
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of diiodoheteroindenes and understanding their Sonogashira cross-coupling selectivity for the construction of unsymmetrical enediynes†‡
Alexander V.
Ponomarev
a,
Natalia A.
Danilkina
 a,
Julia S.
Okuneva
a,
Aleksandra A.
Vidyakina
a,
Ekaterina A.
Khmelevskaya
a,
Alexander S.
Bunev
b,
Andrey M.
Rumyantsev
c,
Anastasia I.
Govdi
a,
Thomas
Suarez
d,
Igor V.
Alabugin
*d and
Irina A.
Balova
*a
a,
Julia S.
Okuneva
a,
Aleksandra A.
Vidyakina
a,
Ekaterina A.
Khmelevskaya
a,
Alexander S.
Bunev
b,
Andrey M.
Rumyantsev
c,
Anastasia I.
Govdi
a,
Thomas
Suarez
d,
Igor V.
Alabugin
*d and
Irina A.
Balova
*a
aInstitute of Chemistry, Saint Petersburg State University (SPbU), Saint Petersburg, 199034, Russia. E-mail: i.balova@spbu.ru
bMedicinal Chemistry Center, Tolyatti State University, 445020 Tolyatti, Russia
cDepartment of Genetics and Biotechnology, Saint Petersburg State University (SPbU), Saint Petersburg, 199034, Russia
dDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306, USA. E-mail: alabugin@chem.fsu.edu
First published on 24th April 2024
Abstract
Electrophile-promoted cyclizations of functionalized alkynes offer a useful tool for constructing halogen-substituted heterocycles primed for further derivatization. Preinstallation of an iodo-substituent at the alkyne prior to iodo-cyclization opens access to ortho di-iodinated heterocyclic precursors for the preparation of unsymmetrical heterocycle-fused enediynes. This general approach was used to prepare 2,3-diiodobenzothiophene, 2,3-diiodoindole, and 2,3-diiodobenzofuran, a useful family of substrates for systematic studies of the role of heteroatoms on the regioselectivity of cross-coupling reactions. Diiodobenzothiophene showed much higher regioselectivity for Sonogashira cross-coupling at C2 than diiodoindole and diiodobenzofuran. As a result, benzothiophene can be conveniently involved in a one-pot sequential coupling with two different alkynes, yielding unsymmetrical benzothiophene-fused enediynes. On the other hand, the Sonogashira reaction of diiodoindole and diiodobenzofuran formed considerable amounts of di-substituted enediynes in addition to the monoalkyne product by coupling at C2. Interestingly, no C3-monocoupling products were observed for all of the diiodides, suggesting that the incorporation of the 1st alkyne at C2 activates the C3 position for the 2nd coupling. Additional factors affecting regioselectivity were detected, discussed and connected, through computational analysis, to transmetalation being the rate-determining step for the Sonogashira reaction. Several enediynes synthesized showed cytotoxic activity, which is not associated with DNA strand breaks typical of natural enediyne antibiotics.
Introduction
Electrophile-promoted cyclization of functionalized alkynes is a valuable tool for the synthesis of halogen-substituted heterocycles.1–7 The reaction is suitable for different nucleophilic functional groups and alkynes. The most recent examples of electrophile-promoted cyclization for “halogen” electrophiles open access to oxazinoindolones,8 3-sulfonamido-4-haloisocoumarins,9 sulfonamido-substituted polyaromatic compounds,10 medium-sized oxacycles,11 iodofuranones,12 iodothiazines,13 highly distorted fused helicenes14,15 and optically active tricyclic piperazine scaffolds through dearomative ipso-cyclization.16 Electrophile-promoted cyclization of various functionalized alkynes has been applied to construct polyheterocycles targeting nucleic acids.17Earlier, we have shown that iodocyclization of ortho-functionalized aryldiacetylenes followed by the Sonogashira reaction is a convenient approach for the construction of unsymmetrically substituted enediyne systems fused to heteroindenes with the highest yields and functional group tolerance in the case of benzothiophene and indole (Fig. 1A).18
 | ||
| Fig. 1 Known examples of the synthesis of unsymmetrical acyclic and cyclic enediyne systems through the “diacetylenic” (A), “stepwise” (B) approaches and the proposed “iodoalkyne” approach (C). | ||
This “diacetylenic approach” was used in the synthesis of 10-membered heterocyclic analogues of enediyne antibiotics.19–22 However, this method has some limitations associated with the possibility of iodination of the second triple bond in the case of benzofuran and isocoumarin derivatives.18,23,24 An alternative way to obtain unsymmetrically substituted enediynes fused to heterocycles could be the regioselective stepwise substitution of different halogens, which has been applied for pyrimidine-fused enediynes (Fig. 1B).25 Here we investigate the applicability of this approach under one-pot conditions for 2,3-diiodoheteroindenes (Fig. 1C).
2,3-Diiodoheteroindenes can be obtained in several ways (Fig. 2). The first method is direct electrophilic iodination which has been applied to indoles,26,27 benzothiophene,28 and 2-iodoindole.29 The second approach is based on the sequences of metalation/iodine exchange steps, which has been reported for diiodoindoles.30 All three compounds can also be obtained by iodinative decarboxylation techniques, either as the desired products (reported for indoles31,32 and benzothiophene33) or as by-products (reported for benzothiophene and benzofuran,34 albeit without the support of spectral data for 2,3-diiodobenzofuran). The highest yield for diiodobenzothiophene is reachable by the synthesis of a 3-iodo-2-trimethylsilyl derivative followed by the Si–iodine exchange using ICl.35,36
 | ||
| Fig. 2 Literature approaches for the synthesis of 2,3-diiodoheteroindenes. | ||
In principle, iodocyclization of ortho-functionalized iodoethynylarenes can be considered a convenient alternative synthetic approach towards diiodoheteroindenes (Fig. 1). However, to the best of our knowledge, the possibility of using electrophile-promoted cyclizations as a route to diiodoheteroindenes has not been studied, despite the fact that electrophile-promoted cyclization has been used as a synthetic tool for benzo[b]thiophenes with different halogen substituents,37 as well as dihalothieno[2,3-b]quinoline and dihaloselenopheno[2,3-b]quinoline.38
The regioselectivity of the CC cross-coupling reaction is an important problem that is being studied both theoretically and experimentally.39 It is known that the halogen atoms in 2,3-diiodo- and 2,3-dibromothiophenes as well as in 2,3-dibromoindoles have different reactivities in cross-coupling reactions.40 Because the C2 position is more reactive than the C3 position, regioselective Sonogashira reactions at the C2 position have been reported for 2,3-dibromothiophene derivatives,41–43 2,3-dibromofuran,43 2,3-diiodothiophenes,44 diiodothienopyridine,45 and 2,3-dibromobenzo[b]thiophene (Fig. 3).46 Even tetrabromothiophene can undergo the regioselective C2,C5 Sonogashira coupling.43 Not surprisingly, the regioselective C2–I Sonogashira coupling has also been shown for C2-iodo/C3-bromo derivatives: 3-bromo-2-iodobenzo[b]thiophene43 and 3-bromo-2-iodoindole,47 where the more reactive halogen was placed at an intrinsically more reactive position. In the case of C2-bromo/C3-iodo derivatives, where the above two factors are in conflict, the C2/C3 regioselectivity is reversed. Thus, C3-iodo regioselectivity has been observed for 2-bromo-3-iodothiophenes48–51 and 2-bromo-3-iodofuran and -benzofuran.52 However, the selectivity of cross-coupling in 2,3-diiodoheteroindenes (benzo[b]thiophene, indole, and benzo[b]furan) has not been previously studied. 2,3-Diiodoindoles were only used in the synthesis of symmetrical diethynylindoles.53
 | ||
| Fig. 3 Known regioselectivity for the Sonogashira reaction of dihalothiophenes, furans and heteroindenes. | ||
In this work, we report on the study of electrophile-promoted cyclization of functionalized iodoethynylarenes and the investigation of the regioselectivity of the Sonogashira reaction for 2,3-diiodoheteroindenes for the development of a new “iodoalkyne” approach towards unsymmetrically substituted enediynes fused to heteroindenes (Fig. 1).
Results and discussion
Study on the electrophile-promoted cyclization of iodoalkynes
The starting 2-(iodoethynyl) derivatives of thioanisole 3a, N,N-dimethylaniline 3b and anisole 3c were obtained from the corresponding TMS-alkynes 1a–c (see the ESI‡) by a stepwise desilylation and iodination of terminal alkynes 2a–c (Scheme 1). | ||
| Scheme 1 Synthesis of o-functionalized (iodoethynyl)arenes 3a–c. | ||
Iodoalkynes 3a–c turned out to be quite sensitive substances. They require mild reaction conditions and should be handled carefully during workup and isolation in order to avoid protodeiodination (see the ESI‡). Thus, the iodoalkynes should not be stored in solution for a prolonged time and the water bath temperature for a rotary evaporator should not exceed 35 °C. With these rules, the key iodoalkynes 3a–c could be obtained in high yields (Scheme 1) and neatly stored in a freezer for several months without any degradation.
Then, we turned our attention towards the study of iodocyclization of iodoalkynes 3a–c under reaction conditions, which gave the highest yields in the case of alkyl-/aryl-substituted ortho-functionalized alkynes.54–56 (Iodoethynyl)thioanisole 3a underwent the iodocyclization smoothly with iodine as an electrophile: 2,3-diiodobenzo[b]thiophene 4 was isolated in an almost quantitative yield (Table 1, entry 1). The cyclization of aniline 3b using iodine gave a complex mixture of products. Switching to N-iodosuccinimide gave indole 5 in 86% yield (Table 1, entry 2). The reaction of anisole 3c with either iodine (under a variety of conditions) or ICl ended with electrophilic addition to the triple bond to form dihalogenated alkenes 6 and 7, respectively (Table 1, entries 3 and 4). When NIS was used, no conversion of the starting (iodoethynyl)anisole was observed even after 6 hours (Table 1, entry 5). However, we could overcome this obstacle using 2-ethynylated phenols instead of anisoles and by switching to NIS/PPh3 as the electrophilic system as described previously by Li et al.57
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Iodoalkyne | X | R | Conditions | Yield, % | Product |
| Conditions: A – I2 (1.05 equiv.), DCM, rt; B – NIS (2 equiv.), DCM, rt; C – I2 (3.00 equiv.), DCM, 40 °C, D – I2 (3.00 equiv.), DCE, 80 °C; E – ICl (1.05 equiv.), DCM, rt. | ||||||
| 1 | 3a | S | H | A | 97 | 4 |
| 2 | 3b | NMe | COOEt | B | 86 | 5 |
| 3 | 3c | O | H | A | 76 | 6 (Y = I) |
| C | 99 | |||||
| D | 77 | |||||
| 4 | 3c | O | H | E | 82 | 7 (Y = Cl) |
| 5 | 3c | O | H | B | — | — |

Interestingly, when optimizing the synthesis of (iodoethynyl)phenol 3d, we inadvertently found that desilylation/iodination of TMS-substituted ethynylphenol 1d under AgF/NIS conditions58 gave the desired 2,3-diiodobenzofuran 8, albeit in a low yield (13%) and along with a mixture of unidentified products. Iodocyclization of (iodoethynyl)phenol 3d under NIS/PPh3 conditions gave 2,3-diiodobenzofuran 8 in 37% isolated yield (Scheme 2).
 | ||
| Scheme 2 Synthesis of 2,3-diiodobenzofuran 8. | ||
Thus, the preinstallation of iodine at the triple bond of ortho-functionalized ethynylarenes enables direct synthesis of 2,3-diiodobenzothiophene and 2,3-diiodoindole. Although iodination of 2-(iodoethynyl)anisole leads instead to the 1,2-diiodination of the triple bond, the target 2,3-diiodobenzofuran can be synthesized using 2-(iodoethynyl)phenol as a starting material and the NIS/PPh3 system as the electrophilic reagent.
Regioselectivity of the Sonogashira coupling in 2,3-diiodoheteroindenes
Next, we explored the regioselectivity of the Sonogashira coupling59,60 for diiodides 4, 5, and 8. The idea of selective substitution of one iodine atom in vicinal diiodoheteroindenes is based on the known regioselective Sonogashira reaction for 2,3-diiodothiophenes44 and 2,3-dibromobenzo[b]thiophene.46 The substitution is known to occur first at C2, and then at the C3 position.From the theoretical point of view, many factors influence the regioselectivity of the oxidative addition step, which is known to be the limiting stage of many Pd cross-coupling reactions.39 An interesting selectivity feature is that the reaction does not prefer the weaker two C–X bonds as indicated by the data shown in Fig. 4 for C–Br and C–I bonds in the two dihalides. Interestingly, the shorter and stronger C2–Hal bonds react first. Such observations are consistent with the earlier suggestion that the rate-limiting step for the Sonogashira cross-coupling of aryl iodides is likely to be not the C–X bond activation via oxidative addition to the catalyst but trans-metalation of Cu-acetylides formed in the initial reaction stage.61
 | ||
| Fig. 4 Energetic and electronic parameters for the two C–Hal bonds in 2,3-dibromo- and 2,3-diiodobenzo[b]thiophenes. | ||
Therefore, we aimed to experimentally compare the regioselectivity of the cross-coupling for all three diiodoheteroindenes 4, 5, and 8 with the goal of evaluating how general the above trend is. From a practical point of view, this information is useful for deciding which heterocycles are appropriate for the synthesis of unsymmetrical enediyne systems under one-pot conditions.
To study the regioselectivity in the Sonogashira reaction of diiodobenzothiophene 4, we chose 3-methoxypropyne 9a as a partner because its regioselective cross-coupling would lead to the corresponding C2-(methoxymethyl)ethynyl derivative, which is an important starting material for the synthesis of cyclic enediynes through the Nicholas reaction (Table 2).19
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | T, °C | Conversion of 4%b | Yield, % | ||
| 10a | 11a | 12 | ||||
| a Reaction conditions: 4 (0.25 mmol, 1 equiv.), 9a (0.30 mmol, 1.2 equiv.), Pd(PPh3)4 (5 mol%), CuI (10 mol%), base (1.00 mmol, 4.00 equiv.), THF (2.50 mL, c = 0.1 M), 24 h. b Conversion and yield were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. c DMF was used as a solvent. d Alkyne 9a was used in an amount of 0.25 mmol, 1.00 equiv. | ||||||
| 1 | K2CO3 | 45 | 94 | 76 | 11 | <1 |
| 2 | K2CO3 | 60 | 70 | 64 | 8 | — |
| 3 | K3PO4 | 45 | >95 | 80 | 9 | 1.5 |
| 4 | KF | 45 | 39 | 45 | 2 | 4 |
| 5c | KF | 45 | 55 | 55 | 2 | 4 |
| 6d | K3PO4 | 45 | 73 | 66 | 7 | 1 |
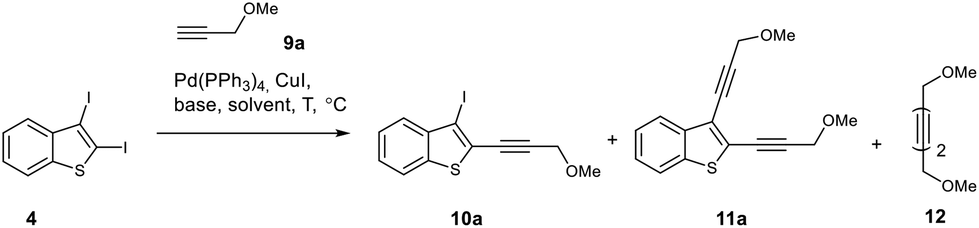
All reactions were carried out using a Pd(PPh3)4/CuI catalytic system, while the nature of the base, solvent, temperature and reagent ratio was varied. The yields of all products and the conversion of diiodide 4 were estimated using 1H NMR spectroscopy of the reaction mixture with 1,3,5-trimethoxybenzene as the internal standard. The Sonogashira coupling of diiodide 4 with alkyne 9a using potassium carbonate as a base at 45 °C gave C2-monosubstituted product 10a in 76% yield. However, a non-desired disubstituted product 11a was also formed in 11% yield (Table 2, entry 1). Raising the temperature from 45 °C to 60 °C decreased the yield of monoethynylated benzothiophene 10a (Table 2, entry 2). Among the various bases tested (Table 2, entries 3–5), the best result was achieved with K3PO4 (80% yield of 10a, entry 3), albeit with the concomitant formation of 9% of enediyne 11a. In order to diminish the formation of undesired enediyne 11a, we reduced the acetylene/diiodide ratio from 1.2 to 1.0 equiv. (Table 2, entry 6) but found that these conditions led to a decrease in the yield of product 10a. Thus, the conditions from entry 3 were chosen for the regioselective synthesis of 2-ethynyl-3-iodobenzothiophene 10a: the reaction should be carried out in THF at 45 °C using K3PO4 as a base and maintaining the starting diiodide 4 to alkyne 9a ratio of 1:1.25.
Importantly, the product of monosubstitution at C3 has never been observed. Thus, the ethynyl substituent at the C2-position of benzothiophene has an activating effect on the rate of cross-coupling at the C3 position. This activation can be associated either with a decrease in electron density at the C3 position for 2-ethynyl-3-iodobenzothiophenes compared to 2,3-diiodobenzothiophene because of the electron-withdrawing properties of Csp atoms, or/and with other factors such as coordination of palladium at the triple bond in the oxidative addition step.
Studies on the regioselectivity of indole 5 and benzofuran 8 derivatives were carried out under optimized conditions for diiodobenzothiophene 4. We found that 2,3-diiodoindole 5 cannot be converted to the corresponding C2-ethynylindoles with the same selectivity as it was observed for 2,3-diiodobenzothiophene 4 (Scheme 3). Although the desired C2-ethynylated indole 13a was the main reaction product (52% yield) of the Sonogashira reaction with alkyne 9a (Scheme 11), enediyne 14a (18%) along with the remaining starting diiodide 5 (87% conversion) was also isolated. Raising the temperature by 10 °C led to the increased formation of symmetrically substituted enediyne 14a, with starting diiodide 5 still present in the reaction mixture.
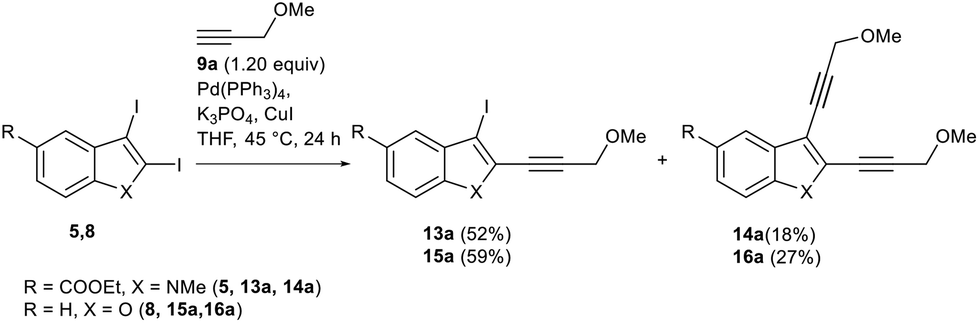 | ||
| Scheme 3 Study of the regioselectivity of the Sonogashira reaction for 2,3-diiodoindole 5 and 2,3-diiodobenzo[b]furan 8. | ||
The Sonogashira coupling of benzofuran diiodide 8 with methyl propargyl ether 9a proceeded mostly through the C2-position with similar regioselectivity to diiodindole 5 of 2/1, with the isolated yields of 59% for 2-ethynylbenzofuran 15a and 27% for enediyne 16.
Moderate yields and less selectivity for monoethynylation in the case of indole 5 and benzofuran 8 compared to benzothiophene 4 against the formation of the diethynylated byproduct can be explained by higher sensitivity of the C3–I position towards additional activation by the initially introduced C2-alkyne moiety for these heterocycles.
Finally, we decided to check the possibility of reversing the regioselectivity for the benzothiophene series from C2–I to C3–I by replacing active halogen (I) with less reactive halogen (Br) at a more activated position (C2). Thus, the corresponding C3–I regioselective Sonogashira reaction has been reported for 2-bromo-3-iodothiophenes (Fig. 3).48–51 2-Bromo-3-iodobenzo[b]thiophene 17 was obtained similarly to its diiodide analogue 4 starting from bromoethynylthioanisole S1 (see the ESI‡). The Sonogashira reaction of 2-bromo-3-iodobenzo[b]thiophene 17 with 3-methoxypropyne 9a under optimized conditions for diiodobenzothiophene 4 (Table 2, entry 3) proceeded at the C3–I bond instead of the C2–Br bond. However, the selectivity for the mono- vs. bis-substitution was significantly lower than the C2-selectivity observed for substitution to diiodide 4. Along with the iodine substitution at the C3 position (product 18a), a significant amount of the disubstituted benzothiophene 11a was formed: the ratio of alkyne 18a to enediyne 11a was found to be 2:1 (Scheme 4). It should be noted that the C2–Br substitution in the starting material was not observed, so the difference in the intrinsic reactivity of C–Br and C–I bonds was more important than the inherent C2 preference. It also indicates once again that even for the C2–Br benzothiophene derivative, a triple bond has an activation effect at the adjacent position.
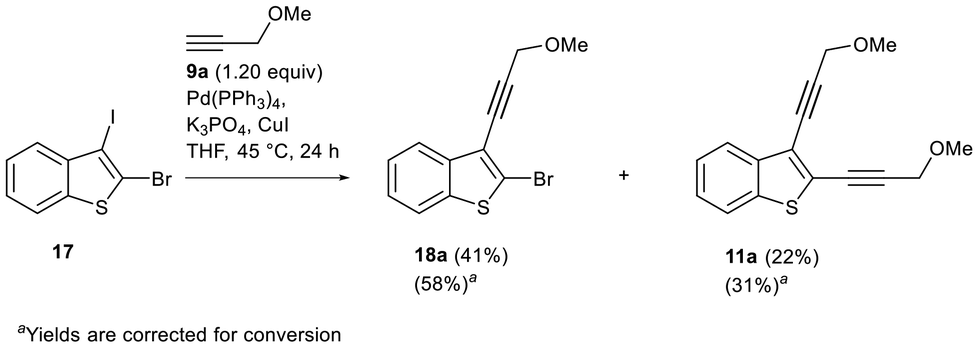 | ||
| Scheme 4 Study of the regioselectivity of the Sonogashira reaction for 2-bromo-3-iodobenzo[b]thiophenes 17. | ||
Additional selectivity studies of dihalobenzothiophenes
To explore the activating influence of the firstly introduced triple bond on the remaining C–Hal in the Sonogashira coupling, we carried out additional experiments. In particular, we studied interactions of the two dihaloheteroindenes with the worst mono- vs. bis-ethynylation selectivity (i.e., 8 and 17) with hept-1-yne 9b, an alkyne without any additional functionalities (Scheme 5).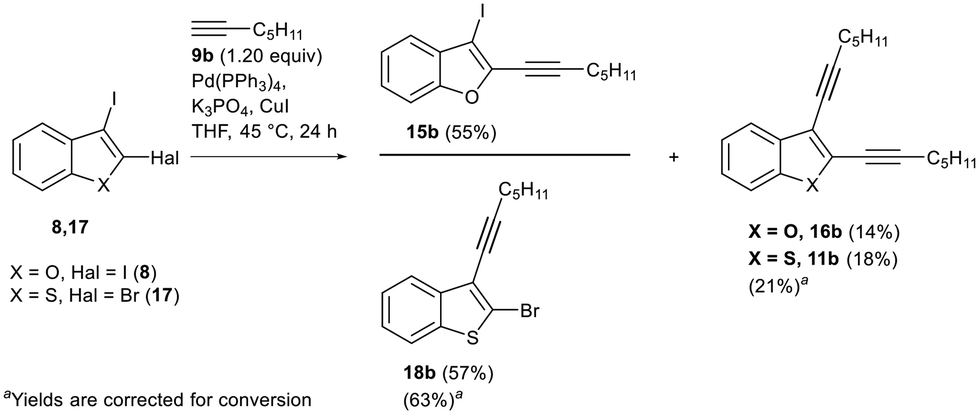 | ||
| Scheme 5 Study of the influence of the alkyne nature on mono- vs. bis-ethynylation selectivity. | ||
In the case of di-I-benzofuran 8, the mono- to bis-ethynylation ratio increased from 2/1 (for methyl propargyl ether 9a) to 4/1 (for hept-1-yne 9b).
An analogous trend was observed for C2-bromo benzothiophene 17: the mono-18b to bis-ethynylation 11b ratio also increased from 2/1 (for methyl propargyl ether 9a) to 3/1 (for heptyne 9b) (Scheme 5). The results are intriguing because the first alkyne is introduced at different carbons (C2 for 8 and C3 for 17) in these Sonogashira reactions.
Exploratory computational analysis
Several interesting observations can be made from the experimental results discussed in the previous sections. First, the introduction of the first alkyne group accelerates the 2nd ethynylation step. Second, the activation effect of the firstly incorporated triple bond is dependent on the alkyne substituent, i.e., the acceleration is less pronounced for heptyne 9b in comparison to that of methyl propargyl ether 9a.In order to understand the electronic consequences of alkyne introduction, we used DFT calculations to evaluate charge distribution in 2,3-diiodobenzothiophene 4 and the two products of Sonogashira mono-substitution at C2 – 2-ethynyl-3-iodobenzothiophens 10a and 10b′ (Scheme 6). At this stage, we limited our computational analysis only to the benzothiophene systems where the experimental data are the most complete. Note that the C2-carbon in the starting diiodide 4 has much more electron density than C3 (natural charges of −0.48 and −0.30e). Upon the introduction of C2-alkyne substituents, the electron density at both C2 and C3 is significantly lowered. This interesting finding illustrates the uniqueness of sp-hybridized carbon and is consistent with the high acceptor ability of alkynes.62,63
 | ||
Scheme 6 The activating effect of the alkyne moiety and πC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C → σ*CO hyperconjugation on the C3 carbon in benzothiophenes. C → σ*CO hyperconjugation on the C3 carbon in benzothiophenes. | ||
The introduction of an oxygen atom at the propargylic position further decreases electron density at the C3 atom. This effect stems from a combination of the inductive effect and πCC → σ*CO hyperconjugation, both of which accentuate the π accepting ability of the sp carbon. If this remote stereoelectronic effect can facilitate a reaction at the remaining C–Hal bond, it can explain the lower mono- vs. bis-ethynylation selectivity for the propargylic ether.
As transmetallation is known to be the rate-limiting step in Sonogashira reactions of aryl iodides,64,65 we evaluated substituent effects on this step with the hope that they will help to explain the observed reactivity trends. Scheme 7 illustrates the differences in the calculated exergonicities for transmetalation. In this context, it is important to remember that, as long as the same elementary processes are compared, exergonicities are known to correlate with activation barriers. Such a connection between reaction thermodynamics and kinetics readily follows from the Marcus theory or the Bell–Evans–Polanyi postulate.66,67 For more recent examples of such connections between kinetics and thermodynamics, see ref. 68 and 69.
 | ||
| Scheme 7 Thermodynamics of transmetallations in relevant benzothiophenes. | ||
The results of these calculations provided three conclusions, all in good agreement with the experimental results. In particular, a comparison of eqn (1) and (2) explains the preference for the reaction at C2 as this process is more favorable. The more exergonic transmetalation at C2 (−19.4 kcal mol−1) is expected to be faster than the less exergonic reaction at C3 (−17.8 kcal mol−1). On the other hand, a comparison of eqn (2) and (3) rationalizes the accelerating effect of the alkyne moiety which complicates reaction designs that rely on the selective formation of mono-alkynylated Sonogashira products from the diiodides. Finally, a comparison of eqn (3) and (4) shows how the propargylic acceptor group at C2 further promotes transmetallation at C3, rendering selective monoalkynylation even more difficult.
Preparation of non-symmetric enediynes
Next, we applied the optimized conditions to the Sonogashira cross-coupling of diiodide 4 with different alkynes 9a–f (Scheme 8). In all cases, the reaction proceeded with the full conversion of diiodide 4 and gave 2-ethynyl-3-iodobenzothiophenes 10a–e in good isolated yields. | ||
| Scheme 8 Synthesis of 2-ethynylbenzo[b]thiophenes 10a–f. | ||
The enediyne by-products were formed in insignificant amounts; therefore, they were not isolated chromatographically. Consequently, 2,3-diiodobenzothiophene shows minimal dependence of regioselectivity on the nature of the alkyne.
In order to demonstrate the utility of the regioselective Sonogashira reactions, we carried out a two-step synthesis of unsymmetrically substituted enediyne systems from 2,3-diiodobenzothiophene 4via two successive cross-coupling reactions with different alkynes under one-pot conditions, i.e., without isolating the product of the first Sonogashira reaction on the C2 atom. It turned out that this approach allowed quick and efficient preparation of the unsymmetrically substituted enediynes 19a–e. In this sequence, the first substitution at C2 was carried out under the optimized conditions (Table 1, entry 3), while the subsequent C3-substitution of iodine was accomplished by adding the second alkyne to the reaction mixture and running the reaction at 60 °C until the complete conversion. In this way, it was possible to obtain a series of unsymmetrically substituted enediynes, including isomeric enediynes 19a/19b and 19c/19d (Scheme 9). Because the resulting enediynes have substituents with different polarity at the triple bonds, chromatographic separation of the target unsymmetrical enediynes 19 from symmetrically substituted by-products 11a and b is straightforward. The isolation of the trace amounts of symmetrical enediyne minor by-products in the syntheses of compounds 19c–e was not carried out.
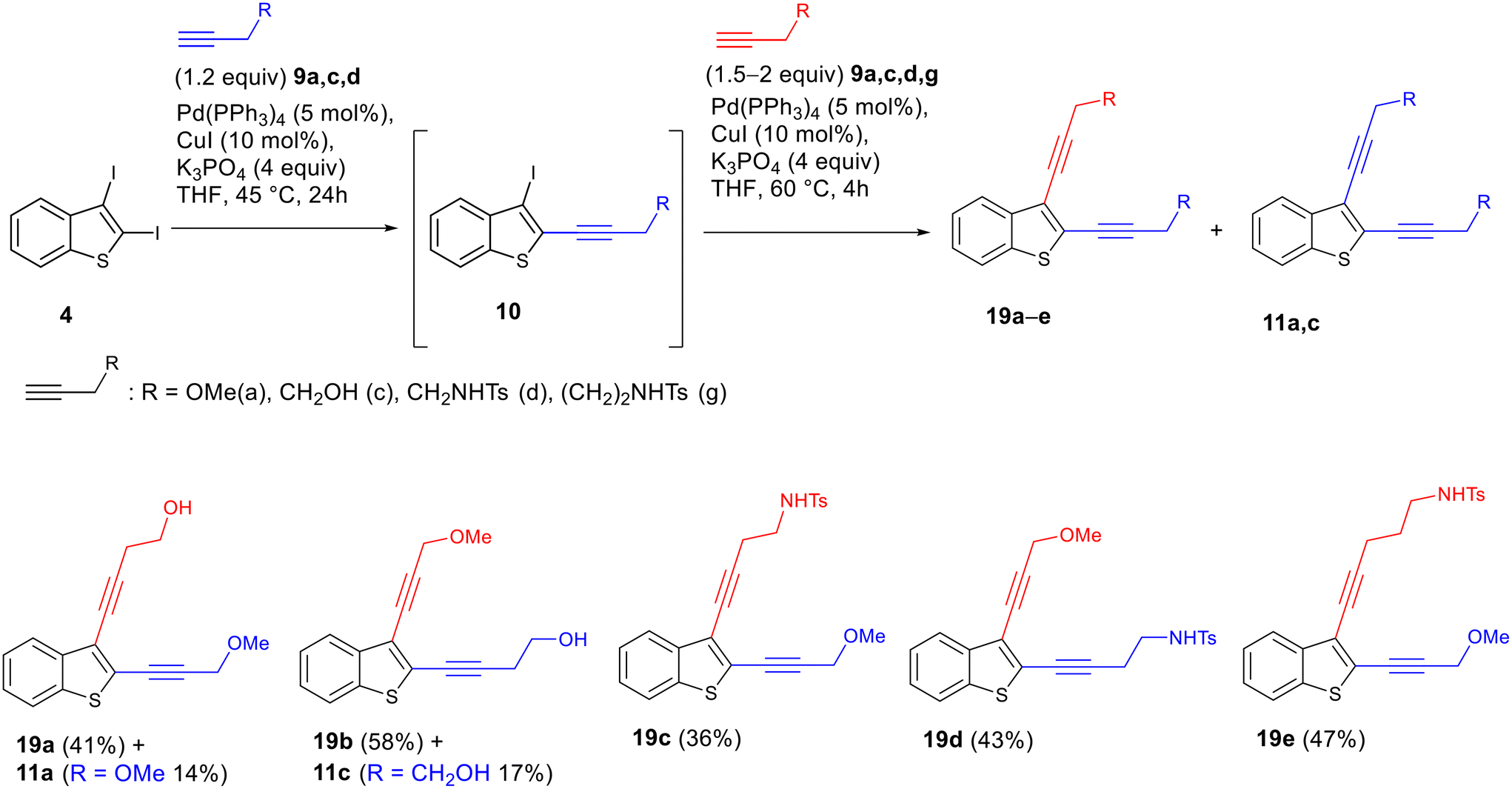 | ||
| Scheme 9 Synthesis of unsymmetrically substituted enediynes 19 using a one-pot approach. | ||
Thus, unsymmetrically substituted enediynes fused to benzothiophene 19 can be synthesized from the easily synthetically accessible 2,3-diiodobenzothiophene 4 within a single synthetic stage using a one-pot procedure involving two subsequent Sonogashira reactions. The advantage of the developed synthetic route compared to the previously described approach based on the iodocyclization of o-(buta-1,3-diynyl)thioanisole followed by the Pd-catalyzed cross-coupling with alkynes is its higher divergence without the need to start from different diacetylenes in the first synthetic steps.
We have compared the overall yield of enediyne 19a synthesized by two different routes – through the “diacetylenic” approach19,24 and via the “iodoalkyne” approach (Scheme 10). The new method gives a somewhat higher overall yield; however, it still requires separating traces of symmetrical enediynes in the last step. Therefore, we would recommend using the developed “iodoalkyne” approach for the synthesis of libraries of unsymmetrical benzothiophene-fused enediynes, while the “diacetylenic” route seems to be more suitable for the gram-scale synthesis of enediynes with an identically substituted triple bond at the C2 position.22
 | ||
| Scheme 10 Comparison of the efficiency of the “diacetylenic” and the “iodoalkyne” routes towards acyclic enediynes fused to benzothiophene. The two routes are defined in Scheme 1. | ||
Preparation of symmetric enediynes
The symmetrically substituted enediynes can be synthesized directly from diiodides 4, 5, and 8 by the Sonogashira coupling at 60 °C. Following this way, several enediynes fused to benzothiophenes 11a–d, indole 14a, and benzofuran 16a were synthesized mostly in good yields (Scheme 11). | ||
| Scheme 11 Synthesis of symmetrically substituted enediynes fused to benzothiophenes 11a–d, indole 14a and benzofuran 16a. | ||
Biological activity of acyclic enediynes
It has recently been shown that several acyclic C6F5S(O)-substituted enediynes exhibit cytotoxic activity unrelated to the Bergman cyclization. It is assumed that cytotoxicity is associated with the disruption of the microtubule network.70 Although the presence of a pentafluoroaryl moiety was thought to be responsible for the binding of enediynes to tubulin through nucleophilic aromatic substitution, an enediyne molecule lacking this group was also active against human pancreatic tumor cells MIAPaCa-2 (IC50 = 15.9 μM). Furthermore, we have independently demonstrated that dialkylaminomethyl-substituted benzothiophene-fused acyclic enediynes show cytotoxic activity against NCI-H460 lung carcinoma.71Therefore, we decided to test cytotoxicity for a series of acyclic enediynes fused to benzothiophene and indole. The list of compounds included enediynes 11a–d and 19a–e obtained in this work, as well as enediynes 19f–h,2219i and j (see the ESI‡), and 2024 synthesized in our group earlier using the “diacetylenic” route. The cytotoxicity of these compounds was tested against NCI-H460 lung carcinoma and WI-26 VA4 lung epithelial-like cell lines using the MTT colorimetric test.72,73 All the enediynes were studied at a concentration of 75 μM. Most of the unsymmetrically substituted enediynes fused to benzothiophene 19 and an indole derivative 20 showed cytotoxic activity against NCI-H460 lung carcinoma cells. Furthermore, enediynes 19b, c, g, and h, and 20 were more cytotoxic towards the NCI-H460 cells than to WI-26 VA4 lung epithelial-like fibroblasts (Fig. 5).
 | ||
| Fig. 5 Screening for the cytotoxicity of the enediynes fused to benzothiophenes 11 and 19 and indole 20. | ||
Earlier, the Hu group showed that acyclic enediynes having a double bond as a part of a maleimide ring and with a σ-acceptor atom at the propargylic position are able to undergo MARACA transformation (a cascade acetylene–allene isomerization) under polar conditions. This reaction cascade leads to enyne allenes, prone to the formation of reactive σ,π-diradicals through the Myers–Saito cyclization that induce DNA breaks.74,75 Taking into account that most of the enediynes tested here have a propargylic σ-acceptor, we decided to check the ability of the acyclic enediynes to induce DNA strand breaks. Enediynes 19a–c, f, i, and j, and 20 were chosen and investigated in the assay with a pBR322 DNA plasmid under the same conditions as those for cyclic analogs of benzothiophene-fused enediynes.20 The obtained data revealed that only enediyne 19i possessed a weak DNA damaging activity: a trace amount of open circular DNA was detected in the electropherogram (Fig. 6). Other enediynes did not show DNA damaging activity. This finding confirms that the cytotoxicity of acyclic enediynes studied here is associated neither with the Bergman cyclization nor with the Myers–Saito cyclization. It may be worth noting that enediynes can also form polar intermediates that can also damage biomolecules.76 Therefore, revealing the mechanism of the cytotoxicity of enediynes requires further studies.
 | ||
| Fig. 6 Electropherogram of the pBR322 plasmid after exposure to 250 μM concentrations of enediynes 19a–c, f, i, andj, and 20. CC – covalently closed circular plasmid; OC – open circular form. | ||
Conclusions
In summary, the iodocyclization of ortho-functionalized (iodoethynyl)arenes along with the regioselective Sonogashira coupling opens a convenient synthetic route towards heteroindene-fused unsymmetrical enediynes. 2,3-Diiodobenzothiophene and 2,3-diiodoindole were easily accessible via iodocyclization. Although (iodoethynyl)anisole underwent iodination of the triple bond instead of cyclization, 2,3-diiodobenzofuran could be prepared, instead, from (iodoethynyl)phenol and NIS/PPh3.The regioselective outcome of the Sonogashira reaction for 2,3-diiodoheteroindenes depends on the nature of the heterocycle, as well as on the nature of alkynes. The Sonogashira coupling for all derivatives follows the C2-regioselectivity rule, without producing a C3 mono-substitution byproduct. However, the regioselectivity suffers from the activating influence of the initially introduced alkyne on the adjacent C–Hal position, resulting in the formation of a disubstituted byproduct. This effect is stronger for diiodoindole and diiodobenzofuran than for diiodobenzothiophene. The ratio of mono- and disubstituted products also depends on the nature of the alkyne. Alkynes with propargylic acceptors at C2 have a greater activating effect at the C3 position towards the subsequent Sonogashira reaction.
As a result, diethynylation does not preclude the synthesis of unsymmetrical benzothiophene-fused enediynes based on a sequential one-pot C2/C3 ethynylation, but limits the use of the one-pot approach for the indole and benzofuran derivatives. In this case, either the “diacetylene” route or the stepwise two-pot substitution of both iodine atoms by the Sonogashira cross-coupling is recommended.
In addition, these experimental selectivity trends provided a tool to probe the mechanistic features of the Sonogashira cross-coupling. A diverse set of DFT data, including C–X bond strength, C–X bond polarizations, electron density distributions, and reaction exergonicities converge on transmetalation, rather than oxidative addition, as the rate-limiting step of this reaction cascade. Computational analysis using various levels of DFT revealed a few significant points about the selectivity and provided insight into the mechanistic basis of the observed selectivities in the Sonogashira coupling.
Importantly, the alkyne moiety is an activating substituent (more than the heavier halogens). Furthermore, a remote (propargylic) acceptor can make this activating effect more pronounced. This observation illustrates once again the unique properties of the alkyne functionality – it is both a significantly electronegative substituent and a perfect conduit for stereoelectronic hyperconjugative effects.77
The acyclic unsymmetrical enediynes showed cytotoxic activity against NCI-H460 lung carcinoma cell lines along with lower cytotoxicity toward WI-26 VA4 lung epithelial-like cell lines. Interestingly, the mechanism of the cytotoxicity is not associated with the induction of DNA strand breaks.
Author contributions
AVP – investigation and writing – original draft; NAD – data curation, methodology, project administration, supervision, visualization, writing – original draft, and writing – review & editing; JSO – investigation; AAV – investigation and validation; EAK – investigation and resources; ASB – investigation and visualization; AMR – investigation and visualization; AIG – investigation and funding acquisition; TS – investigation and writing – original draft; IVA – data curation, methodology, supervision, visualization, writing – original draft, and writing – review & editing; IAB – conceptualization, methodology, funding acquisition, project administration, supervision, visualization, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Russian Science Foundation grant 19-73-10077-II. The study was carried out using equipment from the SPbU Research Park: the Centre for Magnetic Resonance and the Centre for Chemical Analysis and Materials Research. The authors would like to thank Alexander Ivanov (SPbU) and Sergey Smirnov (SPbU) for NMR measurements and Oussama Abdelhamid Mammeri (SPbU) for HR MS measurements. The authors gratefully acknowledge the Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for providing necessary facilities to carry out GC-MS and HRMS-ESI measurements. Computational studies at FSU were supported by the National Science Foundation (NSF) (CHE-2102579).References
- T. Aggarwal, S. Kumar and A. K. Verma, Org. Biomol. Chem., 2016, 14, 7639–7653 RSC.
- B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS PubMed.
- X. Jiang and H. Liu, Compr. Org. Synth. II, 2014, 4, 412–494 CAS.
- S. Singh and S. Chimni, Synthesis, 2015, 47, 1961–1989 CrossRef CAS.
- K. Gilmore, R. K. Mohamed and I. V. Alabugin, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 487–514 CAS.
- T. Khan and S. Yaragorla, Eur. J. Org. Chem., 2019, 3989–4012 CrossRef CAS.
- J. Tummatorn, S. Ruchirawat and C. Thongsornkleeb, Synthesis, 2023, 55, 3073–3089 CrossRef CAS.
- G. Leonel, I. Klann, D. F. Back, B. A. Iglesias, C. W. Nogueira and G. Zeni, Chem. – Eur. J., 2023, 29, e202202847 CrossRef CAS PubMed.
- L. Habert, I. Diachenko, P. Retailleau and I. Gillaizeau, Org. Biomol. Chem., 2021, 19, 6623–6627 RSC.
- T. Okitsu, M. Itoh, A. Inui, E. Muta, R. Nakamoto, Y. Adachi, A. Wada and M. Hatano, Asian J. Org. Chem., 2022, 11, e202200275 CrossRef CAS.
- T. Okitsu, A. Namura, S. Kondo, S. Tada, M. Yanagida and A. Wada, Org. Chem. Front., 2020, 7, 879–884 RSC.
- D. Bag, H. Kour, N. Saha, Kamal, H. Holla, P. V. Bharatam and S. D. Sawant, J. Org. Chem., 2023, 88, 2377–2384 CrossRef CAS PubMed.
- I. Misiūnaitė, R. Bukšnaitienė, J. Pošiūnas, A. Brukštus and I. Žutautė, J. Heterocycl. Chem., 2022, 59, 1712–1722 CrossRef.
- R. K. Mohamed, S. Mondal, J. V. Guerrera, T. M. Eaton, T. E. Albrecht-Schmitt, M. Shatruk and I. V. Alabugin, Angew. Chem., Int. Ed., 2016, 55, 12054–12058 CrossRef CAS PubMed.
- H.-C. Huang, Y.-C. Hsieh, P.-L. Lee, C.-C. Lin, Y.-S. Ho, W.-K. Shao, C.-T. Hsieh, M.-J. Cheng and Y.-T. Wu, J. Am. Chem. Soc., 2023, 145, 10304–10313 CrossRef CAS PubMed.
- T. Okitsu, A. Horike, N. Shimazawa and A. Wada, Org. Biomol. Chem., 2020, 18, 3501–3511 RSC.
- S. Chen, D. L. Priebbenow, J. Somkhit, C. V. Scullino, K. Agama, Y. Pommier and B. L. Flynn, Chem. – Eur. J., 2022, 28, e202201925 CrossRef CAS PubMed.
- N. A. Danilkina, A. E. Kulyashova, A. F. Khlebnikov, S. Bräse and I. A. Balova, J. Org. Chem., 2014, 79, 9018–9045 CrossRef CAS PubMed.
- A. G. Lyapunova, N. A. Danilkina, A. F. Khlebnikov, B. Köberle, S. Bräse and I. A. Balova, Eur. J. Org. Chem., 2016, 4842–4851 CrossRef CAS.
- N. Danilkina, A. Rumyantsev, A. Lyapunova, A. D′yachenko, A. Khlebnikov and I. Balova, Synlett, 2019, 30, 161–166 CrossRef CAS.
- A. G. Lyapunova, N. A. Danilkina, A. M. Rumyantsev, A. F. Khlebnikov, M. V. Chislov, G. L. Starova, E. V. Sambuk, A. I. Govdi, S. Bräse and I. A. Balova, J. Org. Chem., 2018, 83, 2788–2801 CrossRef CAS PubMed.
- N. A. Danilkina, E. A. Khmelevskaya, A. G. Lyapunova, A. S. D′yachenko, A. S. Bunev, R. E. Gasanov, M. A. Gureev and I. A. Balova, Molecules, 2022, 27, 6071 CrossRef CAS PubMed.
- N. A. Danilkina, L. Y. Gurskaya, A. V. Vasilyev and I. A. Balova, Eur. J. Org. Chem., 2016, 739–747 CrossRef CAS.
- N. A. Danilkina, A. S. D′yachenko, A. I. Govdi, A. F. Khlebnikov, I. V. Kornyakov, S. Bräse and I. A. Balova, J. Org. Chem., 2020, 85, 9001–9014 CrossRef CAS PubMed.
- N. Choy, B. Blanco, J. Wen, A. Krishan and K. C. Russell, Org. Lett., 2000, 2, 3761–3764 CrossRef CAS PubMed.
- S. Hamri, J. Rodríguez, J. Basset, G. Guillaumet and M. D. Pujol, Tetrahedron, 2012, 68, 6269–6275 CrossRef CAS.
- J. Kour, P. Khajuria, A. Sharma and S. D. Sawant, Chem. – Asian J., 2022, 17, e202200778 CrossRef CAS PubMed.
- J. Zhang, S. Li, G.-J. Deng and H. Gong, ChemCatChem, 2018, 10, 376–380 CrossRef CAS.
- J. Bergman and L. Venemalm, J. Org. Chem., 1992, 57, 2495–2497 CrossRef CAS.
- Y. Liu and G. W. Gribble, Tetrahedron Lett., 2000, 41, 8717–8721 CrossRef CAS.
- A. Putey, F. Popowycz and B. Joseph, Synlett, 2007, 419–422 CAS.
- H. Hamamoto, Y. Miki, H. Umemoto, M. Umemoto, C. Ohta, E. Fujita, A. Nakamura and T. Maegawa, Heterocycles, 2015, 91, 561 CrossRef CAS.
- Y. Yang, L. Zhang, G.-J. Deng and H. Gong, Sci. Rep., 2017, 7, 40430 CrossRef PubMed.
- G. J. P. Perry, J. M. Quibell, A. Panigrahi and I. Larrosa, J. Am. Chem. Soc., 2017, 139, 11527–11536 CrossRef CAS PubMed.
- S. Mehta and R. C. Larock, J. Org. Chem., 2010, 75, 1652–1658 CrossRef CAS PubMed.
- T. R. Puleo and J. S. Bandar, in Encyclopedia of Reagents for Organic Synthesis, Wiley, 2022, pp. 1–2 Search PubMed.
- T. Kesharwani, C. Kornman, A. Tonnaer, A. Hayes, S. Kim, N. Dahal, R. Romero and A. Royappa, Tetrahedron, 2018, 74, 2973–2984 CrossRef CAS PubMed.
- A. D. Sonawane, D. R. Garud, T. Udagawa and M. Koketsu, Org. Biomol. Chem., 2018, 16, 245–255 RSC.
- J. Lu, S. Donnecke, I. Paci and D. C. Leitch, Chem. Sci., 2022, 13, 3477–3488 RSC.
- M. F. Ibad, D. S. Zinad, M. Hussain, A. Ali, A. Villinger and P. Langer, Tetrahedron, 2013, 69, 7492–7504 CrossRef CAS.
- S. Tšupova, M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem. – Eur. J., 2016, 22, 16286–16291 CrossRef PubMed.
- M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 2593–2598 CrossRef CAS PubMed.
- S. Tšupova, M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem. – Eur. J., 2016, 22, 16286–16291 CrossRef PubMed.
- N. B. Vinh, S. M. Devine, L. Munoz, R. M. Ryan, B. H. Wang, H. Krum, D. K. Chalmers, J. S. Simpson and P. J. Scammells, ChemistryOpen, 2015, 4, 56–64 CrossRef CAS PubMed.
- Z. Szlavik, M. Csekei, A. Paczal, Z. B. Szabo, S. Sipos, G. Radics, A. Proszenyak, B. Balint, J. Murray, J. Davidson, I. Chen, P. Dokurno, A. E. Surgenor, Z. M. Daniels, R. E. Hubbard, G. Le Toumelin-Braizat, A. Claperon, G. Lysiak-Auvity, A.-M. Girard, A. Bruno, M. Chanrion, F. Colland, A.-L. Maragno, D. Demarles, O. Geneste and A. Kotschy, J. Med. Chem., 2020, 63, 13762–13795 CrossRef CAS PubMed.
- G. A. Salman, N. N. Pham, T. N. Ngo, P. Ehlers, A. Villinger and P. Langer, Adv. Synth. Catal., 2017, 359, 1402–1406 CrossRef CAS.
- X. Yin, Y. Li, Y. Zhu, Y. Kan, Y. Li and D. Zhu, Org. Lett., 2011, 13, 1520–1523 CrossRef CAS PubMed.
- Z. U. Levi and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 2796–2797 CrossRef CAS PubMed.
- Y. Morisaki, M. Tominaga and Y. Chujo, Chem. – Eur. J., 2012, 18, 11251–11257 CrossRef CAS PubMed.
- A. Fukazawa, D. Kishi, Y. Tanaka, S. Seki and S. Yamaguchi, Angew. Chem., Int. Ed., 2013, 52, 12091–12095 CrossRef CAS PubMed.
- F. Guo, X. Yin, F. Pammer, F. Cheng, D. Fernandez, R. A. Lalancette and F. Jäkle, Macromolecules, 2014, 47, 7831–7841 CrossRef CAS.
- B.-W. Park, Y.-H. Kwak, S.-Y. Kim, J.-Y. Yun, S.-R. Lee, J.-W. Choi, W.-l. Choi and J.-Y. Lee, 102013214399A1, 2014.
- C. Shu, C.-Y. Shi, Q. Sun, B. Zhou, T.-Y. Li, Q. He, X. Lu, R.-S. Liu and L.-W. Ye, ACS Catal., 2019, 9, 1019–1025 CrossRef CAS.
- D. Yue and R. C. Larock, J. Org. Chem., 2002, 67, 1905–1909 CrossRef CAS PubMed.
- D. Yue and R. C. Larock, Org. Lett., 2004, 6, 1037–1040 CrossRef CAS PubMed.
- D. Yue, T. Yao and R. C. Larock, J. Org. Chem., 2005, 70, 10292–10296 CrossRef CAS PubMed.
- Y.-L. Li, J. Li, S.-N. Yu, J.-B. Wang, Y.-M. Yu and J. Deng, Tetrahedron, 2015, 71, 8271–8277 CrossRef CAS.
- E. J. M. Maduli, S. J. Edeson, S. Swanson, P. A. Procopiou and J. P. A. Harrity, Org. Lett., 2015, 17, 390–392 CrossRef CAS PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- I. Kanwal, A. Mujahid, N. Rasool, K. Rizwan, A. Malik, G. Ahmad, S. A. A. Shah, U. Rashid and N. M. Nasir, Catalysts, 2020, 10, 443 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- C. Hu, J. Mena and I. V. Alabugin, Nat. Rev. Chem., 2023, 7, 405–423 CrossRef PubMed.
- I. Alabugin, E. Gonzalez-Rodriguez, R. Kawade, A. Stepanov and S. Vasilevsky, Molecules, 2019, 24, 1036 CrossRef PubMed.
- X. Wang, Y. Song, J. Qu and Y. Luo, Organometallics, 2017, 36, 1042–1048 CrossRef CAS.
- C. He, J. Ke, H. Xu and A. Lei, Angew. Chem., 2013, 125, 1567–1570 CrossRef.
- R. P. Bell, Proc. R. Soc. London, Ser. A, 1936, 154, 414–429 Search PubMed.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1938, 34, 11–24 RSC.
- I. V. Alabugin and M. Manoharan, J. Am. Chem. Soc., 2005, 127, 12583–12594 CrossRef CAS PubMed.
- G. d. P. Gomes and I. Alabugin, Applied Theoretical Organic Chemistry, World Scientific, Europe, 2018, pp. 451–502 Search PubMed.
- C. Borie, S. Mondal, T. Arif, M. Briand, H. Lingua, F. Dumur, D. Gigmes, P. Stocker, B. Barbarat, V. Robert, C. Nicoletti, D. Olive, M. Maresca and M. Nechab, Eur. J. Med. Chem., 2018, 148, 306–313 CrossRef CAS PubMed.
- A. I. Govdi, S. O. Anisimov, N. A. Danilkina, A. S. Bunev and I. A. Balova, Mendeleev Commun., 2023, 33, 328–330 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- A. Kazantsev, O. Bakulina, D. Dar'in, G. Kantin, A. Bunev and M. Krasavin, Org. Lett., 2022, 24, 4762–4765 CrossRef CAS PubMed.
- M. Zhang, B. Li, H. Chen, H. Lu, H. Ma, X. Cheng, W. Wang, Y. Wang, Y. Ding and A. Hu, J. Org. Chem., 2020, 85, 9808–9819 CrossRef CAS PubMed.
- M. Zhang, H. Lu, B. Li, H. Ma, W. Wang, X. Cheng, Y. Ding and A. Hu, J. Org. Chem., 2021, 86, 1549–1559 CrossRef CAS PubMed.
- P. W. Peterson, R. K. Mohamed and I. V. Alabugin, Eur. J. Org. Chem., 2013, 2505–2527 CrossRef CAS.
- I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, 1–66 CAS.
Footnotes |
| † Dedicated to the 300th anniversary of the Saint Petersburg University Foundation |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00530a |
| This journal is © The Royal Society of Chemistry 2024 |