
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of (R)- and (S)-1,2-diazetidine-3-carboxylic acid derivatives for peptidomimetics†
Matthew
Nutter‡
 ,
Henry
Stone‡
,
Michael
Shipman
and
Stefan
Roesner
*
,
Henry
Stone‡
,
Michael
Shipman
and
Stefan
Roesner
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: s.roesner@warwick.ac.uk
First published on 25th March 2024
Abstract
The stereoselective synthesis of both enantiomers of N-protected 1,2-diazetidine-3-carboxylic acid (aAze) from homochiral glycidol is described. Orthogonal protection of this novel cyclic α-hydrazino acid allows for selective functionalisation at either Nγ or Nδ. This novel peptidomimetic building block was incorporated into the pseudotripeptides Gly-γaAze-Ala and Gly-δaAze-Ala.
Introduction
Cyclic α-hydrazino acids and their derivatives have been shown to possess a diverse set of biological activities.1 They have been isolated from numerous natural sources, including plants, marine organisms and bacteria. Examples include the potent cytotoxins piperazimycin A–C from Streptomyces sp.2 and the antimycobacterial cyclodepsipeptides lydiamycin A–D from Streptomyces lydicus.3 As a class of modified amino acids, cyclic α-hydrazino acids mimic the structural constraints imposed by proline (Pro). Therefore, they have been widely applied in peptidomimetics where they can act as Pro surrogates to modulate the conformational flexibility, bioavailability, proteolytic stability and biological activity of target molecules.4The six-membered cyclic α-hydrazino acid (n = 3), known as piperazic acid (Pip), is a non-proteinogenic amino acid and the only example of a naturally occurring member of this family (Fig. 1). Pip and its derivatives have been identified in over 140 natural products, many of which displaying potent biological activity, ranging from anti-malarial to anti-bacterial properties.5 The five-membered derivative δ-azaproline (aPro, n = 2) has been used as proline mimic in peptidomimetics.6 Detailed conformational studies of aPro containing di- and tripeptides have been performed, which revealed that aPro takes up a cis conformation in simple amides, whereas its carbamate-protected derivative adopts preferentially a trans conformation.6a,7 Tripeptides containing aPro derivatives were found to be stable to proteolytic degradation, whereas native Pro analogues were readily digested.8 Moreover, Sinha described the synthesis of the 5–9 membered cyclic α-hydrazino acids 1 (n = 2–6) using various synthetic strategies.9
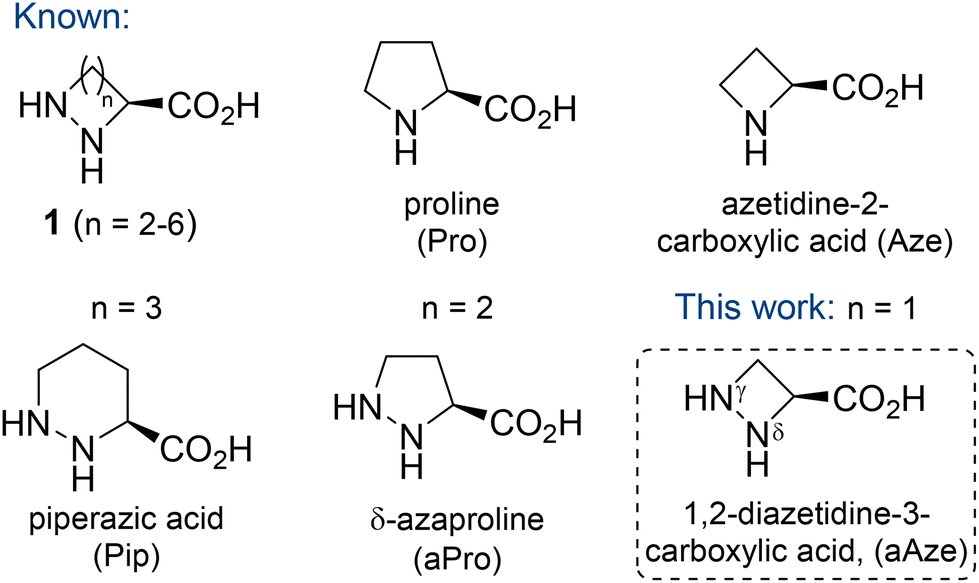 | ||
| Fig. 1 Cyclic amino and α-hydrazino acids. | ||
L-Azetidine-2-carboxylic acid (Aze) was the first example of a naturally occurring azetidine,10 and several natural products containing this non-proteogenic cyclic amino acid have been isolated.11 Theoretical studies have shown that replacement of Pro by its lower homologue Aze, i.e. substitution of a pyrrolidine ring by an azetidine cycle, leads to a reduction of the conformational flexibility due to the quasi-planar geometry of the four-membered ring.12 Similar to aPro-containing peptidomimetics, studies comparing Aze- and Pro-containing peptides have suggested a higher propensity of the amide bond involving the azetidine nitrogen to adopt a cis conformation.13
As part of our efforts in the synthesis of 3-substituted 1,2-diazetidines,14 we were interested to explore the stereocontrolled synthesis of 1,2-diazetidine-3-carboxylic acid (γ-azaazetidine-2-carboxylic acid, aAze, n = 1), the lower homologue of aPro, and its potential application in peptidomimetics (Fig. 1). In particular, we were interested in exploring the structural consequences towards short peptide sequences after incorporation of this unnatural amino acid. Furthermore, we were intrigued if aAze could act as turn-inducing moiety for the cyclisation of short peptide sequences. Previously, we reported the synthesis of C3-functionalised diazetidines by ring opening of enantioenriched epoxides with hydrazine monohydrate, followed by iterative N-protection and Mitsunobu ring closure.14a Taking inspiration from this route, we explored possibilities to install a carboxylic acid in the C3 position of the 1,2-diazetidine ring to give access to the novel unnatural amino acid aAze. Here, we disclose the stereospecific synthesis of orthogonally protected (R)- and (S)-1,2-diazetidine-3-carboxylic acid, aAze derivatives, and their regioselective functionalisation as novel building blocks in peptidomimetics.
Results and discussion
With both glycidol enantiomers being commercially available and inexpensive, we commenced our synthesis with homochiral (S)-glycidol [(S)-2] as a source of chirality. Initially, similar to Williams's asymmetric synthesis of piperazic acid derivatives,15 we masked the hydroxy group in glycidol as TBS (tert-butyldimethylsilyl) ether. However, ring opening of the silyl-protected glycidol with hydrazine hydrate gave inconsistent results with yields varying widely. Thus, we explored the sterically more hindered TIPS (triisopropylsilyl) protecting group. Stereocontrolled ring opening of homochiral epoxide (R)-316 with hydrazine hydrate at the unsubstituted carbon followed by selective protection of the secondary nitrogen gave Boc derivative (R)-4 in 65% yield (Scheme 1).17 This was followed by tosyl protection of the primary amine and azodicarboxylate-mediated Mitsunobu cyclisation to form the four-membered 1,2-diazetidine (S)-5. These steps were easily scalable and could be conducted on multi-gram scale.18 The yield for the ring closure was only moderate as it was accompanied by the formation of elimination product 7 (up to 25%). Interestingly, 7 was selectively formed as the cis alkene as determined by 1H NMR spectroscopy. We hypothesise that the selectivity in this elimination reaction is due to an intramolecular hydrogen bonding between the hydrogen on the tosyl protected amine and the TIPS protected oxygen. Removal of the primary TIPS group in (S)-5 with TBAF followed by oxidation of the resulting primary alcohol with an excess of sodium periodate in the presence of catalytic amounts of ruthenium trichloride and esterification of the resulting carboxylic acid with iodomethane furnished orthogonally protected ester (S)-6 as aAze derivative in 81% yield over the three step sequence.19 Examination of the stereointegrity of (S)-5 and (S)-6 using chiral HPLC revealed that the formation of the 1,2-diazetidine from homochiral (S)-glycidol to (S)-5 and its deprotection/oxidation/esterification to (S)-6 occurred with virtually no racemisation (>98% ee).18 The absolute stereochemical configuration of (S)-5 was confirmed after TBAF-mediated removal of the TIPS group to the primary alcohol and comparison with the literature data.14c Alternatively, following the same reaction sequence starting from (R)-glycidol [(R)-2], (R)-6 was prepared in comparable yield with >98% ee.18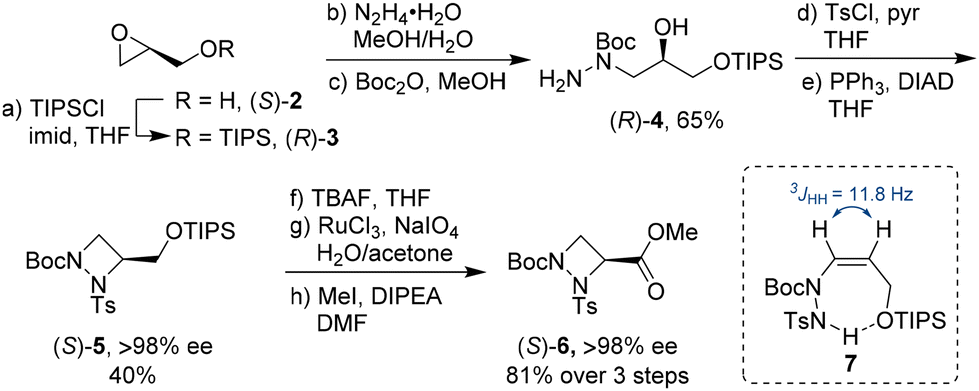 | ||
| Scheme 1 Synthesis of (S)-1,2-diazetidine-3-carboxylic acid building block (S)-6. | ||
Orthogonal protecting groups on Nγ and Nδ allowed for selective functionalisation of the 1,2-diazetidine derivatives 5 and 6 (Scheme 2). The Boc group was selectively removed with trifluoroacetic acid, while the tosyl group was cleaved using magnesium turnings in methanol. The deprotected derivatives could be isolated after removal of either the Boc or tosyl group but were not stable upon storage. To avoid decomposition after deprotection, further functionalisation of the free Nγ and Nδ was directly conducted on the crude material. Trapping of the free amine with phenyl isocyanate gave urea-containing compounds 8, 10, 12 and 14 in 60–73% yield. Alternatively, HATU-mediated coupling with Fmoc-Gly-OH provided pseudodipeptides 9, 11, 13 and 15 in 44–59% yield. These experiments demonstrate that either Nγ or Nδ could be independently functionalised with similar reactivities observed for both nitrogen atoms. This observation stands in contrast to the reactivity of aPro derivatives, where the amide nitrogen was reported to be significantly less reactive.7a We attribute this increased reactivity of Nδ in aAze derivatives to its increased pyramidal character in 1,2-diazetidines, which moves the substituents out of the plane of the four-membered ring to avoid steric interactions.14a Moreover, both nitrogen atoms in the 1,2-diazetidine ring can successively engage in peptide couplings. Starting from 11, removal of the Boc group from Nδ and coupling with Cbz-Ala-OH furnished an orthogonally protected pseudotripeptide.18 In all cases, the C-terminus of silyl protected alcohol 5 and methyl ester 6 were fully compatible during N-functionalisation.
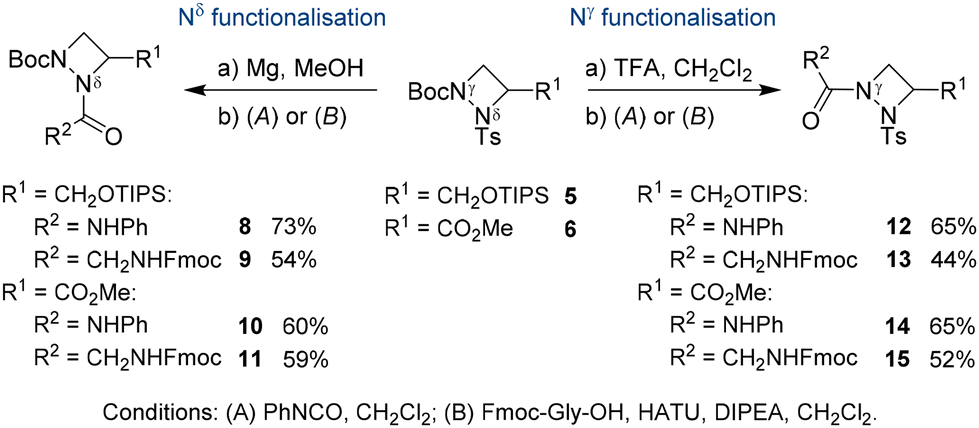 | ||
| Scheme 2 Orthogonal functionalisation of Nγ and Nδ in 5 and 6. | ||
Having established conditions for the selective functionalisation of both nitrogen atoms, we explored the possibility to incorporate the aAza amino acid into pseudotripeptides. Starting from (S)-5, the silyl group was removed, and the free alcohol was oxidised to the carboxylic acid (Scheme 3). Without further purification, HATU-mediated coupling between the carboxylic acid and H-Ala-OMe furnished (S,S)-16 as a single diastereomer in 65% yield over 3 steps. Alternatively, commencing from the enantiomer (R)-5, pseudodipeptide (R,S)-16 was selectively formed proving that the subsequent deprotection, oxidation and amide coupling occurred without any racemisation at the C-terminus.18
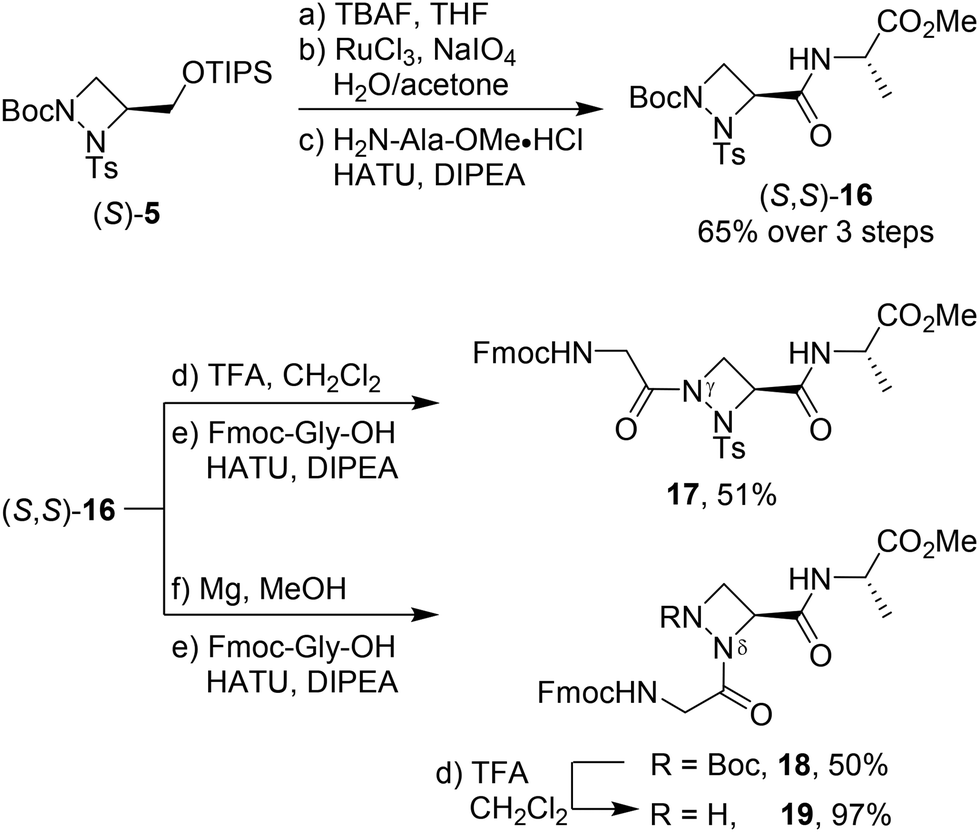 | ||
| Scheme 3 Incorporation of 1,2-diazetidine building block (S)-5 into pseudotripeptides. | ||
With (S,S)-16 in hand, removal of the Boc group from Nγ and coupling with Fmoc-Gly-OH gave pseudotripeptide 17 (Gly-γaAze-Ala) in 51% yield. On the other hand, tosyl deprotection from (S,S)-16 and coupling with Fmoc-Gly-OH gave Gly-δaAze-Ala derivative 18 in 50% yield. While tosyl removal from 17 with magnesium in methanol caused decomposition, the Boc group from 18 was successfully removed with 25% TFA in dichloromethane leading to free amine 19 in nearly quantitative yield after washing with sodium bicarbonate. 1H NMR analysis of pseudopeptides 18 and 19 at 298 K revealed a single set of signals, indicating the presence of only one conformer or a fast equilibrium between conformers in solution.
Conclusions
In summary, a practical synthesis of both enantiomers of orthogonally protected 1,2-diazetidine-3-carboxylic acid, derivates of the novel unnatural amino acid aAze, was developed using readily available homochiral glycidol as a source of chirality. The chemistry is operationally simple and can be performed on a preparative scale. Furthermore, orthogonal protection of 1,2-diazetidine building blocks 5 and 6 allowed for selective functionalisation on either Nγ or Nδ leading to different types of pseudopeptides. Due to the importance of 1,2-diazetidines, these cyclic unnatural amino acid derivatives have potential as novel scaffolds in peptidomimetics and as constrained analogues of natural amino acids. Studies of the structural consequences for the incorporation of aAze into short peptide sequences compared to the higher homologue aPro will be published in due course.Author contributions
M. N., H. S. and S. R.: synthesis, spectral analysis, investigation, and methodology. M. S.: conceptualisation, resources, validation, and writing – review and editing. S. R.: conceptualisation, project administration, supervision, validation, writing – original draft, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Warwick for financial support. S. R. acknowledges funding from the EUTOPIA Young Leaders Academy.References
- (a) G. Le Goff and J. Ouazzani, Bioorg. Med. Chem., 2014, 22, 6529–6544 CrossRef CAS PubMed; (b) C.-H. Küchenthal and W. Maison, Synthesis, 2010, 719–740 Search PubMed.
- E. D. Miller, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Org. Chem., 2007, 72, 323–330 CrossRef CAS PubMed.
- X. Huang, E. Roemer, I. Sattler, U. Moellmann, A. Christner and S. Grabley, Angew. Chem., Int. Ed., 2006, 45, 3067–3072 CrossRef CAS PubMed.
- M. K. Singh and M. K. Lakshman, Org. Biomol. Chem., 2022, 20, 963–979 RSC.
- (a) K. D. Morgan, R. J. Andersen and K. S. Ryan, Nat. Prod. Rep., 2019, 36, 1628–1653 RSC; (b) A. J. Oelke, D. J. France, T. Hofmann, G. Wuitschik and S. V. Ley, Nat. Prod. Rep., 2011, 28, 1445–1471 RSC; (c) M. A. Ciufolini and N. Xi, Chem. Soc. Rev., 1998, 27, 437–445 RSC.
- (a) Y. M. Elbatrawi, K. P. Pedretty, N. Giddings, H. L. Woodcock and J. R. Del Valle, J. Org. Chem., 2020, 85, 4207–4219 CrossRef CAS PubMed; (b) E. Voss, A. Arrault, J. Bodiguel and B. Jamart-Grégoire, Tetrahedron: Asymmetry, 2009, 20, 1809–1812 CrossRef CAS; (c) M. R. Mish, F. M. Guerra and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 8379–8380 CrossRef CAS.
- (a) O. Ovdiichuk, L. Flauder, E. Humbert-Voss, B. Jamart-Grégoire and A. Arrault, Tetrahedron Lett., 2019, 60, 16–19 CrossRef CAS; (b) I. Duttagupta, D. Misra, S. Bhunya, A. Paul and S. Sinha, J. Org. Chem., 2015, 80, 10585–10604 CrossRef CAS PubMed; (c) E. Humbert-Voss, A. Arrault and B. Jamart-Grégoire, Tetrahedron, 2014, 70, 363–370 CrossRef CAS; (d) Y. Che and G. R. Marshall, J. Org. Chem., 2004, 69, 9030–9042 CrossRef CAS PubMed; (e) W.-J. Zhang, A. Berglund, J. L.-F. Kao, J.-P. Couty, M. C. Gershengorn and G. R. Marshall, J. Am. Chem. Soc., 2003, 125, 1221–1235 CrossRef CAS PubMed.
- I. Duttagupta, J. Bhadra, S. K. Das and S. Sinha, Tetrahedron Lett., 2016, 57, 3858–3861 CrossRef CAS.
- (a) I. Duttagupta, K. Goswami, P. Chatla and S. Sinha, Synth. Commun., 2014, 44, 2510–2519 CrossRef CAS; (b) I. Duttagupta, K. Goswami and S. Sinha, Tetrahedron, 2012, 68, 8347–8357 CrossRef CAS.
- L. Fowden, Nature, 1955, 176, 347–348 CrossRef CAS.
- For a review on Aze synthesis, see: F. Couty and G. Evano, Org. Prep. Proced. Int., 2006, 38, 427–465 CrossRef CAS.
- G. Revilla-López, J. G. Warren, J. Torras, A. I. Jiménez, C. Cativiela and C. Alemán, Pept. Sci., 2012, 98, 98–110 CrossRef.
- (a) C. Melis, G. Bussi, S. C. R. Lummis and C. Molteni, J. Phys. Chem. B, 2009, 113, 12148–12153 CrossRef CAS; (b) D. Kern, M. Schutkowski and T. Drakenberg, J. Am. Chem. Soc., 1997, 119, 8403–8408 CrossRef CAS.
- (a) C. Dean, S. Roesner, S. Rajkumar, G. J. Clarkson, M. Jones and M. Shipman, Tetrahedron, 2021, 79, 131836 CrossRef CAS; (b) C. Dean, S. Rajkumar, S. Roesner, N. Carson, G. J. Clarkson, M. Wills, M. Jones and M. Shipman, Chem. Sci., 2020, 11, 1636–1642 RSC; (c) S. Rajkumar, G. J. Clarkson and M. Shipman, Org. Lett., 2017, 19, 2058–2061 CrossRef CAS PubMed; (d) M. J. Brown, G. J. Clarkson, G. G. Inglis and M. Shipman, Org. Lett., 2011, 13, 1686–1689 CrossRef CAS PubMed.
- Y. Aoyagi, Y. Saitoh, T. Ueno, M. Horiguchi, K. Takeya and R. M. Williams, J. Org. Chem., 2003, 68, 6899–6904 CrossRef CAS.
- D. Lainé, M. Fujita and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1999, 1639–1645 RSC.
- (a) A. Oliva, M. Ellis, L. Fiocchi, E. Menta and A. P. Krapcho, J. Heterocycl. Chem., 2000, 37, 47–55 CrossRef CAS; (b) P. Cerea, C. Giannini, S. Dall'Angelo, E. Licandro, S. Maiorana and R. Marchelli, Tetrahedron, 2007, 63, 4108–4119 CrossRef CAS.
- See the ESI† for a detailed description.
- B. Long, S. Tang, L. Chen, S. Qu, B. Chen, J. Liu, A. R. Maguire, Z. Wang, Y. Liu, H. Zhang, Z. Xu and T. Ye, Chem. Commun., 2013, 49, 2977–2979 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data for all new compounds, copies of HPLC traces, 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d4ob00278d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |