Stereocontrolled synthesis of the aconitine D ring from D-glucose†
Received
5th April 2024
, Accepted 2nd May 2024
First published on 3rd May 2024
Abstract
The synthesis of a fully oxygenated aconitine D ring precursor from (D)-(+)-glucose is described. The route features a highly diastereoselective alkynyl Grignard ketone addition and a base-mediated enelactone to 1,3-diketone rearrangement.
Introduction
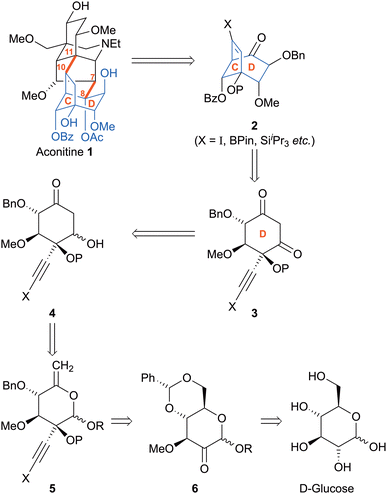
Aconitine 1, a C19 norditerpenoid alkaloid isolated from plants of the genus Aconitum,1 is a potent neurotoxin that exerts its principal pharmacological effect by binding to and activating voltage-gated Na+ channels.2 Its poisonous properties have gained a certain literary notoriety, featuring in the works of authors as diverse as Ovid, Shakespeare, and Arthur Conan Doyle.3 Related diterpenoid alkaloids display significant antagonist activity at the nicotinic acetylcholine receptor4 and cytotoxicity.5 The densely functionalized cage structure, containing 15 stereogenic centres, has attracted interest from the synthetic chemistry community.6 Despite landmark syntheses of simpler related compounds,7 no total synthesis of aconitine has yet been reported, perhaps a reflection of additional stereochemical complexity present throughout the molecule, particularly concentrated in ring D where all six ring atoms are stereogenic centres.8 A retrosynthetic strategy predicated upon disconnections across the C(7)–C(8) and C(10)–C(11) bonds back to a CD subunit (Scheme 1) is appealingly convergent, and has been elegantly showcased by Gin7b and Reisman9 in their respective syntheses of neofinaconitine and talatisamine. Noting that five of the six aconitine D ring stereocentres bear oxygen substituents, we envisioned assembly of this key subunit in enantiomerically pure form from a carbohydrate precursor, by exploiting the potential for Ferrier carbocyclisation10 to convert a pyranose-derived enol ether such as 5 to the corresponding cyclohexanone 4. The additional two carbons necessary to construct the C-ring could then be supplied by addition of an alkynyl metal to ketone 6, setting the stage for ring C closure via Conia-ene intramolecular diketone alkynylation.11 Given that an alkynyl iodide (cf.3, X = I) has been shown to be a competent reactant in this type of cyclisation,11a the potential to access vinyl halide 2 (or an organometallic derivative thereof) and elaborate it towards the target via cross-coupling is apparent. Benzylidene acetal 6 (R = Me) is an attractive substrate given the rigidity imparted by the trans-fused bicyclic scaffold; moreover, selective reduction of the acetal12 would maintain a protecting group on the 2° alcohol whilst revealing the 1° alcohol en route to enol 5.
 |
| | Scheme 1 Retrosynthetic plan. | |
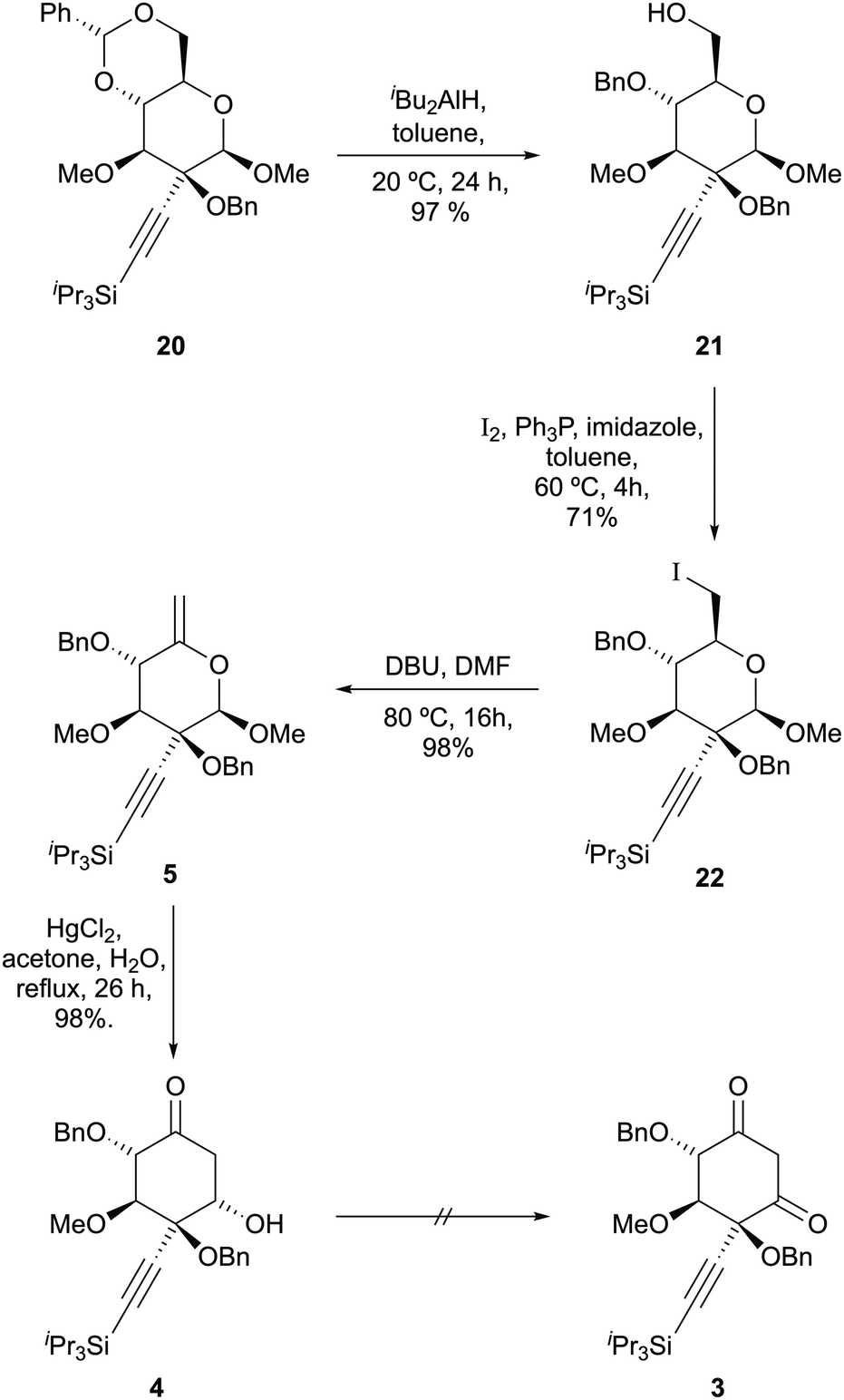
Towards the realization of this concept, we report here a stereocontrolled synthesis of diketone 3 (X = SiiPr3; P = Bn) from (D)-(+)-glucose as a platform for further synthetic studies towards 1.
Results and discussion
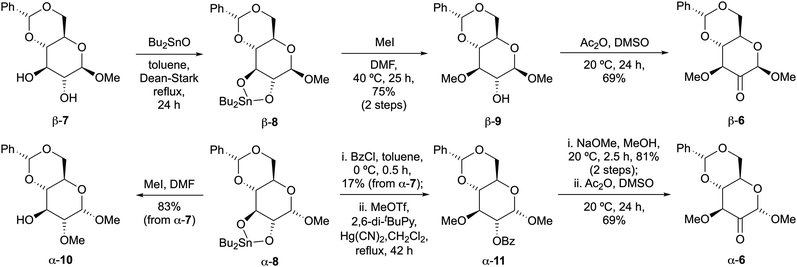
From the outset we recognized that the configuration at the anomeric centre of 6 would influence the stereochemical outcome of nucleophilic attack at the adjacent ketone. Treatment of stannylene acetal β-8 (from benzylidene acetal β-7)13 with methyl iodide gave high selectivity for 2° alcohol β-9, whereupon oxidation under Albright–Goldman conditions delivered the desired 3-OMe isomer β-6 (Scheme 2). On the other hand, methylation of α-8 took place selectively at the 2-position (giving α-10), requiring temporary protection of the 2-hydroxyl group as a benzoate ester to access α-6via α-11 as described by Fuchs.14
 |
| | Scheme 2 Preparation of ketoglycosides α-6 and β-6. | |
With both α-6 and β-6 in hand we turned our attention to the installation of the alkyne moiety at C(2). Mikami and Shin reported that whereas addition of methyl magnesium iodide to analogous ketone 12 proceeded via exclusive axial attack to give 13, nitromethane gave the product of equatorial attack 14 (Scheme 3).15 As the latter outcome would set the correct configuration and install a group capable of manipulation to the alkynyl group of 4, we reacted α-6 with nitromethane under the same conditions but obtained an inseparable mixture of diastereomeric products enriched in the undesired (2R)-isomer 15 (Scheme 4). Under the same conditions, β-6 did give the desired product of equatorial attack 16 as a single stereoisomer, but we were unable to elaborate this intermediate further towards the target.
 |
| | Scheme 3 Stereochemical outcomes of nucleophilic additions to ketone 12 (Mikami & Shin).14 | |
 |
| | Scheme 4 Addition of nitromethane to α-6 and β-6. | |
The high selectivity for equatorial attack on β-6 prompted us to examine addition of alkynyl metals to these substrates. We were delighted to observe complete selectivity for axial attack of bromomagnesium acetylide upon α-6, giving 17 in 84% yield; moreover, the desired product of equatorial attack was obtained as a single diastereomer by exposing β-6 to the same reagent, giving 18 in 85% yield (Scheme 5). These results parallel those of Miljković for sodium borohydride-mediated reduction of these substrates and may be rationalised by minimisation of dipole–dipole interactions in an early transition state.16
 |
| | Scheme 5 Addition of alkynyl Grignards to α-6 and β-6. | |
The relative configuration of these compounds was tentatively assigned with the aid of NOE studies‡ and confirmed for compound 18 by single crystal X-ray diffractometry (Fig. 1). Unsurprisingly, attempted reductive cleavage of the benzylidene acetal of 18 using di-isobutyl aluminium hydride (DIBALH) resulted in hydroalumination of the alkyne, necessitating installation of protecting groups at both the alcohol and alkyne terminus. This was accomplished by treating β-6 with triisopropylsilylalkynylmagnesium chloride and benzylating the resultant 3° alcohol 19 to give 20 (Scheme 5). Treatment of 20 with DIBALH in toluene12 afforded 1° alcohol 21 with complete regioselectivity and no evidence of alkyne reduction. Base-induced elimination of iodide 22 (obtained from alcohol 21via an Appel reaction) gave enol ether 5 (Scheme 6). Gratifyingly, upon exposure to mercury(II) chloride, 5 underwent Ferrier carbocyclisation to give cyclohexanone 4 in 98% yield as a single diastereomer.§ The stereoselectivity observed is in line with that observed by Machado and rationalized by a chelated chair-like transition state.17 A wide range of oxidising agents and conditions was surveyed in an effort to convert 4 to diketone 3, but without success. We therefore considered the possibility that the correct oxidation state at this carbon could be set earlier in the synthesis, envisioning that diketone 3 might be derived directly by rearrangement of enelactone 23,18 accessible in turn from 22via selective O-demethylation and oxidation of lactol 24 (Scheme 7).
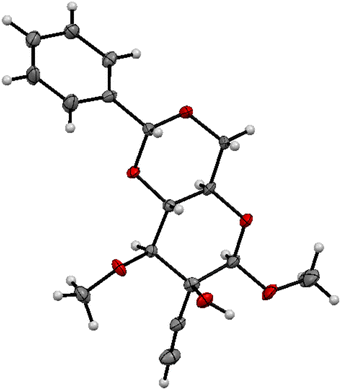 |
| | Fig. 1 ORTEP representation of compound 18 (thermal ellipsoids at 30% probability). | |
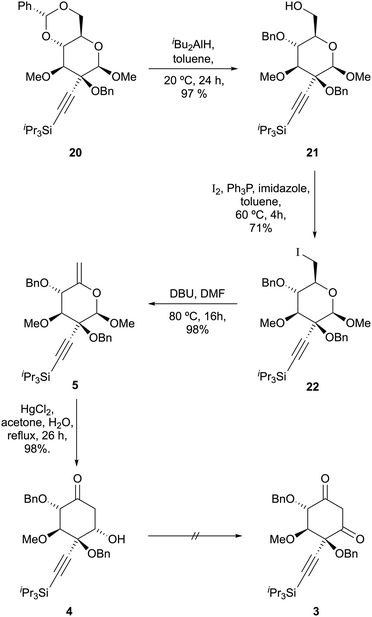 |
| | Scheme 6 Preparation of β-hydroxyketone 4 from glucopyranoside 20 via Ferrier carbocyclisation. | |
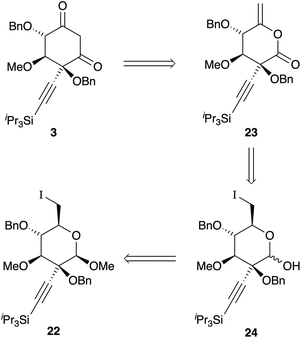 |
| | Scheme 7 Revised retrosynthesis. | |
Unfortunately, we were unable to identify conditions to selectively 1-O-demethylate either 21 or 22. A second-generation route was therefore developed, where the refractory methyl group was replaced with an allyl group. However, this apparently minor structural change resulted in a reversal in O-methylation regioselectivity (cf.Scheme 2), so a more fundamental route redesign was carried out. To circumvent reliance on the capricious stannylene acetal chemistry, 1,2,5,6-di-O-isopropylidene-α-D-glucofuranose 25 (preparable from D-glucose in a single step)19 was chosen as a starting point. Methylation of the single exposed hydroxyl group gave 26,20 which was converted to tetraacetate 27, obtained as an inconsequential 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 mixture of anomers in 63% overall yield. Selective de-acetylation of 27 at the anomeric position with hydrazine,21 followed by treatment with base and allyl bromide22 generated an 87:13 (β:α) ratio of epimeric allyl glycosides, from which the desired β-anomer 28 was isolated in 44% unoptimised yield following methanolysis of the remaining acetate groups. Benzylidene acetal formation gave 2° alcohol 29. Oxidation, alkynyl Grignard addition, and 3° alcohol protection paralleled the methyl glycoside series, giving 30; pleasingly, complete stereoselectivity for equatorial addition to the ketone was also observed (Scheme 8). Following DIBALH reduction of the benzylidene acetal, alcohol 32 was converted to iodide 33a. Palladium-catalyzed deallylation23 and hemiacetal oxidation proceeded smoothly to give lactone 34a in 62% yield; however, base-induced elimination of HI was unsuccessful. Alcohol 32 was therefore converted to selenide 33b using Grieco's protocol24 and processed along the same lines to give lactone 34b. Oxidation of the selenide25 triggered spontaneous elimination to give the desired enelactone 23, which upon treatment with sodium methoxide rearranged cleanly to diketone 3, isolated as a mixture of diketone and ketoenol isomers in 67% yield (Scheme 8).
:15 mixture of anomers in 63% overall yield. Selective de-acetylation of 27 at the anomeric position with hydrazine,21 followed by treatment with base and allyl bromide22 generated an 87:13 (β:α) ratio of epimeric allyl glycosides, from which the desired β-anomer 28 was isolated in 44% unoptimised yield following methanolysis of the remaining acetate groups. Benzylidene acetal formation gave 2° alcohol 29. Oxidation, alkynyl Grignard addition, and 3° alcohol protection paralleled the methyl glycoside series, giving 30; pleasingly, complete stereoselectivity for equatorial addition to the ketone was also observed (Scheme 8). Following DIBALH reduction of the benzylidene acetal, alcohol 32 was converted to iodide 33a. Palladium-catalyzed deallylation23 and hemiacetal oxidation proceeded smoothly to give lactone 34a in 62% yield; however, base-induced elimination of HI was unsuccessful. Alcohol 32 was therefore converted to selenide 33b using Grieco's protocol24 and processed along the same lines to give lactone 34b. Oxidation of the selenide25 triggered spontaneous elimination to give the desired enelactone 23, which upon treatment with sodium methoxide rearranged cleanly to diketone 3, isolated as a mixture of diketone and ketoenol isomers in 67% yield (Scheme 8).
 |
| | Scheme 8 Synthesis of aconitine D ring precursor 3 from D-glucose. | |
Conclusions
The synthesis of diketone 3 from 1,2,5,6-di-O-isopropylidene-α-D-glucofuranose 25 (one step from glucose) was accomplished in 14 steps and 2.5% overall yield (3.9% BRSM). The route features a highly stereoselective alkynyl Grignard addition to a ketopyranose controlled by the configuration at the anomeric centre, and an unusual enelactone to diketone rearrangement. Compound 3 represents a promising staging post in the proposed approach to aconitine, given that β-dicarbonyl moieties have been profitably employed by Toste as the nucleophilic component in gold-catalysed Conia-ene cyclisations (including a 5-endo-dig process to furnish a [3.2.1]-bridged product),11a and replacement of the triisopropylsilyl group with a more tractable iodine substituent is feasible with either alkyne (3) or alkene (2) substrates.26 Moreover, the product of such a reaction would contain carbonyl groups of different reactivity: the ketone embedded in the more strained 5-membered ring would be expected to exhibit greater reactivity towards reducing agents, rendering compound 2 (Scheme 1) potentially accessible via this strategy. Efforts in our laboratory are currently focused on applying these transformations to generate enantiopure CD building blocks as the foundation of a convergent approach to aconitine itself.
Author contributions
JD and IAP carried out all synthetic experimental work under the supervision of DMG and JBS. X-ray crystallography was carried out and analysed by CRR. DMG conceived the ideas and wrote the manuscript. DMG, JD and IAP conceived and designed the reactions. All authors edited and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the Leverhulme Trust (Research Grant RPG-2014-115) and the University of Huddersfield for support of this project.
References
-
(a) A. M. A. Qasem, Z. Zang, M. G. Rowan and I. S. Blagborough, Nat. Prod. Rep., 2022, 39, 460–473 RSC;
(b) F.-P. Wang, Q.-H. Chen and X.-Y. Liu, Nat. Prod. Rep., 2010, 27, 529–570 RSC.
-
(a) A. Ameri, Prog. Neurobiol., 1998, 56, 211–235 CrossRef CAS PubMed;
(b)
M. H. Benn and J. M. Jacyno, in Alkaloids Chemical & Biological Perspectives, ed. S. W. Pelletier, J. Wiley & Sons, Inc., New York, 1983, vol. 1, pp. 153–220 Search PubMed.
-
(a)
P. Ovidius Naso, Metamorphoses, I, 147, Penguin Classics, London, 2004 Search PubMed;
(b)
W. Shakespeare, 2 Henry IV 4.4.49–50, OUP, Oxford, 2008 Search PubMed;
(c) A. K. Chatterjee, Can. J. Health History, 2022, 39, 281–310 CrossRef.
- M. A. Turabekova, B. F. Rasulev, F. N. Dzhakhangirov, D. Leszczynska and J. Leszczynski, Eur. J. Med. Chem., 2010, 45, 3885–3894 CrossRef CAS PubMed.
-
(a) M.-Y. Ren, Q.-T. Yu, C.-Y. Shi and J.-B. Luo, Molecules, 2017, 22, 267–281 CrossRef PubMed;
(b) K. Wada and H. Yamashita, Molecules, 2019, 24, 2317–2338 CrossRef CAS PubMed.
-
(a) R. M. Conrad and J. DuBois, Org. Lett., 2007, 9, 5465–5468 CrossRef CAS PubMed;
(b) C. Dank, R. Sanichar, K.-L. Choo, M. Olsen and M. Lautens, Synthesis, 2019, 51, 3915–3946 CrossRef CAS.
-
(a) K. Wiesner, T. Y. R. Tsai, K. Huber, S. E. Bolton and R. Vlahov, J. Am. Chem. Soc., 1974, 96, 4990–4992 CrossRef CAS;
(b) Y. Shi, J. T. Wilmot, L. U. Nordstrom, D. S. Tan and D. Y. Gin, J. Am. Chem. Soc., 2013, 135, 14313–14320 CrossRef CAS PubMed;
(c) Y. Nishiyama, Y. H. S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2014, 136, 6598–6601 CrossRef CAS PubMed;
(d) C. J. Marth, G. M. Gallego, J. C. Lee, T. P. Lebold, S. Kulyk, K. G. M. Kou, J. Qin, R. Lilien and R. Sarpong, Nature, 2015, 528, 493–498 CrossRef CAS PubMed;
(e) K. G. M. Kou, B. X. Li, J. C. Lee, G. M. Gallego, T. P. Lebold, A. G. DiPasquale and R. Sarpong, J. Am. Chem. Soc., 2016, 138, 10830–10833 CrossRef CAS PubMed;
(f) D. Kamakura, H. Todoroki, D. Urabe, K. Hagiwara and M. Inoue, Angew. Chem., Int. Ed., 2020, 59, 479–486 CrossRef CAS PubMed.
- For alternative approaches to construction of the aconitine D ring, see:
(a) H. Cheng, L. Xu, D.-L. Chen, Q.-H. Chen and F.-P. Wang, Tetrahedron, 2012, 68, 1171–1176 CrossRef CAS;
(b) X.-H. Zhou, Y. Liu, R.-J. Zhou, H. Song, X.-Y. Liu and Y. Qin, Chem. Commun., 2018, 54, 12258–12261 RSC;
(c) G. Yang, M. Zhu, X. Zhao, J. Lei and L. Xu, Tetrahedron Lett., 2021, 69, 152975 CrossRef CAS.
- A. R. Wong, N. J. Fastuca, V. W. Mak, J. K. Kerkovius, S. M. Stevenson and S. A. Reisman, ACS Cent. Sci., 2021, 7, 1311–1316 CrossRef CAS PubMed.
-
(a) M. E. Jung and S. W. T. Choe, J. Org. Chem., 1995, 60, 3280–3281 CrossRef CAS;
(b) R. J. Ferrier and S. Middleton, Chem. Rev., 1993, 93, 2779–2831 CrossRef CAS.
-
(a) S. T. Staben, J. J. Kennedy-Smith and F. D. Toste, Angew. Chem., Int. Ed., 2004, 43, 5350–5352 CrossRef CAS PubMed;
(b) D. Hack, M. Blümel, P. Chauhan, A. R. Philipps and D. Enders, Chem. Soc. Rev., 2015, 44, 6059–6093 RSC.
- N. Tanaka, I. Ogawa, S. Yoshigase and J. Nokami, Carbohydr. Res., 2008, 343, 2675–2679 CrossRef CAS PubMed.
-
(a) P. V. Murphy, J. L. O'Brien, L. J. Gorey-Feret and A. B. Smith III, Tetrahedron, 2003, 59, 2259–2271 CrossRef CAS;
(b) H. Qin and T. B. Grindley, J. Carbohydr. Chem., 1996, 15, 95–108 CrossRef CAS.
- M. Srinivasarao, T. Park, Y. Chen and P. L. Fuchs, Chem. Commun., 2011, 47, 5858–5860 RSC.
- J. Yoshimura, K. Mikami, K.-I. Sato and C.-G. Shin, Bull. Chem. Soc. Jpn., 1976, 49, 1686–1689 CrossRef CAS.
- M. Miljković, M. Gligorijević and D. Miljković, J. Org. Chem., 1974, 39, 2118–2120 CrossRef.
- A. S. Machado, D. Dubreuil, J. Cleophax, S. D. Gero and N. F. Thomas, Carbohydr. Res., 1992, 233, C5–C8 CrossRef CAS.
-
(a) N. P. Shusherina, T. Kh. Gladysheva and R. Ya. Levina, Zh. Org. Khim., 1966, 2, 1801–1805 CAS;
(b) H. Schneider and C. Lüthy, Process for the preparation of cyclic diketones, Eur. Pat. Appl., 2003, 1352901 Search PubMed; H. Tomioka, K. Oshima and H. Nozaki, Tetrahedron Lett., 1982, 23, 99–100 Search PubMed.
- R. N. Mitra, K. Show, D. Barman, S. Sarkar and D. K. Maiti, J. Org. Chem., 2019, 84, 42–52 CrossRef CAS PubMed.
- S. Yan and D. Klemm, Tetrahedron, 2002, 58, 10065–11071 CrossRef CAS.
- G. Excoffier, D. Gagnare and J. P. Utille, Carbohydr. Res., 1975, 39, 368–373 CrossRef CAS.
-
(a) A. S. Henderson, J. F. Bower and M. C. Galen, Org. Biomol. Chem., 2014, 12, 9180–9183 RSC;
(b) A. Lubineau, S. Escher, J. Alais and D. Bonnaffé, Tetrahedron Lett., 1997, 38, 4087–4090 CrossRef CAS.
- J. Zeng, S. Vedachalam, S. Xiang and X.-W. Liu, Org. Lett., 2011, 13, 42–45 CrossRef CAS PubMed.
- P. A. Grieco, S. Gilman and M. Nishizawa, J. Org. Chem., 1976, 41, 1485–1486 CrossRef CAS.
- H. Yamada, M. Adachi and T. Nishikawa, Chem. Commun., 2013, 49, 11221–11223 RSC.
- D. L. Usanov, M. Naodovic, M. Brasholz and H. Yamamoto, Helv. Chim. Acta, 2012, 95, 1773–1789 CrossRef CAS; N. Kerisit, R. Ligny, E. S. Gauthier, J.-P. Guégan, L. Toupet, J.-C. Guillemin and Y. Trolez, Helv. Chim. Acta, 2019, 102, e1800232 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation data for novel compounds, X-ray crystallographic data and NMR spectra. CCDC 2343856. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00561a |
| ‡ The NOESY spectrum for compound 17 displayed a cross-peak corresponding to an interaction between 2-OH and axial 3-H, whereas for compound 18, a cross-peak for 2-OH and axial 4-H was observed. For compound 16, a cross-peak for 2-OH and axial 4-H was also observed, along with one between axial 3-H and one CH2NO2 proton. NOESY spectra of compound 20 and other 2-OBn compounds evidenced interaction between the CH2Ph protons and the –SiiPr3 group but not 4-H, suggesting that these compounds adopt a conformation where the bulky benzyl group is oriented away from the tetrahydropyran ring. |
| § The 1H NMR coupling constants for 2-H [δH 4.31 (d, J = 12.0 Hz)] and 3-H [δH 3.85 (d, J = 12.0 Hz)] of compound 4 suggest that both these protons are axially disposed, whereas that of 5-H [δH 4.10 (t, J = 2.8 Hz)] represents coupling to 6-Heq [δH 2.49 (dd, J = 14.5, 2.8 Hz)] and 6-Hax [δH 2.85 (dd, J = 14.5, 2.8 Hz)] with a J value typical of an equatorial proton. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Julien
Doulcet
ab,
Craig R.
Rice
a,
Julien
Doulcet
ab,
Craig R.
Rice