Development of an efficient and scalable bioprocess for the plant hormone 12-OPDA: Overcoming the hurdles of nature's biosynthesis†
Received
17th February 2024
, Accepted 16th April 2024
First published on 29th April 2024
Abstract
Besides its native biological function as a plant hormone, cis-(+)-12-oxo-phytodienoic acid (12-OPDA) serves as a metabolite for the cellular formation of (−)-jasmonic acid and has also been shown to have an influence on mammalian cells. In order to make this biologically active, but at the same time very expensive natural product 12-OPDA broadly accessible for further biological and medicinal research, we developed an efficient bioprocess based on the utilization of a tailor-made whole-cell catalyst by following the principles of its biosynthesis in nature. After process optimization, the three-step one-pot synthesis of 12-OPDA starting from readily accessible α-linolenic acid could be conducted at appropriate technically relevant substrate loadings in the range of 5–20 g L−1. The desired 12-OPDA was obtained with an excellent conversion efficiency, and by means of the developed, efficient downstream-processing, this emulsifying as well as stereochemically labile biosynthetic metabolite 12-OPDA was then obtained with very high chemical purity (>99%) and enantio- and diastereomeric excess (>99% ee, 96% de) as well as negligible side-product formation (<1%). With respect to future technical applications, we also demonstrated the scalability of the production of the whole cell-biocatalyst in a high cell-density fermentation process.
Introduction
Oxylipins are signal molecules in plants that regulate stress responses. Oxylipins are derived from polyunsaturated fatty acids via oxygenation.1 Among them, cis-(+)-12-oxo-phytodienoic acid (cis-(+)-4, cis-(+)-12-OPDA) has gained increasing interest from a range of directions.2–7 Recent studies revealed that the biological role of 12-OPDA (4) includes that it acts as a plant hormone, but it also has pharmacological activity on mammalian cells,2–4 in addition to being an important biosynthetic key intermediate for (−)-jasmonic acid and its derivatives, altogether called “jasmonates”. They are a type of bioactive oxylipin that plays a role in plant defences against pathogens, wounding, excess light and many other stresses, as well as in plant growth.6,7 Thus, the use of jasmonates in agriculture can reduce the amount of current pesticides and increase the yield of the harvest as well as of specific plant products, such as paclitaxel or camptothecin.8–10 In addition, jasmonate derivatives, such as methyl jasmonate, are important in the fragrance industry due to their characteristic odour.11 For example, the readily available saturated derivative Hedione® is used in many perfumes.11
In order to get access to this biologically active metabolite cis-(+)-12-OPDA (cis-(+)-4), various approaches are possible. The current extremely high price of up to 299 Euros per mg,12 however, indicates that both synthetic concepts applied up to now encounter substantial limitations. To start with classic organic synthetic natural product chemistry, interestingly today's routes are all conceptually based on the formation of the 5-membered ring and subsequent functionalization.13 Such routes, however, represent a tedious multi-step synthesis with many intermediate isolations, resulting in a low overall yield and large waste formation. Thus, such natural product syntheses of 12-OPDA (4) are less practical for even gram-scale synthesis as well as for future production on a technical scale.
An alternative concept is to make use of nature's way to prepare this plant hormone. The biosynthesis of 12-OPDA (4), which is shown in Fig. 1, starts in the chloroplasts through the release of α-linolenic acid (1, α-LA) via phospholipases or acylhydrolases from the chloroplast membrane. In the presence of a 13-lipoxygenase (13-LOX), a H–O–O-substituent is introduced at the C-13 of 1. The resulting hydroperoxide (S)-13-hydroperoxy-octadecatrienoic acid (2, 13-HPOT) is then converted into the allene oxide (9Z,13S,15Z)-12,13-epoxy-octadecatrienoic acid (3, 12,13-EOT) by means of the allene oxide synthase (AOS), a member of the cytochrome P450 (CYP450) oxidoreductase superfamily.14 The allene oxide 3 represents a highly labile intermediate, which has the ability to spontaneously cyclize into racemic 12-OPDA (rac-4) or undergo fast hydrolysis to form α- (5) or y-ketol (6) (Fig. 2). In biosynthesis, an allene oxide cyclase (AOC) converts 12,13-EOT (3) to the enantiomerically pure cis-(+)-12-OPDA (cis-(+)-4) while avoiding such side reactions.15
 |
| | Fig. 1 Biosynthesis of cis-(+)-12-OPDA (cis-(+)-4) in the chloroplasts of plants. | |
 |
| | Fig. 2 Overview of the biocatalytic synthesis of cis-(+)-12-OPDA (cis-(+)-4) with a commercial Gm-13-LOX and recombinant whole-cell catalyst containing AtAOS and AtAOC4. | |
Although being conceptually highly attractive due to the opportunity to get access to 12-OPDA (4) within a one-pot synthesis using only three reaction steps and starting from readily accessible fatty acid 1, attempts to develop a biocatalytic process based on this biosynthetic approach have faced various challenges. In 1979 when Vick and Zimmermann first described this biocatalytic cascade leading to 12-OPDA (4) in vitro by using extracts from tissues of 24 plant species, the product 4 observed was analysed without isolation and purification.16 In 2020, based on further work on the formation and isolation of 4,16,17 the groups of Dietz and Gröger reported the first practical biocatalytic one-pot cascade to prepare 12-OPDA (4) starting from α-LA (1) utilizing a commercially available 13-LOX from Glycine max (soybean, Gm-13-LOX) together with a tailor-made whole-cell catalyst containing recombinant AOS from Arabidopsis thaliana.18 At substrate loadings of up to 2 g L−1, full conversion was achieved in combination with excellent >99% ee for 4. However, in addition to the still low substrate loading, other remaining limitations were the formation of side-product 5 in up to 10% yield as well as 10% isomerized trans-(+)-12-OPDA (trans-(+)-4, Fig. 2) as the opposite diastereomer.18 In addition, the work-up had also not been optimized and required several steps (see also the section “Optimization of the work-up”).
In the following, we report a substantially improved bioprocess, which we developed as a continuation of this previous work18 and in which we succeeded in addressing the remaining challenges of both, the process and work-up stage. It should be added that with this current work we also demonstrate that even complex and labile biosynthetic metabolites can be produced in an efficient manner with technically suitable substrate loadings and by combining multiple biosynthetic steps towards a one-pot cascade. In addition, the presented solutions, which we found to overcome such hurdles, can be considered to have a significant impact beyond the specific product 12-OPDA (4) as they are likely to be applicable for related bioprocesses to prepare other complex natural products in a sustainable manner. In this work, we demonstrate a significant increase of the substrate loading from a maximum of 2 g L−1 in recent studies18 to a technically relevant substrate loading of 10–20 g L−1 with excellent conversion and stereoselectivity. In addition, we optimized the biosynthetic pathway by modulation of the whole-cell catalyst and reaction parameters. This allowed us to avoid side-reactions such as α-ketol (5) formation or cis–trans isomerization of the product cis-(+)-4 completely and enabling a sustainable and efficient work-up. A scalable production of the tailor-made biocatalyst in high cell-density fermentation is also demonstrated.
Results and discussion
Optimization of the whole-cell catalyst
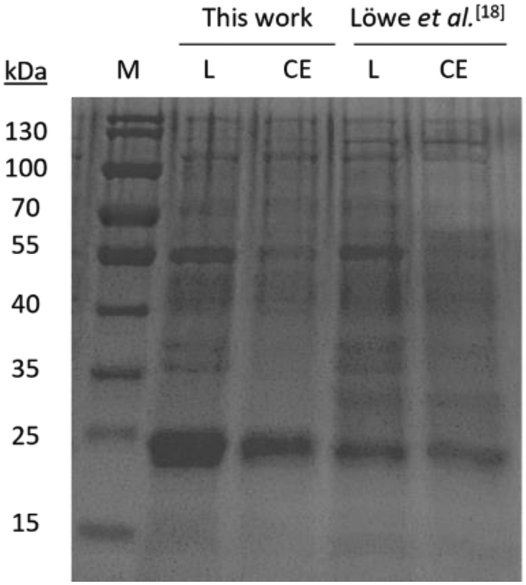
The design of a highly efficient biocatalyst is crucial for any biotransformation when aiming at a high process efficiency. The fatty acid α-linolenic acid (1, α-LA) is the natural substrate for this cascade. Theoretically, no side-product should be formed, but due to the instability of the 12,13-EOT (3), α-ketol (5) is generated. To prevent the formation of side-product 5, the activity of the allene oxide synthase (AOS) must be significantly lower than that of the allene oxide cyclase (AOC). The most efficient way to obtain this goal is to enhance (AOC) or decrease (AOS) the expression levels of the enzymes. In our previous system, we used pET28a and pQE30 vectors. Although being sufficient for the initial process, for further improvement the use of these plasmids raised robustness concerns because the pET28a and pQE30 vectors share the same origin of replication. This makes the realization of the consistent expression ratios of AOS and AOC challenging and can also lead to a varying production of side-product 5. Therefore, the initial focus was on optimizing the whole-cell catalyst (WCC) by changing the two-plasmid system to a DUET-plasmid system, which is commercially available and can accommodate two genes on one vector. This system offers the advantage of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 expression ratio of the genes due to the absence of a terminator after the first gene (Fig. S1, see ESI†). In addition, we substituted AtAOC2 with the more active AtAOC4.19 The resulting construct was transformed into the E. coli strain BL21-CodonPlus(DE3)-RIL, which had already been utilized by us previously.18 The new WCC exhibited a favourable expression ratio between AOS and AOC (AOC ≫ AOS) under our previous expression conditions (30 °C, 1 mM IPTG, 20 h).18 The outcome of test reactions with this expression conditions was already strongly enhanced in comparison to our previous results18 but unfortunately not consistent over different expression batches (data not shown). Thus, the expression system was optimized by reducing the IPTG concentration to 0.05 mM. Changing the expression temperature to 25 °C resulted in an increased expression of AtAOS (Fig. 3). With this optimization we obtained a robust and reproducible WCC, which showed consistent results in every expression. To compare the optimized whole-cell catalyst and expression conditions with the reproduction of our previously reported18 WCC, we conducted an SDS-PAGE analysis with both catalysts. The overexpression of the AOC was significantly enhanced by the optimization steps in this work, whereas the expression of AOS remains at the same level (Fig. 4).
:2 expression ratio of the genes due to the absence of a terminator after the first gene (Fig. S1, see ESI†). In addition, we substituted AtAOC2 with the more active AtAOC4.19 The resulting construct was transformed into the E. coli strain BL21-CodonPlus(DE3)-RIL, which had already been utilized by us previously.18 The new WCC exhibited a favourable expression ratio between AOS and AOC (AOC ≫ AOS) under our previous expression conditions (30 °C, 1 mM IPTG, 20 h).18 The outcome of test reactions with this expression conditions was already strongly enhanced in comparison to our previous results18 but unfortunately not consistent over different expression batches (data not shown). Thus, the expression system was optimized by reducing the IPTG concentration to 0.05 mM. Changing the expression temperature to 25 °C resulted in an increased expression of AtAOS (Fig. 3). With this optimization we obtained a robust and reproducible WCC, which showed consistent results in every expression. To compare the optimized whole-cell catalyst and expression conditions with the reproduction of our previously reported18 WCC, we conducted an SDS-PAGE analysis with both catalysts. The overexpression of the AOC was significantly enhanced by the optimization steps in this work, whereas the expression of AOS remains at the same level (Fig. 4).
 |
| | Fig. 3 SDS-PAGE related to the experiments for optimization of expression conditions. (L = lysate; CE = crude extract) (AtAOS: 56.1 kDa; AtAOC4: 23.6 kDa); for details, see ESI.† | |
 |
| | Fig. 4 Comparison of the expression of previous and current WCC (L = lysate; CE = crude extract) (AtAOS: 56.1 kDa; AtAOC4: 23.6 kDa); for details, see the ESI.† | |
Optimized reaction conditions
The side-product 5 formation was addressed by optimizing the WCC. The reaction conditions, including cosolvent/additive, temperature, and mixing, were optimized to improve the reaction time, substrate loading, and prevent product isomerization. Initially, the new WCC was tested in the same reaction system as before18 (NH4Cl buffer (100 mM, pH 9; 5% EtOH (v/v)); 1 g L−1 α-LA (1); 30 mg L−1 Gm-13-LOX; 3 g L−1 WCC; magnetic stirrer; room temperature). In this system, we observed a poor conversion of α-linolenic acid (1) (77%). The conversion was completed by reducing the temperature to 0 °C and doubling the amount of the catalysts used. The side-product 5 formation was 4%, and we prevented product isomerization. By switching to an overhead stirrer, we were able to achieve the same results at 0 °C, while also benefiting from improved mixing at room temperature. Notably, the temperature had the biggest influence on the result of the reaction (Fig. 5). After determining the optimal reaction settings (overhead stirrer and 0 °C), we compared the commercial Gm-13-LOX (Sigma-Aldrich) used by us previously,18 with the one from TCI, but, no differences were found between these two enzymes (Table S7, see ESI†). To compare the whole-cell catalysts, we used the DUET-plasmid system with AtAOC2 and expressed it under optimized conditions. When comparing the results, a high formation of α-ketol (5) was observed for AtAOC2 (18%), but the diastereomeric excess was excellent (>99%) (Table S7, see ESI†). The comparison of the protein expression levels with SDS-PAGE revealed that the AtAOC2 was expressed less than AtAOC4, which may result in worse selectivity for 12-OPDA (4) (Fig. S4, see ESI†). We chose to use Gm-13-LOX from TCI rather than Sigma-Aldrich, as there was no discernible difference in their activity. In addition, we opted for AtAOC4 instead of AtAOC2 for our WCC due to its higher selectivity in the test reactions. To enhance the substrate loading, we placed the enzyme on a DUET plasmid system with AtAOS and transformed it into the E. coli strain BL21 CodonPlus (DE3)-RIL for further studies.
 |
| | Fig. 5 The impact of mixing and temperature on the 12-OPDA (4) cascade. | |
Optimization of the reaction solution
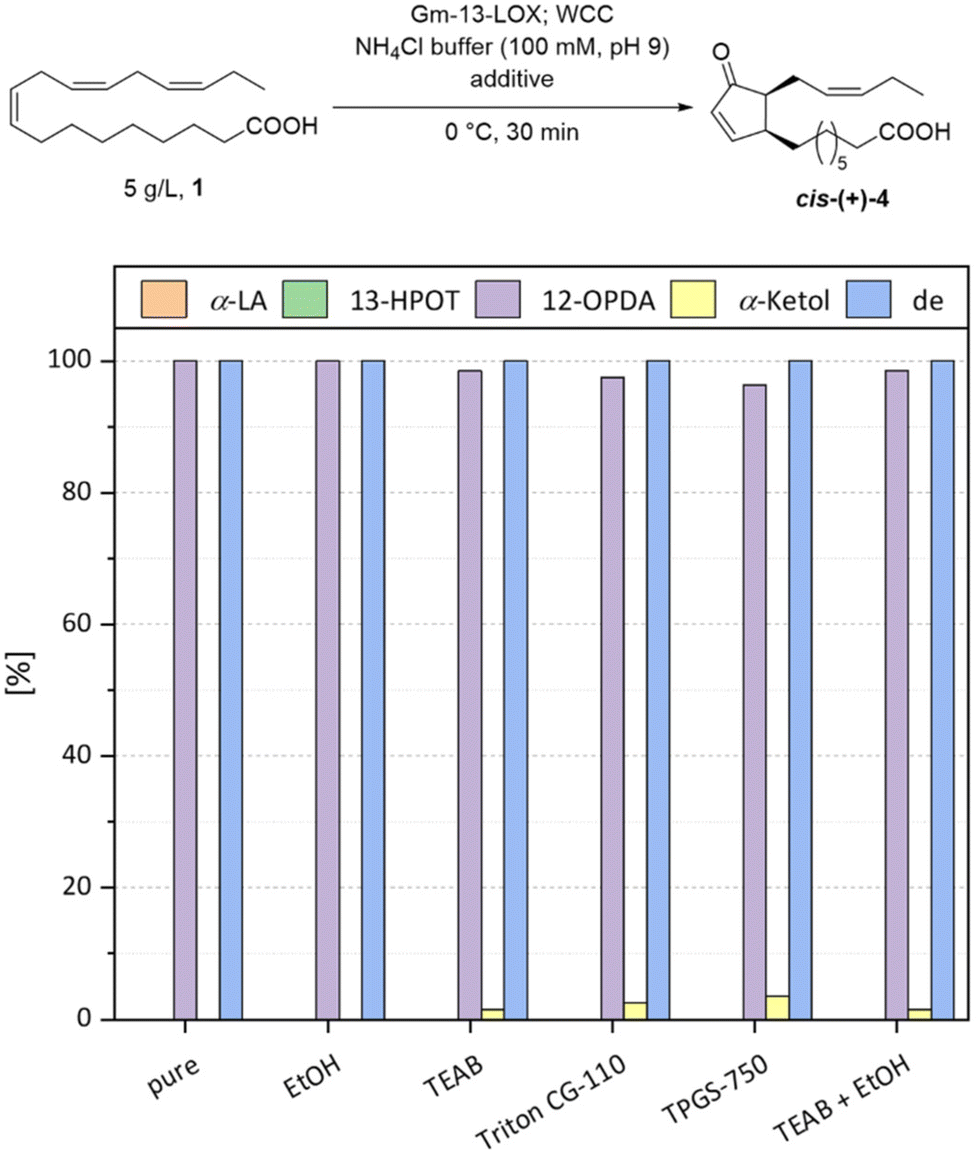
In our previous work,18 we used an aqueous solution with ethanol (5% (v/v)) as a cosolvent because ethanol had a positive effect on the activity of the Gm-13-LOX. In this section we compare this cosolvent with phase transfer catalysts (PTCs) as additives, and the pure buffer. The PTCs used were short-chain ionic tetraethylammonium bromide (TEAB) and the non-ionic Triton® CG-110 and TPGS-750.20 All the reactants were used at 3% (w/w) of the substrate. The first increase in substrate concentration was 5 g L−1 (Fig. 6), and the loading of the catalysts was increased analogously. At this stage, all the reactions were completed after 30 min and no α-linolenic acid (1) or 13-HPOT (2) remained. Reactions with ethanol and pure buffer enabled complete conversion to the desired product 4 (>99%). Addition of the PTCs caused formation of a small amount of side-product (1.5%–3.6%). The TEAB showed the best results among the PTCs that could not be enhanced by the addition of ethanol (2% (v/v)) (1.5%, α-ketol (5)). The diastereomeric excess of all the reactions was excellent (>99%). The results revealed that the PTCs had a small negative effect on the selectivity of the WCC and reduced the activity of the AtAOC4 or accelerated the hydrolysis of the unstable intermediate 12,13-EOT (3). However, we further increased the substrate loading to 10 g L−1 and the amount of catalyst accordingly (Fig. 7). The results confirmed the negative impact of the PTCs on this reaction, but not only on the selectivity of this reaction. The reaction kinetics showed that the long-chain PTCs had a negative influence on the initial rate of the reaction. Because only small amounts of α-ketol (5) were formed and no 13-HPOT (2) accumulated, the negative effect might be related to the Gm-13-LOX. The Gm-13-LOX is a membrane-associated protein in its natural environment and is also feedback inhibited by 12-OPDA (4).21,22 It is therefore unlikely to be very stable in aqueous solution and these long-chain PTCs may denature or inactivate it. The small-chain PTC TEAB showed the least effect on Gm-13-LOX. The initial activity was equal to that of the pure buffer and the percentage of 12-OPDA (4) was very high (95%). The addition of ethanol (2% (v/v)) resulted in a higher initial reaction rate, like the reaction with ethanol (5% (v/v)), but without increasing the yield of 12-OPDA (4) (96%). This increased reaction rate of Gm-13-LOX with ethanol has already been described by us previously.18 The best result was observed by using only ethanol as cosolvent. The reaction was completed after 3 h with a selectivity for 12-OPDA (4) of >99%. The diastereomeric ratio was excellent for all the reactions (>99%). The enhanced reaction time in comparison to the 5 g L−1 scale can be explained by the feedback inhibition of Gm-13-LOX by 12-OPDA (4).22 Nevertheless, we decided to increase the substrate loading to 20 g L−1 to improve our already good results. Based on our recent results, we used only ethanol (5% (v/v)) as co-solvent. In this reaction we never observed a complete conversion of the α-LA (1). After 23 h the conversion was 68% and the initial reaction rate was also lower than that at 10 g L−1 (Fig. 8).
 |
| | Fig. 6 Additive screening at a concentration of 5 g L−1 α-linolenic acid (1). | |
 |
| | Fig. 7 Additive screening at a concentration of 10 g L−1 α-linolenic acid (1). | |
 |
| | Fig. 8 Comparison of 12-OPDA (4) syntheses at 10 and 20 g L−1. | |
However, we assumed that the high substrate loading is not a problem for the WCC because no 13-HPOT (2) was observed in any sample at both 10 and 20 g L−1, but the Gm-13-LOX is affected by 12-OPDA (4) because of feedback inhibition.22 Furthermore, high concentrations of α-LA (1) have a negative influence on the Gm-13-LOX, which is shown by the decreased conversion rate at the beginning of the reaction (Fig. 8). However, the impact of the substrate 1 is lower than the impact of product 4.
To conclude this chapter, our previously reported18 solvent system buffer-ethanol was confirmed to be the most suitable one for this multi-enzymatic cascade. Further improvements have been made in terms of mixing and the reaction temperature, as well as optimization of the whole-cell catalyst, which enabled excellent results in terms of selectivity (>99%, 12-OPDA (4); >99% de) of the reaction at high substrate loading (10 g L−1).
Optimization of the work-up
After optimizing the reaction conditions, we aimed to address the remaining challenges existing for the work-up process. This task involved overcoming various “hurdles” such as, for example, accumulation of 12-OPDA (4) in the cell membrane and lability of 4 against isomerization under formation of the opposite trans-diastereomer of 4 with 10% after isolation in our previous work.18 In addition, methylene chloride being an unfavourable solvent from a sustainability perspective was previously used, and multiple extraction cycles required a high volume of solvent, and, thus, a considerable amount of waste was generated. Furthermore, the extraction cycles and subsequent work-up steps such as treatment of cells with ultrasound resulted in a unfavourably high number of unit operation steps during the work-up.
Thus, in this study we have modified the work-up procedure to a simpler and more sustainable process even if utilizing a higher product loading because the biotransformation was done at a 5 g L−1 substrate loading. Instead of separating the cells from the reaction mixture after the reaction, we added saturated NaCl solution and acidified the whole suspension to pH 1, which disrupted the cells and facilitated the extraction of the product. The mixture was then extracted only once with ethyl acetate as a “green” solvent (1:1 (v/v)). This yielded a yellow-brown oil, which contained ∼55% of the possible 12-OPDA (4) as a crude product, which was, however, substantially contaminated with impurities from the cells (Fig. 9A; for further details, see the ESI†). The brown colour could be removed by filtration over charcoal, but the purity was not increased, and a yellow colouring was still observed (Fig. 9B). However, by means of reversed phase automated column chromatography we were able to obtain a completely purified (>99%) and colourless product (Fig. 9C) with a recovery rate of 64% in this step, thus corresponding to an overall yield of this highly emulsifying product 4 of ∼35%. Because the product was obtained from reverse chromatography with an aqueous eluent, proved to still contain a substantial portion of volatile aqueous components, removal of the water was needed. Although freeze drying represents a general standard method, in our case, due to a long drying procedure, the problem of undesired isomerization exists. This expected limitation of freeze drying was confirmed by observation of strong isomerization overnight (70% de). In order to avoid this undesired isomerization effect, a rotary evaporator and high vacuum was used, which resulted in a dried, colourless product of 12-OPDA (4) with excellent properties, such as a chemical purity of >99% as well as enantio- and diastereomeric excess of >99% ee and 96% de, respectively. Thus, we succeeded in also developing the desired optimized work-up process, which was suitable for higher product loadings (>5 g L−1).
 |
| | Fig. 9 Different stages of purification of 12-OPDA crude product. (A) After extraction with EtOAc; (B) after extraction with EtOAc and filtration over charcoal; (C) after column chromatography. | |
Production of the biocatalyst for 12-OPDA at a 2 L-scale
So far, preparation of the biocatalyst and the bioprocess for 4 was done at the laboratory scale only. Because we anticipate an increased demand for cis-(+)-12-OPDA (cis-(+)-4) due to more applications for this intriguing compound, we were already interested in making the production process for the cost-critical whole-cell biocatalyst more efficient, and to demonstrate the scalability of such a technical fermentation process. Initially, we increased the volumetric productivity of the whole-cell catalyst by using a high-cell density fed-batch fermentation from 0.6 g L−1 h (15 g L−1; shake flask) to 2.5 g L−1 h (100 g L−1; fermenter) (Fig. 10). To achieve this, we increased the IPTG concentration to 1 mM due to the higher cell density during the process. We observed that a lower IPTG concentration (0.15 mM) resulted in a poorer overexpression of the AtAOS and AtAOC4 (data not shown). However, with the altered conditions, the SDS-PAGE showed an excellent overexpression of the AtAOC4 and a moderate expression of the AtAOS (Fig. 10). The activity and selectivity of the WCC obtained from the fermenter were just as good as those of the WCC from the shake flask (Fig. 11).
 |
| | Fig. 10 Overexpression of the fermented whole-cell catalyst. (L = lysate; CE = crude extract) (AtAOS: 56.1 kDa; AtAOC4: 23.6 kDa); for further details, see the ESI.† | |
 |
| | Fig. 11 Chemical and diastereomeric selectivity of the whole-cell catalyst produced in a shake flask or a fermenter (de = diastereomeric excess in %). | |
Conclusion
This work describes the successful optimization of the biocatalytic synthesis of cis-(+)-12-OPDA (cis-(+)-4) in terms of the chemical and diastereomeric selectivity of the whole-cell catalyst and the increase of the substrate loading. We achieved this by changing the whole-cell construction to a DUET-plasmid system, by reducing the reaction temperature, and by changing the stirrer. As a result, we were able to enhance the product formation-related conversion and achieve an excellent diastereomeric excess (>99% each) at a multigram scale (10 g L−1). In addition, it was discovered that the most effective solvent mixture for this reaction cascade is the previously published NH4Cl buffer (100 mM, pH 9) with 5% (v/v) ethanol. The use of phase-transfer catalysts resulted in a decrease in the Gm-13-LOX activity and α-ketol (5) formation. Increasing the substrate loading to 20 g L−1 resulted in a product formation-related conversion of 68%. With every increase of the substrate loading, the reaction time increased. The reasons for this are based on the Gm-13-LOX activity and can be explained by the feedback inhibition by 12-OPDA (4)22 and the substrate inhibition at high concentrations. Therefore, to optimize the process further, it will be beneficial to improve the activity of the Gm-13-LOX at high substrate 1 and product 4 concentrations by mutagenesis or by screening for another 13-LOX with the necessary properties. Furthermore, the foundations for the successful technical production of cis-(+)-12-OPDA (cis-(+)-4) in the future were successfully laid in the fed-batch fermentation of the optimized whole-cell catalyst (WCC) without any loss of chemical or diastereomeric selectivity. In future work, optimization of the column chromatography to enhance the recovery of 12-OPDA (4) and a gentler procedure for drying the product 4 to further avoid isomerization as well as a further increase of the substrate loading are among the major tasks.
Until now, the research and industrial applications of cis-(+)-12-OPDA (cis-(+)-4) were limited by the current high price of this molecule. This work will increase the availability of this attractive chemical cis-(+)-4 and allow a broader use of it in basic research but also in specific fields, e.g., agriculture,8–10 pharmaceuticals,2–4 and fragrances.11 Furthermore, this work might serve as the initial step towards a biocatalytic technical scale production of (−)-jasmonic acid and its derivatives, such as methyl jasmonate, starting from a renewable feedstock. Until now, this important compound is extracted from plants23 or produced by chemical synthesis.11
Experimental section
Materials
The substrate α-linolenic acid (1, 99% purity, ThermoFisher) was used as the raw material for the enzymic cascade, the 13-lipoxygenase from Glycine max (13-LOX; TCI, L0059; lyophilized powder; min. 100000 U mg−1) and all the solvents as well as the chemicals for the buffers used in this study were purchased commercially and used as received.
Enzyme sequence data, biocatalyst preparation and characterization
The genes were obtained from Twist Bioscience, (San Francisco, CA, USA). All the sequences, plasmid maps as well as the microbial procedures for the preparation and characterization of the E. coli whole-cell catalysts are given in the ESI.†
Optimized procedure for expression of the whole-cell catalyst
Terrific broth (TB) medium (1 L) in a 5 L Erlenmeyer flask with kanamycin (50 μg mL−1) and chloramphenicol (34 μg mL−1) was inoculated with an overnight preculture of E. coli BL21-CodonPlus(DE3)-RIL_pET28a-AtAOS-AtAOC4. Cultures were incubated at 37 °C and 180 rpm. With an OD600 of 0.6–0.7, the addition of IPTG to a final concentration of 0.05 mM starts the expression (20 h, 30 °C, 180 rpm). The cells were harvested by centrifugation (4 °C, 4000g, 30 min) and stored at −20 °C.
Optimized procedure for the one-pot synthesis of cis-(+)-12-OPDA (10 g L−1)
Ammonia chloride buffer (100 × 10−3 M, pH 9, 40 mL) with was mixed using a KPG stirrer (500 rpm) while cooling to 0 °C. Afterwards, the commercially available Gm-13-LOX (TCI, L0059, 15 mg) was dissolved in the cold buffer and added to the reaction mixture. The WCC1 (E. coli BL21-CodonPlus(DE3)-RIL_pET28a-AtAOS-AtAOC4, 3 g) was resuspended in ammonia chloride buffer (100 × 10−3 M, pH 9, 10 mL) and added to the flask as well. Ethanol (1.5 mL) was added and α-linolenic acid (1, 500 mg, 1.8 mmol) was dissolved in ethanol (1 mL) and added last. The three-necked flask was closed with septa, and oxygen was supplied to the flask's atmosphere (no bubbling) with a balloon (needle size: inlet = 0.9 × 50 mm; outlet = 0.5 × 40 mm). Control of the reaction was done by 1H-NMR. After full conversion, the reaction mixture was moved to a separating funnel and saturated NaCl solution (25 mL) was added. The mixture was acidified with HCl (2 M, 4 mL). This solution was extracted once with EtOAc (50 mL). The aqueous layer was separated, and the mixed layer was centrifuged (10000g, 10 min) for separation. The organic layer was taken off and MgSO4 was added to remove residual water. The solvent was removed by a rotary evaporator. The yellow-brown oil was further purified using automated reversed-phase column chromatography (MeCN, H2O, AcOH (0.1%)).
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank Nicolai Montua for technical support in the fermentation, and Lukas Hartmann, Sven Hügel, Bjarne Scharkowski and Alisa Maiwald for technical assistance. The authors gratefully acknowledge their financial support from the German Research Foundation (Deutsche Forschungsgemeinschaft; DFG; grant numbers: GR 3461/11-1 and DI 346/22-1).
References
- D.-S. Lee, P. Nioche, M. Hamberg and C. S. Raman, Nature, 2008, 455, 363–368 CrossRef CAS PubMed.
- N. Altiok, H. Mezzadra, P. Patel, M. Koyuturk and S. Altiok, Breast Cancer Res. Treat., 2008, 109, 315–323 CrossRef CAS PubMed.
- N. Taki-Nakano, J. Kotera and H. Ohta, Biochem. Biophys. Res. Commun., 2016, 473, 1288–1294 CrossRef CAS PubMed.
- N. Taki-Nakano, H. Ohzeki, J. Kotera and H. Ohta, Biochim. Biophys. Acta, 2014, 1840, 3413–3422 CrossRef CAS PubMed.
- M. Knieper, L. Vogelsang, T. Guntelmann, J. Sproß, H. Gröger, A. Viehhauser and K.-J. Dietz, Antioxidants, 2022, 11, 855 CrossRef CAS PubMed.
- C. Wasternack and B. Hause, Biol. Unserer Zeit, 2014, 44, 164–171 CrossRef CAS.
- A. Bergey, G. A. Howe and C. A. Ryan, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12053–12058 CrossRef PubMed.
- J. Wang, L. Song, X. Gong, J. Xu and M. Li, Int. J. Mol. Sci., 2020, 21, 1446 CrossRef CAS PubMed.
- Y. Yukimune, H. Tabata, Y. Higashi and Y. Hara, Nat. Biotechnol., 1996, 14, 1129–1132 CrossRef CAS PubMed.
- S. Deepthi and K. Satheeshkumar, Appl. Microbiol. Biotechnol., 2017, 101, 545–558 CrossRef CAS PubMed.
- C. Chapuis, Helv. Chim. Acta, 2012, 95, 1479–1511 CrossRef CAS.
- The product 12-OPDA is available at biomol with a purity of at least 95% (product number 88520, 1 mg: 299 EUR), https://www.biomol.com/de/produkte/chemikalien/lipide/12-oxo-phytodienoic-acid-cay88520-500?pk_campaign=Google-Ads&pk_cid=365745131&pk_kwd=&pk_source=google&pk_medium=cpc&pk_content=645986554986&gclid=Cj0KCQjwnMWkBhDLARIsAHBOftp3-r4NzVu7i32cMmuiP0ye1pJZjZLmc7Bj4R9zbMi6Vnsr2qXTEgYaAiiWEALw_wcB. (accessed: June 2023).
-
(a) M. Ernst and G. Helmchen, Angew. Chem., Int. Ed., 2002, 41, 4054–4056 CrossRef CAS;
(b) P. A. Grieco and N. Abood, J. Org. Chem., 1989, 54, 6008–6010 CrossRef CAS;
(c) D. Maynard, H. Gröger, T. Dierks and K. Dietz, J. Exp. Bot., 2018, 69, 5341–5354 CAS.
-
(a) I. Feussner and C. Wasternack, Annu. Rev. Plant Biol., 2002, 53, 275–297 CrossRef CAS PubMed;
(b) W. Liu and S.-W. Park, Front. Plant Sci., 2021, 12, 724079 CrossRef PubMed.
-
(a) C. Wasternack, Ann. Bot., 2007, 100, 681–697 CrossRef CAS PubMed;
(b) C. Delker, I. Stenzel, B. Hause, O. Miersch, I. Feussner and C. Wasternack, Plant Biol., 2006, 8, 297–306 CrossRef CAS PubMed.
- B. Vick and D. Zimmermann, Plant Physiol., 1979, 64, 203–205 CrossRef CAS PubMed.
-
(a) P. Zerbe, E. W. Weiler and F. Schaller, Phytochemistry, 2007, 68, 229–236 CrossRef CAS PubMed;
(b) D. Maynard, S. M. Müller, M. Hahmeier, J. Löwe, I. Feussner, H. Gröger, A. Viehhauser and K.-J. Dietz, Bioorg. Med. Chem., 2018, 26, 1356–1364 CrossRef CAS PubMed.
- J. Löwe, K.-J. Dietz and H. Gröger, Adv. Sci., 2020, 7, 1902973 CrossRef PubMed.
- M. Otto, C. Naumann, W. Brandt, C. Wasternack and B. Hause, Plants, 2016, 5, 3 CrossRef PubMed.
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed.
- S. A. Tatulian, J. Steczko and W. Minor, Biochemistry, 1998, 37, 15481–15490 CrossRef CAS PubMed.
- D. Maynard, K. Chibani, S. Schmidtpott, T. Seidel, J. Spross, A. Viehhauser and K.-J. Dietz, Int. J. Mol. Sci., 2021, 22, 10237 CrossRef CAS PubMed.
- R. A. Creelman and J. E. Mullet, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4114–4119 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Karl-Josef
Dietz
a,
Karl-Josef
Dietz