
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 3,5-disubstituted isoxazoles by domino reductive Nef reaction/cyclization of β-nitroenones†
Muhammad Ehtisham Ibraheem
Khan
 ,
Tomas Lighuen
Cassini
,
Marino
Petrini
and
Alessandro
Palmieri
*
,
Tomas Lighuen
Cassini
,
Marino
Petrini
and
Alessandro
Palmieri
*
University of Camerino, ChIP Research Center, Via Madonna delle Carceri, 62032 Camerino, MC, Italy. E-mail: alessandro.palmieri@unicam.it
First published on 2nd April 2024
Abstract
β-Nitroenones can be efficiently converted into 3,5-disubstituted isoxazoles by using tin(II)chloride dihydrate and ethyl acetate as a reducing agent and solvent, respectively. Products are obtained in good yields and several functional groups are tolerated thanks to the mild reaction conditions.
Introduction
Isoxazoles constitute one of the most important classes of nitrogen–oxygen containing five-membered heterocyclic systems, as the isoxazole ring is classified as a privileged structure.1 Indeed, their derivatives exhibit an extensive range of biological activities due to the broad spectrum of protein targets by which the isoxazole compounds could interact.2 In particular, the anticancer, antibacterial, antimicrobial, antiviral, and antituberculosis activities are just some of the medicinal properties of isoxazole derivatives (Fig. 1).3 Moreover, the isoxazole scaffold is also a key starting material to access more complex molecular architectures.4 | ||
| Fig. 1 Examples of biologically active isoxazole derivatives. | ||
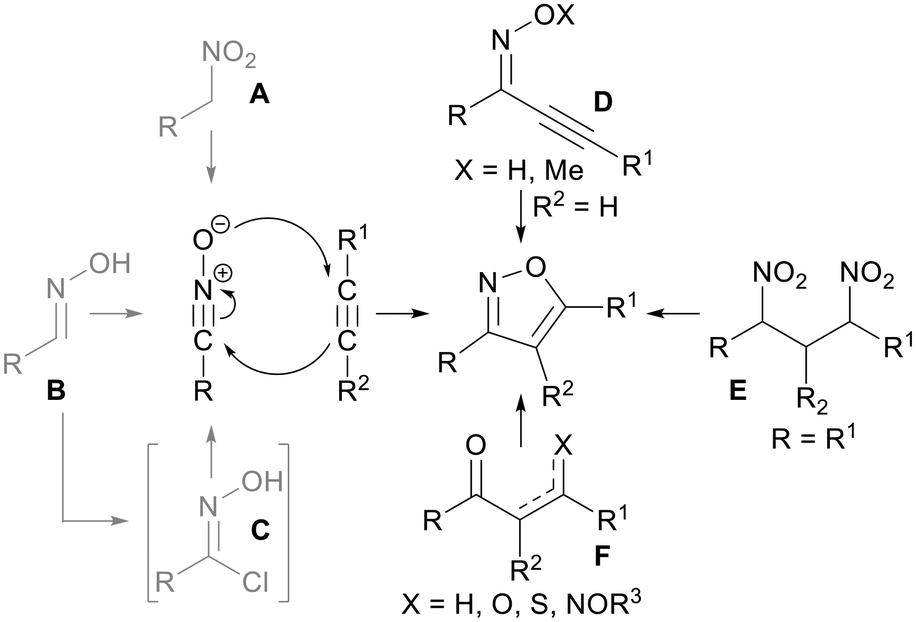
Due to their importance, several procedures for synthesizing poly-functionalized isoxazoles are reported in the literature (Scheme 1).5 The most effective routes are pivoted on 1,3-dipolar cycloaddition reactions of electron-rich alkynes and nitrile oxides, which are generated in situ from nitroalkanes A or oximes B. Although the conversion of A into nitrile oxides is quite effective, it usually requires harsh reaction conditions and/or dangerous and toxic reagents such as POCl3, phenyl isocyanate, PPA and DMTMM.6 On the other hand, B can be directly converted into nitrile oxides by oxidizing agents such as hypervalent iodine reagents, NaOCl and Oxone®,7 or alternatively via imidoyl chlorides C by a chlorination and base-promoted elimination approach.8 Despite the efficiency of this synthetic approach, an appropriate regiochemical control in the isoxazole formation is not always achievable using unsymmetrical alkynes. Alternative methods involving intramolecular cyclization of specific structures were also exploited. In this context, α,β-acetylenic oximes D are suitable for cyclization into isoxazoles by metal catalysis (e.g. AuCl3, CuCl, Pd(OAc)2).9 Similarly dinitro compounds E are easily converted into the isoxazole core under mild basic conditions; nonetheless, R and R1 must be identical in order to prevent the formation of regioisomeric products.10 Finally, 1,3-dielectrophiles F can be used as starting materials for functionalized isoxazoles; nevertheless, even in this case an appropriate selection of the starting scaffold is crucial to prevent regioisomeric issues.8,11
 | ||
| Scheme 1 Main synthetic routes to prepare isoxazoles. | ||
β-Nitroenones are a subclass of nitroolefins featuring the simultaneous presence of the ketone and nitro group in α- and β-positions to the double bond, respectively (Fig. 2).12 The presence of both electron-withdrawing groups dramatically enhances the reactivity and the synthetic versatility of β-nitroenones, making them useful precursors of highly functionalized materials and heterocyclic systems.13
 | ||
| Fig. 2 General structure of β-nitroenones. | ||
Following our ongoing research on the chemistry of β-nitroenones 1 and inspired by the pioneering studies by Wieland at the very beginning of the last century, and by Viel in 1979 (limited to α-acyl β-nitrostilbenes),14 we have now realized a general and efficient conversion of 1 into 3,5-disubstituted isoxazoles 2. The protocol is based on the preliminary reductive Nef reaction of the nitroalkene 1 to the corresponding oxime I,15 which intramolecularly reacts with the carbonyl function generating the five-membered ring II, which upon dehydration finally affords the isoxazole system 2 (Scheme 2).
 | ||
| Scheme 2 Probable reaction mechanism. | ||
Results and discussion
To find the best reaction conditions, we profiled the conversion of 1a into 2a in terms of solvent, temperature, reaction time, stoichiometry and reducing species (Table 1). In particular, the known ability of tin(II) chloride dihydrate to convert nitroalkenes into oximes16 prompted us to test the reaction using 2 equivalents of this salt in different solvents at room temperature (entries a–e). The reaction was completely ineffective when using dichloromethane (DCM) and toluene, while moderate yields of 2a were obtained in dioxane, 2-methyltetrahydrofuran (2-MeTHF) and ethyl acetate. Having identified 2-MeTHF and EtOAc as the most promising solvents for the reaction, we screened different reaction temperatures.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time (h) | Yieldf (%) of 2a |
| a Reaction performed in the presence of 2 eq. of SnCl2·2H2O. b Reaction performed under microwave conditions. c Reaction performed in a sealed vial using a conventional sand bath heater. d Reaction performed in the presence of 1.5 eq. of SnCl2·2H2O. e Reaction performed in the presence of 2.5 eq. of SnCl2·2H2O. f Yield of the pure isolated product. | ||||
| a | 2-MeTHF | r.t.a | 8 | 34 + 1a |
| b | Dioxane | r.t.a | 8 | 8 + 1a |
| c | DCM | r.t.a | 8 | 1a |
| d | Toluene | r.t.a | 8 | 1a |
| e | EtOAc | r.t.a | 8 | 32 + 1a |
| f | 2-MeTHF | 50a | 8 | 42 + 1a |
| g | 2-MeTHF | Refluxa | 2 | 69 |
| h | 2-MeTHF | 90 °Ca,b | 2 | 74 |
| i | EtOAc | 50a | 8 | 44 + 1a |
| j | EtOAc | Refluxa | 2 | 70 |
| k | EtOAc | 90 °Ca,b | 2 | 79 |
| l | EtOAc | 90 °Ca,c | 2 | 73 |
| m | EtOAc | 90 °Cb,d | 2 | 50 |
| n | EtOAc | 90 °Cb,e | 2 | 77 |

At 50 °C, we recorded similar results compared to that when conducting the reaction at room temperature, and in fact 2a was obtained just in a slightly better yield (entries f and i). Successively, we increased the reaction temperature to reflux conditions, observing for both reactions the complete consumption of 1a over two hours and isolating 2a in 69% and 70% yields, respectively (b.p.: 2-MeTHF = ∼80 °C; EtOAc = ∼77 °C; entries g and j). Finally, by means of a Biotage® Initiator microwave synthesizer, we further increased the temperature to 90 °C. Under these conditions, we attained a cleaner reaction mixture and higher yields, and particularly the best yield (79%) was observed when conducting the reaction in EtOAc (entries f–k). This result probably depends on the more efficient energy transfer occurring under microwave irradiation than with the conventional heating technique.17 In this regard, we repeated the reaction at 90 °C in a sealed vial and by means of a sand bath heater; however, 2a was isolated in a lower yield (entry l). It is important to note that the lowering of the amount of tin(II) chloride dihydrate from 2.0 to 1.5 equivalents leads to a significant decrease of the yield, while an increase up to 2.5 equivalents does not improve the yield (entries m and n). Finally, the conversion of 1a into 2a was also attempted using FeCl3, FeCl2·4H2O, CuCl and CrCl2 under the optimized reaction conditions; nevertheless, only chromium(II) chloride led to the isolation of 2a, albeit in a very poor yield (8%).
In order to demonstrate the generality of our protocol we tested the optimized reaction conditions with a number of β-nitroenones 1a–n. In all cases, the target isoxazoles 2a–n were obtained in good to very good yields (55–91%). Moreover, thanks to the mildness of the reaction conditions, a good variety of functionalities, such as chlorine, ester, thiophene, ether, and double and triple bonds, can be embedded in the substrate (Table 2).
|
|
|||
|---|---|---|---|
| R | R1 | Isoxazole 2 | Yielda (%) |
| a Yield of the pure isolated product. b Reaction performed under flow chemical conditions. | |||
| Et | Ph | 2a | 79, 84b |
| MeOOC(CH2)5 | Ph | 2b | 55, 68b |
| CH3(CH2)6 | 3-MeO–C6H4 | 2c | 80, 86b |
| AcO(CH2)3 | 4-MeO–C6H4 | 2d | 85 |
CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)4 CH(CH2)4 |
2-Thienyl | 2e | 78 |
CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(CH2)3 C(CH2)3 |
2-Thienyl | 2f | 79 |
| CH3(CH2)6 | 2-Naphthyl | 2g | 83 |
| CH2CH(CH2)3 |
2-Naphthyl | 2h | 91 |
| PhCH2CH2 | 2-Naphthyl | 2i | 72 |
| Cl(CH2)4 | 4-Me–C6H4 | 2j | 82 |
| CH3(CH2)4 | 4-Me–C6H4 | 2k | 85 |
| Et | 3-(N-Methylindolyl) | 2l | 84 |
| Ph | 4-MeO–C6H4 | 2m | 79 |
| CH3(CH2)4 | t-Bu | 2n | 62 |
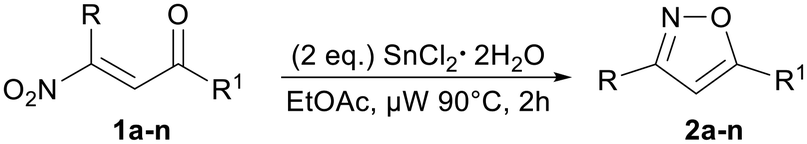
Finally, with the aim of automating the process, we investigated the conversion of β-nitroenones 1a–c under flow chemical conditions by means of a FlowLab™ system of Uniqsis.18 The equipment consists of two HPLC pumps, two reservoirs respectively filled with the ethyl acetate solutions of β-nitroenones 1 (reservoir A) and SnCl2·2H2O (reservoir B), a T-connector T, a heated reactor station equipped with a 10 mL PTFE coil reactor R and a back pressure regulator (BPR) set at 40 psi (Fig. 3).
 | ||
| Fig. 3 Scheme of the flow chemical equipment. | ||
Applying the batch optimized reaction conditions to the flow chemical approach (90 °C and 2 hours as residence time), the target isoxazoles 2a–c were isolated in higher yields than in batch, presumably due to the superior efficiency in the mass and energy exchanges of the flow approach with respect to the batch one.19
Conclusions
In conclusion, we have further demonstrated the usefulness of β-nitroenones as key precursors of privileged structures in medicinal chemistry. In particular, herein we developed a new general and simple protocol for converting β-nitroenones into 3,5-disubstituted isoxazoles under microwave conditions. The protocol allows the preparation of the title compounds in very good yields, under mild reaction conditions compatible with different functional groups. Moreover, the extension of our methodology to flow chemical conditions enables yield improvement and easy process-automation.Experimental
General remarks
1H NMR analyses were performed at 400 MHz on a Varian Mercury Plus 400. 13C NMR analyses were carried out at 100 MHz. IR spectra were recorded with a PerkinElmer FTIR spectrometer Spectrum Two UATR. Microanalyses were performed with a CHNS-O analyzer Model EA 1108 from Fison Instruments. GS-MS analyses were carried out on a Hewlett-Packard GC/MS 6890 N that works with the EI technique (70 eV). Flow chemical reactions were performed by means of a FlowLab™ system of Uniqsis. Microwave irradiation was performed by means of a Biotage® Initiator. Compounds 1a–n were prepared starting from alkyl- and arylglyoxals and nitro compounds by following reported procedures.20Batch general procedure
A solution of the appropriate β-nitroenone 1a–n (1 mmol) and tin(II) chloride dihydrate (2 mmol) in ethyl acetate (13 mL) was irradiated, by means of a Biotage® Initiator microwave oven, at 90 °C for 2 hours. Then, the solution was transferred into a separatory funnel, treated with a 0.5 N aqueous solution of HCl (30 mL), extracted with fresh EtOAc (3 × 30 mL), and the collected organic phase was dried using anhydrous Na2SO4. Finally, after the filtration of sodium sulphate and the evaporation of the solvent under reduced pressure, the crude reaction product 2a–n was purified by flash column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 95:5).
:EtOAc = 95:5).
Flow general procedure
The appropriate β-nitroenone 1a–c (1 mmol) was taken up in ethyl acetate (6.5 mL) and placed in reservoir A, and tin(II) chloride dihydrate (2 mmol) was taken up in ethyl acetate (6.5 mL) and placed in reservoir B. The two solutions were simultaneously pumped with a flow rate of 0.042 mL min−1 for each pump into a T-connector before passing through a 10 mL PTFE coil reactor heated at 90 °C (residence time 2 hours), and the outflow was dropped into a flask containing 30 mL of a stirring 0.5 N aqueous solution of HCl. The two layers were separated, the aqueous one was extracted with fresh EtOAc (3 × 30 mL), and the collected organic phase was dried using anhydrous Na2SO4. Finally, after the filtration of sodium sulphate and the evaporation of the solvent under reduced pressure, the crude reaction product 2a–c was purified by flash column chromatography (hexane:EtOAc = 95:5).
Author contributions
A. P. and M. P. conceived the idea and designed the experiments. M. E. I. K. performed the optimization studies. M. E. I. K. and T. L. C. performed the substrate scope analysis and mechanistic studies. A. P. and M. P. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the University of Camerino. The research network CINMPIS-Bari is also gratefully acknowledged.References
- (a) F. De Sarlo and F. Marchetti, Org. Biomol. Chem., 2023, 21, 7255 RSC; (b) G. N. Pairas, F. Perperopoulou, P. G. Tsoungas and G. Varvounis, ChemMedChem, 2017, 12, 408 CrossRef CAS PubMed; (c) F. M. Cordero, D. Giomi and L. Lascialfari, Five-Membered Ring Systems With O and N Atoms, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elsevier, Radarweg, 2017, 29, 353 Search PubMed.
- J. Zhu, J. Mo, H. Lin, Y. Chen and H. Sun, Bioorg. Med. Chem., 2018, 26, 3065 CrossRef CAS PubMed.
- (a) C. P. Pandhurnekar, H. C. Pandurnekar, A. J. Mungole, S. S. Butoliya and B. G. Yado, J. Heterocycl. Chem., 2023, 60, 537 CrossRef CAS; (b) G. C. Arya, K. Kaur and V. Jaitak, Eur. J. Med. Chem., 2021, 221, 113511 CrossRef CAS PubMed; (c) N. Agrawal and P. Mishra, Med. Chem. Res., 2018, 27, 1309 CrossRef CAS PubMed.
- I. J. Turchi and M. J. S. Dewar, Chem. Rev., 1975, 75, 389 CrossRef CAS.
- (a) S. Das and K. Chanda, RSC Adv., 2021, 11, 32680 RSC; (b) K. S. Kadam, T. Gandhi, A. Gupte, A. K. Gangopadhyay and R. Sharma, Synthesis, 2016, 3996 CAS; (c) W. Chen, B. Wang, N. Liu, D. Huang, X. Wang and Y. Hu, Org. Lett., 2014, 16, 6140 CrossRef CAS PubMed; (d) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed; (e) C. Praveen, A. Kalyanasundaram and P. T. Perumal, Synlett, 2010, 777 CAS.
- (a) J. E. McMurry, Org. Synth., Coll., 1988, 6, 592 Search PubMed; (b) G. Bianchi and M. De Amici, J. Chem. Soc., Chem. Commun., 1978, 962 RSC; (c) C. De Tollenaere and L. Ghosez, Bull. Soc. Chim. Belg., 1997, 106, 677 CAS; (d) A. D. White, C. F. Purchase, J. A. Picard, M. K. Anderson, S. B. Muller, T. M. A. Bocan, R. F. Bousley, K. L. Hamelehle, B. R. Krause, P. Lee, R. L. Stanfield and J. F. Reindel, J. Med. Chem., 1996, 39, 3908 CrossRef CAS PubMed; (e) A. V. Aksenov, N. A. Aksenov, N. K. Kirilov, A. A. Skomorokhov, D. A. Akesenov, I. A. Kurenkov, E. A. Sorokina, M. A. Nobi and M. Rubin, RSC Adv., 2021, 11, 35937 RSC; (f) G. Giacomelli, L. De Luca and A. Porcheddu, Tetrahedron, 2003, 59, 5437 CrossRef CAS.
- (a) C. D. Turner and M. A. Ciufolini, ARKIVOC, 2011,(i), 410 Search PubMed; (b) M. Gutiérrez, M. F. Matus, T. Poblete, J. Amigo, G. Vallejos and L. Astudillo, J. Pharm. Pharmacol., 2013, 65, 1796 CrossRef PubMed; (c) L. Han, B. Zhang, M. Zhu and J. Yan, Tetrahedron Lett., 2014, 55, 2308 CrossRef CAS.
- (a) M. A. P, G. L. Balaji, P. Iniyavan and H. Ila, J. Org. Chem., 2020, 85, 15422 CrossRef PubMed; (b) Y. Ning, Y. Otani and T. Ohwada, J. Org. Chem., 2018, 83, 203 CrossRef CAS PubMed; (c) L. Johnson, J. Powers, F. Ma, K. Jendza, B. Wang, E. Meredith and N. Mainolfi, Synthesis, 2013, 171 CAS; (d) S. Tang, J. He, Y. Sun, L. He and X. She, Org. Lett., 2009, 11, 3982 CrossRef CAS PubMed; (e) M. P. Bourbeau and J. T. Rider, Org. Lett., 2006, 8, 3679 CrossRef CAS PubMed.
- (a) C. Praveen, A. Kalyanasundaram and P. T. Perumal, Synlett, 2010, 777 CAS; (b) M. Duan, G. Hou, Y. Zhao, C. Zhu and C. Song, J. Org. Chem., 2022, 87, 11222 CrossRef CAS PubMed; (c) C. Li, J. Li, F. Zhou, C. Li and W. Wu, J. Org. Chem., 2019, 84, 11958 CrossRef CAS PubMed.
- (a) S. Zen and M. Koyama, Bull. Chem. Soc. Jpn., 1971, 44, 2882 CrossRef CAS; (b) W. M. Best, E. L. Ghisalberti and M. Powell, J. Chem. Res., Synop., 1998, 388 RSC; (c) R. Ballini, F. Bigi, E. Gogni, R. Maggi and G. Sartori, J. Catal., 2000, 191, 348 CrossRef CAS; (d) D. Long, Y. Qin, Q. Wu, X. Zou and Z. Zhou, J. Struct. Chem., 2019, 60, 1339 CrossRef CAS.
- (a) Y. Ning, Y. Otani and T. Ohwada, J. Org. Chem., 2018, 83, 203 CrossRef CAS PubMed; (b) U. S. Sørensen, E. Falch and P. Krogsgaard-Larsen, J. Org. Chem., 2000, 65, 1003 CrossRef PubMed.
- A. Palmieri, Eur. J. Org. Chem., 2020, 4247 CrossRef CAS.
- (a) L. Yuan, L. Kachalova, M. E. I. Khan, R. Ballini, M. Petrini and A. Palmieri, J. Org. Chem., 2023, 88, 4770 CrossRef CAS PubMed; (b) B. Bassetti, R. Ballini, M. Petrini and A. Palmieri, Adv. Synth. Catal., 2023, 365, 13 CrossRef CAS; (c) C. Raviola, C. Carrera, M. Serra, A. Palmieri, G. Lupidi, G. Maestri and S. Protti, ChemPhotoChem, 2021, 5, 871 CrossRef CAS; (d) E. Chiurchiù, S. Xhafa, R. Ballini, G. Maestri, S. Protti and A. Palmieri, Adv. Synth. Catal., 2020, 362, 4680 CrossRef; (e) M. Dell'Aera, F. M. Perna, P. Vitale, A. Altomare, A. Palmieri, L. C. H. Maddock, L. J. Bole, A. R. Kennedy, E. Hevia and V. Capriati, Chem. – Eur. J., 2020, 26, 8742 CrossRef PubMed; (f) E. Chiurchiù, S. Gabrielli, R. Ballini and A. Palmieri, Molecules, 2019, 24, 4575 CrossRef PubMed.
- (a) H. Wieland, Liebigs Ann., 1903, 328, 227 Search PubMed; (b) C. Bellec, D. Bertin, R. Colau, S. Deswarte, P. Maitte and C. Viel, J. Heterocycl. Chem., 1979, 16, 1657 CrossRef CAS.
- For a recent review on the Nef reaction see: R. Ballini and M. Petrini, Adv. Synth. Catal., 2015, 357, 2371 CrossRef CAS.
- (a) J. Bourgeois, I. Dion, P. H. Cebrowski, F. Loiseau, A.-C. Bedard and A. M. Beauchemin, J. Am. Chem. Soc., 2009, 131, 874 CrossRef CAS PubMed; (b) K. Nishide, T. Miyamoto, K. Kumar, S. I. Ohsugi and M. Node, Tetrahedron Lett., 2002, 43, 8569 CrossRef CAS; (c) M. Koóš, Tetrahedron Lett., 2000, 41, 5403 CrossRef; (d) C. Dell'Erba, M. Novi, G. Petrillo and P. Stagnaro, J. Heterocycl. Chem., 1994, 31, 861 CrossRef; (e) G. W. Kabalka and N. M. Goudgaon, Synth. Commun., 1988, 18, 693 CrossRef CAS; (f) R. S. Varma, M. Varma, Y.-Z. Kabalka and W. George, Heterocycles, 1986, 24, 2581 CrossRef CAS.
- C. O. Kappe, A. Stadler and D. Dallinger, in Microwaves in Organic and Medicinal Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2012, vol. 52 Search PubMed.
- https://www.uniqsis.com/paProductsDetail.aspx?ID=FlowLab .
- (a) L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230 RSC; (b) C. Holtze and R. Boehling, Curr. Opin. Chem. Eng., 2022, 36, 100798 CrossRef; (c) C. A. Hone and C. O. Kappe, Chem.: Methods, 2021, 1, 454 CAS; (d) M. Trojanowicz, Molecules, 2020, 25, 1434 CrossRef CAS PubMed; (e) P. Brandão, M. Pineiro and T. M. V. D. Pinho e Mel, Eur. J. Org. Chem., 2019, 7188 CrossRef; (f) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796 CrossRef CAS PubMed; (g) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2 CrossRef CAS.
- (a) R. Ballini, D. Fiorini and A. Palmieri, Tetrahedron Lett., 2004, 45, 7027 CrossRef CAS; (b) A. Palmieri, S. Gabrielli and R. Ballini, Green Chem., 2013, 15, 2344 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00232f |
| This journal is © The Royal Society of Chemistry 2024 |