
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dess–Martin periodinane-mediated oxidation of the primary alcohol of cytidine into a carboxylic acid†
Alexandra R. E.
Serre
a,
Vibhu
Jha‡
b,
Adèle
Rivault
a,
Leif A.
Eriksson
 b,
Goreti
Ribeiro Morais
a and
Robert A.
Falconer
*a
b,
Goreti
Ribeiro Morais
a and
Robert A.
Falconer
*a
aInstitute of Cancer Therapeutics, Faculty of Life Sciences, University of Bradford, Bradford BD7 1DP, UK. E-mail: r.a.falconer1@bradford.ac.uk
bDepartment of Chemistry and Molecular Biology, University of Gothenburg, 405 30 Göteborg, Sweden
First published on 16th April 2024
Abstract
Herein the first example of conversion of alcohols into carboxylic acids by use of the Dess–Martin Periodinane (DMP), which is otherwise routinely employed for the conversion to aldehydes, is reported. This methodology will have significant potential utility in the synthesis of cytidine analogues and other related biologically important molecules.
Cytidine is a nucleoside that plays many roles in biological systems. It is a component of DNA, and also commonly features in enzyme substrates, such as cytidine monophosphate-sialic acid.1 Due to its essential biological importance, multiple analogues have been developed.2 In our endeavors to synthesise analogues of cytidine as potential glycosyltransferase inhibitors, we required introduction of an aldehyde at the 5′-alcohol of ribose as a synthetic intermediate. Based on its wide use as a mild and selective reagent to oxidise such alcohols to aldehydes, we selected DMP as a suitable reagent for this purpose. To our surprise, we found that DMP further oxidised the primary alcohol of cytidine to the carboxylic acid, rather than the expected aldehyde.
Dess–Martin Periodinane (DMP) is routinely used for the oxidation of alcohols to aldehydes and ketones.3 Albeit to a lesser extent, DMP has also been used in other synthetically oxidative transformations, such as: oxidative cascade cyclisation of anilidines with double bonds,4 synthesis of benzothiazole via oxidative cyclisation of thioformanilides,5 synthesis of 2-amino-1,4-benzoquinone-4-phenylimides from anilines,6 α-organosulfonyloxylation of ketones using DMP and organosulfonic acid,7 oxidative allylation of Morita–Baylis–Hillman adducts with allyltrimethylsilane promoted by DMP/BF3·OEt2,8 oxidation of amides and amines,9 oxidation of 2-pyridylseleno to form terminal alkenes,10 one-pot synthesis of trichloromethyl carbinols from primary alcohols,11 oxidation of terminal olefins to methyl ketones,12 oxidative rearrangement for the preparation of α-keto thioesters,13 and synthesis of thiazole-5-carbaldehyde via oxidative cascade annulation of potassium thiocyanate and tertiary enaminones.14 To the best of our knowledge, no oxidation of a primary alcohol to a carboxylic acid has been reported as within the scope of utility of DMP. Commonly, the carboxyl function is introduced at the 4′-position of cytidine and analogues via oxidation with platinum oxide and oxygen15 or iodobenzene diacetate in combination with TEMPO as catalyst.16–18
To avoid oxidation of the secondary alcohols at the 2′- and 3′-positions of ribose, cytidine was orthogonally protected by initial introduction of the tert-butyldimethylsilyl (TBS) group at the primary 5′-O-position, followed by protection of the diol, and subsequent selective hydrolysis of the TBS group. Compound 1a (Fig. 1) was then reacted with an excess of the recommended 1.1 eq. of DMP (2 eq.). This reaction did not proceed to completion, with conversion of alcohol 1a reaching a plateau of 80% after 6 h 30 min of the reaction (Table 1, entry 1 and Table S1A, Fig. S1A†). LCMS analysis also revealed some intriguing information. The expected mass of aldehyde 3a ([M + H]+ = 644) was not observed. Instead, two peaks were observed, both with a mass of 676 amu, which suggested the formation of the stable hemiacetal diastereoisomers 4a (Scheme 1 and Table S1, Fig. S1†). This could be the result of the reaction of the aldehyde function with the acidic methanol solution used as mobile phase during elution from the LCMS column. The conversion of aldehyde 3a into hemiacetal 4a was complete during the LCMS elution and appeared when the analytical sample was prepared both in methanol and acetonitrile (Table S2 and Fig. S2†).
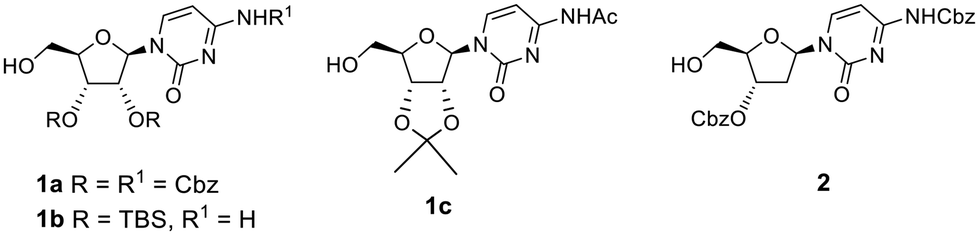 | ||
| Fig. 1 Chemical structures of synthesised cytidine analogues. | ||
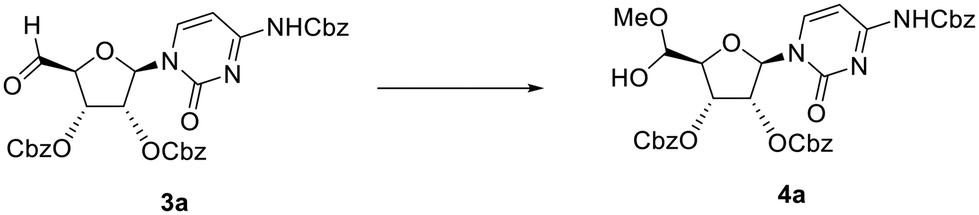 | ||
| Scheme 1 Proposed reaction observed during elution of aldehyde 3a in an LCMS experiment. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | DMP (eq.) | Solvent (mL) | Watera (eq.) | Productb | |
| Aldehyde | Carboxylic acid | ||||
| a All reactions were carried out under an air atmosphere (i.e. non-anhydrous). b Conversion was calculated based on the area under curve (AUC) of compounds in HPLC chromatograms (see ESI Fig. S4†). c Previously opened DMP stored at RT (>3 months). d Previously opened DMP stored at −20 °C (>3 months). e Water added to solvent before reaction. f Anhydrous solvent. | |||||
| 1 | 2c | CH2Cl2 | — | 80% | — |
| 2 | 4d | CH2Cl2 | — | 66% | 31% |
| 3 | 2 | CH2Cl2 | — | 68% | 32% |
| 4 | 4 | CH2Cl2 | — | 20% | 80% |
| 6 | 4d | CH2Cl2 | 100 μL (excess)e | — | — |
| 7 | 4d | CH2Cl2 | 10 μL (10 eq.)e | 80% | 10% |
| 8 | 4d | CH2Cl2 | 1 μL (1 eq.)e | 47% | 53% |
| 9 | 2d | CH2Cl2f | — | 80% | — |
| 10 | 4d | CH2Cl2f | — | 63% | 37% |
| 11 | 2d | CHCl3 | — | 89% | — |
| 12 | 4d | CHCl3 | — | 19% | 81% |
| 13 | 2d | CHCl3e | — | 81% | — |
| 14 | 4d | CHCl3e | — | 26% | 74% |
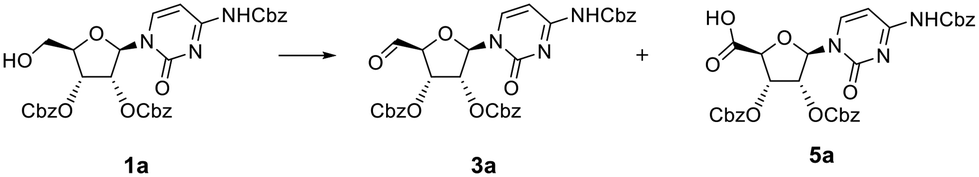
To improve the conversion of 1a to the aldehyde, the equivalents of DMP were increased from two to four (Table 1, entry 2). Surprisingly, while the majority of 1a was converted, the mass of a new peak observed by LCMS was found to be 660 amu (Table S1B and Fig. S1B†). To the best of our knowledge there are no reports using DMP to further oxidise aldehydes to a carboxylic acid. Based on the mass observed, and the increased polarity suggested by LCMS, however, it was hypothesised that the product formed during the oxidation with four eq. of DMP was the respective 5′-carboxylic acid 5a. NMR analysis of this product confirmed the absence of an aldehyde proton.
To confirm the presence of the carboxylic acid function in situ, a simple esterification of this product was carried out by stirring with acidic ion exchange resin (IR-120) in methanol. LCMS analysis indicated that after 4 h (Fig. 2) this compound had been partially converted into a new product with a mass of 674 amu. This conversion into the corresponding methyl ester 6a, as confirmed by 1H NMR, was complete after 72 h (Fig. 2). It is to be noted that the reactions were conducted using DMP reagent stored for several months at room temperature, and that the reaction was carried out using laboratory reagent grade dichloromethane. Concerned that the purity of the DMP material could have led to such reactions, repeats of the oxidation reaction with new batches of DMP – sourced from different suppliers (i.e. Aldrich, Fluorochem, Apollo Scientific) were carried out (Table 1, entries 3 and 4). After 6 h, the addition of four eq. DMP resulted in almost quantitative conversion of the alcohol into the carboxylic acid 5a (Table S1C and Fig. S1C†), whereas two eq. of DMP led to a mixture of aldehyde 3a and carboxylic acid 5a (as for entry 2) (Table S1B and Fig. S1B†). Surprisingly, our results showed that rather than being due to potential impurities, carboxylic acid formation seemed proportional to purity of DMP. To confirm this hypothesis, the purity of the batch of DMP used in this oxidation was evaluated by 1H NMR (Fig. S3†). 1H NMR analysis revealed degradation of DMP over time, with an apparent shift of the protons associated with the aryl moiety, which is perhaps not surprising since DMP is highly sensitive to moisture. Schreiber et al. discussed how the presence of impurities, catalysed by addition of water was sufficient to cause reaction rate acceleration, suggesting that partial hydrolysis of DMP led to a more efficient oxidising agent.19 The presence of water was therefore investigated in light of our findings. In our case, increasing the quantity of water led to reduced conversion to the carboxylic acid (see below), while a large excess of water (100 eq.) prevented the reaction completely (Table 1, entries 6–8 and Fig. S4†).
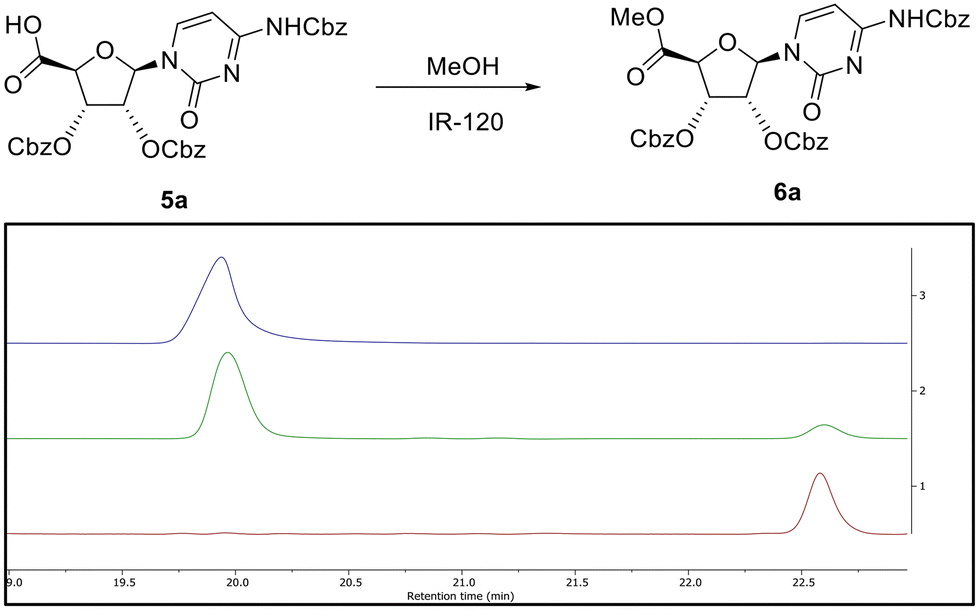 | ||
| Fig. 2 Reaction of the product 5a obtained from the oxidation of 1a with 4 eq. DMP, with acidic ion exchange resin (IR-120) in methanol. Respective HPLC chromatograms of starting material, potential acid carboxylic 5a (purple, upper) at RT = 20 min; reaction after 4 h, appearance of a new peak with mass of 674 amu (green, centre) at 22.5 min; reaction after 72 h, with complete disappearance of starting material (red, lower). | ||
Moreover, we found that LCMS analysis of the oxidation of 1a with a new batch of DMP led to another unexpected peak, albeit in small quantities, with a similar retention time and mass to the corresponding methyl ester 6a (Fig. 2). This additional peak was only observed when the LCMS sample was prepared in methanol (Table S2A and Fig. S2A†), while it was absent if the sample was prepared in acetonitrile (Table S2B and Fig. S2B†), showing that a reaction occurred during preparation of the LCMS sample rather than during sample elution. This additional peak was absent when DMP was previously quenched by extractive work-up (Table S3A and Fig. S5A†). These data indicated that 6a is only formed in the presence of methanol and DMP. Corey and Samuelsson20 demonstrated the one-step conversion of a five-membered carbohydrate furanose (uridine) primary hydroxyl group into carboxylic tert-butyl esters via the formation of a stable hemiacetal. Herein, we propose that a similar mechanism is involved in the formation of the methyl ester 6a in the LCMS vial, where the acetic acid that is released during the first alcohol oxidation acts as the catalyst to allow nucleophilic attack of the methanol on the aldehyde to form the respective hemiacetal, which is then further oxidised into the methyl ester by DMP (or derivative) (Scheme 2A). The formation of 6a, when the LCMS sample contained both DMP and methanol, suggested a similar mechanism for the formation of the 5′-carboxyl cytidine 5avia oxidation by the DMP of a hydrate intermediate (Scheme 2B). It has been demonstrated that water, in the presence of an acid or a base, adds rapidly to the carbonyl group of aldehydes and ketones establishing a reversible equilibrium with an aldehyde hydrate.21 In our case, the mass of the respective cytidine hydrate intermediate ([M + H]+ = 662) could be observed in the LCMS analysis of the oxidation of 1a with two eq. of DMP when the aqueous methanol of the mobile phase was replaced by aqueous acetonitrile (Fig. S6†). In this case, in the absence of methanol, water acts as a nucleophile and the resulting hydrate was stable and visible in the LCMS chromatogram. As stated previously, the reactions were carried out with laboratory reagent grade solvents, and it seemed that to form this stable aldehyde hydrate, water present in the solvent was sufficient.
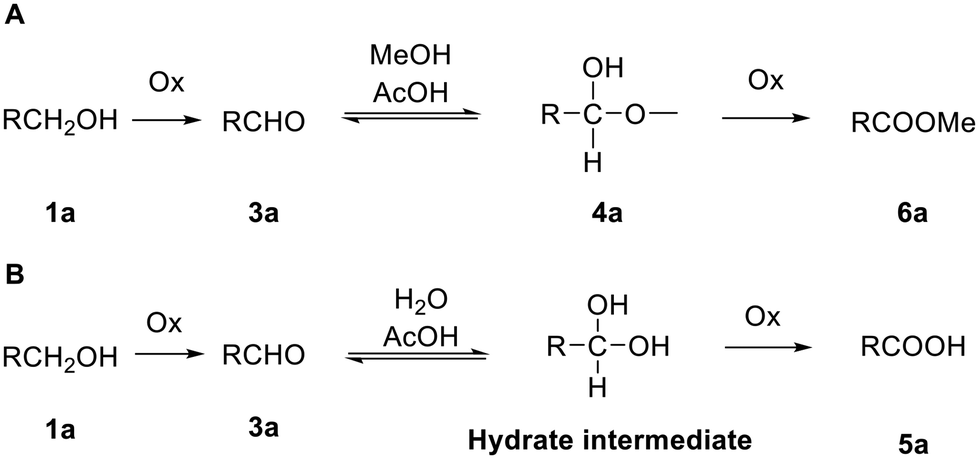 | ||
| Scheme 2 (A) Proposed mechanism for esterification observed (DMP or derivatives oxidise the primary alcohol 1a to form aldehyde 3a, which is in equilibrium with hemiacetal 4a, as observed by LCMS, which can subsequently be oxidised to form 6a); (B) proposed mechanism for formation of carboxylic acid 5a, via oxidation of hydrate intermediate. | ||
Moreover, the reaction was repeated with anhydrous dichloromethane under a normal air atmosphere, using either 2 eq. or 4 eq. of DMP (Table 1, entries 9, 10 and Fig. S4†). The results were similar to those observed with reagent grade solvent, confirming that the moisture present in the air is sufficient to catalyse the reaction (Table 1, entries 1 and 2 compared to entries 9 and 10, Fig. S4†); no glove box or drying reagents were used.
To confirm the proposed mechanism and understand the limitations of this reaction, protected cytidine 1a was reacted with previously opened DMP (4 eq.) with different quantities of water. As discussed earlier, 100 eq. of water prevented any reaction, while 10 eq. of water promoted formation of the aldehyde 3a (seen as hemiacetal diastereoisomers in LCMS) as the dominant product, and 1 eq. of water rendered an approximately equal proportion of carboxylic acid and aldehyde (Table 1, entries 6–8 and Fig. S4†). This showed that the presence of an excess of water in solution (>1 eq.) prevented further oxidation of aldehyde to the carboxylic acid, possibly due to inactivation of the oxidant, though the addition of a small amount of water promoted carboxylic acid formation. However, as Schreiber et al. demonstrated, DMP is most likely converted in situ into acetoxyiodinaneoxide.19 Indeed, in their study 1 μL of water per mL of dichloromethane was sufficient for hydrolysis.
Finally, reactions were repeated in both anhydrous and reagent grade chloroform (Table 1, entries 11–14 and Fig. S4†). DMP degradation by-products commonly precipitate from chloroform, leaving a larger percentage of DMP in solution. Thus, performing the oxidation in chloroform could be quite different than that observed using dichloromethane, particularly when employing aged batches of DMP. In a similar manner to that observed with dichloromethane, the use of laboratory reagent grade or anhydrous chloroform did not impact the outcome of the reaction. As predicted, however, the use of chloroform clearly drove the reaction towards the carboxylic acid, demonstrating the importance of DMP for the over-oxidation.
Altogether, this evidence seemed to corroborate the proposed mechanism of the reaction, in which DMP (or acetoxyiodinaneoxide) most likely has a dual role: it oxidises the alcohol into the aldehyde and then, when combined with acetic acid and residual water, allows further oxidation of the generated hydrate into the carboxylic acid (Scheme 2B).
To investigate which factors (electronic/steric) might stabilise the nucleoside hydrate and influence this unusual finding, computational chemistry was used. Geometries of compounds 1a and the hydrate intermediate of 3a, were optimised in the ground state using density functional theory (DFT) with the M06-2X functional and the 6-31G(d,p) basis set. As shown in Fig. S7,† the low-energy 3D structures of 1a and the hydrate intermediate of 3a show significant changes in their geometries after the DFT calculation was finished, as they do not superimpose at all. In particular, the two phenyl rings from the benzyl carbonate (Cbz)-protected diol of cytidine analogue 1a and the hydrate intermediate of 3a exhibit π–π stacking interactions (Fig. S7C and D†) that are not observed with their initial coordinates. The notable conformational changes and the maintenance of π–π stacking in 1a could correspond to further oxidation of aldehyde to carboxylic acid by the virtue of its hydrate intermediate formation. The presence of conformational changes/π–π stacking within 1a and the hydrate intermediate of 3a are thus hypothesised to drive the oxidation of aldehyde to carboxylic acid. The DFT-optimised coordinates of each of the aforementioned structures were subjected to 200 ns molecular dynamics (MD) simulations, with the purpose of further verifying their conformational dynamics and obtaining useful insights into aldehyde/carboxylic acid product formation. Root mean square deviation (RMSD) was also calculated during the course of the simulation, to measure ligand mobility, starting from the DFT-optimised structures. The Cbz-protected cytidine analogue 1a demonstrated an average RMSD of 2.8 Å, which was in good correlation with the aforementioned conformational changes (Fig. S8†). MD analysis of 1a confirmed the conformational changes identified in the DFT calculations, in particular, the π–π stacking interactions were abundantly observed between the cytidine ring and one of the Cbz-phenyl rings, and between the two phenyl rings of Cbz throughout the simulation (Fig. S9†). Similarly, the hydrate intermediate of 3a showed an average RMSD of 2.8 Å (Fig. S8†) and exhibited conformational changes within the phenyl rings of Cbz, establishing π–π stacking interactions, notably present over the course of simulation (Fig. S10†).
To confirm that the π–π stacking interaction could be responsible for stabilizing the aldehyde hydrate formed, three model compounds 1b, 1c and 2 that lack this interaction were used (Fig. 1). First, we attempted to oxidise the primary alcohol of deoxycytidine analogue 2 (Fig. 1), in which the 3′-hydroxyl was also protected as benzyl carbonate group, but it represented a less hindered analogue with more conformational flexibility. Oxidation of 2 (Table 2, entries 1 and 2) followed the same trend as for compound 1a, and a mixture of the respective aldehyde 7 (detected as methyl hemiacetal 10) and carboxylic acid 8 was obtained with two eq. of DMP (Table S4A and Fig. S11A†). Full conversion into carboxylic acid 8 was observed with four eq. of DMP (Table S4B and Fig. S11B†). We also studied the reactivity of other cytidine analogues in which the diol was protected either as silyloxy (1b) or acetonide (1c) derivatives (Fig. 1). Protection as the acetonide was expected to also induce conformational rigidity in the ribose ring, which would ultimately restrict the flexibility of the primary alcohol of 1c, and its reactivity. Indeed, and contrary to the oxidation of the cytidine 1a, the protection of the diol as an acetonide rendered the analogue less prone to further oxidation; the primary alcohol was completely converted into aldehyde 3b, using two eq. of DMP within the same reaction time (Table 2, entry 3), and no carboxylic acid analogue was identified in the LCMS analysis (Table S5A and Fig. S12A†). The acetonide protecting group also rendered the aldehyde more stable under purification conditions and long-term storage. We observed that although we could see the aldehyde proton in the 1H NMR spectrum of the crude of product 3a, attempts to purify it by column chromatography led to several unidentified decomposition by-products. Aldehyde 3b was successfully purified and characterised, however. Likewise, for aldehydes 3a and 7, LCMS analysis of aldehyde 3c revealed two peaks in the UV chromatogram, both with a mass of 356 amu that corresponded to the respective hemiacetal diastereoisomers, while 1H NMR analysis of the pure compound showed the aldehyde proton at 9.39 ppm (Fig. S13A†).
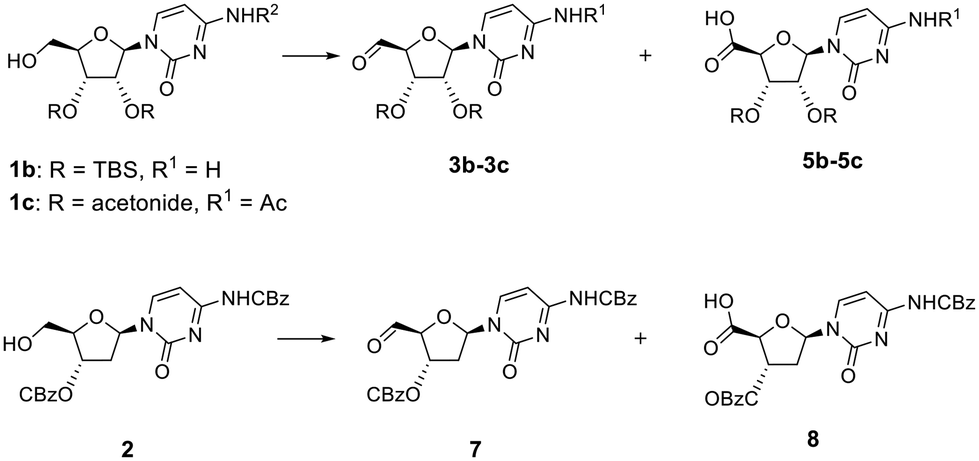
Interestingly, the addition of methanol and silica to the crude sample of oxidation of 1c, followed by solvent concentration at 40 °C and column chromatography led to partial formation of the hemiacetal, which co-eluted with the aldehyde. NMR analysis of this mixture showed that in addition to the aldehyde proton at 9.39 ppm, two more broad peaks were observed at 9.59 and 9.88 ppm, along with two singlets at 3.48 and 3.51 ppm (Fig. S13B†), corresponding to the hydroxyl and methoxy groups of the methyl hemiacetal 4c, respectively. When the amount of DMP was increased to four eq., LCMS analysis showed that oxidation of cytidine 1c led to a mixture of the aldehyde 3c, and the respective carboxylic acid 5b with a mass of [M + H]+ = 340 (Table 2, entry 4 and Table S5B, Fig. S12B†). As shown in Fig. S14,† the initial geometry of cytidine analogue 1c does not change after the DFT-based optimisation as they demonstrated a perfect superposition, which might correlate with a decreased ability to undergo oxidation to the carboxylic acid under the same conditions as compound 1a. When the diol was protected by TBS groups, the addition of two eq. of DMP led to formation of the aldehyde 3b (Table 2, entry 5 and Table S6A, Fig. S15A†), albeit with a slight contamination of the carboxylic acid 5b. However, oxidation of 1b with four eq. of DMP completely favoured the formation of the carboxylic acid 5b (Table 2, entry 6 and Table S6B, Fig. S15B†).
Absence of π–π stacking interactions did not prevent carboxylic acid formation. Computational chemistry also highlighted that unlike the exclusive presence of intramolecular π–π stacking only with 1a and the hydrate intermediate of 3a, the H-bond interaction between the primary alcohol and the cytidine carbonyl consistently existed for 74%, 73% and 86% of the simulation time for 1a, hydrate intermediate of 3a, and 1c, respectively (Fig. S16†). If one hydroxyl of a hydrate intermediate is involved in an intramolecular hydrogen bonding interaction, the nucleophilicity of the geminal hydroxyl would be expected to increase, thereby making it more reactive toward the electrophilic DMP (or derivative). Therefore, methyl ribosides 11 and 12 were chosen as not containing any type of π–π stacking or H-bond interactions. They were prepared and reacted with two and four eq. of DMP for 6 h 30 min (Table 3). Compound 11 was not easily monitored by UV absorbance, so its oxidation was monitored by 1H NMR analysis of the crude product (Fig. S17†) and compared with reported spectra for compound 13 and 14.22,23 The outcome of the reaction was similar to the oxidation of compound 1c, with no 5-carboxylic acid methyl ribose obtained when 11 was reacted with 2 eq. of DMP: total conversion to aldehyde 13 was observed (Fig. S15B†). Comparatively, with addition of 4 eq. of DMP (Fig. S15C†), NMR analysis highlighted the presence of the aldehyde in a 50/50 mixture, similar than the ratio of 42/58 (carboxylic acid/aldehyde) previously reported for oxidation of compound 1c. Oxidation of riboside 12, in which the 2,3-diol is protected as benzyl carbonate, promoted carboxylic acid 16 in also similar percentage with two eq. of DMP (Table S7 and Fig. S18A,† 43%) and four eq. (Table S7 and Fig. S18B,† 81%) compared to compound 1 (Table S1 and Fig. S1,† 32% and 80% respectively).
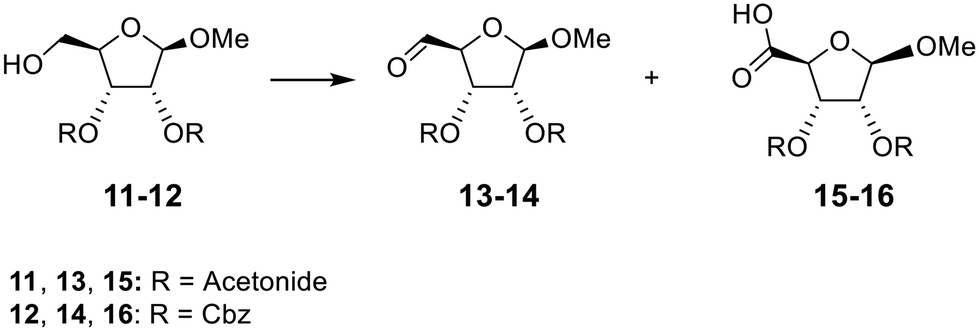
Successful oxidation of compounds 11 and 12 suggested that the π–π stacking and H-bond interactions are not the key factors that stabilise the hydrate, and that some other factors must promote the conversion to a carboxyl group. The particular 5-membered ring conformation would simply allow formation and stabilisation of the hydrate intermediate (which could be observed by LCMS during optimisation of 1a, Fig. S6†), from which the alcohol can further oxidise as classical DMP reaction.
In summary, this methodology affords the 4′-carboxylic acid of cytidine in comparable yields (around 80%) to previously reported methods. However, it poses the advantage that there is no need to use oxygen gas, which is not routinely available in a medicinal chemistry laboratory. Although iodobenzene diacetate has been shown to be an efficient oxidant for the preparation of similar end products, it additionally requires the use of TEMPO as catalyst, whereas in the present study DMP alone as oxidant is sufficient. Also, based on our proposed mechanism we expect that the DMP-mediated oxidation of primary alcohols to carboxylic acids is likely to be more broadly applicable to other nucleosides or compounds in which geminal diols can be stabilised.
To conclude, we observed that DMP fully oxidised the primary alcohol of cytidine into a carboxylic acid depending on 3 factors: (i) the purity of DMP, (ii) the quantity of DMP used in the reaction, and (iii) the protecting group(s) used for the diol of the ribose sugar. This unexpected and not previously reported observation is dependent on the ability of the aldehydes of the carbohydrates to be converted into a stable geminal diol. We have shown the importance of protecting groups that can impact the rate of conversion. We present methodologies to selectively oxidise the primary alcohol of cytidine analogues to either the aldehyde or the carboxylic acid, by careful choice of protecting groups and reaction conditions (see Experimental in ESI†). Given the importance of cytidine in biological systems and in potential therapeutically important molecules, these findings are of importance in the synthesis of cytidine analogues. The findings reported herein provide new opportunities to readily synthesise novel 5′-carboxyl derived cytidine analogues, using mild conditions that overcome the problems seen with traditional oxidation methods.
Author contributions
RAF and GRM supervised the research; RAF, AS and GRM conceived the idea; AS performed the research and data analysis (LCMS, NMR), with AR contributing towards data acquisition; VJ performed the computational chemistry studies, under the guidance of LAE; AS prepared the manuscript with contributions from RAF, GRM, VJ and LAE. All authors approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AS and GRM were supported by the Institute of Cancer Therapeutics Doctoral Training Centre, University of Bradford (RAF). VJ was supported by the Wenner-Gren Foundation. LAE thanks the Swedish Science Research Council (VR; grant number 2019-3684) and the Swedish Cancer Foundation (grant number 211447-Pj) for funding for the computational studies. The National Academic Infrastructure for Supercomputing in Sweden (NAISS) is also acknowledged for allocations of computing time at the National Supercomputing Center (NSC) in Linköping. The authors thank Arianna Dell'Albani, Dr Helen Sheldrake and Dr David Pajtas for useful discussions. Haseeb Ul-Rehman, University of Bradford Analytical Centre, is acknowledged for providing HRMS measurements.Notes and references
- A. Muenster, M. Eckhardt, B. Potvin, M. Muehlenhoff, P. Stanley and R. Gerardy-Schahn, Mammalian Cytidine 5′-Monophosphate N-Acetylneuraminic Acid Synthetase: A Nuclear Protein with Evolutionarily Conserved Structural Motifs, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(16), 9140–9145 CrossRef PubMed.
- R. Szabo and D. Skropeta, Advancement of Sialyltransferase Inhibitors: Therapeutic Challenges and Opportunities, Med. Res. Rev., 2017, 37(2), 219–270 CrossRef PubMed.
- V. V. Zhdankin, Organoiodine(V) Reagents in Organic Synthesis, J. Org. Chem., 2011, 76(5), 1185–1197 CrossRef CAS.
- K. C. Nicolaou, P. S. Baran, Y. L. Zhong and K. Sugita, Iodine(V) Reagents in Organic Synthesis. Part 1. Synthesis of Polycyclic Heterocycles via Dess–Martin Periodinane-Mediated Cascade Cyclization: Generality, Scope, and Mechanism of the Reaction, J. Am. Chem. Soc., 2002, 124(10), 2212–2220 CrossRef CAS PubMed.
- D. S. Bose and M. Idrees, Hypervalent Iodine Mediated Intramolecular Cyclization of Thioformanilides: Expeditious Approach to 2-Substituted Benzothiazoles, J. Org. Chem., 2006, 71(21), 8261–8263 CrossRef CAS PubMed.
- H. C. Ma and X. Z. Jiang, Novel Synthesis of 2-Amino-1,4-benzoquinone-4-phenylimides from Anilines via Dess-Martin Periodinane Oxidation, Synlett, 2007, 1679–1682 CAS.
- U. S. Mahajan and K. G. Akamanchi, A New Application of Hypervalent Iodine (λ5) Reagents with Organosulfonic Acids for Direct α-Organosulfonyloxylation Carbonyl Compounds, Synlett, 2008, 987–990 CAS.
- J. S. Yadav, B. V. Subba Reddy, A. P. Singh and A. K. Basak, One-Pot Oxidative Allylation of Morita-Baylis-Hillman Adducts with Allyltrimethylsilane Promoted by Dess-Martin Periodinane/Boron-Trifluoride-Diethyl Ether Complex, Synthesis, 2008, 469–473 CrossRef CAS.
- K. C. Nicolaou and C. J. N. Mathison, Synthesis of Imides, N-Acyl Vinylogous Carbamates and Ureas, and Nitriles by Oxidation of Amides and Amines with Dess-Martin Periodinane, Angew. Chem., Int. Ed., 2005, 44(37), 5992–5997 CrossRef CAS PubMed.
- T. Andreou, J. Burés and J. Vilarrasa, Reaction of Dess–Martin periodinane with 2-(alkylselenyl)pyridines. Dehydration of primary alcohols under extraordinarily mild conditions, Tetrahedron Lett., 2010, 51(14), 1863–1866 CrossRef CAS.
- M. K. Gupta, Z. Li and T. S. Snowden, One-Pot Synthesis of Trichloromethyl Carbinols from Primary Alcohols, J. Org. Chem., 2012, 77(10), 4854–4860 CrossRef CAS.
- D. A. Chaudhari and R. A. Fernandes, Hypervalent Iodine as a Terminal Oxidant in Wacker-Type Oxidation of Terminal Olefins to Methyl Ketones, J. Org. Chem., 2016, 81(5), 2113–2121 CrossRef CAS PubMed.
- R. Sanichar, C. Carroll, R. Kimmis, B. Reiz and J. C. Vederas, Dess-Martin periodinane oxidative rearrangement for preparation of α-keto thioesters, Org. Biomol. Chem., 2018, 16(4), 593–597 RSC.
- K. Chen, B. Zhao, Y. Liu and J.-P. Wan, Thiazole-5-carbaldehyde Synthesis by Cascade Annulation of Enaminones and KSCN with Dess–Martin Periodinane Reagent, J. Org. Chem., 2022, 87(21), 14957–14964 CrossRef CAS PubMed.
- K.-H. Kim, J.-Y. Kim, K.-H. Lee, M.-J. Noh, Y.-C. Kim and H.-J. Park, Synthesis and biological activity of the new 5-fluorocytosine derivatives, 5′-deoxy-N-alkyloxycarbonyl-5-fluorocytosine-5′-carboxylic acid, Bioorg. Med. Chem. Lett., 2002, 12(3), 483–486 CrossRef CAS PubMed.
- Y. Xu, H. Jin, Z. Yang, L. Zhang and L. Zhang, Synthesis and biological evaluation of novel neamine–nucleoside conjugates potentially targeting to RNAs, Tetrahedron, 2009, 65(27), 5228–5239 CrossRef CAS.
- O. Moukha-Chafiq, R. C. Reynolds, J. C. Wilson and T. S. Snowden, Parallel Solution Phase Synthesis and Preliminary Biological Activity of a 5′-Substituted Cytidine Analog Library, ACS Comb. Sci., 2019, 21(9), 628–634 CrossRef CAS PubMed.
- R. Kumar, R. Nasi, M. Bhasin, N. Huan Khieu, M. Hsieh, M. Gilbert, H. Jarrell, W. Zou and H. J. Jennings, Sialyltransferase inhibitors: consideration of molecular shape and charge/hydrophobic interactions, Carbohydr. Res., 2013, 378, 45–55 CrossRef CAS PubMed.
- S. D. Meyer and S. L. Schreiber, Acceleration of the Dess-Martin Oxidation by Water, J. Org. Chem., 1994, 59(24), 7549–7552 CrossRef CAS.
- E. J. Corey and B. Samuelsson, One-step conversion of primary alcohols in the carbohydrate series to the corresponding carboxylic tert-butyl esters, J. Org. Chem., 1984, 49(24), 4735–4735 CrossRef CAS.
- J. L. Kurz, Hydration of acetaldehyde. I. Equilibrium thermodynamic parameters, J. Am. Chem. Soc., 1967, 89(14), 3524–3528 CrossRef CAS.
- C. Gravier-Pelletier, M. Milla, Y. L. Merrer and J.-C. Depezay, Liposidomycins – Synthetic Studies Towards the Ribosyldiazepanone Moiety, Eur. J. Org. Chem., 2001, 3089–3996 CrossRef CAS.
- L. Huang, N. Teumelsan and X. Huang, A Facile Method for Oxidation of Primary Alcohols to Carboxylic Acids and Its Application in Glycosaminoglycan Syntheses, Chem. – Eur. J., 2006, 12(20), 5246–5252 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00240g |
| ‡ Current address: Institute of Cancer Therapeutics, University of Bradford, UK. |
| This journal is © The Royal Society of Chemistry 2024 |