
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeuroprotective azaphilones from a deep-sea derived fungus Penicillium sp. SCSIO41030†
Weihao
Chen‡
ac,
Jiahui
Jiang‡
b,
Xiaoyan
Pang
a,
Yingying
Song
ac,
Zhiyou
Yang
*b,
Junfeng
Wang
 *acd and
Yonghong
Liu
*acd
*acd and
Yonghong
Liu
*acd
aCAS Key Laboratory of Tropical Marine Bio-resources and Ecology/Guangdong Key Laboratory of Marine Materia Medica/Innovation Academy of South China Sea Ecology and Environmental Engineering, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou 510301, China. E-mail: wangjunfeng@scsio.ac.cn; yonghongliu@scsio.ac.cn
bCollege of Food Science and Technology, Guangdong Provincial Key Laboratory of Aquatic Product Processing and Safety, Guangdong Province Engineering Laboratory for Marine Biological Products, Guangdong Provincial Engineering Technology Research Center of Seafood, Key Laboratory of Advanced Processing of Aquatic Product of Guangdong Higher Education Institution, Guangdong Ocean University, Zhanjiang 524088, China. E-mail: zyyang@gdou.edu.cn
cUniversity of Chinese Academy of Sciences, 19 Yuquan Road, Beijing 100049, China
dSanya Institute of Marine Ecology and Engineering, Yazhou Scientific Bay, Sanya 572000, China
First published on 10th May 2024
Abstract
Ten azaphilones including one pair of new epimers and three new ones, penineulones A–E (1–5) with the same structural core of angular deflectin, were obtained from a deep-sea derived Penicillium sp. SCSIO41030 fermented on a liquid medium. Their structures including absolute configurations were elucidated using chiral-phase HPLC analysis, extensive NMR spectroscopic and HRESIMS data, ECD and NMR calculations, and by comparing NMR data with literature data. Biological assays showed that the azaphilones possessed no antitumor and anti-viral (HSV-1/2) activities at concentrations of 5.0 μM and 20 μM, respectively. In addition, azaphilones 8 and 9 showed neuroprotective effects against Aβ25–35-induced neurotoxicity in primary cultured cortical neurons at a concentration of 10 μM. Azaphilones 8 and 9 dramatically promoted axonal regrowth against Aβ25–35-induced axonal atrophy. Our study indicated that azaphilones could be promising lead compounds for neuroprotection.
Introduction
Azaphilones are an enormous family with diverse structures of fungal secondary polyketide metabolites, which possess a highly oxygenated isochroman scaffold containing a pyrone–quinone bicyclic core and a quaternary carbon center. According to the latest review on azaphilones,1,2 up to now, over 625 azaphilones have been discovered from more than 20 genera of fungi, such as Penicillium,3Hypoxylon,4Pestalotiopsis,5Chaetomium,6 and so on. Besides, most azaphilone molecules exhibit a wide range of significant biological activities including enzyme inhibition,7 antimicrobial,8 cytotoxic, antiviral, antioxidative, and anti-inflammatory activities,2 due to which they attract lots of attention continuously from chemists and pharmacologists. Recently, during our ongoing search for bioactive molecules from marine-derived fungi,9,10 a chemical investigation of a deep-sea derived Penicillium sp. SCSIO41030, fermented on a rice medium, resulted in the isolation and identification of two new p-terphenyl derivatives with antiviral activity against herpes simplex virus and one novel 4,5-diphenyl-2-pyrone.11 And further study of the strain SCSIO41030 fermented on a liquid medium led to the discovery of ten azaphilones with the same structural type of angular deflectin,2 including one pair of new epimers and three new compounds, penineulones A–E (1–5), along with five known analogues. Herein, the details of the isolation, structural identification and biological activities of compounds 1–10 are described.Results and discussion
After the purification of the EtOAc extract from the culture broth of Penicillium sp. SCSIO41030, the 13C NMR data of compound P1 were found to possess two sets of similar carbon shift signals (Fig. S1†). Therefore, P1 was subsequently subjected to separation on a chiral column of DAICEL CHIRALPAK IC to afford enantiomeric compounds penineulone A (1) (tR = 12.5 min) and penineulone B (2) (tR = 16.1 min) in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8, using ethanol/n-hexane (v/v: 55/65, flow rate 1 mL min−1) with 0.1% trifluoroacetic acid (TFA) as the mobile phase. Compounds 1 and 2 were isolated as a light-yellow oil and have the same molecular formula C19H24O6 with eight degrees of unsaturation deduced using the HRESIMS data and 13C NMR data (Table 1). The 1D NMR data along with the HSQC experiment of 1 showed the presence of three aromatic/olefinic methines at δC/H 146.2/9.36 (s, CH-1), 107.5/6.01 (s, CH-3), and 103.7/5.41 (s, CH-4); three sp3 methines at δC/H 54.7/4.30 (d, J = 7.7 Hz, CH-10), 48.6/2.66 (q, J = 6.9 Hz, CH-12), and 45.5/3.65 (d, J = 7.7 Hz, CH-7); five methyls at δC/H 53.0/3.76 (s, CH3-19), 21.7/1.14 (s, CH3-16), 19.4/2.14 (s, CH3-15), 16.6/1.18 (d, J = 6.9 Hz, CH-18), and 11.4/0.86 (t, J = 6.5 Hz, CH-14); and one methylene at δC/H 25.3/1.77 and 1.39 (m, CH2-13). Besides the above twelve corresponding hydrogen-bearing carbons, seven more carbons were present in the 13C NMR spectrum, including three carbonyls (one ester group), three olefinics (one oxygenated), and one oxygenated tertiary carbon. The aforementioned NMR data resembled those of chermesinone B (8),12 an azaphilone derivative obtained from a mangrove endophytic fungus Penicillium chermesinum (ZH4-E2). The main differences were one additional methoxyl group [δC/H 53.0/3.76 (s, CH3-19)] and one less degree of unsaturation in 1, which indicated that the angular γ-lactone ring of the azaphilone skeleton was opened. The above deduction was further verified using key HMBC signals from H3-19 and H-10 to C-17. Detailed analysis of the COSY and HMBC data (Fig. 2) allowed the assignments of all carbon and proton resonances in 1.
:0.8, using ethanol/n-hexane (v/v: 55/65, flow rate 1 mL min−1) with 0.1% trifluoroacetic acid (TFA) as the mobile phase. Compounds 1 and 2 were isolated as a light-yellow oil and have the same molecular formula C19H24O6 with eight degrees of unsaturation deduced using the HRESIMS data and 13C NMR data (Table 1). The 1D NMR data along with the HSQC experiment of 1 showed the presence of three aromatic/olefinic methines at δC/H 146.2/9.36 (s, CH-1), 107.5/6.01 (s, CH-3), and 103.7/5.41 (s, CH-4); three sp3 methines at δC/H 54.7/4.30 (d, J = 7.7 Hz, CH-10), 48.6/2.66 (q, J = 6.9 Hz, CH-12), and 45.5/3.65 (d, J = 7.7 Hz, CH-7); five methyls at δC/H 53.0/3.76 (s, CH3-19), 21.7/1.14 (s, CH3-16), 19.4/2.14 (s, CH3-15), 16.6/1.18 (d, J = 6.9 Hz, CH-18), and 11.4/0.86 (t, J = 6.5 Hz, CH-14); and one methylene at δC/H 25.3/1.77 and 1.39 (m, CH2-13). Besides the above twelve corresponding hydrogen-bearing carbons, seven more carbons were present in the 13C NMR spectrum, including three carbonyls (one ester group), three olefinics (one oxygenated), and one oxygenated tertiary carbon. The aforementioned NMR data resembled those of chermesinone B (8),12 an azaphilone derivative obtained from a mangrove endophytic fungus Penicillium chermesinum (ZH4-E2). The main differences were one additional methoxyl group [δC/H 53.0/3.76 (s, CH3-19)] and one less degree of unsaturation in 1, which indicated that the angular γ-lactone ring of the azaphilone skeleton was opened. The above deduction was further verified using key HMBC signals from H3-19 and H-10 to C-17. Detailed analysis of the COSY and HMBC data (Fig. 2) allowed the assignments of all carbon and proton resonances in 1.
 | ||
| Fig. 1 Structures of compounds 1–10. | ||
 | ||
| Fig. 2 Key COSY, HMBC and NOESY correlations of 1–4. | ||
| Pos. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δ C type | δ H mult. (J) | δ C type | δ H mult. (J) | δ C type | δ H mult. (J) | |
| a Recorded in chloroform-d. b Recorded in DMSO-d6. | ||||||
| 1 | 146.2, CH | 6.91, s | 147.7, CH | 7.39, s | 145.1, CH | 7.27, s |
| 2 | 158.7, C | 158.6, C | 158.7, C | |||
| 3 | 107.5, CH | 6.01, s | 107.7, CH | 6.01, s | 104.7, CH | 6.38, s |
| 4 | 103.7, CH | 5.41, s | 103.3, CH | 5.37, s | 110.6, C | |
| 5 | 198.3, C | 197.9, C | 190.3, C | |||
| 6 | 73.3, C | 73.2, C | 82.5, C | |||
| 7 | 45.5, CH | 3.65, d (7.7) | 45.2, CH | 3.43, d (2.7) | 43.3, CH | 3.77, d (12.0) |
| 8 | 118.1, C | 116.8, C | 113.7, C | |||
| 9 | 148.6, C | 148.8, C | 139.2, C | |||
| 10 | 54.7, CH | 4.30, d (7.7) | 54.9, CH | 4.45, d (2.7) | 54.8, CH | 4.56, d (12.0) |
| 11 | 207.1, C | 208.9, C | 170.1, C | |||
| 12 | 48.6, CH | 2.66, q (6.9) | 47.2, CH | 2.72, q (6.8) | 206.9, C | |
| 13 | 25.3, CH2 | 1.77, m | 26.0, CH2 | 1.69, m | 47.1, CH | 2.81, h (7.0) |
| 1.39, m | 1.38, m | |||||
| 14 | 11.4, CH3 | 0.86, t (6.5) | 11.7, CH3 | 0.84, t (6.4) | 24.0, CH2 | 1.61, m |
| 1.29, m | ||||||
| 15 | 19.4, CH3 | 2.14, s | 19.5, CH3 | 2.14, s | 11.5, CH3 | 0.79, t (7.4) |
| 16 | 21.7, CH3 | 1.14, s | 22.4, CH3 | 1.13, s | 19.1, CH3 | 2.15, s |
| 17 | 169.4, C | 169.8, C | 9.6, CH3 | 1.72, s | ||
| 18 | 16.6, CH3 | 1.18, d (6.9) | 16.5, CH3 | 1.16, d (6.8) | 22.9, CH3 | 1.40, s |
| 19 | 53.0, CH3 | 3.76, s | 52.7, CH3 | 3.75, s | 15.3, CH3 | 0.92, d (7.0) |
The NMR data of compound 2 (Table 1) showed great similarity to those of 1, suggesting that they shared the same planar structure based on the same molecular formula. The remarkable distinction was the proton coupling constants between H-7 and H-10 (7.7 Hz in 1vs. 2.7 Hz in 2), indicating that they possessed different relative configurations at C-7 and C-10 (threo configuration for 1 and erythro configuration for 2).13 In addition, the optical rotation values of 1 and 2 were [α]25D +21.3 and +28.2, respectively, as well as their electronic circular dichroism (ECD) spectra were also almost the same (Fig. 3), indicating that both 1 and 2 were a pair of epimers. Considering the limited contribution of the flexible side chain to the ECD spectrum and the lack of NOESY signals of H-7 and H3-16, two truncated models a/b were used for the ECD calculations. The ECD spectrum of 6R,7S-b (Fig. 3) showed the best agreement with experimental curves, which led to the determination of the 6R,7S absolute configuration for 1 and 2. The NMR calculations of the four candidate diastereoisomers (1a/1b and 2a/2b) were then carried out using the gauge independent atomic orbital (GIAO) strategy at the B3LYP/6-31+G(d,p) level of theory with an IEFPCM solvent model of chloroform using the ORCA 5.0.3 program.14 The calculated chemical shifts of 1a/1b and 2a/2b were compared with the experimental values, respectively, applying total absolute deviation (TAD), mean absolute error (MAE), and DP4+ probability analysis as all three methods have been widely applied in addressing the stereochemical assignment of isomeric compounds.15 The lower TAD and MAE results showed that 1a and 2a are the most probable stereoisomers, and these results were further strongly supported by the DP4+ probability analysis with a high confidence level of 100% (both 1H and 13C data) (Fig. S37†). Finally, the absolute configurations of 1 and 2 were defined as (6R,7S,10S,12S) and (6R,7S,10R,12S), respectively, which confirmed that they were indeed a pair of epimers at C-10.
 | ||
| Fig. 3 (A) Experimental ECD spectra of 1 and 2 and (B) calculated ECD spectra of a and b. | ||
Compound 3 was obtained as a light-yellow oil and has the molecular formula C19H22O5 as deduced from HRESIMS data, suggesting nine degrees of unsaturation. The 1D NMR data of 3 shown in Table 1 reveal the same azaphilone skeleton with an angular lactone ring as that of the known analogue chermesinone B (8).12
The obvious differences were that the 1H NMR signal of H-4 (δH 5.34) was absent and the 13C NMR resonance of C-4 showed an upfield shift (δC 110.6 vs. 106.1), while an additional methyl signal (δC/H 9.6/1.72, s, CH3-17) was observed. Further analysis of HMBC signals from H3-17 to C-4, C-5, and C-9 (Fig. 2) indicated that the additional methyl was located at C-4. Consequently, the planar structure of 3 was established as shown. The relative configurations at C-6, C-7, C-10, and C-12 in 3 were determined by a combination of a NOESY experiment and 1H NMR coupling constant analysis. The NOESY correlations of H-10/H3-18 and H-10/H3-19 indicated that H-10, H3-18, and H3-19 were on the same side of the molecule. A large coupling constant between H-7 and H-10 (JH-7/H-10 = 12.0 Hz) and the lack of a NOE between H-7 and H3-18 revealed the trans configurations of H-7/H-10 and H-7/H3-18. Thus, considering the limited contribution of the flexible side chain to the ECD spectrum, two truncated models c and d were employed for the ECD calculations. As shown in Fig. 4A, the Boltzmann-weighted ECD spectrum of 6R,7S,10S-d showed the best agreement with the experimental curve of 3, which led to the determination of the 6R,7S,10S absolute configuration in 3. The same as for 1 and 2, NMR calculations of the two candidate diastereoisomers 3a/3b were then carried out to determine the absolute configuration at C-13 using the GIAO strategy at the B3LYP/6-31+G(d,p) level of theory with an IEFPCM solvent model of DMSO. As a result, the calculated NMR data with lower TAD and MAE values and a higher DP4+ probability of 100% (both 1H and 13C data) showed 3a to be the most probable stereoisomer (Fig. 4B). Therefore, the absolute configuration of 3 was defined as 6R,7S,10S,13S and it was named penineulone C.
 | ||
| Fig. 4 (A) Experimental ECD spectra of 3 and calculated ECD spectra of c/d; (B) DP4+ probability, TAD, and MAE analysis of 3a/3b. | ||
Compound 4 was also obtained as a light-yellow oil and has the molecular formula C17H24O4 as determined from the HRESIMS ion peak at m/z 293.1675 [M + H]+ (calcd for C17H25O4, 293.1646), suggesting six degrees of unsaturation. The 1D NMR data of 4 shown in Table 2 reveal the presence of two olefinic methines CH-1 (δC/H 147.9/6.39, s) and CH-4 (δC/H 116.3/5.48, s), three sp3 methines CH-2 (δC/H 72.6/4.09, m), CH-7 (δC/H 40.9/3.00, d, J = 9.1 Hz), and CH-12 (δC/H 46.6/2.62, p, J = 6.6 Hz), three methylenes CH2-3 (δC/H 34.4/2.55, m; 2.46, m), CH2-10 (δC/H 37.7/2.86, dd, J = 18.0, 2.7 Hz; 2.76, dd, J = 18.0, 9.1 Hz) and CH2-13 (δC/H 25.5/1.59, m; 1.35, m), and four methyls at δC/H 11.3/0.79, t, J = 7.4 Hz CH3-14; 20.3/1.30, d, J = 6.2 Hz, CH3-15; 20.2/1.01, s, CH3-16 and 15.7/0.99, m, CH3-17. Besides, the signals of the remaining five tertiary carbons including two carbonyls (δC 200.8 and 212.8), two olefinics (δC 112.2 and 150.5), and one oxygenated sp3 tertiary carbon (δC 74.5) were observed in the 13C NMR spectrum. The above characteristics of the NMR data indicate the same azaphilone skeleton as that of the co-isolated known analogue chermesinone A (9).12
| Pos. | 4 | 5 | ||
|---|---|---|---|---|
| δ C type | δ H mult. (J in Hz) | δ C type | δ H mult. (J in Hz) | |
| 1 | 147.9, CH | 6.39, s | 153.1, CH | 8.63, s |
| 2 | 72.6, CH | 4.09, m | 159.2, C | |
| 3 | 34.4, CH2 | 2.55, dd (13.6, 2.8) | 108.1, CH | 6.49, s |
| 2.46, d (13.6) | ||||
| 4 | 116.3, CH | 5.48, s | 103.9, CH | 5.29, s |
| 5 | 200.8, C | 189.5, C | ||
| 6 | 74.5, C | 87.4, C | ||
| 7 | 40.9, CH | 3.00, d (9.1) | 166.0, C | |
| 8 | 112.2, C | 110.5, C | ||
| 9 | 150.5, C | 144.5, C | ||
| 10 | 37.7, CH2 | 2.86, dd (18.0, 2.7) | 122.2, C | |
| 2.76, dd (18.0, 9.1) | ||||
| 11 | 212.8, C | 168.0, C | ||
| 12 | 46.6, CH | 2.62, p (6.6) | 199.8, C | |
| 13 | 25.5, CH2 | 1.59, m | 44.2, CH | 3.36, q (6.5) |
| 1.35, m | ||||
| 14 | 11.3, CH3 | 0.79, t (7.4) | 25.5, CH2 | 1.72, m |
| 1.29, m | ||||
| 15 | 20.3, CH3 | 1.30, d (6.2) | 11.6, CH3 | 0.88, t (7.4) |
| 16 | 20.2, CH3 | 1.01, s | 18.9, CH3 | 2.20, s |
| 17 | 15.7, CH3 | 0.99, m | 24.8, CH3 | 1.59, s |
| 18 | 15.6, CH3 | 0.93, d (7.0) | ||
The obvious difference between 4 and 9 was that the double bond Δ2,3 was reduced as one methine and one methylene, which was further verified by COSY correlations of H-2 and H2-3 along with the HMBC signals from H3-15 to C-2 and C-3 and from H2-3 to C-4, C-8 and C-9 (Fig. 2). Detailed analysis of the other 2D NMR signals in Fig. 2 confirmed that the planar structure of compound 4 was a reduced product of chermesinone A (9).
The absolute configuration at C-12 was determined as S, due to the same chemical shifts at CH-12 of 4 and 9 recorded in the same solvent DMSO-d6 (δC/H 46.6/2.62 for 4vs. 46.6/2.65 for 9). In addition, CH3-16 and CH2-10 were deduced to be cofacial in compound 4 through a NOESY experiment (Fig. 2), which indicated the relative configuration of 6R,7S or 6S,7R. Subsequently, the four possible diastereoisomers, (2R,6R,7S,12S)-4a, (2S,6R,7S,12S)-4b, (2R,6S,7R,12S)-4c, and (2S,6S,7R,12S)-4d, of 4 were subjected to TDDFT ECD calculations in the MeOH model. However, the calculated ECD spectra of 4a–4d were nearly the same and none of them showed the best agreement with the experimental curve (Fig. 5A). The TAD and MAE analysis of NMR calculations for the four candidate diastereoisomers (4a–4d) led to the deduction that 4 adopted the absolute configuration of (2R,6R,7S,12S)-4a with a DP4+ probability (both 1H and 13C data) of 81.11% (Fig. 5B). Therefore, the absolute configuration of 4 was defined as 2R,6R,7S,12S as shown in Fig. 1 and it was named penineulone D.
 | ||
| Fig. 5 (A) Experimental ECD spectra of 4 and calculated ECD spectra of 4a–4d; (B) DP4+ probability, TAD, and MAE analysis of 4a–4d. | ||
Compound 5 was obtained as a light-yellow oil and has the molecular formula C18H18O5 as deduced from HRESIMS data, suggesting ten degrees of unsaturation. The 1D NMR data of 5 shown in Table 2 exhibit a slight difference compared with those of the co-isolated analogue 8,11-didehydrochermesinone B (6),16 which is produced by an endophytic fungus Nigrospora sp. of Aconitum carmichaeli. In the same solvent of DMSO-d6, the most obvious differences between 5 and 6 were the chemical shifts of C-12 (199.8 vs. 200.1), CH2-14 (δC/H 25.5/1.29, 1.72 vs. 25.7/1.25, 1.53), and CH3-18 (δC/H 15.6/0.93, d, J = 7.0 Hz vs. 14.5/1.05, d, J = 6.7 Hz), which enlightened us that 5 and 6 may be a pair of epimers at C-13. Similar ECD spectra of 5 and 6 indicated that they were indeed not the same compound and the calculated ECD spectra of the four candidate diastereoisomers (5a–5d) exhibited that the ECD spectra of 5 and 6 were governed mainly by the C-6 chirality center as the 6R absolute configuration (Fig. 6). Consequently, the only explanation of different chemical shifts around C-13 in 5 was that 5 was a C-13 epimer of 6, while they shared the identical 6R absolute configuration. Finally, compound 5 was determined as a new azaphilone with the 6R,13R absolute configuration and named penineulone E. To our knowledge, 5 represents the third azaphilone with an R configuration at the side chain chirality center linked to a methyl group,17,18 while most azaphilones have been discovered with an S configuration.1,2
 | ||
| Fig. 6 Experimental and calculated ECD curves of 5, 6 and 5a–5d. | ||
Additionally, the other five known compounds were identified as 8,11-didehydrochermesinone B (6),16 chermesinone C (7), chermesinone B (8), chermesinone A (9),12 and anishidiol (10)19 by using X-ray diffraction crystal structures or comparison of their NMR data with those reported in the literature. The colorless crystals of 8 and 9 were obtained from a mixed solvent of CHCl3 and CH3OH (v:v, 1:1). Crystallographic data for structure 8 were first reported and crystallographic data of 8 and 9 have been deposited with the Cambridge Crystallographic Data Centre under supplementary publication numbers CCDC 2214106 and 2214107† (Fig. 7).
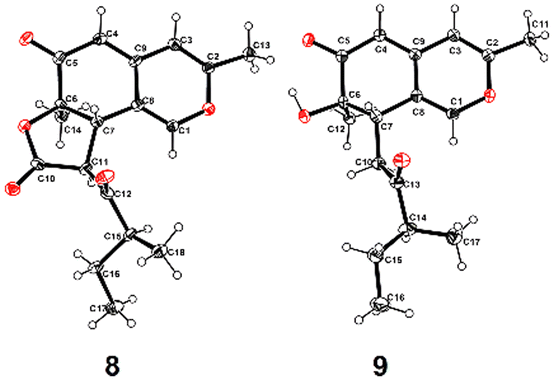 | ||
| Fig. 7 X-ray diffraction crystal structures (ORTEP drawing) of 8 and 9. | ||
The isolated compounds (1–10) were evaluated for their antitumor (C42B, H446, and H69 cell lines) and anti-viral (HSV-1/2) activities. However, the cell viabilities of the three human tumor cell lines were all over 80% after treatment with compounds 1–10 at a concentration of 5.0 μM for 72 h. And the CPE assay result showed no inhibition of 1–10 against HSV-1/2 at a concentration of 20 μM.
Neuroprotective effects of azaphilones against Aβ25–35-induced neurotoxicity were assessed in primary cultured cortical neurons.20 Among the ten tested compounds, 8 and 9 showed significant neuronal protective effects against Aβ25–35-induced neuronal death at a concentration of 10 μM (Fig. 8). In addition, the axonal regrowth effects of 8 and 9 were evaluated (Fig. 9A). As a result, compared with the control group, the Aβ25–35-treatment significantly reduced the length of axons in primary cultured cortical neurons. However, treatment with compounds 8 and 9 dramatically increased the length of pNF-H positive axons at both concentrations of 1 and 10 μM (Fig. 9B and C).
 | ||
| Fig. 8 Neuroprotective effects of 8 and 9 on Aβ25–35-induced neuronal death in primary cultured cortical neurons. The error bars represent SEM. *p < 0.05, ***p < 0.001 vs. Veh, one-way ANOVA post hoc Dunnett's test (n = 3 repeated experiments). | ||
 | ||
| Fig. 9 Axonal regenerative effects of 8 and 9 on Aβ25–35-induced axonal atrophy in primary cultured cortical neurons. A. Experimental schedule. B. Statistical results. C. Representative photos of pNF-H-positive axons and MAP2-positive neurons. The error bars represent SEM. ***p < 0.001 vs. Veh, one-way ANOVA post hoc Dunnett's test. | ||
Experimental section
General experimental procedures
Optical rotations were acquired using a PerkinElmer MPC 500 (Waltham) polarimeter. UV spectra were recorded on a Shimadzu UV-2600 PC spectrometer (Shimadzu). ECD spectra were recorded using a Chirascan circular dichroism spectrometer (Applied Photophysics). IR spectra were recorded using an IR Affinity-1 spectrometer (Shimadzu). NMR spectra were recorded using a Bruker DRX-700 spectrometer (Bruker BioSpin, Fallanden, Switzerland) at 700 MHz for 1H-NMR and 175 MHz for 13C-NMR using TMS as the internal standard and chemical shifts were recorded as δ-values. HRESIMS spectra were recorded using a Bruker miXis TOF-QII mass spectrometer (Bruker). X-ray diffraction intensity data were collected using a CrysAlis PRO CCD area detector diffractometer with graphite-monochromated Cu Kα radiation (λ = 1.541 78 Å). TLC and column chromatography (CC) were performed on plates precoated with silica gel GF254 (10–40 μm) and over silica gel (200–300 mesh) (Qingdao Marine Chemical Factory, Qingdao, China), and on Sephadex LH-20 (Amersham Biosciences, Uppsala, Sweden), respectively. Spots were detected on TLC under 254 nm UV light or by heating after spraying with 5% H2SO4 in EtOH. All solvents used were of analytical grade (Tianjin Fuyu Chemical and Industry Factory, Tianjin, China). Semipreparative HPLC was performed using an ODS column (YMC-pack ODS-A, 10 × 250 mm2, 5 μm, 2 mL min−1).Fungal material
The fungal strain SCSIO41030 was isolated from the sediment collected at a depth of 1509 m in the South China Sea and was the same strain as reported previously.11Fermentation and extraction
A large-scale fermentation of the fungal strain SCSIO41030 was carried out at room temperature in 1 L conical flasks containing the liquid medium (300 mL per flask) composed of mannitol (20 g L−1), yeast extract (3 g L−1), sodium glutamate (10 g L−1), glucose (10 g L−1), maltose (20 g L−1), corn steep liquor (1 g L−1), MgSO4·7H2O (0.3 g L−1), KH2PO4 (0.5 g L−1) and tap water after adjusting its pH to 7.5. After static cultivation for 30 days, the whole fermented broth (20 L) was filtered through cheesecloth to separate it into filtrate and mycelia. The filtrate was extracted three times with EtOAc to yield an EtOAc solution, while the mycelia were cut into small pieces, soaked in EtOAc, and sonicated for 20 min to yield another EtOAc solution. Both EtOAc solutions were combined and concentrated under reduced pressure to obtain a black extract (16.2 g).Isolation and purification
The EtOAc extract was subjected to vacuum liquid chromatography on a silica gel column using step-gradient elution with MeOH–CH2Cl2 (0–100%) to separate it into six fractions based on TLC properties. Fraction 2 was divided into three parts (Frs 2-1–2-3) followed by Sephadex LH-20 column elution with MeOH. Compounds 7 (7.2 mg, tR = 18.5 min) and 8 (10.5 mg, tR = 23.5 min) were obtained from Fr. 2-1 purified by semi-preparative HPLC (45% CH3CN–H2O, 2 mL min−1). Fr. 2-2 was then purified by semi-preparative HPLC (45% CH3CN–H2O, 2 mL min−1) to yield P1 (3.1 mg, tR 16.9 min). P1 was subsequently subjected to a chiral column of DAICEL CHIRALPAK IC to afford enantiomeric compounds penineulone A (1) (2.2 mg, tR = 12.5 min) and penineulone B (2) (1.76 mg, tR = 16.1 min), using ethanol/n-hexane (v/v: 55/65, flow rate 1 mL min−1) with 0.1% trifluoroacetic acid (TFA) as the mobile phase. Fr. 2-3 was separated by semi-preparative HPLC (52% CH3CN–H2O with 0.1% TFA, 2 mL min−1) to afford 5 (4.2 mg, tR 35.9 min), 6 (6.3 mg, tR 20.0 min), and 9 (9.0 mg, tR 18.2 min). Fraction 3 was separated into two parts by ODS column and Sephadex LH-20 column elution with MeOH. Subfraction 3-2 was further purified by semi-preparative HPLC (56% CH3OH–H2O, 2 mL min−1) to yield 3 (3.1 mg, tR 14.8 min) and 10 (8.2 mg, tR 22.5 min). Subfraction 4-2, obtained from Sephadex LH-20 column separation of fraction 4, was also subjected to semi-preparative HPLC with 45% CH3OH–H2O (0.1% TFA) elution to give 4 (5.3 mg, tR 18.5 min, 2 mL min−1).
Penineulone A (
1
): light yellow oil; [α]25D +28.3 (c 0.1, MeOH); UV (MeOH) λmax (logε) 352 (3.88), 225 (3.76) nm; for 1H and 13C NMR data (DMSO-d6, 700/175 MHz), see Table 1; HR-ESI-MS m/z 349.1638 [M + H]+ (calcd for C19H25O6, 349.1646).
Penineulone B (
2
): light yellow oil; [α]25D +21.7 (c 0.1, MeOH); UV (MeOH) λmax (logε) 351 (3.98), 226 (3.96) nm; for 1H and 13C NMR data (DMSO-d6, 700/175 MHz), see Table 1; HR-ESI-MS m/z 349.1637 [M + H]+ (calcd for C19H25O6, 349.1649).
Penineulone C (
3
): light yellow oil; [α]25D −6.5 (c 0.1, MeOH); UV (MeOH) λmax (logε) 349 (3.75), 225 (3.58) nm; for 1H and 13C NMR (DMSO-d6, 700/175 MHz) data, see Table 1; HR-ESI-MS m/z 331.1467 [M + H]+ (calcd for C19H23O5, 331.1469).
Penineulone D (
4
): light yellow oil; [α]25D +30.1 (c 0.1, MeOH); UV (MeOH) λmax (logε) 348 (3.85), 226 (3.78) nm; for 1H and 13C NMR (DMSO-d6, 500/125 MHz) data, see Table 2; HR-ESI-MS m/z 293.1675 [M + H]+ (calcd for C17H25O4, 293.1646).
Penineulone E (
5
): light yellow oil; [α]25D −12.0 (c 0.1, MeOH); UV (MeOH) λmax (logε) 349 (3.68), 225 (3.98) nm; for 1H and 13C NMR (DMSO-d6, 500/125 MHz) data, see Table 2; HR-ESI-MS m/z 314.1167 [M + H]+ (calcd for C18H18O5, 314.1159).
ECD and NMR calculations
The preliminary conformational distribution search was performed using the Confab algorithm in OpenBabel software. The conformers with Boltzmann population of over 1% (the relative energy within 10 kcal mol−1) were reoptimized using density functional theory (DFT) at the B97-3c level under vacuum using the ORCA 5.0.3 program.14 After that, frequency calculations following the geometry optimizations were performed to verify that all the structures correspond to energy minima and have no imaginary frequency. The overall theoretical calculation of ECD was conducted in MeOH using time-dependent density functional theory (TDDFT) at the PBE0 def2-TZVP level for the stable conformers (the relative energy within 5 kcal mol−1). Rotatory strengths for a total of 35 excited states were calculated. The ECD spectra of different conformers were generated using the Multiwfn program21 with a half-bandwidth of 0.5–0.6 eV, according to the Boltzmann-calculated contribution of each conformer after UV correction. Solvent effects of methanol solution were evaluated at the same DFT level by using the CPCM method.In the case of conformationally flexible compounds, the conformational search was done in the gas phase using the MMFF force field. All conformers within 5 kcal mol−1 of the lowest energy conformer were subjected to further reoptimization and frequency calculations at the B3LYP/6-31G* level of theory. NMR calculations of all candidate diastereoisomers were then carried out using the gauge independent atomic orbital (GIAO) strategy at the B3LYP/6-31G*(d,p) level of theory with an IEFPCM solvent model in the ORCA 5.0.3 program.
Assay of neuroprotection effects
To detect neuronal viability, primary cortical neurons were cultured for 3 days and treated with Aβ25–35 (Sigma, A4559) for 0.5 h, followed by treatment with 8 and 9 (1 and 10 μM) for 48 h. Afterwards, the cell counting kit (CCK8, APEXBIO, K1018) reagent (10 μL) was added to each well for 3 h according to the manufacturer's instructions. The absorbance values of CCK8 test were measured using a 96-well ELISA microplate reader (Biotek, VT, USA) at 450 nm. Aβ25–35 was dissolved in dH2O to a final concentration of 5 mM and was incubated at 37 °C for 4 days for aggregation.
To test the axonal regenerative effects of 8 and 9 on Aβ25–35-induced axonal atrophy, after culturing neurons for 3 days, 10 μM Aβ25–35 was pretreated for 0.5 h and then cocultured with 8 and 9 (1 and 10 μM) for 4 days. The neurons were then fixed with 4% paraformaldehyde (PFA; Solarbio, Beijing, China) at 25 °C for 1 h and immunostained for visualizing phosphorylated neurofilament H (pNF-H, 1:500; Covance, SMI-35R, CA, United States) and microtubule-2 associated protein 2 (MAP2, 1:2000; Abcam, ab32454). Alexa Fluor 594-conjugated goat anti-mouse IgG (1:300, Abcam, ab150116) and Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:300, Abcam, ab150081) were used as secondary antibodies. 4′,6-Diamidino-2-phenylindole (DAPI) (1 μg mL−1, Biomol, Hamburg, Germany) was used for nuclear counterstaining. A fluorescence microscope system (Echo Revolve, ECHO, CA, United States) was used for image capture at a size of 480 × 640 μm2. Ten images of each group were captured and analyzed using ImageJ (NIH), with the Neurite Tracer plugins. The average lengths of pNF-H positive axons per neuron were measured.
Conclusions
In conclusion, a further study of a deep-sea derived Penicillium sp. SCSIO41030 that produced p-terphenyl derivatives revealed ten azaphilones including one pair of new epimers and three new ones, penineulones A–E (1–5) with the same structural core of angular deflectin, which were obtained from the strain fermented on a liquid medium. Extensive NMR spectroscopic and HRESIMS data and ECD and NMR calculations were used to determine the structures of new compounds. In addition, azaphilones 8 and 9 were found to possess neuroprotective effects against Aβ25–35-induced neurotoxicity at a concentration of 10 μM and significantly promoted axonal regrowth in primary cultured cortical neurons. To our knowledge, this is the first report of azaphilones with neuroprotective effects.Author contributions
W. Chen: investigation, data curation, visualization, and writing – original draft; J. Jiang: investigation, data curation, and writing – original draft; X. Pang and Y. Song: investigation; Z. Yang: methodology, supervision, and resources; J Wang: project administration, funding acquisition, supervision, resources, and writing – review & editing; Y Liu: funding acquisition, supervision, and resources. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Guangdong MEPP Funds (No. GDNRC [2024]28), the Hainan Provincial Joint Project of Sanya Yazhou Bay Science and Technology City (2021CXLH0013, 2021JJLH0097), the National Natural Science Foundation of China (U23A201140 and 42376124), the Guangdong Local Innovation Team Program (2019BT02Y262), the Guangzhou Science and Technology Project (202201010678), the Key-Area Research and Development Program of Guangdong Province (2023B1111050008), and the Key Science and Technology Plan Projects in Nansha District (2023ZD010). We are grateful to ZH Xiao, AJ Sun, X Ma, XH Zheng, and Y Zhang in the analytical facility at SCSIO for recording spectroscopic data.References
- J. M. Gao, S. X. Yang and J. C. Qin, Chem. Rev., 2013, 113, 4755–4811 CrossRef CAS PubMed.
- C. Chen, H. Tao, W. Chen, B. Yang, X. Zhou, X. Luo and Y. Liu, RSC Adv., 2020, 10, 10197–10220 RSC.
- Y. Shao, H. Yan, T. Yin, Z. Sun, H. Xie, L. Song, K. Sun and W. Li, J. Antibiot., 2020, 73, 77–81 CrossRef CAS PubMed.
- E. Kuhnert, F. Surup, E. B. Sir, C. Lambert, K. D. Hyde, A. I. Hladki, A. I. Romero and M. Stadler, Fungal Diversity, 2015, 71, 165–184 CrossRef.
- F. Cao, Z. H. Meng, P. Wang, D. Q. Luo and H. J. Zhu, J. Nat. Prod., 2020, 83, 1283–1287 CrossRef CAS PubMed.
- W.-Y. Zu, J.-W. Tang, K. Hu, Y.-F. Zhou, L.-L. Gou, X.-Z. Su, X. Lei, H.-D. Sun and P.-T. Puno, J. Org. Chem., 2021, 86, 475–483 CrossRef CAS PubMed.
- C. Huo, X. Lu, Z. Zheng, Y. Li, Y. Xu, H. Zheng and Y. Niu, Phytochemistry, 2020, 170, 112224 CrossRef CAS PubMed.
- M. Chen, N.-X. Shen, Z.-Q. Chen, F.-M. Zhang and Y. Chen, J. Nat. Prod., 2017, 80, 1081–1086 CrossRef CAS PubMed.
- W. Chen, C. Chen, J. Long, S. Lan, X. Lin, S. Liao, B. Yang, X. Zhou, J. Wang and Y. Liu, J. Antibiot., 2021, 74, 156–159 CrossRef CAS PubMed.
- X. Pang, W. Chen, X. Wang, X. Zhou, B. Yang, X. Tian, J. Wang, S. Xu and Y. Liu, Front. Microbiol., 2021, 12, 730807 CrossRef PubMed.
- W. Chen, J. Zhang, X. Qi, K. Zhao, X. Pang, X. Lin, S. Liao, B. Yang, X. Zhou, S. Liu, J. Wang, X. Yao and Y. Liu, J. Nat. Prod., 2021, 84, 2822–2831 CrossRef CAS PubMed.
- H. Huang, X. Feng, Z. Xiao, L. Liu, H. Li, L. Ma, Y. Lu, J. Ju, Z. She and Y. Lin, J. Nat. Prod., 2011, 74, 997–1002 CrossRef CAS PubMed.
- V. A. Cao, B.-K. Choi, H.-S. Lee, C.-S. Heo and H. J. Shin, J. Nat. Prod., 2021, 84, 1843–1847 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS PubMed.
- S.-P. Zhang, R. Huang, F.-F. Li, H.-X. Wei, X.-W. Fang, X.-S. Xie, D.-G. Lin, S.-H. Wu and J. He, Fitoterapia, 2016, 112, 85–89 CrossRef CAS PubMed.
- P. S. Steyn and R. Vleggaar, J. Chem. Soc., Perkin Trans. 1, 1986, 1975 RSC.
- C. Chen, J. Wang, H. Zhu, J. Wang, Y. Xue, G. Wei, Y. Guo, D. Tan, J. Zhang, C. Yin and Y. Zhang, Chem. Biodivers., 2016, 13, 422–426 CrossRef CAS PubMed.
- T. Hosoe, N. Mori, K. Kamano, T. Itabashi, T. Yaguchi and K. Kawai, J. Antibiot., 2011, 64, 211–212 CrossRef CAS PubMed.
- J. Deng, X. Feng, L. Zhou, C. He, H. Li, J. Xia, Y. Ge, Y. Zhao, C. Song, L. Chen and Z. Yang, Food Res. Int., 2022, 158, 111576 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra of all new products, 2D NMR spectra of selected compounds, UV and MS data, and energies of all calculated conformers. CCDC 2214106 and 2214107. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00586d |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |