
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu nanoparticle-catalyzed double hydrosilylation of nitriles by diethylsilane†
Maria Ioanna
Karapanou
,
Dimitra
Malliotaki
and
Manolis
Stratakis
 *
*
Department of Chemistry, University of Crete, Voutes, 71003, Heraklion, Greece. E-mail: stratakis@uoc.gr
First published on 6th June 2024
Abstract
We present the first example of Au-catalyzed reduction of nitriles into primary amines. In contrast to monohydrosilanes which are completely unreactive, diethylsilane (a dihydrosilane) is capable of reducing aryl or alkyl nitriles into primary amines under catalysis by Au nanoparticles supported on TiO2, via a smooth double hydrosilylation pathway. The produced labile N-disilylamines are readily deprotected by HCl in Et2O to form the hydrochloric salts of the corresponding amines in very good to excellent yields. The catalyst is recyclable and reusable at least in 5 consecutive runs.
Introduction
The reduction of nitriles to amines1 can be been achieved employing pyrophoric metal hydrides such as borohydrides and aluminum hydrides,2 with such processes often suffering from serious safety issues,3 or by the direct metal-catalyzed hydrogenation.4 Apart from these traditional reducing conditions, several catalytic protocols were developed employing hydroboranes5 or hydrosilanes. Particularly, the reaction between hydrosilanes and nitriles has gained significant attention, with four distinct pathways being observed depending on the catalytic conditions (Scheme 1). In the less common, hydrodecyanation6 or complete reduction of the cyano functionality into a methyl group may occur.7 The most common pathways are monohydrosilylation leading to N-silylated imines,8 or depending on the catalyst, an additional hydrosilylation may occur leading to N-disilylamines. N-Silylamines9 can be readily deprotected to primary amines. The double hydrosilylation pathway has been so far reported under catalysis by oxo-rhenium complexes,10 a Fe(III) complex,11 by Cp*Ir(III)-based iridacycles,12 by Co(OPiv)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13a or Co2(CO)8,13b by tetrabutylammonium fluoride (TBAF),14 by Fe3(CO)12/InCl3 using a significant excess of nitrile,15 by Mn(III) complexes,16 by Ti(OR)4,17 and by t-BuOK applicable only to alkoxysilanes.18 Mono versus double hydrosilylation can also be controlled by the appropriate use of the amount of hydrosilane using B(C6F5)3,19a [(C6F5)3PF][B(C6F5)4],19b or a Ru complex19c as catalysts. So far there is no reported example in the literature employing Au catalysts for the reduction of nitriles. Given that hydrosilanes and ammonia borane complex (NH3BH3) can be readily activated by supported Au(0) nanoparticles and other nano Au-based materials (Au NPs),20 we attempted to study their ability to reduce nitriles in the presence of Au/TiO2, a readily available and widely used catalyst in our lab. So far, hydrosilanes have been used under Au(0)-catalysis conditions as reducing agents of aldehydes/ketones,21 imines,22 amides,23 nitro compounds,24 quinolines,25 and diazo compounds,26 while ammonia borane for nitro compounds,27 α-diazocarbonyl compounds,28 and more recently to azoarenes.29
13a or Co2(CO)8,13b by tetrabutylammonium fluoride (TBAF),14 by Fe3(CO)12/InCl3 using a significant excess of nitrile,15 by Mn(III) complexes,16 by Ti(OR)4,17 and by t-BuOK applicable only to alkoxysilanes.18 Mono versus double hydrosilylation can also be controlled by the appropriate use of the amount of hydrosilane using B(C6F5)3,19a [(C6F5)3PF][B(C6F5)4],19b or a Ru complex19c as catalysts. So far there is no reported example in the literature employing Au catalysts for the reduction of nitriles. Given that hydrosilanes and ammonia borane complex (NH3BH3) can be readily activated by supported Au(0) nanoparticles and other nano Au-based materials (Au NPs),20 we attempted to study their ability to reduce nitriles in the presence of Au/TiO2, a readily available and widely used catalyst in our lab. So far, hydrosilanes have been used under Au(0)-catalysis conditions as reducing agents of aldehydes/ketones,21 imines,22 amides,23 nitro compounds,24 quinolines,25 and diazo compounds,26 while ammonia borane for nitro compounds,27 α-diazocarbonyl compounds,28 and more recently to azoarenes.29
 | ||
| Scheme 1 Known reactivity pathways in the catalyzed reaction between hydrosilanes and nitriles. | ||
Results and discussion
Our exploration commenced with aryl-substituted benzonitrile (1a). To our disappointment, although NH3BH3 is capable of achieving reduction of nitriles under certain metal-catalyzed conditions,30 it failed to provide any reaction in the presence of Au/TiO2 (entry 10, Table 1). Additionally, common monohydrosilanes, such as Et3SiH or PhMe2SiH, did not react with 1a even under prolonged refluxing conditions in the presence of Au/TiO2 (entries 1 and 2, Table 1). Similar non-product forming results were observed for benzyl-substituted 2-(p-tolyl)acetonitrile (1o). Then we turned to diethylsilane (Et2SiH2). This specific dihydrosilane has shown in the recent past a surprising reactivity and unprecedented reaction pathways if activated by Au(0) nanoparticles (Scheme 2). For example, in contrast to monohydrosilanes which in the presence of Au/TiO2 add to alkynes or allenes (hydrosilylation pathway), Et2SiH2 provides unique dehydrogenative 1,2-disilylation pathways.31,32 In addition, aldehydes/ketones or imines33 are rapidly reduced by diethylsilane, whereas monohydrosilanes perform the reduction at orders of magnitude lower rates. | ||
| Scheme 2 The unique reactivity of Et2SiH2 with several functionalities under Au/TiO2 catalysis conditions. | ||
|
|
|||||
|---|---|---|---|---|---|
| Reductant | Catalyst | Solvent | Time | Conversion | |
| a 70 °C. b Identical results were obtained in toluene instead of benzene. c Benzylamine. | |||||
| 1 | Et3SiH | Au/TiO2 | Benzene | 5 ha | — |
| 2 | PhMe2SiH | Au/TiO2 | Benzene | 5 ha | — |
| 3 | Et2SiH2 | — | Benzene | 5 h | — |
| 4 | Et2SiH2 | Au/TiO2 | Benzeneb | 0.5 h | >99% |
| 5 | Et2SiH2 | Au/TiO2 | DCE | 0.5 h | 85% |
| 6 | Et2SiH2 | Au/TiO2 | THF | 0.5 h | 12% |
| 7 | Et2SiH2 | Au/Al2O3 | Benzene | 0.5 h | 8% |
| 8 | Et2SiH2 | Au/ZnO | Benzene | 0.5 h | 28% |
| 9 | (HMe2Si)2O | Au/TiO2 | Benzene | 2 ha | 12%c |
| 10 | NH3BH3 | Au/TiO2 | MeOH | 5 ha | — |
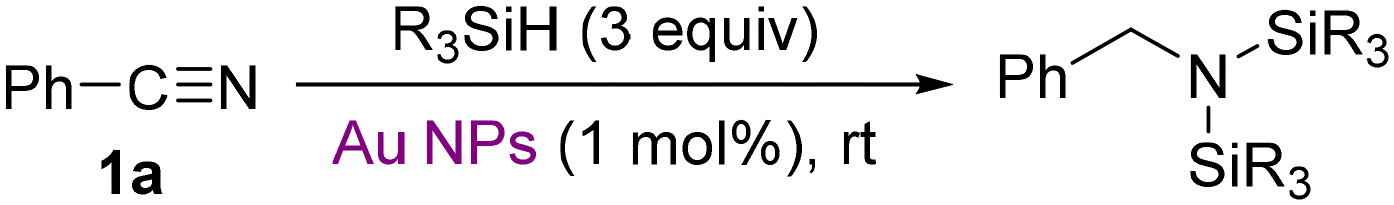
To our delight, in the presence of 1 mol% Au/TiO2, 1a underwent at room temperature double hydrosilylation with 3 molar equivalents of Et2SiH2, forming 2a within 30 min (entry 4, Table 1). Suitable solvents are primarily benzene and toluene or 1,2-dichloroethane (DCE), and they must by as dry as possible. The only side-products in the crude reaction mixture (see ESI†) were oligosiloxanes from the partial dehydrogenative hydrolysis of the excess of diethylsilane by traces of moisture. Di-adduct 2a is unstable and cannot be purified by column chromatography. Yet, after evaporation of the supernatant solvent from the crude reaction slurry we managed to characterize it properly (see ESI†). Upon treatment of 2a with 1 M HCl/Et2O, the hydrochloric salt of benzylamine (3a) was isolated as a solid in 87% yield. Labile disilylamine 2a has been previously reported by Chang and co-workers19a as an intermediate in their B(C6F5)3-catalyzed silylative reduction of nitriles, but no spectroscopic data were provided. Au/Al2O3 or Au/ZnO are also active, but less efficient catalysts. Finally, 1,1,3,3-tetramethyldisiloxane (TMDS), a highly reactive reducing agent of carbonyl compounds in the presence of Au/TiO221c resulted to merely 12% product after 2 h at 70 °C.
The smooth double hydrosilylation of benzonitrile by diethysilane urged us to explore the scope and limitations of the Au/TiO2-catalyzed reaction. The results are collectively presented in Fig. 1, and were performed on an approximately 0.15–0.20 mmol scale of nitriles. Aryl, heteroaryl or alkyl nitriles react smoothly within 30 min to 2 h affording the corresponding amines as hydrochloric acid salts in isolated yields >75%. Labile disilylamines from double hydrosilylation are formed in all cases and can be seen by in situ NMR, however, following a generalized protocol, after the reaction was complete (TLC or GC), the slurry was immediately filtered with the aid of DCM through a short pad of Celite, the solvents were evaporated, and the residue was treated with 1 M HCl in Et2O. The hydrochloric salts of the amines precipitate and can be isolated via filtration. The catalyst is recyclable and reusable in five consecutive runs at 0.2 mmol scale without any obvious deterioration of its activity. After each run, the catalyst was recovered by supernatant solution decantation, washed, dried in the oven at 100 °C for 1 h, and then reused.
 | ||
| Fig. 1 Reduction of nitriles with Et2SiH2 catalyzed by Au/TiO2. | ||
In the case of co-existence of a second formally reducible group in reacting nitriles, the following selectivity modes were observed (Scheme 3). In nitro-substituted benzonitrile 1v, the nitro functionality was selectively reduced relative to the cyano. There are several reports in the literature concerning the competing chemoselectivity of such reductions. Generally, treatment of cyano and nitro bearing substrates with specific boron hydrides,34 and primarily in the metal-catalyzed reduction with hydrosilanes,8a,17,19a the cyano group is selectively reduced. The opposite selectivity, such as under our conditions, occurs performing catalytic hydrogenation,4 or employing hydrosilanes with Au/Fe3O424 or Cu(OTf)235 as catalysts. The reduction of ester-bearing benzonitrile 1w provides exclusively product 3w in which only the cyano was reduced. In contrast, metal hydrides react unselectively with 1w, reducing both functional groups,36 although under several modifications only the ester can be reduced.37 Our observed chemoselectivity has precedents in the literature, primarily via catalytic hydrogenation4 or hydroboration,34b and is complementary to metal-catalyzed protocols employing hydrosilanes as reducing agents, in which the ester functionality is reduced over cyano.14,38 Another chemoselective reduction is that of cinnamonitrile (1x) in which the double bond remains unaffected. In reactions involving hydrosilanes as reductants, the Fe(III)-catalyzed process11 provides similar selectivity to our protocol, while the B(C6F5)3-catalyzed reaction of dihydrosilanes with conjugated nitriles affords Michael-type addition of silyl group on the double bond.39 Apart from these selective reductions, we observed some limitations, with indole-carbonitrile 1y and 4-cyanopyridine (1z) being completely unreactive. We postulate that the strong coordination of their sp2-N on Au NPs disfavors reduction of the cyano functionality.
 | ||
| Scheme 3 Chemoselectivity issues and limitations in the reduction of functionalized nitriles. | ||
In Scheme 4, we propose a simplified mechanism of the transformation catalyzed by Au NPs (denoted as Aun). The first hydrosilylation provides a N-silyl imine followed by a second addition. Since imines are rapidly reduced by Et2SiH2 in the presence of Au/TiO2,33 we reasonably postulate that the second hydrosilylation occurs much faster than the first one. In fact, no silyl imines were detected in the crude reaction mixture, interrupting the progress of the reaction at any stage.
 | ||
| Scheme 4 Proposed mechanism of the double hydrosilylation of nitriles catalyzed by Au nanoparticles (Aun). | ||
Conclusions
In conclusion, we report the first example of reduction of nitriles to amines under Au-catalysis conditions. The process occurs at ambient conditions via double hydrosilylation of the cyano functionality in the presence of recyclable and reusable Au nanoparticles supported on TiO2 as catalyst. The only hydrosilane that is capable of reacting is diethylsilane, which is proven once more a unique reagent under Au NP catalysis conditions.31–33 The initially formed labile disilylamines undergo deprotection with etherated HCl and the amines are isolated as their hydrochloric salts in very good to excellent yields. These results exemplify once more the unique potency of supported Au nanoparticles in catalyzing organic transformations of high synthetic utility, but also the superior activity of a dihydrosilane (Et2SiH2) as a reducing agent,40 relative to simple monohydrosilanes.Experimental section
The reactions were monitored by thin-layer chromatography (TLC) carried out on silica gel plates (60F-254). Benzene was passed though silica gel and kept over 4 Å molecular sieves. NMR spectra were recorded on a Bruker Avance-500 instrument. HRMS were recorded with a Q-Exactive Plus Orbitrap MS from Thermo Scientific, with a direct infusion of the corresponding product. GC-MS analyses were performed on a Shimadzu GC-MS 2030 model equipped with a 30 m MEGA-5 HT capillary column. Flash column chromatography for the purification of compounds was carried out on SiO2 (silica gel 60, particle size 0.040–0.063 mm). The Au catalyst is commercially available and has a gold content of 1% w/w. Its nanoparticles have an average crystallite size of 2–3 nm. The catalyst was ground in a mortar to become a fine dust and was kept in the dark. Diethylsilane was obtained from commercial suppliers. Apart from 1a, 1i–1l, 1o–1q and 1z which were obtained from commercial suppliers, all aryl or heteroaryl nitriles were prepared from the corresponding aldehydes via the triflic acid-mediated Schmidt reaction,41 while the alkyl ones via treatment of aldehydes with hydroxylamine hydrochloride in DMSO.42 The spectroscopic data of synthesized nitriles (see ESI†) are in agreement with those of the commercially available substances. The only nitrile that is a new compound in the literature is 1n.4-Bromofuran-2-carbonitrile (1n)
White solid (45 mg, 35% yield). 1H NMR (500 MHz, CDCl3): 7.59 (d, J = 0.5 Hz, 1H), 7.12 (d, J = 0.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): 145.6, 127.3, 124.3, 110.2, 100.7. HRMS (ESI-Orbitrap) m/z: [M + H]+ calcd for C5H2BrNO + H, 171.9393; found 171.9391.General procedure of the Au/TiO2-catalyzed double hydrosilylation of nitriles
To a properly dried vial containing a solution of 0.15 mmol of nitrile in dry benzene (0.5 mL) are added 0.45 mmol of Et2SiH2 followed by the addition of 29 mg of Au/TiO2 (1.0 mol% in Au) at room temperature for a certain period of time (30 min to 2 h depending on the substrate), until the reaction was complete (TLC or GC-MS). The slurry is filtered with the aid of dichloromethane under a low pressure through a short pad of silica gel and the filtrate is evaporated. The residue was treated at 25 °C with a 1 M solution of HCl in Et2O (1 mL) for 1 h. The hydrochloric salts of the amines precipitate and are isolated in pure form as white solids via filtration. Apart from the 0.15–0.20 mmol scale in which the reduction experiments of Fig. 1 were performed, the double hydrosilylation was also achieved on a larger scale using 103 mg (1.0 mmol) of parent benzonitrile (1a), 390 μL of diethylsilane (3.0 mmol), 3 mL of benzene and 197 mg of Au/TiO2 (2.0 mg of Au, as the catalyst contains 1 w/w% Au). After 30 min at 25 °C the reaction was complete. The crude filtrate was evaporated at reduced pressure and the residue was treated with 6 mL of a 1 M solution of HCl in Et2O. The precipitated hydrochloric salt 3a was isolated via filtration and was dried in the oven for 12 h at 100 °C (130 mg, 91% yield).Spectroscopic data of products
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the HRMS facility of the Department of Chemistry, University of Crete, for obtaining the HRMS spectra of the unknown compounds.Notes and references
- (a) R. C. Larock and A. A. Pletnev, Formation of amines from nitriles, in Comprehensive organic transformations, ed. R. C. Larock, 2018. DOI:10.1002/9781118662083.cot05-012; (b) S. Das, J. Maity and T. K. Panda, Chem. Rec., 2022, 22, e202200192 CrossRef CAS PubMed; (c) J. Wua and C. Darcela, Adv. Synth. Catal., 2023, 365, 948 CrossRef.
- Selected examples: (a) R. F. Nystrom, J. Am. Chem. Soc., 1955, 77, 2544 CrossRef CAS; (b) R. Kumar, R. K. Meher, J. Sharma, A. Sau and T. K. Panda, Org. Lett., 2023, 25, 7923 CrossRef CAS PubMed; (c) M. Ding, J. Chang, J.-X. Mao, J. Zhang and X. Chen, J. Org. Chem., 2022, 87, 16230 CrossRef CAS PubMed; (d) J. Peng, Y. Song, Y. Wang, Z. Liu and X. Chen, Org. Chem. Front., 2022, 9, 1536 RSC.
- I. V. Taydakov, ACS Chem. Health Saf., 2020, 27, 235 CrossRef CAS.
- Recent selected examples: (a) K. Tokmic, B. J. Jackson, A. Salazar, T. J. Woods and A. R. Fout, J. Am. Chem. Soc., 2017, 139, 13554 CrossRef CAS PubMed; (b) D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883 CrossRef CAS; (c) Q. Lu, J. Liu and L. Ma, J. Catal., 2021, 404, 475 CrossRef CAS; (d) S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809 CrossRef CAS PubMed; (e) Z. Liu, F. Huang, M. Peng, Y. Chen, X. Cai, L. Wang, Z. Hu, X. Wen, N. Wang, D. Xiao, H. Jiang, H. Sun, H. Liu and D. Ma, Nat. Commun., 2021, 12, 6194 CrossRef PubMed.
- Recent selected examples: (a) N. Gautam, R. Logdi, P. Sreejyothi, N. M. Rajendran, A. K. Tiwari and S. K. Mandal, Chem. Commun., 2022, 58, 3047 RSC; (b) S. H. Doan and T. V. Nguyen, Green Chem., 2022, 24, 7382 RSC; (c) G. S. Kumar, R. Kumar, A. Sau, V. Chandrasekhar and T. K. Panda, J. Org. Chem., 2023, 88, 12613 CrossRef CAS PubMed; (d) M. Afandiyeva, X. Wu, W. W. Brennessel, A. A. Kadam and C. R. Kennedy, Chem. Commun., 2023, 59, 13450 RSC; (e) S. H. Doan, B. K. Mai and T. V. Nguyen, Org. Lett., 2023, 25, 8981 CrossRef CAS PubMed.
- (a) Fe-catalyzed: H. Nakazawa, K. Kamata and M. Itazaki, Chem. Commun., 2005, 4004 RSC; (a) Rh-catalyzed: M. Tobisu, R. Nakamura, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 3174 Search PubMed; (a) Ni-catalyzed: T. Patra, S. Agasti, Akanksha and D. Maiti, Chem. Commun., 2013, 49, 69 Search PubMed.
- Y. Peng and M. Oestreich, Org. Lett., 2022, 24, 2940 CrossRef CAS PubMed.
- (a) R. K. Sahoo and S. Nembenna, Inorg. Chem., 2023, 62, 12213 CrossRef CAS PubMed; (b) D. V. Gutsulyak and G. I. Nikonov, Angew. Chem., Int. Ed., 2010, 49, 7553 CrossRef CAS PubMed.
- V. Verma, A. Koperniku, P. M. Edwards and L. L. Schafer, Chem. Commun., 2022, 58, 9174 RSC.
- I. Cabrita and A. C. Fernandes, Tetrahedron Lett., 2011, 67, 8183 CrossRef CAS.
- S. Das, H. S. Das, B. Singh, R. K. Haridasan, A. Das and S. K. Mandal, Inorg. Chem., 2019, 58, 11274 CrossRef CAS PubMed.
- M. Hamdaoui, C. Desrousseaux, H. Habbita and J.-P. Djukic, Organometallics, 2017, 36, 4864 CrossRef CAS.
- (a) A. Sanagawa and H. Nagashima, Org. Lett., 2019, 21, 287 CrossRef CAS PubMed; (b) T. Murai, T. Sakane and S. Kato, Tetrahedron Lett., 1985, 42, 5145 CrossRef.
- C. Bornschein, S. Werkmeister, K. Junge and M. Beller, New J. Chem., 2013, 37, 2061 RSC.
- M. Ito, M. Itazaki and H. Nakazawa, ChemCatChem, 2018, 8, 3323 CrossRef.
- N. Bandopadhyay, K. Paramanik, G. Sarkar, S. Chatterjee, S. Roy, S. J. Panda, C. S. Purohit, B. Biswas and H. S. Das, New J. Chem., 2023, 47, 9414 RSC.
- S. Laval, W. Dayoub, L. Pehlivan, E. Metay, A. Favre-Reguillon, D. Delbrayelle, G. Mignani and M. Lemaire, Tetrahedron, 2014, 70, 975 CrossRef CAS.
- J. A. Clarke, A. van der Est and G. I. Nikonov, Eur. J. Org. Chem., 2021, 4434 CrossRef CAS.
- (a) N. Gandhamsetty, J. Jeong, J. Park, S. Park and S. Chang, J. Org. Chem., 2015, 80, 7281 CrossRef CAS PubMed; (b) M. Perez, Z.-W. Qu, C. B. Caputo, V. Podgorny, L. J. Hounjet, A. Hansen, R. Dobrovetsky, S. Grimme and D. W. Stephan, Chem. – Eur. J., 2015, 21, 6491 CrossRef CAS PubMed; (c) S. Wubbolt and M. Oestreich, Synlett, 2017, 2411 Search PubMed.
- Selected review articles: (a) M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469 CrossRef CAS PubMed; (b) T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636 RSC; (c) B. S. Takale, M. Bao and Y. Yamamoto, Org. Biomol. Chem., 2014, 12, 2005 RSC; (d) T. Jin, M. Terada, M. Bao and Y. Yamamoto, ChemSusChem, 2019, 12, 2936 CrossRef CAS PubMed; (e) M. Stratakis and I. N. Lykakis, Synthesis, 2019, 2435 CAS.
- (a) A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Angew. Chem., Int. Ed., 2007, 46, 7820 CrossRef CAS PubMed; (b) B. S. Takale, S. Wang, X. Zhang, X. Feng, X. Yu, T. Jin, M. Bao and Y. Yamamoto, Chem. Commun., 2014, 50, 14401 RSC; (c) E. Vasilikogiannaki, I. Titilas, C. Gryparis, A. Louka, I. N. Lykakis and M. Stratakis, Tetrahedron, 2014, 70, 6106 CrossRef CAS.
- B. S. Takale, S. M. Tao, X. Q. Yu, X. J. Feng, T. Jin, M. Bao and Y. Yamamoto, Org. Lett., 2014, 16, 2558 CrossRef CAS PubMed.
- Y. Mikami, A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. – Eur. J., 2011, 17, 1768 CrossRef CAS PubMed.
- S. Park, I. S. Lee and J. Park, Org. Biomol. Chem., 2013, 11, 395 RSC.
- (a) M. Yan, T. Jin, Q. Chen, H. Ho, T. Fujita, L.-Y. Chen, M. Bao, M.-W. Chen, N. Asao and Y. Yamamoto, Org. Lett., 2013, 15, 1484 CrossRef CAS PubMed; (b) A. Louka, C. Gryparis and M. Stratakis, ARKIVOC, 2015, iii, 38 Search PubMed.
- M. Kidonakis and M. Stratakis, Org. Lett., 2018, 20, 4086 CrossRef CAS PubMed.
- E. Vasilikogiannaki, C. Gryparis, V. Kotzabasaki, I. N. Lykakis and M. Stratakis, Adv. Synth. Catal., 2013, 355, 907 CrossRef CAS.
- M. Kidonakis and M. Stratakis, Nanomaterials, 2021, 11, 248, DOI:10.3390/nano11010248.
- E.-M. Zantioti-Chatzouda, D. Malliotaki and M. Stratakis, Adv. Synth. Catal., 2023, 365, 2982 CrossRef CAS.
- W. Zhao, H. Li, H. Zhang, S. Yang and A. Riisager, Green Energy Environ., 2023, 8, 948 CrossRef CAS.
- I. Saridakis, M. Kidonakis and M. Stratakis, ChemCatChem, 2018, 10, 980 CrossRef CAS.
- M. Kidonakis, V. Kotzabasaki, E. Vasilikogiannaki and M. Stratakis, Chem. – Eur. J., 2019, 25, 9170 CrossRef CAS PubMed.
- A. Louka, M. Kidonakis, I. Saridakis, E.-M. Zantioti-Chatzouda and M. Stratakis, Eur. J. Org. Chem., 2020, 3508 CrossRef CAS.
- Selected examples: (a) G. Amberchan, R. A. Snelling, E. Moya, M. Landi, K. Lutz, R. Gatihi and B. Singaram, J. Org. Chem., 2021, 86, 6207 CrossRef CAS PubMed; (b) B. Sieland, A. Hoppe, A. J. Stepen and J. Paradies, Adv. Synth. Catal., 2022, 364, 3143 CrossRef CAS; (c) Ref. 2b and c.
- M. Sutter, L. Pehlivan, R. Lafon, W. Dayoub, Y. Raoul, E. Metay and M. Lemaire, Green Chem., 2013, 15, 3020 RSC.
- Selected examples: (a) M. E. Belowich, C. Valente, R. A. Smaldone, D. C. Friedman, J. Thiel, L. Cronin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 5243 CrossRef CAS PubMed; (b) Ref. 5d.
- Selected examples: (a) C. P. Prasanth, E. Joseph, A. Abhijith, D. S. Nair, I. Ibnusaud, J. Raskatov and B. Singaram, J. Org. Chem., 2018, 83, 1431 CrossRef CAS PubMed; (b) Y. Kamitori, M. Hojo, R. Masuda, T. Inoue and T. Izumi, Tetrahedron Lett., 1983, 24, 2575 CrossRef CAS.
- S. Das, K. Moller, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 7414 CrossRef CAS PubMed.
- N. Gandhamsetty, J. Park, J. Jeong, S.-W. Park, S. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 6832 CrossRef CAS PubMed.
- While this manuscript was under reviewing process, the different reactivity of Et2SiH2 relative to a structurally similar monohydrosilane (EtMe2SiH) in their Pd-catalyzed reaction with ketones has been also reported: E. W. Roos, K. Basemann and M. R. Gagne, Organometallics, 2024, 43 DOI:10.1021/acs.organomet.4c00104.
- B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2012, 77, 5364 CrossRef CAS PubMed.
- S. T. Chill and R. C. Mebane, Synth. Commun., 2009, 39, 3601 CrossRef CAS.
- K. Murugesan, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2019, 58, 5064 CrossRef CAS PubMed.
- M. Heilmann and K. Tiefenbacher, Chem. – Eur. J., 2019, 25, 12900 CrossRef CAS PubMed.
- S. Pradhan, R. V. Sankar and C. Gunanathan, J. Org. Chem., 2022, 87, 12386 CrossRef CAS PubMed.
- A. Bach, D. Pizzirani, N. Realini, V. Vozella, D. Russo, I. Penna, L. Melzig, R. Scarpelli and D. Piomelli, J. Med. Chem., 2015, 58, 9258 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR of reactants and products (PDF). See DOI: https://doi.org/10.1039/d4ob00534a |
| This journal is © The Royal Society of Chemistry 2024 |