
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalysed additive-free alkoxycarbonylation of indoles: 2,4,6-trimethylbenzoic acid-based carbonate anhydrides as a versatile alkoxycarboxyl source†
Hirotsugu
Suzuki
 *a,
Yuki
Ito
b,
Kentaro
Yabe
b,
Yosuke
Takemura
b and
Takanori
Matsuda
*b
*a,
Yuki
Ito
b,
Kentaro
Yabe
b,
Yosuke
Takemura
b and
Takanori
Matsuda
*b
aTenure-Track Program for Innovative Research, University of Fukui, 3-9-1 Bunkyo, Fukui-shi, Fukui 910-8507, Japan. E-mail: h-suzuki@u-fukui.ac.jp
bDepartment of Applied Chemistry, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: mtd@rs.tus.ac.jp
First published on 27th March 2024
Abstract
We report a CO-free approach to indole-2-carboxylic esters: rhodium-catalysed C(2)-alkoxycarbonylation of indoles with 2,4,6-trimethylbenzoic acid-based carbonate anhydrides. Selective C–O bond cleavage of the anhydrides facilitates the introduction of various alkoxycarbonyl groups. Control experiments suggest that merging a rhodium catalyst and KI promotes the in situ formation of the RhI species.
Indole-2-carboxylic acid esters and their derivatives are prevalent structural motifs in numerous biologically active compounds and pharmaceuticals and serve as versatile intermediates in organic synthesis (Scheme 1A).1–4 Given their structural importance, diverse synthetic methods toward indole-2-carboxylic acid esters have been developed. For instance, an intramolecular or intermolecular cyclisation forming a pyrrole ring is essential for synthesising these esters.5–14 However, the requisite multi-step synthesis to prepare prefunctionalised starting materials can consume substantial resources and time, posing significant drawbacks for practical applications. As an alternative, transition-metal-catalysed C(2)–H carbonylation of indoles has been explored as an atom- and step-economical approach for synthesising these molecules.15,16 Among them, carbon monoxide (CO) serves as a cost-effective and readily available C1 carbonyl source (Scheme 1B).17,18 However, its toxicity and inflammability restrict its utility, demanding special handling techniques and equipment. Moreover, the alkoxycarbonylation using CO requires a stoichiometric amount of an oxidant such as a copper salt, diminishing the atom economy of the reaction and complicating reaction operation. Therefore, the development of a complementary carbonyl source is highly desirable for enhancing safety and synthetic efficiency. Additionally, in recent decades, investigation of CO surrogates has become a crucial point of focus in C–H carbonylation reactions of arenes.19,20 Consequently, exploring safe, easy-to-handle and CO-free C(2)–H alkoxycarbonylations of indoles is warranted.
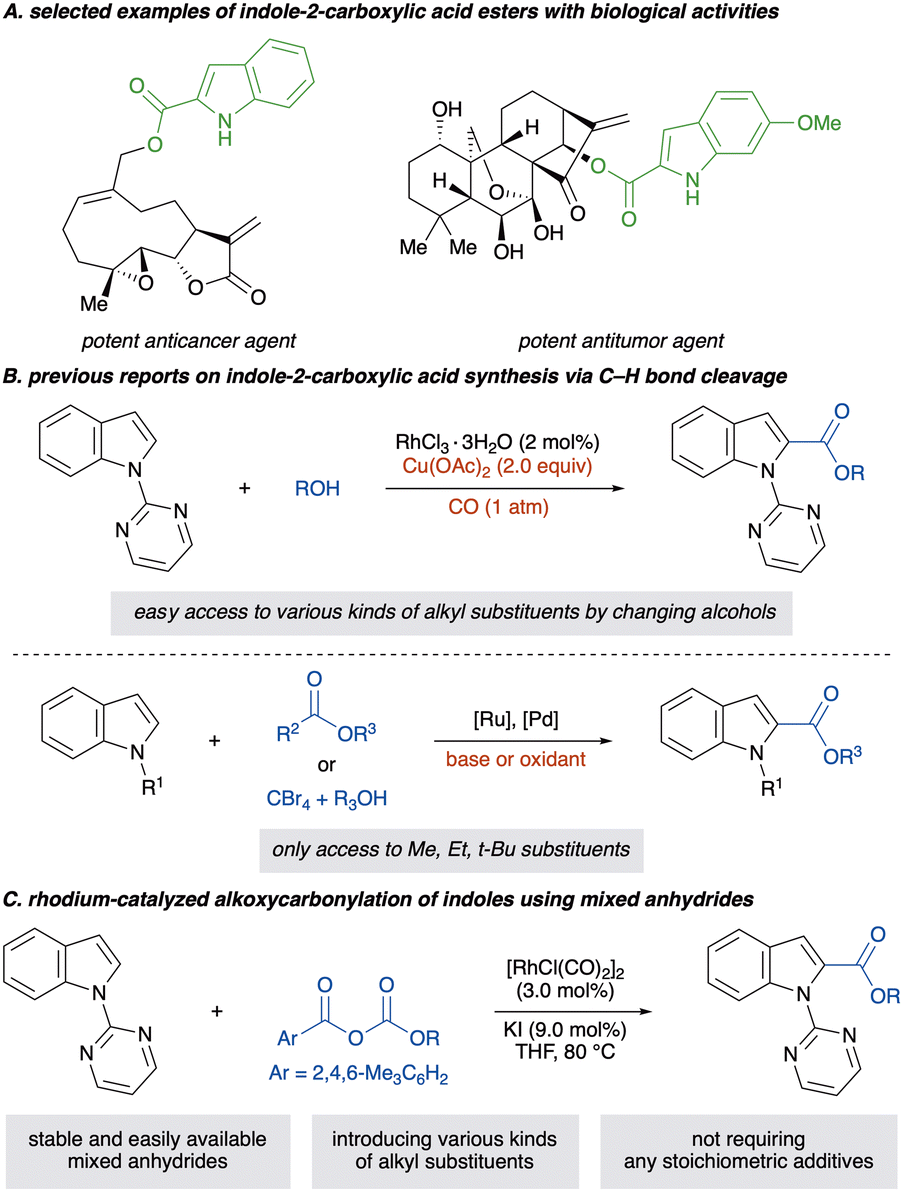 | ||
| Scheme 1 Previous C(2)–H alkoxycarbonylations of indoles and this work. | ||
The investigation on carboxylating reagents has garnered considerable attention in the past decade for their environmentally benign CO-free process, resulting in various reagents, such as azodicarboxylates,21–28 chloroformates,29–31 oxaziridines,32 glyoxylates,33 α-keto esters,34 dicarbonates,35–38 formates,39 potassium oxalates,40 carbazates,41–43 bromodifluoroacetates44 and tetrabromomethanes45–48 (Scheme 1B). However, these reactions necessitate the use of stoichiometric amounts of additives, such as oxidants and bases, and alkoxycarbonylations without any stoichiometric additives have rarely been reported.32,36–38 Furthermore, introduced substituents on alkoxycarbonyl moieties are restricted to simple alkyl chains such as methyl, ethyl and tert-butyl groups, likely due to the structural limitations of available carboxylating reagents, even though the structure of the ester moiety crucially affects biological activities.2,3
Recently, Dong et al. reported the palladium-catalysed Catellani reaction using a novel carbonate anhydride as a carboxylating reagent.49,50 Inspired by these reports49,50 and our previous studies,36–38,51,52 we hypothesised that rhodium carboxylate species possessing various alkoxycarbonyl groups could be generated by the selective C–O bond cleavage of a carbonate anhydride, thereby promoting C(2)-selective C–H activation of indoles without any stoichiometric additives. The key challenges, however, are (1) how to control C–O bond cleavage due to unsymmetrical anhydrides possessing the two weak C–O bonds and (2) how to promote selective C–H activation by rhodium carboxylate species in the absence of an oxidant and a base. Herein, we report a rhodium-catalysed alkoxycarbonylation of indoles with mixed carbonate anhydrides without stoichiometric amounts of additives, yielding indole-2-carboxylic acid esters with various ester substituents (Scheme 1C). The easily accessible and stable mixed carbonate anhydrides make alkoxycarbonylation more straightforward and operationally simple.
Our study commenced by investigating the C(2)-selective alkoxycarbonylation of 1-(pyrimidin-2-yl)-1H-indole (1a) with the mixed carbonate anhydride 2 in the presence of [RhCl(CO)2]2 (5.0 mol%, 10 mol% Rh) and potassium iodide (KI, 15 mol%) in THF at 80 °C (Table 1). After 18 h, the desired indole-2-carboxylic acid ester 3a was obtained in 78% yield from the mixed carbonate anhydride 2a, synthesised from 2,6-dimethylbenzoic acid and methyl chloroformate in a single step. However, a small amount of 2-aroylindole 4 was also formed as a byproduct (entry 1). Encouraged by this result, other mixed carbonate anhydrides derived from 2,6-disubstituted benzoic acids were examined. The yield of 3a was further improved using 2d (entry 4), although other mixed anhydrides gave lower yields (entries 2 and 3). Employing methyl chloroformate instead of 2 resulted in no product formation, indicating the importance of the carboxylate moiety for C–H bond activation (entry 5). Among the iodide sources examined, KI was found to be optimal, affording 3a in 89% yield(entries 4 vs. 6–8). Only [RhCl(CO)2]2 exhibited catalytic activity, and other rhodium(I) complexes were found to be ineffective (entries 9–12). Moreover, this alkoxycarbonylation reaction was not affected by atmospheric air conditions (entry 13). On the other hand, adding a small amount of water decreased both the yield and the chemoselectivity, probably due to the decomposition of 2d by water (entry 14). Lowering the catalyst loading to 3.0 mol% gave a comparable yield of 3a while suppressing byproduct formation (entry 15). Control experiments revealed that [RhCl(CO)2]2 is crucial for the reaction to proceed, and the addition of KI significantly improved the yield and chemoselectivity (entries 16 and 17). Therefore, the additive is considered to assist the formation of RhI species by exchanging halogen ions in situ.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Anhydride | Additive | Rh catalyst | Yield of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
3:4 |
| a Reaction conditions: 1a (0.30 mmol), 2 (0.45 mmol), Rh catalyst (10 mol% of [Rh]) and additive (15 mol%) in THF (0.75 mL) at 80 °C for 18 h under an Ar atmosphere. b Yields were determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. Value in parentheses indicates isolated yields. c Methyl chloroformate was used instead of 2. d The reaction was performed under an air atmosphere. e 2.0 equivalents of H2O were added. f [RhCl(CO)2]2 (3.0 mol%) and KI (9.0 mol%) were used. TBAI: tetrabutylammonium iodide; cod: 1,5-cyclooctadiene; and nbd: 2,5-norbornadiene. | |||||
| 1 | 2a | KI | [RhCl(CO)2]2 | 78 | 8.7:1 |
| 2 | 2b | KI | [RhCl(CO)2]2 | 87 | 12:1 |
| 3 | 2c | KI | [RhCl(CO)2]2 | 70 | >20:1 |
| 4 | 2d | KI | [RhCl(CO)2]2 | 89 | 15:1 |
| 5c | — | KI | [RhCl(CO)2]2 | 0 | — |
| 6 | 2d | NaI | [RhCl(CO)2]2 | 81 | 9.0:1 |
| 7 | 2d | LiI | [RhCl(CO)2]2 | 25 | 1:2.0 |
| 8 | 2d | TBAI | [RhCl(CO)2]2 | 0 | — |
| 9 | 2d | KI | [RhCl(cod)]2 | Trace | — |
| 10 | 2d | KI | [RhCl(nbd)]2 | 0 | — |
| 11 | 2d | KI | RhCl(PPh3)3 | Trace | — |
| 12 | 2d | KI | RhCl(CO)(PPh3)2 | Trace | — |
| 13d | 2d | KI | [RhCl(CO)2]2 | 90 | 13:1 |
| 14e | 2d | KI | [RhCl(CO)2]2 | 52 | 7.4:1 |
| 15f | 2d | KI | [RhCl(CO)2]2 | 91 (87) | >20:1 |
| 16 | 2d | KI | — | 0 | — |
| 17 | 2d | — | [RhCl(CO)2]2 | 50 | 5.0:1 |
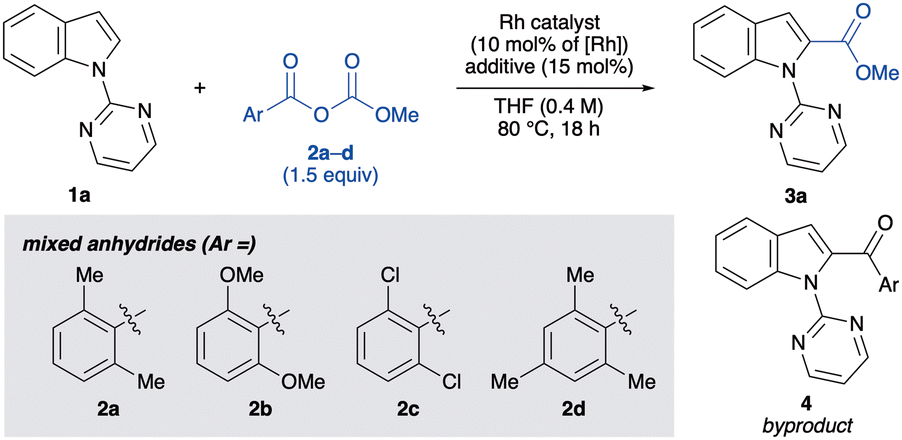
With the optimised reaction conditions in hand, we examined the generality of the alkoxycarbonylation (Scheme 2). Initially, we tested the model reaction on a large scale.
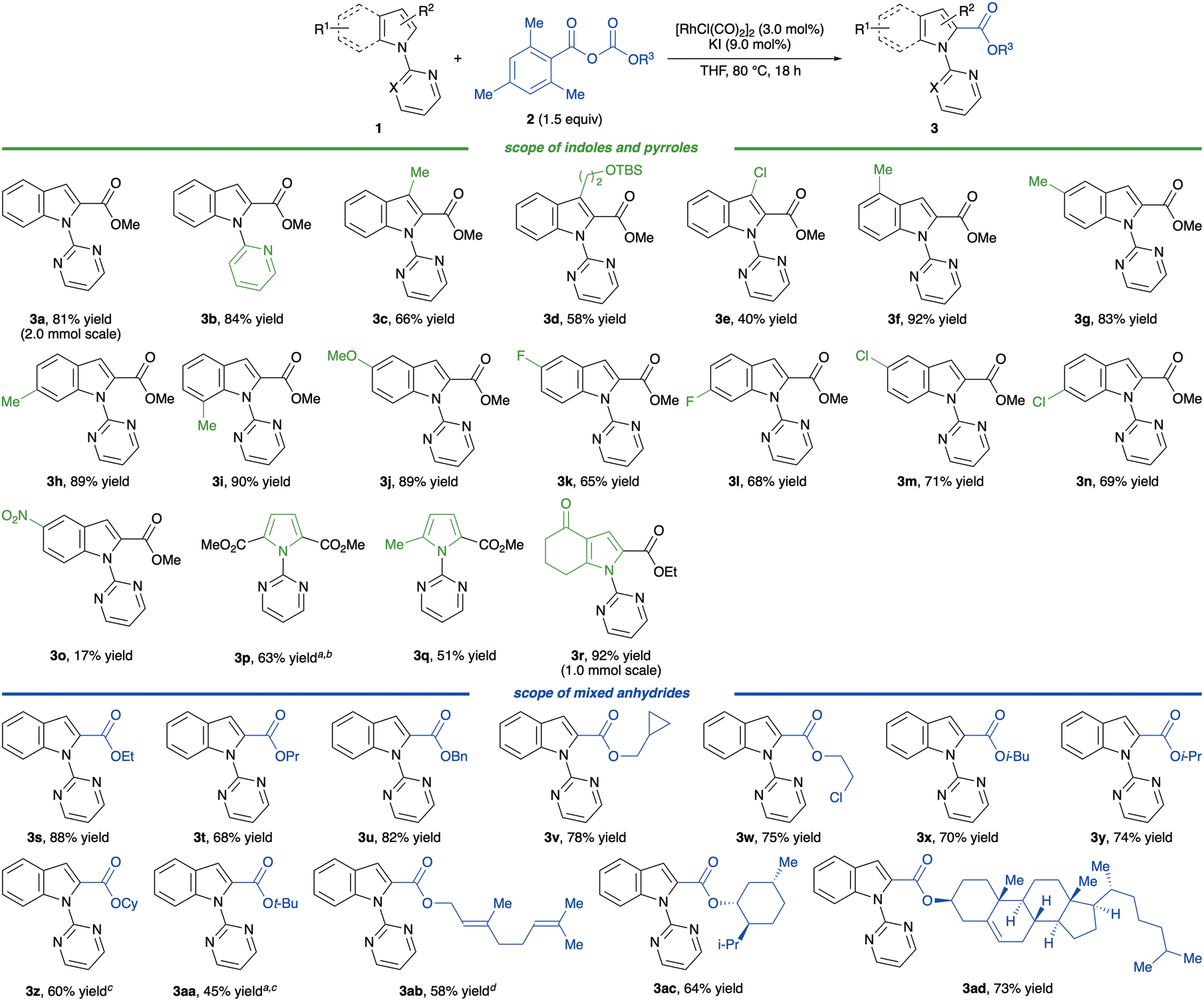 | ||
| Scheme 2 Substrate scope. Reaction conditions: 1 (0.30 mmol), 2 (0.45 mmol), [RhCl(CO)2]2 (3.0 mol%), and KI (9.0 mol%) in THF (0.75 mL) at 80 °C for 18 h under an Ar atmosphere. a[RhCl(CO)2]2 (5.0 mol%) and KI (15 mol%) were used. b4.0 equivalents of 2 were used. c2.0 equivalents of 2 were used. dThe reaction was conducted with 1.2 equivalents of 2 at 100 °C. | ||
Subjecting 2 mmol of 2a to the optimised reaction conditions gave indole 3a in 81% yield. A pyridyl directing group could function in this reaction, affording 3b in 84% yield. Introducing a substituent at the 3-position of indoles resulted in a slight decline in the reactivity; however, products 3c–e were still obtained in 40–66% yields. Notably, a 3-chloroindole, which did not provide any product in previous CO-mediated alkoxycarbonylations,17 furnished the corresponding ester 3e in 40% yield. A methyl group on the benzenoid moiety did not affect the yield, affording the desired esters 3f–i in 83–92% yields. An electron-rich indole bearing a methoxy group at the 5-position was well tolerated (3j). In contrast, indoles attached with a weak electron-withdrawing group slightly decreased the yields but still yielded 3k–n in 65–71% yields; thus, the yield of a nitro-substituted indole 3o was insufficient. The results indicated that this alkoxycarbonylation was likely to be sensitive to the electronic properties of indoles. Pyrrole substrates were also good coupling partners, affording products 3p–r in 51–92% yields.
We then tested the effect of a substituent on the ester group. Linear alkyl groups, such as ethyl, propyl, benzyl, cyclopropylmethyl, 2-chloroethyl and isobutyl groups, gave the esters 3s–x without compromising on reactivity and selectivity. Moreover, a mixed carbonate anhydride possessing an α-branched alkyl group such as isopropyl and cyclohexyl groups also produced 3y and 3z in 74 and 60% yields, respectively. A more sterically hindered tert-butyl group diminished the yield of 3aa to 45% due to the formation of a significant amount of the undesired product 4. To demonstrate the broad applicability of this reaction, mixed carbonate anhydrides possessing a natural product moiety, geraniol, menthol and cholesterol, were tested, and fortunately, all reacted smoothly to afford products 3ab–ad in 58–73% yields. Thus, these results indicate that this alkoxycarbonylation demonstrates good functional group tolerance and potential for late-stage functionalisation.
Taking into account the importance of indole-7-carboxylic acid esters and their derivatives in pharmaceutical science,53–55 we next investigated the C(7)-alkoxycarbonylation of indole derivatives (Scheme 3). Unfortunately, applying the standard reaction conditions to 2-substituted indole 5 did not afford the desired alkoxycarbonylated indole 6. Interestingly, omitting KI from the conditions enhanced the reactivity, furnishing product 6 in 59% yield (Scheme 3A). Encouraged by this result, alkoxycarbonylations of indoline 7 and carbazole 9 were tested, and both reactions afforded the corresponding products (Scheme 3B and C). Thus, our protocol was extended to functionalise benzenoid moieties, proving the versatility of this methodology for a site-selective alkoxycarbonylation of indoles.
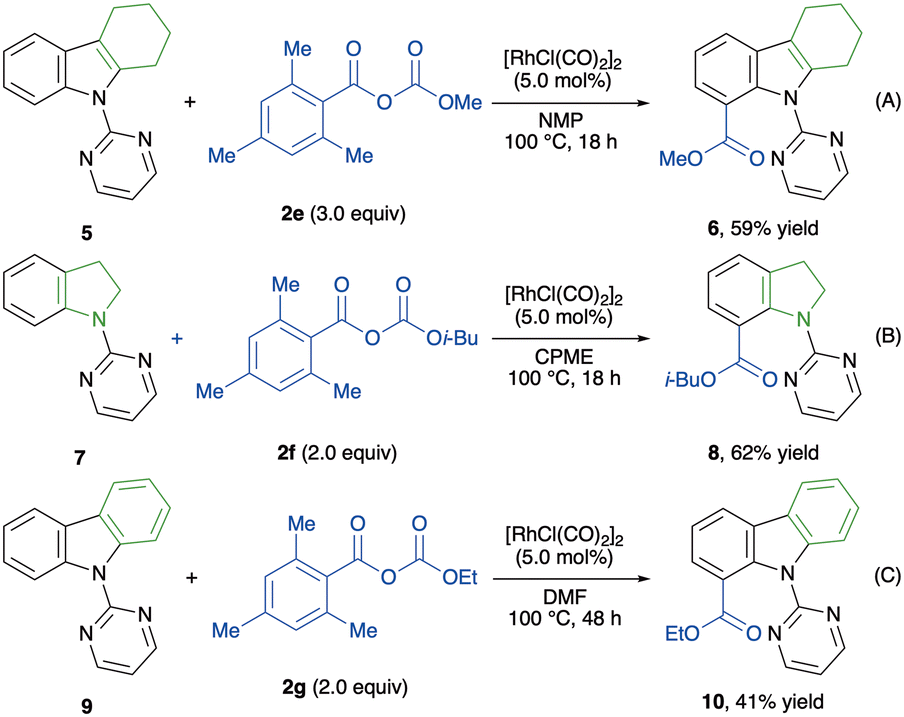 | ||
| Scheme 3 C(7)-alkoxycarbonylations of indoles and their derivatives. | ||
To demonstrate the utility of this alkoxycarbonylation, further transformation of the products was examined (Scheme 4). Subjecting indole-2-carboxylic acid ester 3s to the basic reaction conditions enabled the smooth deprotection of the pyrimidyl directing group, providing the unprotected product 11 in 79% yield (Scheme 4A). Similarly, the N-pyrimidyl pyrrole 3r underwent deprotection to afford the free NH-pyrrole 12 in 62% yield (Scheme 4B). These transformations indicate the possibility of further application of this methodology to the synthesis of biologically active molecules.
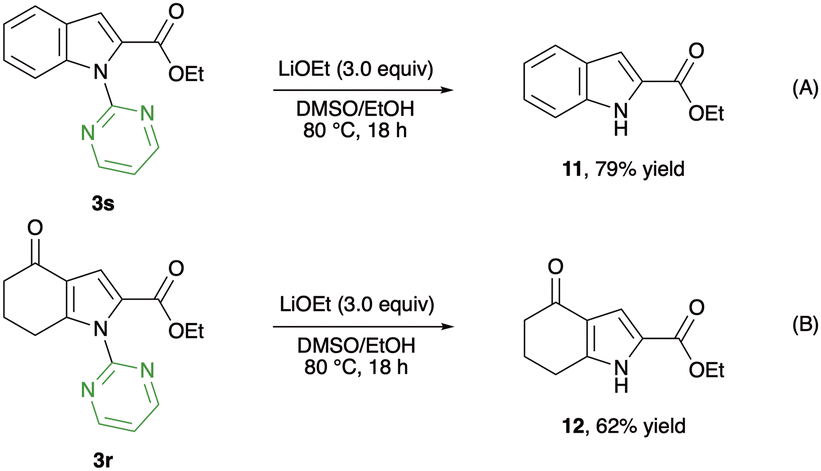 | ||
| Scheme 4 Deprotection of the pyrimidyl directing group. | ||
To elucidate the reaction mechanism, several control experiments were conducted (Scheme 5). Initially, H/D scrambling experiments were performed by adding 5.0 equivalents of D2O to the reaction of 1a; over 80% deuterium incorporation at the C(2) and C(3) positions occurred, indicating the reversibility of the C–H activation step (Scheme 5A, right). Interestingly, removing KI from the reaction conditions promoted C(7)–H activation, which was not observed in a [RhCl(CO)2]2 and KI system (Scheme 5A, left). This reactivity difference might explain the result of the C(7)-alkoxycarbonylation of an indole and its derivatives. Deuterium incorporation of 1a was also observed even when 2a was employed with EtOD (Scheme 5B). Next, a competition experiment was conducted by reacting electron-rich and -deficient indoles with mixed anhydride 2a in the same vessel (Scheme 5C). A larger amount of 3j was formed than 3m, implying that the C–H activation step could proceed via an electrophilic mechanism. In addition, kinetic isotope effect experiments were conducted (Scheme 5D). Parallel experiments showed a KIE of 0.9, indicating that the rate-determining step is not involved in the C–H activation step. Finally, we synthesised [RhI(CO)2]2 to confirm the active catalyst species.56 When [RhI(CO)2]2 was employed in the reaction of indole 1a with anhydride 2d, 3a was formed in a yield comparable to that obtained under the optimised reaction conditions (Scheme 5E). Therefore, we assume the active species of this transformation is [RhI(CO)2]2.
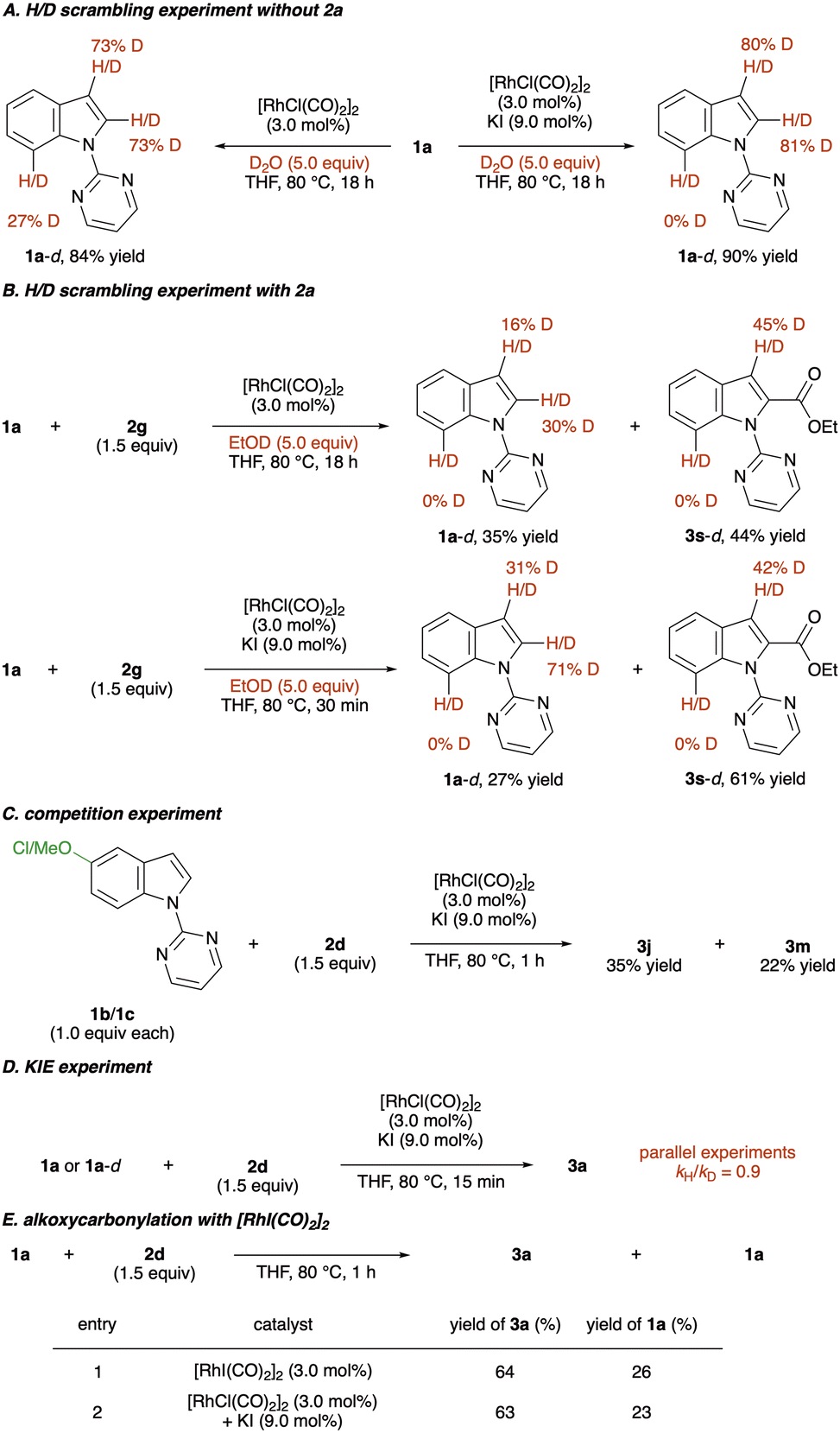 | ||
| Scheme 5 Mechanistic investigations. | ||
Based on the mechanistic investigations and previous reports on rhodium-catalysed C–H functionalisation with anhydrides,36–38,51,52 we propose the reaction mechanism for the C(2)-alkoxycarbonylation of indoles, as depicted in Fig. 1. First, an anion exchange between [RhCl(CO)2]2 and KI yields the catalytically active [RhI(CO)2]2. Then, selective C–O bond cleavage of mixed carbonate anhydride 2a, controlled by the steric hindrance of a mesityl group, generates the rhodium carboxylate B. A subsequent pyrimidyl-directed C–H bond activation, which might proceed by an electrophilic mechanism, gives the five-membered rhodacycle intermediate C. Finally, reductive elimination of C leads to the formation of the indole-2-carboxylic acid ester 3a and the regeneration of the RhI species A.
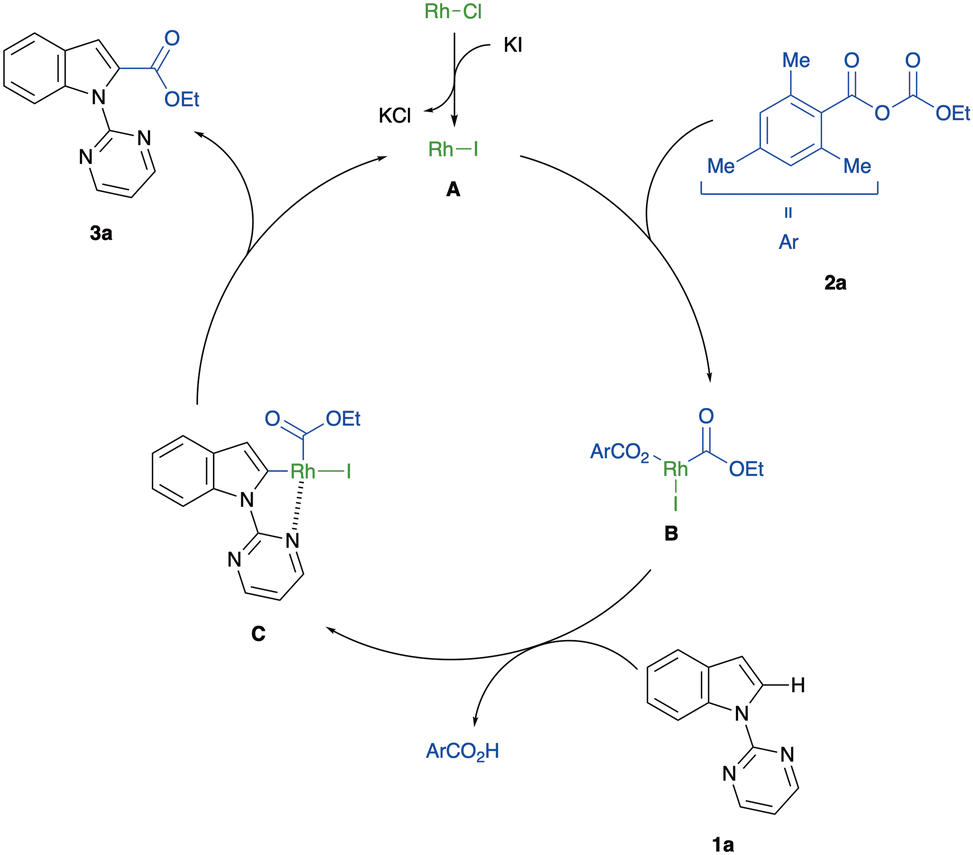 | ||
| Fig. 1 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed a concise synthesis of indole-2-carboxylic acid esters using 2,4,6-trimethylbenzoic acid-based carbonate anhydrides as carboxylating reagents. Preliminary mechanistic investigations revealed that [RhI(CO)2] generated from [RhCl(CO)2] and KI accelerates the C(2)–H bond cleavage of an indole while suppressing the undesired C(7)–H bond cleavage. The C(2)-alkoxycarbonylation of indoles proceeds selectively to afford the indole-2-carboxylic acid esters in up to 92% isolated yield. Removing KI from the standard reaction conditions enables the C(7)-alkoxycarbonylation of 2-substituted indole derivatives, indoline and carbazole, by facilitating C(7)–H bond cleavage. We anticipate that this novel alkoxycarbonylation using mixed anhydrides will open a new avenue for C–H alkoxycarbonylation with CO surrogates and expand the scope of this field.Author contributions
Conceptualisation: H. S.; supervision: H. S. and T. M.; investigation: Y. I., K. Y., and Y. T.; resources: T. M.; writing – original draft: H. S.; and writing – review and editing: H. S. and T. M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (grant numbers JP23K13743 and JP21K05061).References
- M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC.
- S. Bommagani, J. Ponder, N. R. Penthala, V. Janganati, C. T. Jordan, M. J. Borrelli and P. A. Crooks, Eur. J. Med. Chem., 2017, 136, 393–405 CrossRef CAS PubMed.
- Q.-K. Shen, H. Deng, S.-B. Wang, Y.-S. Tian and Z.-S. Quan, Eur. J. Med. Chem., 2019, 177, 15–31 CrossRef PubMed.
- G. Cui, F. Lai, X. Wang, X. Chen and B. Xu, Eur. J. Med. Chem., 2020, 188, 111985 CrossRef CAS PubMed.
- C. Barberis, T. D. Gordon, C. Thomas, X. Zhang and K. P. Cusack, Tetrahedron Lett., 2005, 46, 8877–8880 CrossRef CAS.
- B. J. Stokes, H. Dong, B. E. Leslie, A. L. Pumphrey and T. G. Driver, J. Am. Chem. Soc., 2007, 129, 7500–7501 CrossRef CAS PubMed.
- A. G. O'Brien, F. Lévesque and P. H. Seeberger, Chem. Commun., 2011, 47, 2688–2690 RSC.
- C. Bolm and J. Bonnamour, Org. Lett., 2011, 13, 2012–2014 CrossRef PubMed.
- Z. Zhu, J. Yuan, Y. Zhou, Y. Qin, J. Xu and Y. Peng, Eur. J. Org. Chem., 2014, 511–514 CrossRef CAS.
- X. Xiao, T.-Q. Chen, J. Ren, W.-D. Chen and B.-B. Zeng, Tetrahedron Lett., 2014, 55, 2056–2060 CrossRef CAS.
- D. Formenti, F. Ferretti and F. Ragaini, ChemCatChem, 2018, 10, 148–152 CrossRef CAS.
- A. Baykal and B. A. Plietker, Eur. J. Org. Chem., 2020, 1145–1147 CrossRef CAS.
- M. A. Gouad, F. Ferretti, D. Formenti, F. Milani and F. Ragaini, Eur. J. Org. Chem., 2021, 4876–4894 Search PubMed.
- M. A. Fouad, F. Ferretti and F. Ragaini, J. Org. Chem., 2023, 88, 5108–5117 CrossRef CAS PubMed.
- M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed.
- R. A. Jagtap and B. Punji, Asian J. Org. Chem., 2020, 9, 326–342 CrossRef CAS.
- K. Zhao, R. Du, B. Wang, J. Liu, C. Xia and L. Yang, ACS Catal., 2019, 9, 5545–5551 CrossRef CAS.
- R. Du, K. Zhao, J. Liu, F. Han, C. Xia and L. Yang, Org. Lett., 2019, 21, 6418–6422 CrossRef CAS PubMed.
- K. Mondal, P. Halder, G. Gopalan, P. Sasikumar, K. V. Radhakrishnan and P. Das, Org. Biomol. Chem., 2019, 17, 5212–5222 RSC.
- Z. Chen, L.-C. Wang and X.-F. Wu, Chem. Commun., 2020, 56, 6016–6030 RSC.
- W.-Y. Yu, W. N. Sit, K.-M. Lai, Z. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2008, 130, 3304–3306 CrossRef CAS PubMed.
- Y. Huang, G. Li, J. Huang and J. You, Org. Chem. Front., 2014, 1, 347–350 RSC.
- N. Xu, D. Li, Y. Zhang and L. Wang, Org. Biomol. Chem., 2015, 13, 9083–9092 RSC.
- R. Sang, Y. Zheng, H. Zhang, X. Wu, Q. Wang, L. Hai and Y. Wu, Org. Chem. Front., 2018, 5, 648–652 RSC.
- M. Usman, X.-W. Zhang, D. Wu, Z.-H. Guan and W.-B. Liu, Org. Chem. Front., 2019, 6, 1905–1928 RSC.
- J. Ni, J. Li, Z. Fan and A. Zhang, Org. Lett., 2016, 18, 5960–5963 CrossRef CAS PubMed.
- T. T. Nguyen, L. Grigorjeva and O. Daugulis, Chem. Commun., 2017, 53, 5136–5138 Search PubMed.
- F. Ling, C. Ai, Y. Lv and W. Zhong, Adv. Synth. Catal., 2017, 359, 3707–3712 CrossRef CAS.
- T. Kochi, S. Urano, H. Seki, E. Mizushima, M. Sato and F. Kakiuchi, J. Am. Chem. Soc., 2009, 131, 2792–2793 CrossRef CAS PubMed.
- G. Liao, H.-M. Chen and B.-F. Shi, Chem. Commun., 2018, 54, 10859–10862 RSC.
- Y. Shi, F. Yang and Y. Wu, Org. Biomol. Chem., 2020, 18, 4628–4637 RSC.
- X. Peng, Y. Zhu, T. A. Ramirez, B. Zhao and Y. Shi, Org. Lett., 2011, 13, 5244–5247 CrossRef CAS PubMed.
- S. Wang, Z. Yang, J. Liu, K. Xie, A. Wang, X. Chen and Z. Tan, Chem. Commun., 2012, 48, 9924–9926 RSC.
- W. Zhou, P. Li, Y. Zhang and L. Wang, Adv. Synth. Catal., 2013, 355, 2343–2352 CrossRef CAS.
- X. Hong, H. Wang, B. Liu and B. Xu, Chem. Commun., 2014, 50, 14129–14132 RSC.
- H. Suzuki, Y. Liao, Y. Kawai and T. Matsuda, Eur. J. Org. Chem., 2021, 4938–4942 CrossRef CAS.
- H. Suzuki, F. Sasamori and T. Matsuda, Org. Lett., 2022, 24, 1141–1145 CrossRef CAS PubMed.
- H. Suzuki, Y. Ito and T. Matsuda, Chem. Lett., 2022, 51, 775–777 CrossRef CAS.
- J. Wu, J. Lan, S. Guo and J. You, Org. Lett., 2014, 16, 5862–5865 CrossRef CAS PubMed.
- Z.-Y. Li and G.-W. Wang, Org. Lett., 2015, 17, 4866–4869 CrossRef CAS PubMed.
- Y. Gao, W. Lu, P. Liu and P. Sun, J. Org. Chem., 2016, 81, 2482–2487 CrossRef CAS PubMed.
- L.-Y. Xie, S. Peng, T.-G. Fan, Y.-F. Liu, M. Sun, L.-L. Jiang, X.-X. Wang, Z. Cao and W.-M. He, Sci. China: Chem., 2019, 62, 4660–4464 Search PubMed.
- G. R. Y. Kumar and N. S. Begum, Eur. J. Org. Chem., 2020, 4698–4704 CrossRef.
- N. Tao, J. Wang, C. Yuan, R. Zeng and Y.-S. Zhao, Org. Lett., 2019, 21, 8607–8610 CrossRef CAS PubMed.
- Q.-Q. Yang, M. Marchini, W.-J. Xiao, P. Ceroni and M. Bandini, Chem. – Eur. J., 2015, 21, 18052–18056 CrossRef CAS PubMed.
- C. Sen, T. Sahoo, H. Singh, E. Suresh and S. C. Ghosh, J. Org. Chem., 2019, 84, 9869–9896 CrossRef CAS PubMed.
- T. Sahoo, C. Sen, H. Singh, E. Suresh and S. C. Ghosh, Adv. Synth. Catal., 2019, 361, 3950–3957 CrossRef CAS.
- S. Kumar, S. Pradhan, S. Roy, P. B. De and T. Punniyamurthy, J. Org. Chem., 2019, 84, 10481–10489 CrossRef CAS PubMed.
- J. Wang, L. Zhang, Z. Dong and G. Dong, Chem, 2016, 1, 581–591 CAS.
- Z. Dong, J. Wang, Z. Ren and G. Dong, Angew. Chem., Int. Ed., 2015, 54, 12664–12668 CrossRef CAS PubMed.
- H. Suzuki, Y. Kawai, Y. Takemura and T. Matsuda, Org. Biomol. Chem., 2022, 20, 2808–2812 RSC.
- H. Suzuki, Y. Takemura and T. Matsuda, Synlett, 2023, 34, 1894–1898 CrossRef CAS.
- D. D. Miller, P. Bamborough, J. A. Christopher, I. R. Baldwin, A. C. Champigny, G. J. Cutler, J. K. Kerns, T. Longstaff, G. W. Mellor, J. V. Morey, M. A. Morse, H. Nie, W. L. Rumsey and J. J. Taggart, Bioorg. Med. Chem. Lett., 2011, 21, 2255–2258 CrossRef CAS PubMed.
- J. K. Kerns, J. Busch-Petersen, W. Fu, J. C. Boehm, H. Nie, M. Muratore, A. Bullion, G. Lin, H. Li, R. Davis, X. Lin, A. S. Lakdawala, R. Cousins, R. Field, J. Payne, D. D. Miller, P. Bamborough, J. A. Christopher, I. Baldwin, R. R. Osborn, J. Yonchuk, E. Webb and W. L. Rumsey, ACS Med. Chem. Lett., 2018, 9, 1164–1169 CrossRef CAS PubMed.
- F. L. Gonzalez, S. R. Wisniewski, K. Katipally, J. M. Stevens, V. Rosso, B. Mack and T. M. Razler, Org. Process Res. Dev., 2019, 23, 1143–1151 CrossRef CAS.
- D. H. Nguyen, N. Lassauque, S. Vendier, S. Mallet-Ladeira, C. L. Berre, P. Serp and P. Kalck, Eur. J. Inorg. Chem., 2014, 326–336 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00205a |
| This journal is © The Royal Society of Chemistry 2024 |