ene-VDCPs (for details, see page S4 in the ESI†). We also investigated substrates 1p–1s bearing electron-donating and electron-withdrawing groups at different positions of the benzyl ring, and found that the reactions took place successfully and products 2p–2s were obtained in good yields ranging from 80% to 92%. (E)-ene-VDCPs 1t and 1u bearing a naphthalene and a thiophene ring were also compatible, providing 2t and 2u in 88% and 90% yields, respectively. Subsequently, we investigated the R2 substituents located at the all-carbon quaternary center and found that R2 could be an ethyl and a phenyl substituent, affording the desired products 2v and 2w in 92% and 80% yields. In addition, we examined the R3 substituent of the ester group in this reaction as well, and both ethyl and benzyl substituents gave the corresponding products 2x and 2y in 90% yields. Using N-SO2Ph as an N-protecting group (1z) afforded the corresponding product 2z in 90% yield. However, substrate 1aa with an ortho-substituted methyl group did not give the desired product under the standard conditions, probably due to steric hindrance. Lengthening the carbon chain as substrate 1ab did not provide the target product as well perhaps due to the spatial issue of the cis-bicyclo[5.3.0] skeletons.
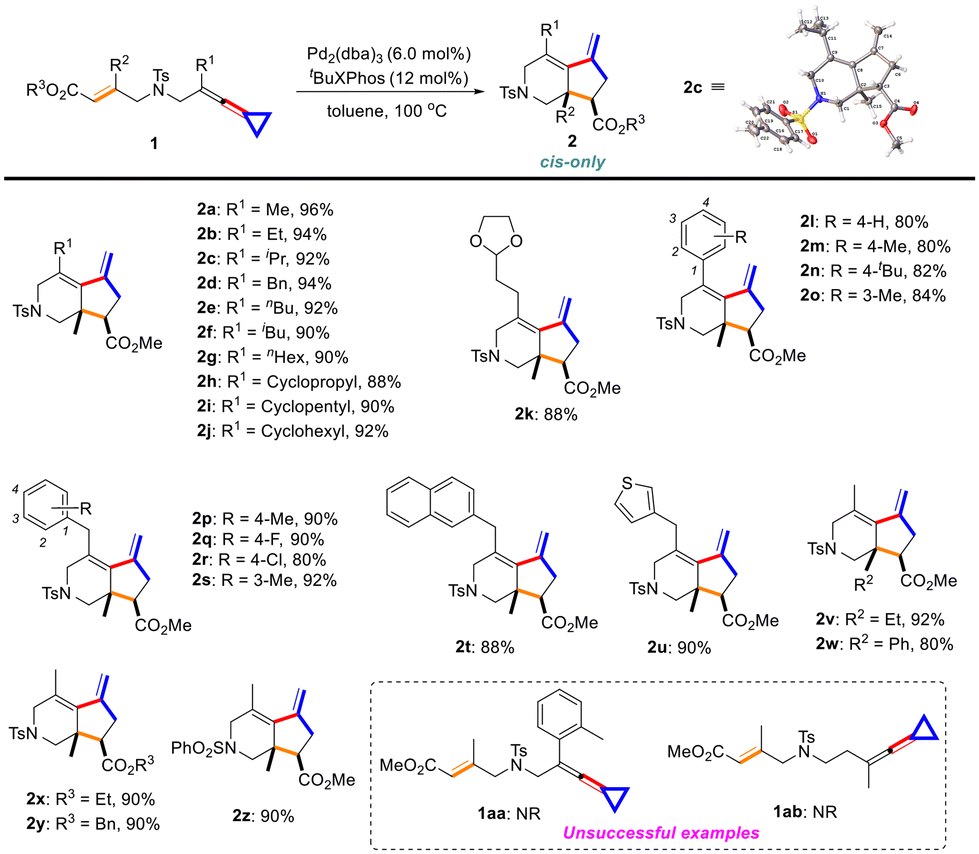 | ||
| Scheme 3 Substrate scope of (E)-ene-VDCPs. | ||
Next, we turned our attention to investigate the substrates (Z)-ene-VDCPs 3 (Scheme 4). For (Z)-ene-VDCPs 3, the reaction temperature should be increased to 120 °C to promote the progress of the reaction (for details, see Table S1 in the ESI†). Using the (Z)-ene-VDCPs 3a–3g as substrates, in which R1 was a different alkyl or cycloalkyl group, a phenyl and an acetal group, provided the corresponding trans-bicyclic products 4a–4g in good yields ranging from 70% to 96%. In addition, introducing a methyl, Cl and F substituents into the benzyl group delivered the target products 4h–4j in 88% to 90% yields. The crystal structure of the trans-bicyclic product 4i is shown in Scheme 4. Similarly, the thiophene moiety in (Z)-ene-VDCP was also tolerated, giving the corresponding product 4k in 80% yield. Introducing a phenyl group at the quaternary carbon center gave 4l in 70% yield and changing NTs to NSO2Ph furnished the desired product 4m in 90% yield.
 | ||
| Scheme 4 Substrate scope of (Z)-ene-VDCPs. | ||
To explore the synthetic applicability of the [3 + 2] cycloaddition reaction, we performed a gram-scale reaction using 3.0 mmol (1.1 g) of 1a under the standard conditions and found that 2a was obtained in 92% yield (Scheme 5a). Then, we conducted several transformations of 2a (0.1 mmol scale) and the results are shown in Scheme 5b. The Pd/C catalyzed hydrogenation of 2a produces 5 as a diastereomeric mixture in 96% yield. The olefinic moiety of product 2a could be oxidized to yield ketone 6 in 96% yield. We also carried out the reduction of the ester group in 2a with LiAlH4 to give product 7 with an alcohol fragment at the bicyclic skeleton in 96% yield. Furthermore, we examined the synthetic utility of the bicyclic products as the N-tosyl group of 8 derived from 2d was removed upon treating with Mg and NH4Cl in MeOH at 80 °C, affording the desired product 9 in 56% yield (Scheme 5b).
 | ||
| Scheme 5 Gram-scale experiment and synthetic transformations. | ||
To further understand the mechanism, we attempted to clarify the influence of temperature on the reaction result and stereochemical issue. All calculations have been performed at the SMD(toluene)/M06/6-311 + G(d,p)/Lanl2dz//B3LYP/6-31G(d)/Lanl2dz level with the Gaussian 16 program. All transition states were characterized by only one imaginary frequency pertaining to the desired reaction coordinate. The intrinsic reaction coordinate (IRC) calculations were carried out at the same level of theory to further authenticate the transition states (Scheme 6). We investigated the reaction pathways using (E)-ene-VDCPs 1a (black letters) and (Z)-ene-VDCPs 3a-Me (blue letters) by DFT calculations, respectively (for more details, see Table S2 in the ESI†). The palladium complex coordinated with the distal C–C bond to produce 2-Int1via a slightly exothermic process (ΔG = −0.8 kcal mol−1). In the same manner, 4-Int1 is also generated through a slight exothermic process (ΔG = −0.2 kcal mol−1). 2-Int1 undergoes an oxidative cyclometallation to give the palladacyclic intermediate 2-Int2 through 2-Ts1 with an energy barrier of 6.5 kcal mol−1. Similarly, 4-Int1 undergoes an oxidative cyclometallation to give the palladacyclic intermediate 4-Int2 through 4-Ts1 with an energy barrier of 5.6 kcal mol−1. In this process, the free energy between 2-Ts1 and 4-Ts1 produced by the E & Z substrates is very small (0.3 kcal mol−1). The 2-Int2 then isomerizes to 2-Int3via2-Ts2 with an energy barrier of 20.2 kcal mol−1. The corresponding process via4-Ts2 has a higher energy barrier by 6.2 kcal mol−1. Subsequently, olefinic moiety insertion occurs via2-Ts3 with an activation free energy of 15.7 kcal mol−1 to give the palladacyclohexane intermediate 2-Int4. The E- & Z-substrates control the stereoselectivity of the reaction during migratory insertion. For (E)-ene-VDCPs 1, the ester group is on the same side with the methyl group and this results in the cis products 2 during the migration of the olefin moiety. Conversely, (Z)-ene-VDCPs 3 produced the trans products 4. Kinetically, the reaction of (Z)-ene-VDCP should overcome an energy barrier that is higher than that of (E)-ene-VDCPs by 4.1 kcal mol−1. Thus, the reaction of (Z)-ene-VDCPs is conducted at a higher temperature in the experiment. Then, reductive elimination converts 2-Int4 to 2a + PdL (via2-Ts4, 5.4 kcal mol−1) and 4-Int4 to 4a-Me + PdL (via4-Ts4, 5.6 kcal mol−1), respectively.
 | ||
| Scheme 6 Proposed mechanism for the production of 2a and 4a-Me. | ||
In conclusion, we have developed an efficient palladium-catalyzed intramolecular [3 + 2] cycloaddition reaction for (E) & (Z)-ene-vinylidenecyclopropanes, which stereoselectively provides the corresponding fused cis & trans-bicyclo[4.3.0] products in moderate to good yields with a broad substrate scope and good functional group tolerance. We realized the reaction on a gram scale and performed interesting transformations of the products. The rational reaction mechanisms have been proposed on the basis of DFT calculations. Further studies are underway in our laboratory to synthesize biologically active molecules using this synthetic approach.
Author contributions
C. N. contributed to the experimental work; Z. Q. Y. and Y. W. contributed to the computational work. C. N., Y. W. and M. S. contributed to ideation and writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R & D Program of China (2023YFA1506700), the National Natural Science Foundation of China (21372250, 21121062, 21302203, 20732008, 21772037, 21772226, 21861132014, 91956115 and 22171078) and the Fundamental Research Funds for the Central Universities 222201717003.References
- (a) Y.-L. Kuo, M. Dhanasekaran and C.-K. Sha, J. Org. Chem., 2009, 74, 2033–2038 CrossRef CAS PubMed; (b) J. E. Wilson, J. Sun and G. C. Fu, Angew. Chem., Int. Ed., 2010, 49, 161–163 CrossRef CAS PubMed; (c) J. Ye and S. Ma, Angew. Chem., Int. Ed., 2013, 52, 10809–10813 CrossRef CAS PubMed; (d) I. Felker, G. Pupo, P. Kraft and B. List, Angew. Chem., Int. Ed., 2015, 54, 1960–1964 CrossRef CAS PubMed; (e) N. Eddy and P. Ichalkaranje, Molecules, 2016, 21, 1358 CrossRef PubMed.
- S. Aoki, M. Sanagawa, Y. Watanabe, A. Setiawan, M. Arai and M. Kobayashi, Bioorg. Med. Chem., 2007, 15, 4818–4828 CrossRef CAS PubMed.
- J. Shiina, R. Obata, H. Tomoda and S. Nishiyama, Eur. J. Org. Chem., 2006, 2006, 2362–2370 CrossRef.
- S. Bernasconi, M. Ferrari, P. Gariboldi, G. Jommi, M. Sisti and R. Destro, J. Chem. Soc., Perkin Trans. 1, 1981, 1994 RSC.
- (a) Y. Yamamoto, Chem. Rev., 2012, 112, 4736–4769 CrossRef CAS PubMed; (b) Y. Shibata and K. Tanaka, Synthesis, 2012, 323–350 CAS; (c) A. Lledó, A. Pla-Quintana and A. Roglans, Chem. Soc. Rev., 2016, 45, 2010–2023 RSC; (d) G. Domínguez and J. Pérez-Castells, Chem. – Eur. J., 2016, 22, 6720–6739 CrossRef PubMed; (e) A. Roglans, A. Pla-Quintana and M. Solà, Chem. Rev., 2021, 121, 1894–1979 CrossRef CAS PubMed; L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 16260–16265 Search PubMed; (f) S.-H. Hou, X. Yu, R. Zhang, L. Deng, M. Zhang, A. Y. Prichina and G. Dong, J. Am. Chem. Soc., 2020, 142, 13180–13189 CrossRef CAS PubMed; (g) Z. Ding, Y. Wang, W. Liu, Y. Chen and W. Kong, J. Am. Chem. Soc., 2021, 143, 53–59 CrossRef CAS PubMed; (h) Y. Wang, W. Liao, Y. Wang, L. Jiao and Z.-X. Yu, J. Am. Chem. Soc., 2022, 144, 2624–2636 CrossRef CAS PubMed.
- (a) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213–1270 CrossRef CAS PubMed; (b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed; (c) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913–924 CrossRef CAS PubMed.
- (a) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883–5913 CrossRef CAS PubMed; (b) S. Yang and M. Shi, Acc. Chem. Res., 2018, 51, 1667–1680 CrossRef CAS PubMed.
- (a) F. Verdugo, L. Villarino, J. Durán, M. Gulías, J. L. Mascareñas and F. López, ACS Catal., 2018, 8, 6100–6105 CrossRef CAS; (b) F. Verdugo, E. Da Concepción, R. Rodiño, M. Calvelo, J. L. Mascareñas and F. López, ACS Catal., 2020, 10, 7710–7718 CrossRef CAS; (c) F. Verdugo, R. Rodiño, M. Calvelo, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2022, 61, e202202295 CrossRef CAS PubMed.
- (a) P. A. Evans and P. A. Inglesby, J. Am. Chem. Soc., 2008, 130, 12838–12839 CrossRef CAS PubMed; (b) S. Mazumder, D. Shang, D. E. Negru, M.-H. Baik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569–20572 CrossRef CAS PubMed; (c) P. A. Inglesby, J. Bacsa, D. E. Negru and P. A. Evans, Angew. Chem., Int. Ed., 2014, 53, 3952–3956 CrossRef CAS PubMed; (d) P. A. Evans, D. E. Negru and D. Shang, Angew. Chem., Int. Ed., 2015, 54, 4768–4772 CrossRef CAS PubMed; (e) K.-H. Rui and M. Shi, Org. Chem. Front., 2019, 6, 1816–1820 RSC.
- (a) E. Da Concepción, I. Fernández, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2021, 60, 8182–8188 CrossRef PubMed; (b) X. Xiao and Z. Yu, Chem. – Eur. J., 2021, 27, 7176–7182 CrossRef CAS PubMed.
- (a) S. Komagawa and S. Saito, Angew. Chem., Int. Ed., 2006, 45, 2446–2449 CrossRef CAS PubMed; (b) L. Saya, G. Bhargava, M. A. Navarro, M. Gulías, F. López, I. Fernández, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2010, 49, 9886–9890 CrossRef CAS PubMed; (c) S. Saito, K. Maeda, R. Yamasaki, T. Kitamura, M. Nakagawa, K. Kato, I. Azumaya and H. Masu, Angew. Chem., Int. Ed., 2010, 49, 1830–1833 CrossRef CAS PubMed; (d) L. Saya, I. Fernández, F. López and J. L. Mascareñas, Org. Lett., 2014, 16, 5008–5011 CrossRef CAS PubMed.
- K.-H. Rui, S. Yang, Y. Wei and M. Shi, Org. Chem. Front., 2019, 6, 2506–2513 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data. CCDC 2306250 and 2328425. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00607k |
| This journal is © The Royal Society of Chemistry 2024 |